
QHAPrE - I - Shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/68845/7/07_chapter 1.pdf · It is...
76
QHAPrE - I CHEMICAL COI'm'ITUEITS OF JIARSILEA IIIUTA LINI
Transcript of QHAPrE - I - Shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/68845/7/07_chapter 1.pdf · It is...

QHAPrE - I
CHEMICAL COI'm'ITUEITS OF JIARSILEA IIIUTA LINI

1
CHEMICAL CONSTITUENTS OF MARSILEA M!NUTA LINI
Ma:rsilea min uta Linn 1-' (Ma:rsileaoeae > is a genus of aquati o
or aubaquatio ferne widely distributed in the tropical and temperate
regions of the world. About nine species are found in India. Many
of the Indian floras refer th1 s common Indian species to Marsilea
guadrifolia Linn4• The plant has long and thin stem and with
creeping rhizome and quadrifoliolate leaves. It grows by the side
of the ponds and irrigation ohanaele throughout the greater pa:rt of
India. It can be propagated by divisions of the rhizome or by spores.
!he stalks and leaves of the plant are eaten as pol-herb, especially
in times of scarcity.
It is a pOpular drug in folk medioiae. Leaves and whole plants
are used for sedation and in insomnia as it has been mentioned in
Ayurvedio medicine to induce sleep in insomic persona.
The hypnotic activity of Marsilia minuta was tirst studied
experimentally in 1961 by M.L. Chatterjee et al5 at the school of
Tropical Medicine, Calcutta. An alcoholic extract of the leaves was
given orally to white mice (20-30) through a syringe fitted with a
special type o:t needle. Doses varying trom 5 to 100 mg/kg contained
in a five volume o:t 0.2 ml of the suspension were used. Investigations
were made (a' on the action of Nare1lea minuta given alone and (b) on
its effec~ on the hypnosis induced by pentobarbital sodium (50 mg/k:g)
intraperitoneally administered 30 minutes later. Initially a control
study was also made With vehicle (gum acacia sol.) along with that o!

. 2
pentobarbital sodium and extract of Jfareilea llinuta. The tranquilising
effect with the extract of Marsilea minuta was found to be most
effective in doses between 5-10 mg/Kaorally in white mice. Lack of
significant differ enoe in the effect w1 th 5 ,1 0 and 20 mg/kc d osee
wae found which may probably be due to the crude preparation of the
extract.
Ohatter jee et a16 in 1963 however worked with Kareilea Jlinuta
Id.nn with the object of isolating the active principle present in
the plant. They isolated the only compound deeignated as 118Xsilin
c29Hs 40 (M = 418 from mass spectrometric analysis and by Raet method) 30.
m.p. 85-86•. (oe]]) l O• (CHC13) from the ~hloroform. extract of the
leaves. They observed the following che~cal 8l'ld physical properties
of the compound. It did not give any teet tor sterols or triterpenes '
and did not reduce Fehling' a solution or Toll en• s reagent a. Tetra-
ni tromethane had no action on this compound. The infrared epectrum
showed peak absorption fOr carbonyl at 5.76 p (in KBr diek) but
fails to .produce any oxime or semicarbazone •. Lithium aluminium
hydride reduction of the compound afforded a product, c291fs60, m.p.
80•, showed peak absorption for hydroxyl at ,.1 ;U. Nuclear JI.Bgnetio
resonance spectrum showed a sharp signal at 1 .26 which they accounted
for the pr esenoe of one -c-CH3 funct1 on. From analysis an4 the above
ohemioal and physical studies they designated marsilin as a
macr ocyolic ketone. They performed pharmacal ogioal test of mar silin
and :from their obs.ervati one they described marsilin as the sedative
and the anticonvulsant principle of Yarailea m1nuta Linn. It has been
said that it is specially effective against electro and ohemooonvuleive
agent and it may be a promising drug in the treatment of epilepsy and

.. a
in various mental disorders. The sedative action of marsilin was 7-10 subsequently described by other workers as well· • They olai11.ed
the confirmation of their views by clinical trial on some epileptic
patients.
While searching for saponin bearing plant the fern Marsilea
minuta :U.nn was brought to our notice as the aqueous solution of
the leaves of the plant ga'fe a stable froth indicating the presence
of saponin in it. As such a detailed chemical exam1nat1 on was
undertaken •12
The air dried powdered leaves and stems were extracted successively
with.petrolium ether (60-80°), chloroform and ethanol in a eoxblet.
Each fraction was studied separately and as critically as possible.
Treatment of Petroleum ether extract 1
The residue from petroleum ethe:r extract waa subjected to
chromatographic separation over Brockmann alumina (Table-5-experimental}.
156 (A1-A156 ' t:ractions were collected from this chromatography and
3 major components were obtained. Their purity was assayed by TJ.C. It
is worthwhile mentioning that while working with the higher aliphatic
hydrocarbon, ketone and alcohol the TLC often fails to give true
picture. Ther! are some inseparable mixtures whioh can only be detected
by GLC.
Each fraction from A1-A20 was examined separately by T U! when a
single strip was obtained. These fractions ehaNed no absorption at
1720-1740 om-1 or 3600-3400 cm-1 in the IR (Pig. 16> confirming the e
absence of carbonyl and hydroxy containing cOJtpounds. GLC indicated a
mixture of normal hydrooatbons ranging from o27 to c33
with o27 , c29
,

LL.J Cl) ~ a Q. (/')
lJJ 0::
0: a 1-u lJJ 1-lJJ t:::l
~solvent
II ii
II /I
I
L ..... ... -······· ................. .
10 20
TIME IN MINUTES
-Mixture of alkanols from C5 -CB
--···Hydroxy Ketone (!) from 886-8105
I
21 23 25 27 29
CARBON NUMBER--._
o Standard compounds 0 Hydroxy ketone (I)
D. Alkano!s from c5-c8 o Synthetic Hentriacontan-16- of
...
30
31
.· . . ·. .. . .
FIG. 1. GL C OF ALI<ANOLS AND HYDROXY KETONE (I)
40

o31 and o33 predominating.
Fractions A21 -A45 gave only a very small quantity of aaterial
and was found to be mixture by TLO.
Fractions A46-A110 gave one elongated diatinct strip in TLC.
GLC r ~vealed that it is a mixture of several component e. These
fractions were mixed together and chromatographed over Argentized
eilicage1 14• 15 .120 (B1-B120) fractions were collected troa this
chromatography (Table-6).
:Fr aoti on s B11 -B20 gave a solid, mp. 9'3•, and having a o abeorpti on
for =0=0 or -OH 1 n the IR and it was found to be a mixture of' .
hydr ocerbons by GIC.
Fractions B21 -B80 gave a solid, mp. 95° IR absorption at '3600--1 '
3400 em showed the presence of hydroxyl containing compound and
GLC revealed that it was a mixture. It was then rechromato1raphed
over A.rgentized silica gel (Table-7). 2'3 (o1-c23) fractions were
collected from t hie chr omat ogr aphy.
Fractions B86-B105 eluted with benzene and petroleum ether
(40-60°' was found to be a single component by TLC and GLC (Fig. 1)
after crystallisation from petroleum ether (40~0°) and be1zene
mixture. On elemental an.alysis and molecular weight deter111nati on
t~e oo~pound · ~· was found to have molecular tormla c3c)B60o2 (452,Jf"),
mp. 102°, [oe] ~5.-9° (cHol3).
Anal. Found I O, 79·7~ 1 H, 13.01~ I
Calc. for c3cJI60o2:o, 79.64"1 H, 13·2~ 1

i ...
WAVELENGTH (MICRONS) '3 7 B 9 10 12. 13 5 6 4 14 11 100--~--_J~ __ _L ____ ~ __ _j ____ ~ ____ L_ __ ~-----L----L---~----~--~ 15
so ,......_
~ 0
~ 60 ltJ l..> <: ~ ~ 40 ~ (/)
<: ~
~ 20
0 400~0~~~~----~~------~~------_L ________ _L ____ ~--------L---------~--~ '3000 BOO 700 12.00 1'500 1000 900 2000
CM -f
Fig 2. I R curve of Hydroxy-l<etone I .

5
It did not give any oolouration with tetranitr omethane. It
also did not correspond to the teet for sterols and triterpenes.
The functions of the two oxygen atoms in the molecule were
revealed from IR spectrum of the compound wbioh showed absorptions
at 3210 and 1700 cm-1 (Pig. 2) assignable to a hydroxyl and a
ketonic group respectively. The significant absorption discernible
in the IR spectra are at 1455 om""1 (-cH2 bending), 1'75 011-1
(-o-aH3 bending), 1066 om-1 (-c-o stretching of secondary alcohol), -1 \ doublet 727 and 718 om - (CH2,n bending(n >4) vibration due to
straight chain methylene gx oups. Moreover, it furniehed a mono
acetyl derivative and a mono-oxime. consequently the presence of
a hydroxyl and a ketonic group in thi molecule was ascertained.
Oxime of the gy~r oxy Ketone 1
Oxime of the hydroxy ketone was prepared by hydroxyl aDd.ne
hydrochloride in the usual WfJN and the oxime thus formed had . .
Anal • Found 1 I, 3~ 1
Calc. fOJ! c,oH61N02 IN, :5·1,. I
Acetate of the Hydroxy Ketone 1
The hydrox.yketone was acetylated with acetic anhydride and
pyridine in the usual way and the acetate thus formed had mp .ga• [ oC):o; :5 • ( CH013)
Anal. Found a C, 79 ·7~ J H, 12 .51" J
Calc • for c32B620, 1 0, 79. 76~; H, 12 .55" 1
The structure of the hydroxyket one was determined mainly from
its mass speot:ral fragmentation pattern. The absolute identification

+
d m;e 200 b m;e 252
Scheme-l
c m;e 310
Scheme- II
----------- - - ---- .... , 423 \
I I
--- - - - --- - -- .. \ I
393 \ l I 1
CH3 (CH2)tB CO (CH2) 7-: CHOH ~C2H5
I I 29 1 \ I 1
l 59
.... ___ ------
. ..... _______________ _
Scheme-m:
Chert I- Mess Fragmentation Patrern of Hydroxy- Ketone (I)
..

100
80
60
40
20
1 ·~
60
40
20 244
I I I
240
(I)
59
29
2.95
i· !
310 c
127
393
260 280 300 320 '340 390 410
Fig. 3. Mass spectrum of Hydroxy-l<etone (I)
200 d
IB6
220
430 450

6
of higher aliphatic ketone has given great trouble on acco•nt of •
very similar physical properties, IR and !MR spectra. The fragmenta
tions obtained in mass spectra have been of immenee help in
elucidating the structure of higher aliphatic ketone, both symmetri
cal and unsymmetrical19-23. The moet intense peaks are oi'tained by
-ex:: -fission and by (.3 -fission which is accompanied by Me-Lafferty
rearrangement with the addition of plus one (Chart-!). In the mase
spectrum of the compound, no significant (M-15'+ peak was observed
and the ratio of {M-15)+ /M+ was close to zero. This indicated that
the compound is a straight chain24 • Ma_ss spectrum showed peak at
0~1 )+ which is characteristic of unsymmetrical ketone25 •
Besides the molecular ion peak at e/m 452 (M+, 25~) the other
significant peaks desoernible were the following (Fig. 3)
42:5 ( oC -fission to the -OH group, 10)
393 { oC -fission to the -OH group, 12)
337 ( 6 -fission to the •C•O group, 5) -
323 ( 'l -fission to the =0=0 group, 9)
310 (fragment c, (? -fission to the •0•0 group - .
Mc-Lafferty plus one ion, 100)
296 ( .£-0~, 14)
295 (.Q. -oH, or oC -fission to the cC:zO group, 66)
292 (.Q.-H20, 17)
250 (.Q.-Hz0-3CH2,14)
244 (.!!-H2o, _20 ' 238 (~CH2 , 17,
237 (~C~ , 6 )
227 ( o -fission to the •0=0 group, 7)

CH3 (CH2>18 .co(CH2)7
. CHOH CzHs
1 1 Huar:g- Minion r~duction CH 3 (CH2) 26 . CHOH . CzHs
IT l Cr03/AcOH
][
+/H 0 I
C2.H 5 . c II CH2
n rnje 364 m m;e· 72
Chart II- Synthesis of compound ill and its Mass Fragmentation Pattern

.:
~ Q
t::::: -;::..., ......
'!I)
t::::: (l) ..... -S
IU -:::... ·--en -~
Q:
+o/H I
c2 H 5 - c II CH7.
m m;e 72
54
57
2.9
n m/e 364
72 m
364 !1
Fig. 4. Mass spectrum of the Ketone (m)
..
407

213 { Y-fission to the =0=0 group, 9)
200 ( fragment ~. @-fission to the =C=O group, Mc-
Ie.fferty plus one ion, 100)
186 {.!-CH2, 33)
185 (,!-ca, or oe-:f'ission to the =0=0 group, 33)
18~ (~-H20, 22)
167 (A-H20~0H3 , 18,
127 (!-OH3 ~ 25)
126 (!-CH2, 18,
59 ( oC - fission to the -OH group, 44)
29 ( oC -fission to the -QH group, 23)
From the above mass fragmentation data coupled with other
physical and chemical evidences the structure of the hydroxy ketone
may be represented by (I) 0 II
Me.(CH2)17 .cH2.o(cH2)7.cHOH.cH2Me
I
In order to ascertain further the position of the -OH group
in (I) it was reduced (Chart-II) to (II) by Huang-Minloa reduction26
having mp .gao, molecular formula, c30u62o. This on oxidation with
chl:omium trioxide gave a monoketo compound (III) having mp.92•,
molecular formula, c30H60~. The mass spectrum (Fig. 4) o:f' (III)
showed the mol~cular ion peak at m/e 436 (II'", 15~). The other
significant peaks appeared were the following•
422 (M--CH!,10)
408 ( M-2CH2
, 12)

,....... ~ 0 ....__
lU \..)
<: ~
lOO
80
60
:: 40 ~ U)
<: "<:(
n: 1-- 20
3 4 WAVELENGTH (MICRONS)
5 6 7 8 9 10 11 12 13 14 15
~
0----~----------~----------~--------~----------~------~--------~----------~--~ 4000 3000 2000 1500 1200 1000 9000
CM-t 800 700
Fig. 5. IR curve of the hydroxy compound (II}

WAVELENGTH (MICRONS) 3 4 5 6 7 8 9 10 11 12 13 14 15
100 .-~----~------~----~----~------~----~------L-----~----~------~-----L----~
80
'""" ~ 0 '-......
60 ltj (J
2: ;: !::: 40 ~ (J)
<::. "'(
0::. 1-- 20
0~--~----------~--------~----------~--------~------~--------~----------~--~ 4000 3000 2000 1500 1200 1000
CM- 1 900 800 700
Fig. 6. !R curve of fhe Ketone (ill)

407 (oe -fission to the =C=O group, 22)
394 (rf-3CH2, 8)
379 ( oC -fission to the =C=O group, 8'
364 (fragment !• 15)
72 (t.ragment m, (3 -fission to the =C•O groJ.p, -Mo-Le.fferty plus one ion 100)
58 <.a-ca2 , 42)
57 ( oc-fissi on to the =C•O group, 58)
54 (!-H20, 60) . 29 ( o<:-fissi on to the =0•0 group, 18)
The mass fragmentation pattern of the compound (III) is
quite in conf'orm:l. ty w1 th the structure asaigoed for it. Consequently
the original hydroxy ketone may reasonably be represented as (I).
Reduction of' the hydroxyketone (I) to {II) by Huans-Minlon reduction 1
The hydroxy ketone (I) was reduced by hydrazine hydrate in
alkaline sol uti on using diethylene glycol sol vent. The product
obtained after the reduction had mp. 98•, IR 3220 011-1 (Jig • 5)
Anal. Fou.nd t c, 82.14~ 1 H, 14·09~ J
Calc. for c3QB62o 1 C, 82.19~ J H, 14.12~ J
Ohromiu11 trioxide oxidation of compound (II) to oompoun4 (III) a
The compound (II) was oxidieed by chromium trioxide in acetic
acid and worked up in the usual way. The product formed had mp.92•,
IR 1700 cm-1 (Fig. 6)

WAVELENGTH (MICRONS) 3 4 5 6 7 8 9 10 11 12. 13 14 15
1--~----~----~------~----~----~'------~'----~~----~'------~-----L------~----~ 100 I '
......._ ~ ~
80
lJJ 60 '-.)
<: ~ I-
~ 40 (/)
<: oq: 0::: 1-
20
OL_~------~---~-----~~--~~~--~~---~~ 4000 3000 1500 2000 1000 1200
CM- 1 900 800 700
Fig. 7. I R curve of a/kano/s from c5 -c10 fractions

WAVELENGTH (MICRONS) 3 4 5 6 7 8 9 10 11 f2 1'3 14 15
,I
100 ,_~----~------~----~----~------~-----L----~~----J-----~------L------L----~
80
"'""' ~ a '-..J
l1j 60 LJ < ~ I--~ 40 U)
<: ~ ct: 1-
20
0 -
4000 3000 2000 1500 1000 1200 CM -f
·goo ' 800 700
Fig. 8. /R curve of ester from CHC/ 3 extract of M. m. Linn.

Anal. Found a o, 82.36~ a H, 13.72~ a Calc. for o3QH60o a c, 82.56~; H, 13.76~~
All the foregoing evidences have led in support of the structure (I)
for the hydroxy ketone.
An other major o omponent in the :tr acti on• c5-c1 0 was obt aintd from
chromatography over argentized silica gel and was found to be a
single component by TLC. lR spectrum {Fig, 7' gave absorption at
3220 cm-1 assignable to a hydroxyl group, It did not giYe any
colouration with tetra-nitro-methane. It also did not correspond to
the test for sterols and triterpenes. However GLC (rig. 1) revealed
that it was a mixture of 5 components with carbon content ranging froa
027 to 031 with 027,029' 0,1 predominating, only' minute traces Of
compounds with even number were present, These compounds were supposed
to be a mixture of alkanole as one of the compounds wa1 identical
with hentriaoontan-16-ol17 and was compared with the synthetic compound
by GLC study where both of them have been found to contain ea11e
carbon number with the same retention value. Hentria-oontan-16-ol
was prepared from palmi tone 18 by sodium borohydride reduction.
Treatment of the ohlorofo.rm extract 1
The residue from the ohl or oform extract which had a fishy odour
on being heated with dilute alkali gave profuse quantity of methyl
amine. Chromatographic separation of the neutral moiety over alumina
gave @-sitosterol, mixture of hydrocarbons and a very small amount
of ester which Showed carbonyl absorption in the IR at 1740 om- 1 (~
disc) (Fig. 8). TLC of the ester showed two spote-oae very weak. The , ..
major spot ·.had R:t 0.60 (5~ ether to pet:r oleum ether) corresponding

100
80
.....-... ~ -...__,
l..t.J 60 u <: ~ 1---~ 40 U)
<: ~ a: ~ 20
3 4 WAVELENGTH (MICRONS)
5 6 7 B 9 10 11 1'2. 13 14 15
·~
ol_~~----L-----~----~~----~~~~--~~--~~~ 4000 3000 2000 1500 1000 1200 900 800 700
CM -l
Fig. 9. I R curve of Marsileaqenin A

lO
to esters of high molecular weight. The ket ODe mp.85-86• isolated
from this fraction by Chatterjee et a16 could not be traced..
Treatment of the ethanolio extract a
The ethanol! c extract gave copious froth with water ther ebJ
indicating the presence of a saponin in it~ On removing the solYeDt
a orude saponin was obtained aDd it was extracted repeatedly with
Butanol. :Butanol was distilled off under reduced pressure when the
mixture of saponin was obtained as a dark brown gummy mae a. !he
tongue loses -the taste !or sugar when the saponin is put over it.
This effect exists from half an hour to one hour. In this respect 16 it resembles the saponin of Gymnemagenin •
The crude saponin was hydrolysed by re!luxing with ~ ethanolic
hydrochloric acid for 10 hours. The alcohol was evaporated ott
keeping the acid strength more or lea·s constant on frequent addition
of water. The crude sapogenin was filtered and the residue was
washed repeatedly with water until free from acid, d:ried at 60•. The
crude genin on :repeated chromatography over Brockmann alumina aDd
subsequent erystallisati on from ethanol yielded a new tri terpene 2.S•
called ' marsileageninA', mp. 332-33•, [oc]D + 48• (ethanol), IR
spectrum (Fig. 9) of the compound showed bands at 3500 (hydroxyl
group) and 820, 830 om_, ( trisubsti tut ed double bond).
The hydrolysis of the saponin with ethanolic sulphuric acid was
avoided due to the fact that the crude sapogenin obtained after
hydrolysis becomes a tar-like material and it was dif!ioult to isolate
the pure genin from this product.
\

WAVELENG TI-l (MICRONS) 3 4 5 6 7 8 9 10
100 I
80 ...-....._ ~ 0
'--
lJj 60 C.> <: ~ 1--~ 40 U)
2: ~ 0:: t--
20
0 I
4000 3000 2.000 1500 1200 1000 CM -f
11
900
Fig. 10 IR curve of Marsi/eagemn A Hexaacetate
_,
12 13 1·:
I
800 700

~ .. ll
Molecular formula 1
Analytical sample of marsileageninA was analysed tol carbon and
hydrogen. Difficulties were realised in detecting the &JOleculal
weight from mass spectroscopy as it was not volatile upto 320•, but
the molecular weight was d ed uoed from the molecular weight of its
hexaacetate. The data given below clearly indicated a molecular
formula o3oa50o6 for marsileageninA.
Name of the compound
Mar sileageni nA
Found
colour reaction a
71.23
71·11
9 ·9
9·95
Kol. wt.
506
506
MarsileageniDA gave a dark red colouration with concentrated
sulphuric acid. It showed pink oolouration in the Id.ebermann-Burchard
test indicating it to be a triterpene. It also gave a yellow colour
with tetra-nitro-methane indicating the presence of unsaturation.
MarsileageninA Hexaaoetate 1
MarsileageninA was acetylated with acetic anhydride and pyridine 2.50
in the usual way and the product thus formed had mp. 285•,~] +35·5• D
(CHcl3), molecular formula c4~62o12 • It showed m bands at 1740 (br),
1242 (br) 011-1 (acetate carbonyl) and 830 c11.-1 (trisubstituted double
bond) (Fig. 10).

Thus the above results showed that all the six oxygen aio11e
in marsileageninA are present as hydroxyl :f'unct1o1us.
Monotrityl derivative of MarsileageninA 1
MarsileageninA was heated with trityl chloride 27 in pyridine
and the product formed had mp. 205-7°, molecular fcn=mula, o49H64o6•
The result showed the presence of one primary hydroxyl group
in marsileageninA.
Detection o! number Of double bonds in marsileageninA 1
standard chloroform solution o:f' marsileageninA hexaacetate
was treated w1 th an excess of standa:rd per benzoic ac1d28 solution.
From the amount of consumption of perberizoic acid per mole o!
marsileageninA hexaacetate it was found that marsileageninA contains
one double bond,
Detection of number of oe-glyool systems in marsileyeninA 1
MarsileageninA was found to consume two moles o.t periodic acid
per mole of the compound. The products obtained after periodic acid
oxidation29 ''0 was found t~ give yellow oolouration with tet:ra
nitro-methane and hence the double bond of the compound remained
intact during periodic acid oxidation.
The results from the above experiment showed the presence of
two oe-glycol systems in the compound.
Preparation of 11-keto marsileagenin! hexaacetate a
MarsileageninA hexaacetate was heated under reflux with chromium
trioxide fn glacial acetic acid and the 11-keto compound thus .formed

239 mJ-1 (logE 4·tz)
2.40 250 260
A cO
270
:A max
A cO
280 290 300
Fig.11. UV Curve of 11-l<eto Marsileagenin A Hexaacetate
oAc
-- oAc
CHz.oAc
oAc
310
I b ·-U)
t::: Q)
L:::l
-Q:l \..) -a_
a

100
80 ......... ~ 0 ~
Lu 60 \._)
<: ~ ~
~ 40 U)
<. ""l:: 0: ~
20
' WAVELENGTH (MICRONS) 3 4 5 6 7 8 9 10 11
0 v
4000 2000 1500 1200 t 1000 900 eM-
3000
Fig. 12 IR curve of /1-l<eto Marsileagenin A Hexaacetate
12 13 14 15 I
800 700

1a
was worked up in the usual way. The molecular :formula of the compound
was found to be c42H60o13 , mp. 287°, UVmax 239 nm (-log.E 4.12)
(lig. 11 ), IR ~"uJol 1735, 1240 (acetate carbonyl), 1670 ( oC,@ mo..x
flneaturated ketone), 830 om-1 (tr1substitute4 double bond) (Fig.12).
The above reaction indi~ated the presence o:r a 1211' double
bond in marsileageninA. The :following Tables (1 and 2) shows the
UV absorption maxima o:r some 11•keto .. compound belonging to 18~ and
18@ oleanane and ur sane series.
TABLE - 1
UV Maxima of 180C and 18@ oleanane epimere
!fame of the compound
3@ -Acetoxy-12-bromo-11-oxoolean-12-ene
3@ -Aoet oxy-11-oxoolean-12-ene
3@ -Acet oxy-11-oxoolean-12-ene-30 oic acid
2 -Bromo-3, 11-dioxoolean-12-ene-30 oio acid
3, 11-Dioxoolean-12-ene-30 oio acid
., @ -Hydro:xy-11-o:xoolean-12-ene
3@ -Hydroxy-11-oxoolean-12-ene oic acid
269
250
248
250
250
245
248
Methyl-3oC-aoet oxy-11-oxoolean-12-ene-24-oate 248
Methyl-3@ -acet oxy-11-oxolean-12-ene-28-oate 250
Methyl-3~ -aoetoxy-11-oxoolean-12-en-30-oate 248
Methyl-3~ -hydroxy-11-oxoolean-12-en-30-0ate 248
max ( nm) R e:f.
18@
267 31
245 32, .,,
244 34,35
245 36
245 36
243 .,7
242 34,35
244
248
243
243
38
35,39
34,40
,4,35

lt
TABLE - 2
UV Maxima of 180C and 18@ oleanane epimere and the corresponding
Vrsane derivatives
lame ot the oom.pound ~A. max (am)
'3@ -Aoet oxy-11-oxour s-12-ene 251
3@ -Aoetoxy-11-oxoolean-12-ene 250
3~ -Aoetoxy-11-oxo-18oC-olean-12-ene 245
Methyl-'3 ~ -acet o:xy-11-oxours-12-en-28-oate 250 '
Methyl- '3@ -aoet oxy-11-o:x:oolean-12-en-26-oate 250
Ref
41
32,33
42
4'3
35,39
Methyl-'3@ -aoet oxy-1 1-oxo-180C -olean-12-en-28-oate 248 42
5 ,8,14-Tri methyl-11-oxo-18~-novoleana-9( 10),
12-diene 256,287 42
5,8,14-Trimet~yl-11-oxo-novurea-9( 10), 12 diene 256,290 43
5 ,a, 14-Trimethyl-11-oxo-18oC-novoleana-9( 10 ),
12-diene-28-0io acid
5,8,14-Trimethyl-11-oxo-novursa-9(10)-12-diene
28-oio acid
5 ,a, 14-Tr1methyl-11-oxo-18~novoleana-9( 10)
12-diene-30-0ic acid
2-Benzylidiene-3,11-dioxoolean-12-ene
2-:Ben eylidiene-.,, 11-dio:x:ours-12-ene
3,11 ,ai oxoolean-12-ene
3,11,dioxours-12-ene
257,288 44
259,292 43
254,287 44
231,255,294 45
232,255,293 45
294
293
45
45

Relative Intensity in % --~
~
~ IJ1 0
\.() U1 0
- 0 ~
s:: cu (/) (J)
(JJ \:)
(1) C) ........ .., t:: 3' Cl \.11
- 1.)1
::X: (1)
X co I
Cl I ~ () I"(} .....
"<:: --1
~ tll ..,
0
(J) 0'1 :::.. 0 (1:) 0 1:\)
1..0 (t)
:::J -· ::::!
.h
I- 4 58 ( M+ - 5 A c 0 H)
~ 476 (M+-4AcOH-C 2HzO)
~
518 Uvt+ 4AcOH) t---
~---
,...._ 536 ( M+-3Ac01-i-Cz/-/zO)
1-
578 (M+-3AcOH) .
~---
~
F. ::::::. .::. . ::==._ -t-···--- 63B (M+-2AcOH)
.
1--- 683 ( f1+ -AcOH -C H3)
698 (M+-AcOH)
3 (13'
(1' 0
r--- /87 ( ?_ -·2 AcOH) !=-- ---
197 (Q 3AcOH-CH2 0Ac)
~ 1\J F" 1'\J 0
I=-r-- ;
t'V =--(Jl F 247 (~-AcOH) 0
t'V ~
(] r--- 270 ( Q- '3AcOH)
()j
302 (b-AcOH-CHzOAc-CH3) 0 0 ...=.-
r--
F- 3!5(Q-2Ac0H-CH3)
1::-
1-- 375 (g-AcOH-CH3) t::.=:
1--
1---- :::\qn f h -A~OH)
f:=:-
~
~
445 (M+ -4Ac0H-CH2.~Ac)

---~ -
241 m_}J(/ogE4·43) 250 m_)J (logE 4·46)
259 m_}J (logE 4·33)
\/ I t oAc
-- oAc I I
CH2oAc b ·-{I) oAc t:::::
AcO --/ ~ ~~~ Q)
C)
-A cO ___(__ ~ / I OJ \.) ·--/ '\. I Q_
0
230 240 250 260 270 280 290 300 310
A max
fig. 13 UV Curve of 611
:12
' 13
·'8 diene Marsi/eegenin A Hexaacetate

.• 1 s
The 11-keto compound of marsileageninA hexaacetate displayed
UV abeorpti on maximum at 239 nm against usual position at about
249 nm. This hypsochromic shift is reasonably attributed to the
presence of a number of acetoxyl groups in the molecule.
Selenium dioxide O:xidation of mare1leagen1n.A. hexaacet ate '
The hexaacetate in glacial acetic acid was refluxed with .
selenium dioxide and the prOduct was worked up in the us•al way.
The molecular formula of the compound was found to be c42H60o12,
mp • 285-86• X mo.)f. ~41, 250 and 259 nm (logE 4.43, 4.46 and 4. 38 ·
respectively) (Fig. 13).
The triple UV maxima of the selenium dioxide oxidation product
of marsileageninA hexaacetate is characteristic of trana heteroannular 46-48 12 49 diene system and is a characteristic of b.- oleanenes • It
should be mentioned here that tri-tetpenes of the ursane series
having 12t 13 double bond yields above short o! dienes only on heating
at a higher temperature (200°) with Se02 in benzylal.cohol.
Mass spectrum of MarsileageninA Hexaacetate J
The mass spectrum of the hexaaoetate (Jig. 14' of marsileageninA
showed the characteristic retro Diels-Alder fragmente50 involving
12t13 double bond of the oleanenes or the lll!senes.
+ . 01\.~
01\e.
--~
Ac.O
ReO
A eO
AeO
Cl
mje. 307
b
mfe 450
+ .

The other significant peaks appeared at m/e a
6$3 (lt -AcOH-cH3)
6:58 (It--2Ao0H'
578 (rf-3Ao0H)
536 (wr-3AcOB-c2H5o' 516 (I+ -4Ao0H)
476 (x+-4AcOH-o2H5o)
458 (M+ .. 5Ac0H)
445 (M+-4AoOH-cH20Ao)
385 ( Jt -5!cOH-oH20Ac}
247( a-Ao'OH) -187(,!-2Ao0R)
- 16
390(b-Ac0H) -375(~Ao0B-CH3 )
315 (,k-2AcOB-cH3)
270( b-:5Ao0H) -
The mass spectrum of the hexaaoetate supports the presence of i2.
/J -oleanane skeleton. The mass spectrum did not show the moleoula't
ion peak, but it showed the rt -AoOH peak at m/e 698 due to the
facile loss of a molecule of acetic aoid in the 1onisin1 chamber
whiCh is a phenomenon of common occurrence. The mass spectrum showed
the retro Diels-Alder fragments a at m/e 307 and b -AcOH at m/e·390 . - -indicating that two hydroxyl groups are present in the part containing
rings A/B and four hydroxyl groups are present in the part containing
rings D/E in marsileageninA. The mass fragmentation pattern suggests
the primary acetoxyl group to be at C-28 as there is pronounced 7'
unit loss from fragment l• As has been mentioned previously,
marsileageninA contains two oe -glycol systems. Jaw only four hydroxyl
groups are present· in the part containing rings D/E of which one

17
primary hydroxyl group is present at c28 • It follows, therefore,
that one oc -glycol system and another secondary hydroxyl are
present in this part. The presence of a ·«:: -glycol eystea at
a15 ,c16 was ruled o~t, because it has been mention,ed by "Yarioue 5 ~
workers 1 that hydroxyl group at c15
, whether a.xial or equatorial
is resistant to acetylation under ordinary condition. But
marsileageninA furnished a hexaacetate under normal condition.
consequently the p1esence of~ oe-glycol system at o21
,a22
seems
to be certain52 • The presence of a 16~- hydroxyl is re"Yealed :troa
NE spectrum of the hexaacetate and corroborated by the :tact that
the hexaacetate is· easily formed, whereas the 16oC-hydroxy is
reported to be highly hinde~ed53 • Furthermore, the secoad oC -glycol
system seems to be present at c2, c3
• The presence of a hydroxyl
group at c3 is more probable from b1ogenatic point of view and
as there are two hydroxyl groups in the part containiag rings ~B,
constituting a oe•glycol system, the .other hydroxyl group must be
present at c2. The stereochemistry of the hydroxyl group is e'Yideat from. the
NMR spectrum of the hexaacetate •
signals ara shown in Table-3.
TA:BLE - 3
Tertiary methyls
. The eigni!i oant NMR
0 ·91 (311, e)
1 .02 ( 6H,e)
1 .11 (9B, e)
1. 35 (3H, e)

18
TABLE- 3 (Contd.)
ocooa, 2.0 (6H,s)
2.10 (6H,a)
2.14 (6H,a>
c17-cH20Ae 3 .g6 ( 2H, br s)
C-2H 4 .ea < 1H .. m)
c-3H ' 4.84 (1H,d,Ja10Hs)
C-21H 5.0 (1H,d,J=11 Hz)
o-22H 5.39 (1H,4,J=11 Hs)
C-12H 5 ·41 ( 1H,a)
C-16~! 5.75 ( 1H,t-like)
The lOR data discloses the existence of seven tertiary methyls . 12 (eliminating the possibility o! ~ ursene skeleton). One olefinic
hydrogen and six acetoxyl functions, uong which one is attached to
primary and the othe~ five are connected to secondary }Qdroxyls. The
broad singlet at b 3.96 is assigned to methylene prot on a ot C-28
acetoxymethyl group. The AB quartet at 5.0 and .at.5.39 is better
ascribed to the trane-diaxial prot one c:£ to acetylated 112 glycol
system at c-21 and c-22 rather than C-15 and c-16. The presence ot
triplet like multiplet at 5·75 is attributed to C-16 axial proton54. \
The downf'ield shift is due to the deshielding effect by 1t3 inter
action with the 0-27 methyl groups. Diequatorial relationship of
the glyo ol eyst em at c2 and ~3 i e revealed by the appeaJ: ance ot
signals at 4.98 (1H,m) and 4.82 (1H,d,J=10 Hz) ascribable to diaxial

OH HO
I Barrigenol R1
III Theasapogenol A
HO
\ \
\
' '
\ \
V Gymnemagenin
'
OH
-- OH
CH 20H
'OH
OH
-- OH
i
HO~
Ho--
HO
I I
H CH20H
II Protoascigenin
W Tang(nol
;<1 oH
-- OH
C H'2. OH
TI Marsi/eagenin A
Chart ill Naturally occuring Hexahydroxy Triterpenes

Name of the
compound
Barrigenol R1
P.r ot oascigenin
Theasapogen olA
Tanginol
Gymnemagenin
llareileageninA
[oe]D & mp.
+ '37° (dioxane)
'308-10•
+ 31·5•(dioxane)
'31 0°
+14° (py.)
301-'3°
_,.go (EtOH)
28'3-4•
+ 53.1• (MeOH)
329-'31 °
+ 48• ( BtOH)
'332-3•
I
TABLE - 4
Acetate
mp. & [oe]D
Hexaacetate -28°(CHC~)
186-87.5°
Hexaacet ate -3 .5o ( CHol3 '
140-41•
Pentaacetate + 29°( CHC13 )
174-178°
Hexaacetate -63• ( OHC13 '
150-3•
source
Tea. ~yngium
Aesculus
turbinata seed
Thea sinensis
seed
'Barr ingt oni a
acutangula
Hexaacetate + 35 .5• (CHC~) Gymnemaeylveetre
290-1•
Hexaaeetate + 37.5• (CH013 ) Marsilea minuta
285• !d.nn
Re:C.
55
56
57.58
59,60
16.61
13

19
protons at c2 and c, respectively.
All the foregoing evidences have led to the conelueion that
marsileageninA may b.e formulated as olean-12-en-2 «: , 3<?, 16 ~ ,
21 @ , 22oC , 28-hexol.
Hexabydroxy triterpenes· isolated from ti:t!erent plute are
presented in the Table-4 and Chart-III.
Identification of sugars s
:For identit:ioation of sugars in the saponin obtaiaed :trom
ethanolio extract of marailea minuia, a portion of the ethanolio
extract of the plant was b¥drolyeed with ~ ethanolic sulphuric
acid for ten hours under refiux. The alcohol was evaporated off
keeping the acid strength more or less oonat ant on tr equent
addition of water. !hie was filtered and the filtrate wae neutrali
sed with barium carbonate. The barium sulphate thus precipitated
and then excess of barium carbonate was added and :til tered off.
The filtrate was deioniaed, dried aad examined :tor its sugar
constituents by descending paper partition chromatography using
n-ButanolsP.yridineaWater (6a4a3) as solvent at the upper layer and
aniline oxalate as the developing agent. The sugars thus identified
were D-glucoae, D-galaotoee, D•xylose, D-arbinose, ~rbamnos~ by
comparison with their authentic specimens.
After ten long years, however, Ike. A· Chatterjee et al
corrected 11 that marsilin isolated :from Marailea minuta Itnn was
not a ma.orooyolio ketone as reported ea:rlier 6• It has bee1 claimed
by them that marsilirl has e.lso been isol.a.ted trom another plan-t

• Ipomoea :!istulosa tt but the molecular formula wae changed trom
~29Hs4o to c56H112o2 • They corrected that mareilin is a high
molecular ester. The m spectrum of the compound here showed peak
tor carbonyl absorption at 1735 c11-1 and l:r.fi 1peotrum of the compound
gave signals at 'r 9·1 (6H}, 8.75 [ H of (OB2)11
] , 7·75 (2H,OH2-c.o), 5 ·9 (2H,OR2-o·'· Behaviour tower~s chemical reagents wae e111ilar as
described in earlier literature. In this later communication it has
been reported to be an ester of triaoontanol and hexacoeanoio acid
which were obtained by the hydrolysis of the compound with (21)
potassium hydroxide solution. This compound wae synthesised froa ita
hydrolysed products and also from erotic acid and 1-triacontanol.

100
80 ....-.._ ~ 0
'-
Lt.! 60 l.l <: ~ 1--~ 40 ~-(/)
<::: "'(
0::: 1- 20
3 WAVELENGTH (MICRONS)
4 5 6 7 8 9 10 11 12 13 14 15 I
OL---~-----------L----------~--------~----------~------~~------~----------~--~ 4000 3000 2000 1500 1QOO 1000
CM - 1 900 800 700
Fig. /6 . I R curve of the hydrocarbons

21
EzyERIMEITAL
The stems and the leaves of Marsilea minuta Linn was
out irrt o pieces, air dried and powdered. !he material ( 640 g)
was euocessi vely extracted in a one litre sexhlet extract~ w1 th
petroleum ether (60-80°) tor 50 hours, with chloroform te.r 30
hours and with ethanol (95~) toz 50 hours.
Treatment of the petrolium-ether Extract a
The petr olium ether extract Ol'l removal of the solvent and
proper d:y·ing yielded a dark green solid (5 g). The solid was
die solved in 50 ml of petroleum ether ( 40-60•) containing about
6 m.l of benzene and chromatographed over a column of neutral
Brockmann alumina (200 g), the elution being carried out eucoessivel1
w1 t h petroleum ether ( 40-60° ) , Petl: oleum ether c Benzene ( '11 ) . .
(1s1), Benzene, Benzene·Ether C5a1) (111), Ether, Ether-Kethanol
( 111) and Methanol. Each fraction contains 10 ml of eluent. !he ,
reeults of the chromatography are shown in table-5.
Each fraction from A1-A20 ( 105 ~) •as tXBllined separately by
TLC when a single strip was obtained. These !racti one showed no
absorption at 1720-1740 011.-1 or 3400....3600 011.-1 in the m apectrua
(Fig. 16) coofirming the absence of carbonyl and cydroXJl containiag
compounds. These tractions did not give any colouratioa with
tetranitr ometbane indicating the absenoe o:t any unsaturation in the
compound. Moreover, they did not respond to Liebermann-Burohard
reaction eliminating the possibility of any eteroids or tri-terpene.

22
TABLE - 5
Fractions Eluent a llatW!e o! the products Remarks
· 11-A2o Petroleum Ether White solid, a.p.S0-81• Mixture of
( 40-60°) 105 mg hydrocarbons
bJ m & GLC
report
A21-A45 -do- White solid, m.p. 88°, Mt.xture 15 mg
A46-Aao Petroleum Ether- White solid, m..p.91• Jlixture 'by
Benzene (3a1) 150 rag TLO & GLC
A~u-.&.110 Petroleum Ether- White solid,m.p.91• -do-
Benzene ( 111) 105 mg
A111-A1,0 Benzene :tlil '
A131•A140 Benzenes Ether Little .yellow solid ( ,, 1) 8 mg m.p. 73•
A141-A147 Ben Eenes Ether Very little yellow aolid
( h 1' 5 mg, m.p. 75•
A148-A150 Ether Nil
A151•A153 Ether : Methanol Little oily liquid, 12 mg
A154-A156 Methanol Little Oily liquid, 8 llg
These fractions were then mixed together and examined with the help
o! GLC when it was :found. that they contain mixture o:f seYen bydrOca!boru

', i I, 'I
I I
lr
I'
~so.' vent
10
I IJ
I I I I I
20
7/ME IN 'M(NUTES --..::;,.
27 2 9 31 33
CARBON NUMBER --~ ~:;. Standard compounds J o Hydrocarbons
FIG. /7. GLC OF THE HYDROCARBONS
3C

23
:from o2rc33 with c27,o29 ,o,1 and o33 predominating (Fig. 17). These
were examined with three standard hydroca:bons contatninl c27,o29 and o31 ca:r bon at oms.
Fraoti one A21-A45 ( 15 mg) gave only a Vely small quantity at
material and was f'oond to be a mixture by TLC.
Fractions A4g-A110 eluted with petroleum et~er (40-60•) and
benzene gave one elongated distinct strip in TLC. GLC reYealed that
it was a mixture Of several components. !heee f:raotione were mixed
together and ohromatographed over .A.rgentized silica ae described
in Table.-6.
TABLE - 6
Fractions Eluent a Nature Of the products Remarks
B,-:s,o Petroleum Ether Nil
B11-B20 Petroleum ethera White eo114, m.p. g,o, Jrtl. :xt ur e o! Benzene ( 3a1) 40 Jig hydrocarbon by
IR & GLC repor-t
B21Bso -do- 'fbi te solid, m •. p.95° lixture of
120 1lg bJdr OXJ C Ollp OUDd
by IR & GLC•
Bef .. %5 Petroleum. ether 1 111 Benzene ( 1 a1 )
Bso-B105 Benzene Whi t'e solid,a.p.102-10'• GLC indicated 40 mg oae c omponen1
o out a1 ni ng :50 car 'bon at oae, Ill showed t~ preeence o! H
B -B Ben zenea Ether 1'11 &•O=Ogr. 106 115 (1a1)

24
Preparation of Silver nitrate Impregnated Silica sel 1
A 2C),C solution ·of silver nitrate (80 ml) wae heated on a steam
bath with silica gel (40 gm) !or hal! an hOUI• cooled and filtered.
fhe adsorbant was dried at 120• tor 16 hrs. and cooled ira a deei-
coat or.
Reobromatograpby of the fractions A40A110 over .Argent1ze4 silica t
Fracti one A40A110 (255 mg) was d1ssolyed in 1 ral ot pe"trolium
ether containing a few drops of Benzene and adsorbed in 5 gu o!
argentized silica gel. The adsorbed material was cbroma:tcrcraphed
over a column of argenti 2:ed silica gel ( 15 ga) and elute4 w1 th
different solvents (Table-6) and each traction collected in 10 ml
portions.
Fractions B11 -B20 (40 mg) eluted with petrolium ether (40-60•)
and benzene C5:1) gave a solid m.p. 93°. No peak for carbonyl or
hydroxyl group was found in the IR spectrum and 1 t was found to be
a mixture of hydrocarbon by GLC. These hydrocarbons are the same as
found in A1-'2o• Fr acti one ~1-Bao eluted w1 t h petr olium ether ( 40-60•) and
benzene (h1) gave a solid m.p. 95°0. IR (Fig. 7) absorption at -1 :5600-3400 c• shows the presence of hydro;ql c out ainin1 c om.pound e
and GLC revealed that it is a mixture of ee-.eral components. It was
then rechromatographed over .Argentized eilica gel as deseri 'bed in
Table-7.

25
Jraotione Ja6-B105 eluted with benzene aDd pet~olium ether
(40-60•' was found to be a single component b7 TLC and GLO (F1g.1).
The product obtained !rom these :t'racti ons after oryetallieati on
from petrolium ether and benzene (1s3) yielded a white crystalline
solid m.p. 102• [oc.]:s. 9o(CBC1') Jt 452. !hie compound 4td n'"
give any colouration with tetranitromethane aor it ga'Ye any teet
far eter ols and tri-terpene.
Infrared spectrum (Nujol)(Fig. 5) of the compound showed
absorption at 3210 cm-1 (..OH stretching due to polymeric association), . .
-1 -1 -1 1700 om (ac-0 oaxbonyl ), 1455 ca ( -c~ bending), 1375 CJl ( -c-cH3 bending), 1066 cpt1 (·C-0 stretching of secondary alcOhol), doublet
727 and 718 cm-1 •( OH2 )0 bending ( n > 4) vi bl ati on due to 1tr sight
chain methylene groupe.
Anal. Found. o, 79-70,CJ
B.echromatography of the fraction J21 -B80 over a:rgentized 1ilica gel 1
Fractions B21 -B80 (140 mg) was dissolved in 1 ml o! lenzene and
adeatbed i~ .3 gms of argentized silica gel and dried in a 'Yacuum
desiccator. The adscr'bed material was cbromatographed O"fe! a.J:geatized
silica gel (12 g) and eluted with different solvents (!able-7) aid
each fraction collected in 10 m1 portions.

'f!BLE - I
26
TLC Report
One c ompoaen"t
Two components in
equal amoun"t
Two components. one in trace amoun-t
One c ornponent
Two components, one
1 n trace amount
One component
Fractions c5-c10 were found to be a single component by TLC.
However GLC (Fig. 1) revealed that it was a mixture of 5 components
w1. th carbon content ranging from c27-c 31 1 odd number of oar bon at 0118
prevailiag, only minute traces of compound w1 th e'YeD number were
present. The mixture of these compounds did not give, any coloutati on
with tetra-nitromethane and did not give a1JY test for sterols or

27
-1 triterpenes. IR spect~a showed absorption at 3510 oa thereby
indicating the presence or eydroxyl group in the compounds. Jlo.reover
one of the compounds was seen to be identical with hentria-oontan-. .
16-ol17 in the GLC (Fig. 1) report. Hentriaoontan-16-ol was prepared
synthetically from Palmitone18 by sodium borobydride reduetion, Other
.tour components were supposed to be alkanols ot this k1D4 as it has
been :t'ound that certain natural plant generally contains a symmetri
cal class ot compounds.
Synthesis of di-n-tet~adecll ketone (Palmitoae) 1
Palmi tone was prepared by the method o! Kippings ( 1890). 2 gas
of pentadeoylic acid (Dlp 52°' was taken in a 100 ml. beaker. fhe
beaker was kept in a metal bath. The temperature of the bath was
kept at 205-210°. Phosphorous pentaoxide (;g) was added to it
slowly in small portions with constant stirring !oz about 5 minutes.
The molten mass was then poured into water to decompose the excess of
phosphorous pentaoxide. An excess of sodium hydroxide solution was
then added to 1 t to convert the excess pentadeoyl acid to soap and the . temperature of the mixture was then raised to its boiliDI pOint. On
cooling the product sol1d~t.ie4 at the surface as a waxy cake. It was
then taken out and washed well w1 th water. The product ( 1.5 g) waa
dried and then dissolved in 2 ml of petroleua ether {40-60•)-Benzene
( 111) and ads~rbed in neutral Br OokllaDD alullina (5 g) and dried iD
the usual wq. The adsorbed material wae ohrom.atographed oYer neutral

Brockmann alumina (20 g' and eluted with dit.terent sol'YeDts
(Table-8) end each :t:raoti on collected in 10 ml portions.
TABLE •' 8
I
Z8
Eluent a Fractions Wt. of the leaa:rka materials
Petrolium Ethe:r a Benzene , .. , 0.18 g Brown seal solid
( 1011'
Petrolium Ether a Benzene 4-8
( 4a1)
Petroleum ether t Benzene 9-16
(1a1)
Benzene
Benzene a Chloroform
(1t1)
Chloroform
17•22
23-30
,,_,, Chloro:tormamethanol { 111) 3•-'a Methanol 39-42
0.21 g
0.95 g
0.15 g
0.06 g
111
trace
o.oa
'lhit e solid, mp 82•
Brown semi solid
Gummy
Br own g\tll'JIY
The product obtained :trom the .traoti on 9-16 were llixed together
and crystallised from Benzene. TUl gaYe a single apo~.
IR 1705 om-1, mp. 82-8,•.
Anal. Found I C, 82 .65~ J H, 1:5 ·7~ J
Calc. tor c31B62o, c, 82.66- 1 H, 13.77~ 1

Z9
Preparation of Hentriaoontan-16-ol 1
Palmitone (80 mg) was suspended in 10 ml of methanol and to
this sodium borohydride ( 100 mg) was added. Tbe mixture was kept
!or 15 hours at room temperature. Hydrochloric acid (0.7 Ill) and
water (10 ml) was then added to it and the reaction llixtYre was
warmed on the steam bath !or 10 minut ee and then dllut ed w1 th
water. The solid was collected by filtration and washed with water,
dried, dissolved ~n ~nimum volume of benzene and ads~ bed in 5 gms.
o:t Brookma:nn alumina. The adsorbed material was chromatogre.phed over
a column o:t Brockmann alumina (15 g) and eluted With different
solvents (Table-9) and each :traction collected in 10 ml portions
TABLE ... 2
Eluent a Jtaoti one Wt .o:t the Reaarke residue
Petroleum ethe% (40-60°) 1-4 5mg gua
Petroleum ethersBenzene 5-12 50- ·White solid ( :511)
Petroleum ethersBenzene 1,·16 10 Ill Brown solid (1a1)
Benzene 17-20 111
:Ben zene• Ether (1t1) 21-23 l'il
Ether 24-25 Nil
Chloroform 26-28 trace
Methanol 29-30 trace

The fraction a 5-12 was recrystallised from petroleum ether 1
Benzene ( 1t1' as white plates and showed a single spot on T LC on
silica gel G. IR 3400-3200 cm-1 mp. 78•
Aaal. Found. 1 O, 82.'5" 1 B, 14.18" I
Calc. f~ o31H64o, c, 82·3~ 1 H, 14.15~ 1
P:eparati on of OXime of the Rydrog ketone a
HYdroxy ketone (50 mg), hydroxylamine hydrochloride (100 D!),
ethanol (3 ml) and pyridine (0.3 ml) were heated on a water bath
under renux condition for one hour. The alcohol was e'Yaparated,
diluted with water, eol>led in ice and filtered. !he residue wae
washed with water and d:ried and then c:rystellieed troa 80" ethanol
yield 30 mg,mp. 106°.
Anal. Found 1 1, ~·J
Calc. for o,oft61N02 I I, 3·1" C
Preparation of Acetate of the PYdr oxy ketone t
The hydroxy ketone (50 mg' was taken With aceti o anhydride ( 0.8
ml) and two dxope of py:idine ·and the mixture was heated 01 water bath
foz one hour. Working up in the usual way the product was cxystal:1eed
:!% om a mixture of chloro:torm and ethanol. Yield. 40 mg
mp. ga• [ae J ~o + .,. ( oHol3'
Anal. Pound 1 c, 79·70faH, 12.51" J
Calc. far e32! 62o, t 0, 79.76~ 1 H, 12.55" 1

31
Reduction of the &drozy ketone (I) to (II) bx H•ans-M:l.nln Reduction 1
100 mg of the hydroxyke~one (I), 200 mg of KOH, 1 Dll of diethylene
glycol and 1 ml of 85~ hydraziDe hydrate were refluxed for one hour.
Water was then drained of! from the condenser and the te~~perature
was allowed to rise to 200•. Refiuxing was continued for .2 hours more.
The reaction mixture was cooled, diluted with water and poured slowly
into 6lf HCl. Working up in the usual way the product obtained was
crystallised from petroleum ether-Benzene (1a1). GLC gave a single . . .
peek. Yield 80 mg, mp. 98°, IR 3220 cm-1 •
Anal. Found 1 c, 82 ·14~J H, 14 ·09" J
Calc. for c,o~~620 a C, 82·19" J H, 14.12" 1
Chromium trioxide OXidation of compound (II} to compoual (III.) 1
compound II (50 mg) was taken in glacial acetic acid ( 10 Til) am
wae heated to refiu:x and then ch:romium trioxide (50 11g) 4issolved in
acetic acid (85", 5 ml' was added gradually over a period of one hour •.
:Re!luxing was oont .inued for another one houx and the prOduct was then
diluted With water. The precipitate was filtered sad washed well with
water and dried. It was crystallised from Benzene as plates. GLC ga'fe
a single peak. Yield 35 mg, mp. 92°, IR 1700 ca - 1•
Anal. Found a c, 82.3~ J H, 13·7~ J
Calc. for c3oH60o a c, 82.56", H, 13·7'",

32
1reatmen't ot the Ohlorotorm Ext:raot 1
letpyl amine from Chloroform extract 1
The chloroform extract on removal of the solvent and proper
drying yielded a deep green· solid ( 30 g) which posses !isl!ly odour •
The solid ( 10 g) obtained from chloroform -extract wae heated
with 5" sodium hydroxide (100 ml) in a round bottom flask. !he
issuing gas was absorbed into 21 hydrochloric acid. When the issuing
of the gas is complete, the acid was completely removed ua4er reduced
pressure. The residue was then crystallised fl011 water ae needles.
Yield 0.3 g mp. 224°
Anal. :round a 1, 20.6~ 1
CH6J01 requires N, 20.74" 1
Pzeparation o! 2a4-dinitromet&l aniline derivative a
The hydrochloride (0.2 g) obtained from the above experiment is
taken in 5 co of ethaDOl and 5 oo ethanolic eoluti on of 2a4
dinitrochlorobenzene (10}C) was added and warmed a little on a water
bath. Solid obtained on cooling was filtered, washed witlt. a little
water and reoryetallieed :f'rom ethanol as needles, yield 0.1 g,mp.
174-75°.
Anal. :round 1 1, 21 .45" J
Calc. for C-r!:5H704 t ll, 21 .32~ 1

The solid {20 g) obtained fro the chloroform extract was
dissolved in minimum volume of ohl roform and adsorbed in 25 gaa
33
of neutral Brockmann alumina and ied. The adsorbed material was
chromatographed over a column of neutral alumina (80 g) 8M eluted
with different solvents (Table 10) and each fraction oolleeted ia
50 m1 portions.
TABLE - 10
Eluente Fractions Wt, Of the Remarks :residues
Petroleum ether 1 111
(40-60°) 2 111
'3 0.15 g White gua
4 0.0'3 g " 5 111
Petroleum ether 1 6 trace Yellow gum
Benzene ( 1t1)
7 0.24 tt
8 o.og • 9 0.06 White gua
.10 trace • 11 111

TABLE-10 (Contd.)
Eluent s Fractions Wt. of the Remark• residues
Benzene 12 o.og colourless gum
13 0,15 col our lees gum + White oryst ale
14 0.45 White crystals
15 0.24 .. 16 0.15 .. 17 0.12 " 18 o.Og " 19 t:raoe
Benzene s ether ( 1s1) 20 111
21 111
Ether 22 111
Ether 1 Methanol(90a1 0) 23-24 111
25 111
Methanol 26-27 trace Brownish gum
Ieolati on of @ -si tost er ol 1
Fraction nos. 14-18 were mixed together. It responded to
Li ebe:rmann Bur chard test indicating the presence Of et er ol aDd on
crystallisation from methanol afforded colourless needles (0.8 g)
34
· 2.5 nuJol o! @-eitoseterol mp. 135-136• [oe]l>-'.55° (CHcl3),IR ~"'o.x3400 ca·1•
No depression in melting point was observed on admixture w1 th

35
eD authentic specimen of @-sitosterol
Anal. Found 1 c, 83.85~ ' B, 12.26~ '
Oalc • tor o24H; 00 1 C, 8,.99~ J H, 12.15" J
Acetzlation of <!-si toaterol 1
~-sitosterol (50 mg) in pyridine (5 Ill) was heat ... oD a
steam 'bath atte't addition of acetic anhydride (0.5 ml) fer 2 brs.
Working up in the usual way the product was o:ryetellieed f:roa
ohl or of orm-11et han ol ( 111 ) yielded ~ •a1 t ost er ol ac et at • 1 n fiat . . . 2S
needles (35 mg), mp. 125-26•, [o<] I> -36° (oHol3)
Anal. Found 1 o, 81 .4~ J H, 11 .. 4~ ;
Calc. far c31 ~~; 2o2 t c, 81.5~ 1 H, 11.48~ ;
Ben zQ1lati on Of @ -ai t oat er ol t
@-sitosterol (50 mg) was dissolved in pyridine (1.2 11.1) and
benzoyl chloride (0.25 ml) was added to it. The result inc ldxture
was stoppered and left over night. Working up in the usual wq the
prOduct on crystallisation trom ethanol afforded a benzoate. 2.5
mp. 142-143°, {~]I>_,,, (ohl~ro:tora)
Anal• :Pound a O, 8., .36~ f B, 10;.59" I
calc .• tO% o,6Bs 4o2 a o, a., ·39~ , H, 10 ·4~ '
Treatment o:t Ethanolio Extract ·•
The ethanolic extract on removal of the solvent and p:rope:r
drying yielded a dark browa gummy substance (80 g). It ga'Ye auoh
troth when a small portion of it was shaken with water indicating

36
the presence of a saponin in the extract. '.rhe saponin ia auch
hygroscopic. f.he solid waa repea:tedly extracted with Butaaol. Butanol
was distilled off under. reduced pressure when the oru4e eaponin was
obtained as a brown solid mass (60 g).
Bldrollsis of the Ethanolic EXtract t
'.rhe ethanolio extract (55 g) was dissol~ed in 6~ ethanolio
bydtoohlorio acid ('380 ml) in a 500 m1 round aottom flux aad it
was refluxed for 16 hours. After the hydrolysis is oyer alcohol
was distilled off on a water bath keeping the volume of the distilling . '
product same on .frequent add.ition of water. It was then .filtered,
washed well W1 th water till free· of acid and then the resitue was
dried. The dried resid'ae (30 g) was then dissolved in 20 ml o.f
ethanol and mixed up w1 th Brockmann alumina ( 30 g) and dried. !rhe
adsorbed aaterial was ch:tomatographed over a column ot neu-tral alumina
(600 g) and eluted with different eolYents (Ta~le-11) aDi eaoh
fraction collected in 100 ml portions.
The .fraction numbers 16-25 obtained from the above ohromatograpey
were mixed together. It was brown in cOlour. It was charcoalieed by
dissolving in methanol and crystallised from this eolveat. A. faint
brown colour still remained in the product and T~ ga"fe the eXistence
of th:zee components in this product With the pr'edominanoe of one and
the other two are in minor quantities. The material (3.2 g) was then
dissolved in 5 ml of ethanol and mixed up w1 th Brockmann alullina
(10 g). It was then rechromatographed on a column of neu:tral Brockmann
alumina ( 100 g). Elution being carried out with ohl oro form aad methanol
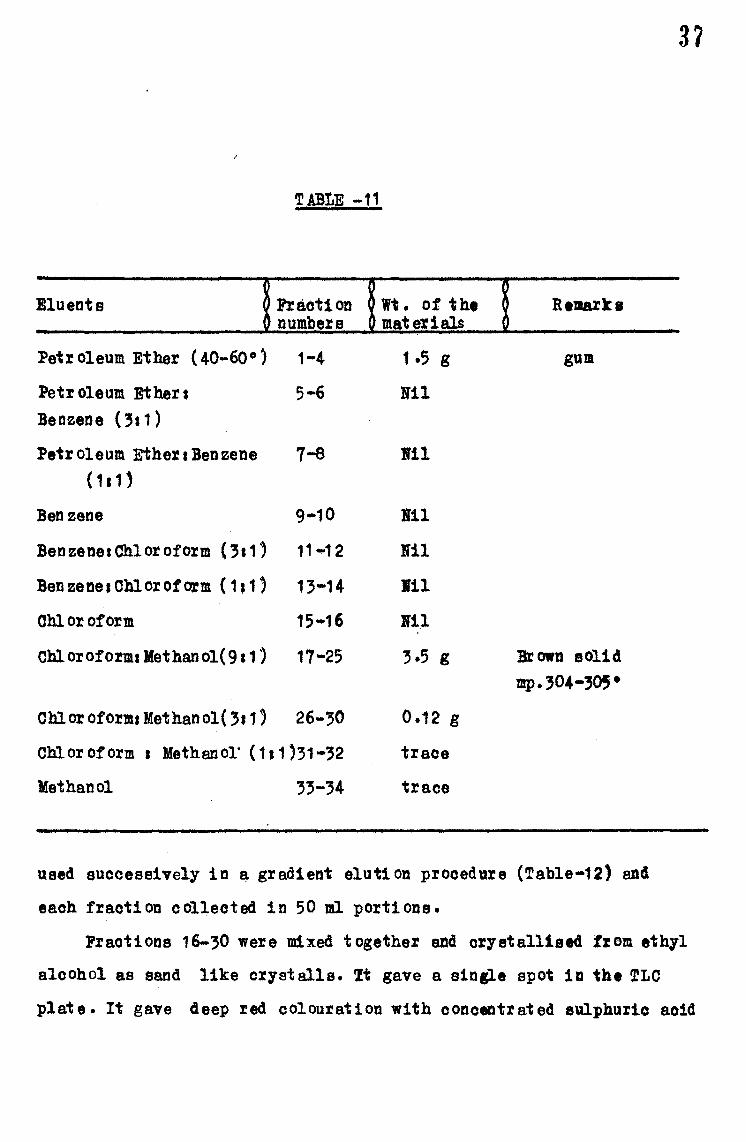
TABLE -11
Eluents Fraction it. of the Re11ark1 numbers materials
Petroleum Ether (40-60°' 1-4 1·5 g gum
Petroleum ~her• 5-6 Nil Beozene ( 3a 1)
Petroleum Ether a Benzene 7-8 111
(1•1)
Benzene 9•10 111
Benzene a Ob.l or oform ( 3a1' 11•12 Nil
Ben zenea Chloroform ( 1a 1) 13•14 111
Chloroform 15-16 Ni:l.
Chloroforma Methanol(9a1) 17-25 3·5 g Brown solid mp. 30-4-,05.
Ohl.or oforma Methanol( 3J 1' 26-30 0.12 g
Cbl or of orm 1 Methanor (1a1)31-'32 trace
Methanol '33-34 trace
used successively in ~ gradient elution procedure (Table•12) and
each fraction collected in 50 ml portions.
3?
Fractions 16-30 were mixed together and crystallised from ethyl
alcohol as sand like cryatalls. 'It gave a sin&].e spot in the TLO
plate. It gave deep red colouration with oonceatr at ed eulpb.uric acid

38
TABLE - 12
Eluente Fraction Wt. o! the Reaarks numbers residues
Cbl. or otor m 1-2 10 mg White gua
Chl or o:f'orma Methanol 3-5 30 mg White eol14 mp.180•
(98t2)
-do- 6-9 20 mg White solid mp.165•
Ohl or ot or ma Methanol 10-15 15 JJg White solid mp ·320-25•
(95 15'
-do- 16-30 1·5 g White solid mp.330-31•
Ohl or ofornu let han ol 31-34 20 Jig Brown soli4 11p .225-28•
(911'
Chloro!ormaMethanol 35-40 25 Jig Jr on ••mi.-solid
< ''1' Ohl oro tor taaMet han ol 41-42 trace
( h 1'
Methanol 43-44 111
and gave pink to violet oolouration in Id.eberaann Burchard test, .2. !5.
mp. 332-33° (DL)D + 48~(Et0R}
Anal. Found a 0~ 71.23~ J H, 9.91~ J
Calc. for a301fs0o6 a c, 71.11~1 H, 9·95",
Preparation o! marsileageninA hexaacetate a
MarsileageninA (300 mg) was suspended in acetic anhydride (6 ml)
and pyridine (3 ml) was added to it. The mixture was heate4 on a wate:r:

39
bath for 4 hrs. under reflux condition. After the reaotiOI was over
the mixture was dropped into ioe-cold water. The solid product was
washed well w1 th water. The solid was then extracted with ether and
the etherial layer was washed successively with dilute sodium bicar
bonate solution and then with water. The etherial solution was then
dried by adding anhydrous sodium sulphate. Etherial solution was
separated from sodium sulphate and. ether was removed. The residue
(2.45 g) was dissolved again in dry ether (5 ml) and· then mixed llp
with Brockmann alumina (10 g). It was then chromatographed Ol'er a
column of Brockmann alumina (50 g). Elution was carried out wiih
petroleum ether (40-60° ), Benzene, chloro!ora and methanol uaed
successively in a gradient elution procedure (Table-13). Each
fraction of the eluents contains 75 m1 of solvent.
TABLE - 1' Eluents Fraction Wt. o! the Remarks
numbers residues
Petroleum ether (40-60o) 1-2 trace gum
Petroleum ether sBen zene 3-4 lil
( 311'
Petroleum ether a Benzene 5-6 llil
(1a1)
Benzene 7-8 111
Benzene 1 Chloroform 9-10 Nil (9515'
Benzene' Chlor o.form ( 9 a1) 11-12 :111

fABLE - 13 ( contd.)
Eluents Fraction 'It. of the :Rem.a:rks numbers residues
Benr;eneaOhloro:torm ( 3&1) 13-16 50 mg White aol14 :ap .2,5•
-do- 17-18 111
Benzene a Chloroform 19-28 210 mg White eolid mp.284•
( 111)
Chloroform 29-30 Nil
Chl or of orm 1 Methanol 31-32 Nil
(9a1')
Chloroform a Methanol 33-34 lfil
( 311'
Ohl or of orm 1 Methanol 35-36 trace Brown o111 residue
{1J1)
Methanol 37-'38 111
Thi fractions obtained !rom 19-28 was crystallised .trom aethanol
as needle shaped crystals. It gave single spot on Ttc plate. mp.285•,
lR 1740 (br,, 1242 (br) cm·1 (acetate carbonyl) and 830 c•-1 (tri-7.50
substitute~ double bond~ [c,L]J> +_35~5o (CHcl3).·
Anal. Found 1 c, 66.;9~ J H, 8.26~ 1
Oalo. for o42a62o12 a Ot 66.4~; H, 8.24~ 1
Kydrolzeis of mareileageninA hexaacetate 1
MareileageninA hexaaoetate (50 mg} was taken in 5~ methanolio
XOH (5 m1' and the mixture was kept under refiux on a water bath for

1rsileagenin A He xaacetate
oAc
--oAc
oAc
oAc
--OAC
OAC
AcO __
AcO --
AcO
1 oAc
. -- oAc
, CH20AC
l oAc
I R i) max 1735, 1670 em-'
UV :\max 239 nm
'oAc
UV A max 241 ,250,259 nm
+ AcO __
A cO
RDA >
a
b
mje 450
+ '-':::: c Hz
mje :307
+ ~oAc
.
~--OAc
OAC

41
one hour. Then the product was dropped in water and eolid obtained
wae filtered and washed well with water till it wae oomple'tely free
of alkali • It was then orystalli sed !:rom ethanol when sand like
crystals were obtained. It gave single spot on TUJ. axed melting
point with previous sample of marsileageninA remained undepressed ,_s·
mp • ,,,. • [ oC J » .,. 48 ° (EtOH)
, Anal. Pound 1 Ot 71 ·2~ ' I, 9·93- I
oalc. for c3oa50o6 • o, 71.11- 1 H, 9·95~,
Preparation of c£·,@ unsaturated ketone from aareileyenia! hexaaoetate t
The hexaacetate (50 mg} was taken in glacial aoetic acid ( 10 al)
and was heated to reflux and then chrOilium trioxide (50 mg) dissolved
in acetic acid (85", 5 ml) was added gradually over a period of one
hour. Re:fluxing was continued tor another one hour and the pr educt
was then diluted wi'th water. The precipitate was filtered and waehecl
well with water and dried. The solid was clissolvecl in 0.5 ml of methanol
and mixed up with alumina ( 2 g) and ohr ome.t ogr aphed over Br octunn
alumina (10 g' eluating euooess1Yely with different solvents and eaoh
fraction oolleoted in 10 ml por'ti.ons (!able-14) •
Fractions 10-18 were orystallieed t.rom methanol as colourless 2-So
tleedles and was found to be a single eompound by TLC mp. 287• [c£]D + 20•
UV (EtOH) 239 nm (log f 4·12) \. nt.tJol 1 IR ')) 1735, 1240 011- (acetate carboayl>
!'\'lo-x
1670 oa-1 ( ae, ~ uosaturated lcetoae)
830 cm-1 (tri-subetituted dou~le boad)
A.nal. Found 1 o, 65 .28" 1 H, 7. 79"'
Calc. tor o42860o13 1 o, 65.2~ J H, 7·8~ 1

100
so --
" ~ 60 '--l!j '-> <: ;:s 40 1--~ {/)
<: ~ 20 1'-
3 '
WAV£LENGT!-i (MICRONS) 4 5 6 7 8 9 10 11
I
0 v v
\J
4000 3000 2000 1500
Fig.t5.~:::.'t:i2 ,13: re
1200 CM- 1
1000 900
diene Marsileagentn A hexaacetate
12 13 14 15
.-~-
800 700

42
TABLE - 14
I
Eluents Fraction Wt. of the Reaarks
numbers Residues
Petr oleu~ ether (40-60•) 1-2 l{il
Petroleum ether a Benzene 3-4 lil
( 3a1)
Petroleum ether:Benzene 5-6 Nil
( 1 11)
Benzene 7-9 tract
Ben zenea Ether ( 4a1' 10-18 35 mg fbi te etlid
Ben zen ea Ether ( 1 a1) 19-20. 5. -do-
Ether 21-22 111
Ethers Chloroform. ( 111 ) 23-24 5mg Brown semi solid
Chloroform 25-27 trace Brown oily residue
Chl or otorma Methanol ( 111' 28-30 trace -do-
Methanol 31-32 111
Selenium dioxide oxidation of MarsileageninA hexaacetate 1
Marsileagen1nA hexaacetate (50 mg) was taken in glacial acetic
acid (3 ml) and mixed up with freshly subliaed selenium dioxide (50 mg)
The solution was then refluxed for 4 hours. It was then filtered hot
and the filtrate was diluted with water when a solid product wa1
eepar at ed • It was filtered and washed well w1 th water and dried • The

43
sOlid was dissolved in 0.5 m1 ot methanol and mixed up w11)1. Brookmana
alumina (2 g). It was then ohromatographed OYer a colum.a •t Brockmann
alumina ( 10 g) and eluted with different eolyente (Table-15). Each
fraction contains 10 ml of solvents.
tABLE - 15
Eluent a
Petroleum ether ( 40-60°)
Petroleum ether 1 Benzene ( 1a 3)
Petroleum ether 1 Benzene ( 111)
Benzene
Benzene a Ether ( 4s1'
Benzene 1 Ether (1•1'
Ether
Ether s Ohl or oform ( 311 )
Ether 1 Chloroform ( 111)
Chloroform
Chloroform 1 Methanol (3a1'
Chloroform s Methanol ( 1a1)
Methanol
Fraction Wt. ot the Remarke 1 numbers residue •
1-2
3-4
5-8
9-16
17-20
21-22
23-24
25-26
27-28
29-30
31-32
33-34
35-36
111
Nil
5mg
trace
111
lil
lfil
Nil
111
.11
Nil
Wh1 te gum+
crystal
White crystal
-do-
gua
Fractions 9-16 were mixed together and orystallieed from •ethanol
as colourless needle shaped crystals. It was :round to be a single

'O'V (EtOH) 241 nm (log E
250 nm (log f
259 nm (logE
Anal. Found t c, 66.57~ 1 R, 7·~ J
Oalc. for c4iB60o12 1 o, 66.44~ 1 H, 7·99- 1
Acetylation of Ma.rsileageninA ,at room temperature a
44
MareileageninA (50 ~) was treated With acetic anhydride and
two drops of pyridine. The mixture w8 s kept for 24 hours at rooa
temperature. The mixture was poured into water and t~e solid which
separated, was worked up in the usual wq and was or7etallieed two
times from methanol when a single spot was obtained 111 TUl. mp. 2.5
285• ~ [a<:) D + 35 ·5°
No dep~eeeion in melting pOint was observed on admixture w1 th
marsileageninA hexaacetate prepared earlier. . .
Anal. Found a C, 66 .45~ J H, 8 .22~ J
Oalo. for o42ft62o12 a c, 66.46~ J H, 8.24~1
ll:eparation of mono-trityl derivative o! M'a:reileyeninA 1
KareileageninA (50 mg' was dissolved in pyridine (0.5 ml) and
tri tyl chloride ( 150 mg) was added to 1 t. !he mixture was retluxed
on a water bath tor :5 hours. It was dropped in c•ld wa'ter, filtered
and washed. The residue was crystallised !roa etayl alcohol. It gaYe
single spot on TIC plate arp. 205 -7•

45
Anal. Found 1 o, 78·5~ J H, 8.51~ 1
Calc. for c49H6406 I c, 78.6~' B, 8.55~ •
Estimation of number of double bonds in marAt.leaseninA pexaaoetate 1
Marei~eageninA hexaaoetate (50 mg) was dissolved in 10 ml
chloroform. To this 5 ml of perbenzoic acid solution dissolved in
chloroform was added to it. A blank s.olution was .similarly prepared
by taking 10 ml of chloroform and 5 ml of perbenzOic acid in a flask •
• Both the !laske were shaken well and kept at 0°C in the refrigerator
for 15 days. Then the flask containing the mareileageninA hexaacetate
was taken out and to this solution sodium iodide ( 1 gm in 25 ml of water
and acetic acid (2 m1 ' were added. The solution was ehaken and kept
s:toppered in the dark for 5 minutes. The liberated iodine was titrated
with standard (1/100) sodium thiosulphate solution. The blank solution
was similarly titrated fOllOWing the same procedure. !he result of the
titrations is given below.
Amount of MarsileageninA hexaacetate = 50 mg
strength of sOdium tbiosulphate solution= (N/100)
Volume of sodium thi oeulphate solution required to titrate the
liberated iodine by unreacted per benzoic acid in the flask containing
mareileageninA hexaacetate = 87·3 ml.
Volume of sodium thiosulphate solution required to 'titrate the
liberated iodine by per benzOic acid in the blank flask:100 111.
Amount of perbenzoic acid oonsume6 by 50 mg of mazaileageninA
hexaaoetate ::: 100-87 ·3 ::: 12.7 ml of (1/100) sodium thiosulphate
solution;; 12.7 ml. or (N/100) perbenzoio acid solution.

46
Calculated amount of (N/100) perbenzo:l.o acid solution requi:t:ed
to r eaot w1 th one double bond in 50 mg ot mar eileageni nJ. hexaaoet at e•
13.2 ml
Found t 12.7 ml ot (1/100) per benzOic acid solutio•.
The value indicates the presence of one double bond in
me.t"SileageninA.
Eetimation of number of o<: -glycol system in maJa±leageninA s
Me.rsileageninA (50 mg' was dissolved in methanol ( 10 ml) and
periodic ac~d solution (3 ml, 0.5 K aqueous m.ethanolic eolution) as
added to it~ The mixture was shaken and kept for 24 hour• at r ooa
temperature. A blank solution was similarly kept by takiag aethanol
(10 ml' and periodic acid solution (3 ml, 0.5 11 aqueous aethanolio
solution). After 24 hours the solution containing marsileageninA
was mixed up with an excess of sodium arsenite solution (5 ml of 0.21). I.
Sodium bicarbonate ( 1 g' and water ( 25 ml) in presence of few crystals
of :potassium iodide were added and the fiask was .haken and kept for
fifteen minutes. !he excess of arsenite was back titrate4 with 0.01
(N) iodine solution using starch as indioato:. Blank solution was
similarly titrated. From the difference in titre values ),etween the
blank and compound. reaction mixture the periodate uptake was
calculated. The result o! the ti trations is given below.
Amount of marsileageninA • 50 mg
strength of iodine sol uti on = (ll/100)
Volume of iodine solution required to titrate e:xoeae of sodiu11
arsenite in the solution containing maxsileageninA = 15,.4 ml.

41
Volume of iodine solution required to titrate exeeaa sodiua
arsenite in .the blal'lk flask:: 110.4 ml
Difference in the volume of iodine solution • 15J.4•110.4c
43.0 ml
Calculated difference in the volume of iodine solution :f'o: oae
oe -glycol system = 19.7 ml.
Calculated difference in the volume of iodint solution !o:r two ce..
glycol syet em = 39 .4 ml.
!he above result shOWed the presence Of two d:. -glycol system
in mareileageninA.
Hydrolysis of,the saponin mixture from ethanolic extract aDd
identification of suga:re t
The crude saponin (5 g' obtained from ethanolic extract was
refluxed with 5~ methanolic sulphuric acid (50 ml) on a eteam bath
for 10 hours. Methanol was evaporated off in the usual wq by repeated
addition of water 1 keeping the acid strength more or leal constant,
It was then filtered and the filtrate was nutrallised by llhaldng
successively with barium carbonate to pH 5.5-6.0 using uniTersal pH
indicator paper and filtered. The f~ltrate was deionieed w1 th Amber lite
m-120 (H) and Amberli te IR-45 ( OH'. The deiontsed filtrate was
reduced to a smaller volume (4 ml} under reduced pressure and
lypholieed. The syrup thus obtained was exald.ned for sugars by
descending method of paper chromatography with the apparatus uae4
by Hough & Jones on Whatman Jo. 1 chromatography papez. !he follOWing
solvent systems (v/v) were used for the ohromat ography

A62 = Ethyl aoetateaPyridineaWater; (8a2s1 h
B = 1!,-ButanolaPyridinetWatera (6a4a:5);
o63 = s.-Butan ol' acetic aoidtWateJq (4t h5 h n64 = .!!-Butanolabenzeneapyridineawater;(5a1a3z3h
(R:r values wexe measured using solvent B)
48
!he solvent was allowed to run :tor 18 b%s. The paper was then
dried in air and sprayed with saturated solution of anil1•• oxalate. 65
The paper was then kept at 110-20• in a hot air oven !or 10 minutes.
The sugars present were identified to_be D-gluooee (R! 0.19),
D-galaotose (R:r 0.16), D-Iyloee (R:r 0.23}, D-arabinose (!.t 0.29),
L-rhamn~se (R:r 0.39) by direct ~omparison with respective authentic
samples.

49
REFERENCES
1. IC. 1:. Gupta, Mareilea, Botanical Monograph lo. 2, pw'bliehed
by CSIR, New Delhi, India. ( 1962).
2. IC. P. Biewae and A. K41 Ghosh, Bha.ratya Bonouahadi (Jengali),
Calcutta University, ,l, 622 (1952).
3. SUpplement to Glossary of Indian Medicinal Plants b7 Chopra
and Verma, publication & Information Dixeotorate (CSIR),
New Delhi, P• 65 (1969).
4• The Wealth of India, Council of Scientific & Industrial
Research, lew Delhi, Vol. VI ( L-M), p 306. . .
5 • M. r.. Chatterjee 4: s. Pal, Bulletin of the Calcutta School
of Tropical Medicine, Vol. IX, lfo. 3, p. 123, Jul.J (1961 ).
6. A. Chatterjee, c. P. Dutta, B. Choudhury, P. K. Dlf, C. D. Dey,
C. Chatterjee and s. R. Mukherjee, Science & Cul tu:e, ~.
619 (1963). . . .
1. P. K. Dey, Mira Chatterjee and Chitralekha Chatterjee.
Nattttewisaenschaften, 22, 693 (1963). -8. Chitralekha Chatterjee, P. x. Dey aad o. D. Dey, Xaturwissens
ohaften, 21• 411 {1964).
9. Ohitralekha. Chatterjee and p. I. Dey, laturwieaensolla!ten
il· 466 ( 1964).
10. c. Datta and P. K. 'Dey, India J. Exp. B1.ol • .2_, '6 (1967).
11 • 0. s. Rukmani Iyer, G. D. Shah and A· Ohatt er j ee, Indian J. ot
Ohem., ll• 281 (1974).

12· D. Chakravarti and JJ. B. Debnath, J. Indian Chem Soo,, iJ.,
260 (1974}.
14. L. J. Morris, Chem. and Ind., 1239 ( 1962).
15. B. Devries, Chem. and Ind., 1049 (1962).
16. Von W. stocklin,Helv. Chim. Acta, 2_g, 365 (1969)•
17. · s. Wakayama and s. Namba, Nippon Kagaku Zasshi, iQ, 1160 (1969),
18. H. _J. Obannon ~nd A. c. Chibanall,- Biochem. J. (London),
_u, 168 { 1929'. .
.. 19. N. Hayashi and H. Komae, J. Indian Chem. Soc., J!, 218 (1971)
20. v. Wollrab, pbyt oohem., ~. 623 ( 1969).
21. Mass spectrometry for Organic Chemists, by R.A.W. Johnstone,
Cambridge University Press, P. 83 (1972). . . .
22. H. Bud nkiewicz, C. Djeraes1 and D. M. Williams, " Interpret a--
tion of Mass Spectra of Organic Compou.n4e •, Holden. DeJ, Inc.,
san Francisco ( 1965).
23. s. B. Mahato, 1. p. Sahu, A. larayanswami, R. N. Chakravarti
and Debi Chakrabarti, J. Ind. Chem. Soc. ll• 626 ( 1975).
24. B. st oianova-Ivanova and P. Hadjieva, Pbyt ochem.., !• 1549 ( 1969) •
25. J. H. Bey on, G. R. Lester, K. A. saunders and A· E· 'lilliaae,
Tran~. Faradat Soo., ll• 1259 ( 1961 ).
26. H~ang-Minl?n,_J. Amer._Chem. Soc., !!• 2487 (1946).
27• I. Yoeioka, A. Matsuda, K. Im.ait T. Nishimura and I. Kitagawa.
Chem. Pharm. Bull .(Tokyo) .12. (6), 1200 (1971).
28. R. AdaDlS, Organic synthesis, Vol. 1, P• 4'31
29. P. Fleury and J. Lange, J. Pharm. Chim. Jl(S), 107 (19,'3)·
\

51
30. !. K. Barua, s. P. Dutta a: B. c. Das, Tetrahedron, ,gj,C5), 1113,
(1968).
31. o. P. Arya and R. c. cookson, J ..• Chem. soc., 972 ( 1957).
'32· R. Budziarek, w" Mans on and F. s. Spring, J. Chem.. Soc.,
3336 ( 1951 '.
c. w. Picard and.F, s. Spri·ng, J. Ohem. soc •• 1196 ( 1940).
J. M. Beaton and F. s. Spring, J. Chem. Soc. 2417 (1956).
35. J. •• Beaton and F. s. Spring, J. Chem. soc., 3126 (1955)
36. F. Aauria, Gazz. Ohim. Ita.l., 89, 685 (1~59). . r
37. L. Ruzicka, G. Muller and H. Schellenberg, Helv. Ohia. Aota,
~. 758 ( _1939 '.
38. G. G •. Allan: at'J~ C, S. ChOpra, Phy~ ochem1stry, .12• 13'3 ( 1971) •
39. G. G. Allan, J.D. Johnston and F. s. Spring, J. Ohea. soc.,
1546 (1954).
40. o. Djerassi and c. M. Foltz, J. Amer. Chem. Soc., 1,!, 4085 (1954).
41. E.· s. EWen and F. s. Spring, J, Chem. Soo., 1196 (1940).
42· G. G. Allan and F. s. Spring, J. Ohem. Soc., 2125 (1955).
43. G, G. Allan, F. s. Spring, R. stevenson and w. s. straohan,
J. Chem. Soc., 3371 (1955).
44• G~ G. Allan, M. B. E. Fayez, F. s. Spring and R. Stevenson,
J. Chem. Boo., 456 ·(1956). " "
45. D. H. R. Barton, A. J. Heal and P. J. M'q, J. Chem.. soo.,
935 ( 1957). " " "
46. D. B. R. Barton, H. T. Cheung, p. J. L. Daniele, K. G. Lewis
and J. F. MCGhie, J. Ohem. soc., 5163 (1962).

52
47• . P. Dietrich and 0. Jeger, Helv. Cbim. Acta_u, 711 {1950),
48· D. R. R. Barton and c. J. w. Brooks, 257 (1951).
49. L. Ruzicka, G. Muller and H. sohellenierg, Hely. Cbim. Acta.
'z.g,, 767 (1939).
50. H. Bud zikiewioz, J. M. Wilson and C. Djeraesi, J. Am. Chem •
. soc. ~. 3688 ~ 1963).
51. c. Djerassi, 0• H. Robinson and D. B. fhomas, J. All. Chem. Soc.,
78, 5685 (1956). -51. G. Subba Rao and J. E. Sinsheimer, Chem. comm. 1681 (1968).
53. J. Simonsen and W.o. J. Rose, The Terpenes, Vol •. 2.:• P• 286
Cambridge University PreAs (1951)•
54. M, Shamma, R .. E. Glick and R. 0. Mumma, J. Org. Chea., £1, 4512 (1962').
55. s. G. Errington, D. E. White and M. w. Fuller, Tetrahedron
Lett. 14, 1289 (1'967)· . .... . .
56. I· Yoeioka, T. Nishimura, A. Matsuda and I. Kitagawa
Tetrahed.ron Lett. 7, 6'37 ( 1967) • . . .....
57. I. Yosioka, T. Nishimura, A. Matsuda and I. Kitagawa
Tetrahedron_ Lett, ~. 5979 ( 1966).
58. s .Ito and T •. Ogin o. Tetrahedron Lett., .Jl• 1127 ( 1967) •
59 • L. Ramaohandr a Tow and 0 • s. P% akas Sastry, Indian I. Che:m. . .
1(7), 322 (1963).
60. L~ Ra.m.aohandr a Tow and c. s. Pr ak:ae Sastry, T etr ahedr on ,D( 9),
3837 ( 1967). . .
61. W. Stooklin, E. Weise and T. Reichetein, Helv. Chim.. Acta,
2.2 (55), 474 (1967).

62. J. x. Hamilton and N .. s. fhompeon, J. Am. Ch••· soc., .72.· 6464 ( 1~57).
63. s. M. Part!idge and R. G. Westall, !to. Oaea. J, !&• 238 (1948)
64. G. 0. Aspinall and R. J. Ferrier, J. Chem. Soc. 4188 ( 1957).
65. R. H. Horrocks and G. B. fla.nning, Lancet, Vol. 2561 1042 ( 1949) •










![Multicomponent reactions (MCRs) of acetylenic …...When a-hydroxy carbonyl compounds were used, the intramolecular Wittig reaction of the ylide intermediate afforded oxygen heterocycles.[92-93]](https://static.fdocuments.net/doc/165x107/5f355a90eb6d38496e329123/multicomponent-reactions-mcrs-of-acetylenic-when-a-hydroxy-carbonyl-compounds.jpg)







