
Qavi ppt epileptic syndromes of neonate and infancy (2)
84
EPILEPTIC SYNDROMES OF NEONATE AND INFANCY PRESENTER: DR ABDUL QAVI ( NEUROLOGY)
-
Upload
qavi786 -
Category
Health & Medicine
-
view
31 -
download
2
Transcript of Qavi ppt epileptic syndromes of neonate and infancy (2)

EPILEPTIC SYNDROMES OF NEONATE AND
INFANCY
PRESENTER: DR ABDUL QAVI ( NEUROLOGY)

OUTLINE
NEONATAL EPILEPTIC SEIZURE EPILEPTIC SYNDROMES IN NEONATE FEBRILE SEIZURES EPILEPTIC SYNDROMES OF INFANCY TAKE HOME MASSEGE

Neonatal epileptic seizures
Occurs from birth to the end of the neonatal period. This is the most vulnerable of all the other periods of life for the
development of epileptic seizures, particularly in the first 1 or 2 days from birth.
They may be short-lived events lasting for just a few days, but they often signify serious malfunction or damage of the immature brain, and constitute a neurological emergency that demands urgent diagnosis and management.
Prevalence of neonatal seizures is 1.5% and overall incidence approximately 3/1000 live births.
80% occur in the first 1 or 2 days during the first week of life.

Clinical manifestations- Neonatal seizures are paroxysmal, repetitive and stereotypical
events. They are usually clinically subtle, inconspicuous and difficult to
recognize from the normal behaviours of the inter-ictal periods or physiological phenomena.
There is no recognizable post-ictal state. Generalized tonic–clonic seizures (GTCSs) are exceptional. Five main types of neonatal seizure:-1. subtle seizures (50%)2. tonic seizures (5%)3. clonic seizures (25%)4. myoclonic seizures (20%)5. non-paroxysmal repetitive behaviours

Subtle seizures- They are described as subtle because the clinical manifestations
are frequently overlooked.
Subtle seizures manifest with-1. Ocular movements2. Oral–buccal–lingual movements (sucking, chewing, smacking and
tongue protrusions).3. Progression movements (rowing, swimming, pedalling, bicycling,
thrashing or struggling movements).4. Complex purposeless movements (sudden arousal with episodic
limb hyperactivity and crying).

Motor seizures- Clonic seizures are rhythmic jerks that may localize in a small part of
the face or limbs, axial muscles and the diaphragm, or be multifocal or hemiconvulsive.
Tonic seizures manifest with sustained contraction of facial, limb, axial and other muscles. They may be focal, multifocal or generalised, symmetrical or asymmetrical.
Myoclonic seizures are rapid, single or arrhythmic repetitive jerks. They may mimic Moro reflex and startling responses and are more frequently in pre-term than in full-term infants indicating, if massive, major brain injury and poor prognosis.
Spasms producing flexion or extension similar to those of West syndrome are rare.

Autonomic ictal manifestations-
Occurs in 37% of subtle seizures. These are paroxysmal changes of heart rate, respiration and
systemic blood pressure. Apnoea Salivation and pupillary changes are common.
Duration of neonatal seizures- Usually brief (10 s to 1–2 min) and repetitive with a median of 8
min in between each seizure.

Classification of Neonatal Seizures ELECTROENCEPHALOGRAPHIC SEIZURE
CLINICAL SEIZURE COMMON UNCOMMON
Subtle +*Clonic Focal + Multifocal +Tonic Focal + Generalized +Myoclonic Focal, multifocal + Generalized +---------------------------------------------------------------------------------------------------------------*Only specific varieties of subtle seizures are commonly associate with simultaneous Electroencephalographic seizure activity.
Volpe JJ.Neonatal Seizures:Neurology of the Newborn.4th ed.

Jitteriness Versus Seizure
CLINICAL FEATURE JITTERINESS SEIZURE
Abnormality of gaze or eye O + movementMovements exquisitely stimulus + O sensitivePredominant movement Tremor Clonic jerking
Movements cease with passive + O flexionAutonomic changes O +------------------------------------------------------------------------------------------------------------------

Aetiology• Hypoxia-ischemia ( 32%)• Hemorrhage and intra-cerebral infarction(17% & 7%)• Trauma• Infections(14%)• Metabolic( 3%)• Malformation of cerebral development• Neuro-cutaneous syndrome• Drug withdrawal and toxic (1%) • Inadvertent injections of local anaesthesia during
delivery

0 10 20 30 40 50
Hypoxia-ischemia
Hemorrhage
Trauma
Stroke
Meningitis
Hypocalcemia
Hypoglycemia
Malformation
Incidence (%)
19701987
Comparison of prominent etiologic diagnoses of seizures in the newborn period. (Data modified from
Mizrahi and Kellaway, 1987; Rose and Lombroso, 1970)
Fanaroff A, Martin R.Neonatal seizures. In:Neonatal and Perinatal Medicine, Diseases of the Fetus and Infant,6th ed.

Ferriero D. N Engl J Med 2004;351:1985-1995

Diagnostic procedures- CT brain scans - They can accurately detect haemorrhage,
infarction, gross malformations, and ventricular and other pathological conditions. Sensitivity is low in malformations.
MRI is the superior modality for normal developmental and maturational states of neonates and infants.
Electroencephalography- Only 10% of neonates suspected of having seizures have EEG
confirmation. Clonic movements have the highest yield of 44%, but only 17% for
‘subtle’ movements.

Ictal EEG Consist of repetitive waves with a predominant beta, alpha, theta
and delta range, or a mixture of all, which may accelerate or decelerate in speed or both.
They consist of spikes, and sharp, saw-tooth or sinusoidal waves (monomorphic or polymorphic), ranging in amplitude from very low to very high.
The patterns may be synchronous or asynchronous, focal or multifocal and, less frequently, generalized.
They may appear and disappear suddenly.


Zip-like electrical discharges

Inter-ictal EEGCertain inter-ictal EEG patterns may have diagnostic significance- Electro-cerebral inactivity of a flat or almost-flat EEG of severe
brain damage the burst-suppression pattern of neonatal epileptic
encephalopathies. persistently focal sharp or slow waves in localized lesions multifocal pattern in neonatal herpes simplex encephalitis inter-hemispheric or intra-hemispheric abnormalities Post-ictal EEG Usually returns to the pre-ictal state immediately . Transient slowing or depression of EEG activity may occur after
frequent or prolonged seizures.

Management- Accurate aetiological diagnosis and treatment of the cause of the
seizures. Neonatal seizures of metabolic disturbances need correction of the
underlying cause and not antiepileptic medication. The drug treatment of neonatal seizures is empirical. Phenobarbital and phenytoin are equally effective. If either drug is
given alone, the seizures are controlled in less than half of neonates.
Intravenous benzodiazepines such as diazepam, lorazepam, clonazepam and midazolam for acute seizure.

Drug Therapy For Neonatal Seizures
Standard Therapy
AED Initial Dose Maintenance Dose RoutePhenobarbital 20mg/kg 3 to 4 mg/kg per day lV, lM,
POPhenytoin 20 mg/kg 3 to 4 mg/kg per day lV, POªFosphenytoin 20 mg/kg phenytoin 3 to 4 mg/kg per day lV, lM
equivalents Lorazepam² 0.05 to 0.1 mg/kg Every 8 to 12 hours lVDiazepam²´ 0.25 mg/kg Every 6 to 8 hours lV
AED= andtiepileptic drug; lV= intravenous; lM= intramuscular; PO= oralªOral phenytoin is not well absorbed.²Benzodiazepines typically not used for maintenance therapy.³Lorazepam preferred over diazepam.

Duration of Anticonvulsant Therapy GuidelinesNeonatal PeriodIf neonatal neurological examination becomes normal, discontinue therapyIf neonatal neurological examination is persistently abnormal, consider the cause and obtain an EEG.In most such cases: Continue phenobarbital Discontinue phenytoin Reevaluate in 1 month At 1 Month After DischargeIf neurological examination has become normal, discontinue phenobarbitalIf neurological examination is persistently abnormal, obtain an EEG.If no seizure activity is noted on the EEG, discontinue phenobarbita Volpe, Neurology of the Newborn, 5th ed. 2008

Neonatal epileptic syndromes
Epileptic syndromes in neonates are rare. The following four syndromes have been recognized by 1989
ILAE classification-1. Benign neonatal familial convulsions2. Benign neonatal convulsions3. Early myoclonic encephalopathy4. Early infantile epileptic encephalopathy with suppression burst

Benign Familial Neonatal Seizures Rare autosomal dominant channelopathy. Charecterized by frequent brief seizures within the first days of life
mainly on the second or third day. Boys and girls are equally affected. Clinical manifestations- Seizures mainly occur in otherwise normal neonates after a normal
pregnancy and delivery, and with no precipitating factors. Seizures are brief, 1 or 2 min duration, as frequent as 20–30 per day. Most seizures start with tonic motor activity and posturing with
apnoea, followed by vocalizations, ocular symptoms, other autonomic features, motor automatisms, chewing and focal or generalized clonic movements.
Post-ictal state is brief and inter-ictally the neonates are normal.

Aetiology- autosomal dominant channelopathy with a high degree (about 85%)
of penetrance. Mutations (conventional, deletions or duplications) in the voltage-
gated potassium channel subunit gene KCNQ2 on chromosome 20q 13.3
Small proportion of families have mutations in the associated gene KCNQ3 on chromosome 8q.24. Mutations in the sodium channel subunit gene
SCN2A specific to ‘benign familial neonatal–infantile seizures’, which is a newly described, clinically intermediate variant between benign familial neonatal seizures and benign familial infantile seizures .

Electroencephalography-
The ictal EEG commonly starts with a synchronous and bilateral flattening of 5–19 s coinciding with apnoea and tonic motor activity.
This is followed by bilateral and often asymmetrical discharges of spikes and sharp waves with a duration of 1 or 2 min, which coincide with vocalizations, chewing and focal or generalized clonic activity.
The inter-ictal EEG may be normal or discontinuous, have focal or multifocal abnormalities.

Prognosis & Management-
Seizures remit between 1 and 6 months from onset. 10–14% may later develop other types of febrile (5%) or afebrile
seizures. Convulsions usually remit spontaneously without medication. The use of AEDs does not influence the eventual outcome. Prolonged seizures may be shortened or terminated with
benzodiazepines and phenytoin.

Benign neonatal seizures (non-familial)
Constitute a short-lived and self-limited benign epileptic syndrome.
Manifest with a single lengthy episode of repetitive clonic seizures, which constitute clonic status epilepticus.
Each seizure lasts for 1–3 min, repeating at frequent intervals. The whole seizure–status event lasts from between 2 hours and 3
days, with a median of about 20 hours. It is far more common (90%) between days 4 and 6, for which the
syndrome’s synonym ‘fifth day fits’ was coined. Boys (62%) are affected slightly more than girls.

AetiologyProbably environmental attributed to:- acute zinc deficiency detected in the cerebrospinal fluid of
affected neonates. viral illness, mainly rotavirus There is no genetic background. The ictal EEG Consists of rhythmic spikes or slow waves mainly in the rolandic
regions, although they can also localize anywhere else. The duration of the ictal discharges is 1–3 min. The prognosis is commonly excellent with normal development
and no recurrence of seizures. AEDs should be discontinued soon after the seizures subside.

Inter-ictal EEG in a neonate with benign neonatal seizures (non-familial

Benign (non-familial) neonatal seizures versus benign familial neonatal seizures
Benign (non-familial) neonatal seizures
Benign familial neonatal seizures
Main seizure type Mostly clonic Tonic-clonic
Onset Fifth day of life Second or third day of life
Duration of seizure Status epilepticus (median 20 hrs) Repetitive isolated seizure
Main cause Unknown, probably environmental Autosomal dominant
Subsequent seizures Practically nill ( 0.5%) Relatively high (11%)
Psychomotor deficit minor Practically non-existant
Ictal EEG Usually localized spikes Usually generalized flattening

Early myoclonic encephalopathy Dreadful but rare epileptic encephalopathy. Starts in first days of life, sometimes immediately after birth. More than 60% start before 10 days of age and rarely after the
second month. Boys and girls are affected equally.
Manifests with a triad of intractable seizures-1. Erratic or fragmentary myoclonus appears first,2. followed by simple focal seizures and 3. later by tonic epileptic (infantile) spasms

Aetiology-
Multi-factorial disease with a high incidence of familial cases. Some families there is an autosomal recessive inheritance. Inborn errors of metabolism are the most common causes
include non-ketotic hyperglycinaemia, propionic aciduria, methyl malonic acidaemia, sulphite and xanthine oxidase deficiency, Menkes’ disease and Zellweger syndrome, and molybdenum co-factor deficiency.

MANAGEMENT Brain imaging is usually normal at the onset of the disease but
progressive cortical and periventricular atrophy often develop.
Inter-ictal EEG consists of a repetitive burst suppression pattern
The bursts of high-amplitude spikes and sharp-and-slow waves last for 1–5 s and alternate with periods of a flat or almost flat EEG, lasting 3–10 s.
There is no effective treatment.

33

343 months later

Ohtahara syndrome (early infantile epileptic encephalopathy
with suppression burst) Rare and devastating form of severe epileptic encephalopathy. First 10 days of life, sometimes within the uterus or up to 3 months
after birth. There may be a slight male predominance. Manifests with clinico-EEG features of mainly tonic spasms and
burst suppression EEG patterns that consistently occur in the sleeping and waking states.
Tonic spasms usually consist of a forward tonic flexion lasting 1–10 s, which is singular or in long clusters 10–300 times every 24 h.

Aetiology & DIAGNOSIS-
Malformations of cerebral development such as hemimegaloencephaly, porencephaly, Aicardi syndrome, agenesis of mamillary bodies, cerebral dysgenesis and focal cortical dysplasia.
Between 2 and 6 months of age. Brain imaging usually shows severe abnormalities and
malformations of cortical development Metabolic screening is mandatory if brain imaging is normal.
An age-related evolution of clinical and EEG patterns from Ohtahara syndrome, first to West syndrome and then to Lennox–Gastaut syndrome.

MANAGEMENT The EEG burst-suppression pattern has a pseudo-rhythmic
periodicity, is continuous during wakefulness and sleep. Appears at the onset of the disease and disappears within the first
6 months of life. This is a devastating syndrome associated with high mortality and
morbidity. Half the patients die within weeks or months of onset and the
others soon develop permanent severe mental and neurological deficits.
There is no effective treatment. Zonisamide and vigabatrin had some beneficial effect in single
case reports. Neurosurgery in focal cerebral dysplasia is sometimes beneficial.

Ohtahara syndrome ( severe neonatal epilepsy cerbral cortical atrophy)

Unilateral suppression burst (ohtahara) hemimegalencephaly

40
Fp1-F3
F3 –C3
C3 – P3
P3 – O1
Fp2 F4
F4 – C4
C4 – P4
P4 – O2
Fp1 –F7
F7 – T3
T3 – T5
T5 – O1
Fp2 F8
F8 – T4
T4 – T6
T6 – O2
EKG

Ohtahara (dysgenesis corpus callosum)

Ohtahara syndrome versus early myoclonic encephalopathyOhtahara syndrome Early myoclonic
encephalopathy
Main seizures Tonic spasms Erratic myoclonias, focal seizures, cluster of spasm
Main aetiology Malformations of cerebral development
Genetic and metabolic
Burst suppression pattern Pseudo-rhythmic & continuous in sleep & awake, starts early & shorter duration
Probably accentuated by sleep & may not occur in wakefulness, start later & last longer
Paroxysmal burst Longer (2-6 sec.) Shorter (1-5 sec.)
suppression Shorter (3-5 sec.) Longer (3-10 sec.)
Transformation to west syndrome
As a rule Common but transient

Epileptic seizures and syndromes in infancy

Febrile seizures Febrile seizure are due to an age-related and predominantly
genetic benign susceptibility to epileptic fits, precipitated by fever without evidence of intracranial infection or other cause.
Aged from 6 months to 5 years, who is otherwise neurologically normal.
Boys slightly (60%) predominate. Prevalence is about 3% of children. A rectal temperature level of at least 38°C and a rapid rise of
fever. The majority (78%) of febrile seizures occur within the first day of
the onset of fever.

Clinical manifestations Causes of fever vary and include upper respiratory tract infection
or pharyngitis (38%), otitis media (23%), pneumonia (15%), gastroenteritis (7%), roseola infantum (5%) and non-infectious illness (12%).
Viral diseases are more common. Generalized tonic–clonic seizures (GTCSs) are by far the most
common seizure type (80%). Tonic (13%), atonic (3%), unilateral or focal onset tonic–clonic
seizures (4%) may occur in the remaining 20%. Repetitive seizures in the same febrile illness occur in 16% of
patients. Febrile seizures are categorized into1- simple and 2- complex febrile seizures

Simple febrile seizures versus complex febrile seizuresSimple febrile seizures Complex febrile seizures
Prevalence among febrile seizures 70% 30%
Neurologically normal Inclusion criterion Included but not necessary
With neuro-developmental abnormalities
Exclusion criterion 3.5–7% of all febrile seizures
Age range 6 months to 5 years 6 months to 5 years
Duration <15 min <15 min Included but not necessary
Duration >15 min or febrile status epilepticus
Exclusion criterion 8% of all febrile seizures
Once in a 24-hour period of a febrile illness
Inclusion criterion Included but not necessary

Cont.-Simple febrile seizures Complex febrile seizures
Repetitive in clusters of two or more in a 24-hour period of a febrile illness
Exclusion criterion 11–16% of all febrile
Generalized-onset tonic–clonic seizures(80% of all febrile seizures)
Inclusion criterion Included but not necessary
Focal onset or focal epileptic seizures Exclusion criterion 4% of all focal seizures
Post-ictal hemiparesis Exclusion criterion 0.4% of all febrile seizures
Risk of subsequent epilepsy Low (1%) High (6–49%)

Aetiology- Febrile seizures are often familial with a genetic predisposition. Children with siblings or parents who have a history of febrile
seizures are at a four to five fold higher risk than the general population.
Concordance rate is as high as 70% in monozygotic and 20% in dizygotic twins.
DIAGNOSIS- Clinical diagnosis. The EEG and brain imaging are unhelpful and should therefore be
discouraged.

MANAGEMENTACUTE- Diazepam intravenously at a dose of 0.25–0.5 mg/ kg, or in
rectal preparations at a dose of 0.5 mg/ kg, is probably the first choice.
Lorazepam administered intravenously (0.1 mg/kg), which is less likely to cause respiratory depression.
Midazolam administered by buccal (0.4–0.5 mg/kg) or intranasal (0.2 mg/kg) application has superior efficacy to diazepam.
Antipyretic treatment during febrile illnesses does not reduce the recurrence rate.

Prophylactic management- The best treatment with a first febrile seizure is education and
reassurance for the parents Prophylactic treatment may be desired if a child has one or, mainly, a
combination of the following features:I. complex febrile seizuresII. neurological abnormalitiesIII. age <1 yearIV. frequent recurrences Treatment may be continuous or intermittent - Continuous treatment consists of daily administration of, mainly,
phenobarbital (which at a blood level of 15 μg/ml) Intermittent treatment at the time of a febrile illness, mainly with
rectal or oral diazepam There is a small reduction in the recurrence risk with a dose of
diazepam 0.3 mg/kg.


Epilepsy with febrile seizures plus
start earlier (from first month to a mean of 1 year) than the classical febrile seizures.
They are often multiple and continue beyond the age of 5 years, usually remitting by mid childhood (median 11 years)
Inheritance considered to be generally autosomal dominant with incomplete penetrance.
Two loci described on chromosome 19q (GEFS+) and 2q (GEFS2). Mutations were found in the SCN1A, SCN1B and SCN2A genes
(encoding voltage-gated sodium channel subunits) and the GABRG2 gene.

Clinical manifestations- Heterogeneous clinical phenotypes. Typical febrile seizures and FS+ are the most common clinical
phenotypes that may occur alone (75% of affected patients) . other types of seizures, including:-
brief non-febrile generalized convulsions
other generalized seizures, such as absences, myoclonic jerks, tonic seizures and, more frequently, myoclonic–atonic seizures
focal seizures of mainly frontal or temporal lobe origin may occur in approximately 13% of affected individuals.

Diagnosis & Management-
Brain MRI is normal. The EEG findings depends on clinical phenotype. Half of the patients have normal EEGs.
The most common EEG abnormality is sparse and brief generalized polyspike–wave discharges (GPSWD) or generalized spike–slow-wave discharges (GSWD) that might require sleep EEGs for their detection.
considered benign and self-limited. Repetitive febrile seizures or FS+ may require prophylactic
treatment.

Benign infantile seizures Watanabe–Vigevano syndrome Is familial and non familial, A benign self-limiting age-related idiopathic syndrome of infancy. The seizures are focal in otherwise normal infants. Age at onset is from 3 to 20 months with a peak at 5 or 6 months. Seizures characteristically occur in clusters of five to ten per day for
1–3 days and may recur after 1–3 months. The seizures are focal, predominantly diurnal and brief (0.5–3 min). motor arrest, impairment of consciousness with decreased
responsiveness, staring, eye and head deviation occurs. The ictal EEG demonstrates focal discharges of fast activity mixed
with spikes . Prognosis is usually good. Seizures remit within 1 to 2 years of
onset.

Benign infantile seizure

Myoclonic epilepsy in infancy ( MEI ) Between 6 months and 3 years Boys are twice as likely to be affected as girls. Myoclonic seizures are the predominant and often the only type of
fits in MEI, brief (1 or 2 s). Mainly affect the head, eyeballs, upper extremities and the
diaphragm. Manifest clinically with head nodding and, more rarely, flexion or
extension of the body. Precipitated by-clinical and EEG photosensitivity (20%) &
unexpected acoustic or tactile stimuli, with better prognosis. History of epilepsy or febrile seizures is present in 30% of cases

Diagnosis & Management- All tests other than EEGs are normal. Ictal EEG during jerks shows GPSWD or GSWD with a duration of
1–3 s. Frequently, ictal EEG discharges are limited to the rolandic and
vertex regions. Remission usually occurs between 6 months and 5 years of onset. Response to AED treatment is usually excellent. With valproate, 80% of patients become seizure free. Clonazepam is more effective than valproate in controlling
myoclonic jerks levetiracetam significantly suppresses photosensitivity.

A typical patient with MEI with myoclonic jerks only and excellent prognosis

Dravet syndrome Severe myoclonic epilepsy in infancy is a rare progressive epileptic
encephalopathy. Onset within first year of life, with a peak age at 5 months. Twice as many boys are affected. Charecterized by a tetrad of seizures:I. early infantile febrile clonic convulsionsII. myoclonic jerksIII. atypical absencesIV. complex focal seizures. Convulsive, myoclonic or absence status epilepticus are frequent.

• There are three periods of evolution with Dravet syndrome.1. The first period is relatively mild (the pre-seismic period)-lasts for
2 weeks to 6 months with febrile clonic convulsions2. The second period is relentlessly aggressive (the seismic period)
with the emergence of other multiple-seizure types and severe neuro-cognitive deterioration.
3. The third period is static (the post-seismic period). seizures may improve, but serious mental and neurological abnormalities are irreversible.
Seizure-precipitating factors are hyperthermia Photic and pattern stimulation, movements and eye closure.
Genetically determined, mutations in the voltage gated sodium channel gene SCN1A were found in a high percentage (range 35–100%)

Diagnosis & Management- Genetic testing: a severe SCN1A gene defect is strongly supportive. Brain CT and MRI scans are either normal or show mild cerebral or
cerebellar atrophy. EEG shows a similar progression to that of the clinical state. Inter-ictal EEG may initially be normal, but 20% show generalised
photo-paroxysmal responses. Diagnosis is nearly certain if intractable myoclonic jerks and mental
retardation appear within 1-2 yrs of onset. Early death occurs in 15% of patients. Valproate, benzodiazepines, phenobarbital (in convulsive seizures),
ethosuximide (in absence and myoclonic seizures) are beneficial. Carbamazepine, Lamotrigine and phenytoin are contraindicated. Of the newer AEDs, topiramate, stiripentol, zonisamide and mainly
levetiracetam, early ketogenic diet have been found to be useful.

Dravet syndrome (nonepileptic status)

Dravet syndrome(absence seizure)

DRAVET SYNDROME (complex partial seizure)

Myoclonic encephalopathy in non-progressive disorders
Non-progressive myoclonic epilepsy in infancy charecterized by- a fixed, non-progressive encephalopathy recurrent episodes of
prolonged and erratic atypical myoclonic-absense status epilepticus. Onset is from day 1 of life to 5 years of age (peak at 12 months) All patients have pre-existing neuropsychological deficits of a fixed
encephalopathy charecterized by severe axial hypotonia, ataxia, continuous jerky movements, tremor, and severe cognitive and learning abnormalities.
Half of cases suffer from chromosomal disorders, mainly Angelman and 4p syndromes.
20% of patients have prenatal brain anoxia–ischaemia or malformations of cortical development.
The ictal EEG shows continuous brief bursts of diffuse slow spikes and waves.
• Prognosis is poor

West syndrome An age-related specific epileptic encephalopathy charecterized by
a unique type of seizure called epileptic (infantile) spasms and gross EEG abnormalities of hypsarrhythmia.
Onset between 3 and 12 months (peak at 5) Males (60–70%) predominate. Usually starts insidiously with mild epileptic spasms occurring two
or three times in succession. The full-blown features develop in a few weeks with spasms
typically occurring in clusters of 1–30 per day, with each cluster having 20–150 attacks.
The epileptic spasms are clusters of sudden, brief (0.2–2 s), bilateral tonic contractions of the axial and limb muscles.
They are slower than myoclonic jerks and faster than tonic seizures.

Cont.- Spasm is often followed by motionlessness and diminished
responsiveness lasting up to 90 s. Spasms may be-1- Flexor spasms are common (40%) and synonyms are ‘salaam
spasms’, ‘jackknife spasms• There is abrupt flexion of the neck and the trunk, the arms raise
forwards or sideways with flexion at the elbows, and the legs are elevated with flexion at the hips and knees.
2-Extensor spasms are less frequent manifesting with sudden backwards movements of the head, hyperextension of the body, and extension and abduction of the limbs similar to the Moro reflex.
3-Flexor–extensor spasms- most common spasms (50%), and combine sudden contraction of both flexor and extensor muscles with flexion of the neck, trunk and arms, but extension of the legs.

Aetiology-1-Symptomatic West syndrome most common (80%). Several pre-, peri- and postnatal insults are responsible from
hypoxia–ischaemia, infections, trauma and ICH, malformations of cortical development, neuro-cutaneous diseases, genetic and chromosomal abnormalities and, less often, inborn errors of metabolism.
2-Probably symptomatic (cryptogenic) West syndrome may have a prevalence of 10–15%.
3-Idiopathic West syndrome, with normal pre-morbid development and possible hereditary predisposition family history of epilepsy, febrile seizures or EEG genetic patterns, constitutes 5–30% of all cases.
Idiopathic West syndrome may have a good prognosis

Diagnosis & Management- Brain CT scan and, more specifically, MRI are indicated shows
apparent atrophy. PET of brain glucose metabolism is highly sensitive in detecting
focal cortical abnormalities.Inter-ictal EEG pattern of Hypsarrhythmia occurs in two-thirds of
patients. chaotic mixture of giant abnormal, arrhythmic and asynchronous
biological brain electrical activity of slow and sharp waves, multi-focal spikes and poly-spikes.
Hypsarrhythmic pattern of West syndrome gradually becomes more organized, fragmented and disappears with age.
Ictal EEG patterns brief duration (1–5 s), and it consists ofI. a high-voltage, generalized slow wave, II. episodic, low-amplitude fast activity andIII. marked diffuse attenuation

Epileptic spasms of a child with Down syndrome who developed West syndrome

Tuberus sclerosis( subependymal nodule presenting with west syndrome)

Typical hypsarrhythmic pattern seen at standard and reduced sensitivity


Cont.- ACTH and less often corticosteroids or vigabatrin are the drugs of
choice, controlling the epileptic spasms in two-thirds of patients. Lamotrigine, levetiracetam, pyridoxine, topiramate, valproate and
zonisamide are also used as adjunctive medications when ACTH and vigabatrin fail.
No treatment has been conclusively shown to improve the long-term intellectual development.
Prognosis depends upon etiology, 60% develop other types of seizure that are usually resistant to
treatment. Half of the patients have permanent motor disabilities, and two-
thirds have, usually severe, cognitive and psychological impairment, Autistic behaviour, hyperkinetic syndrome and psychiatric disorders.

West syndrome (hypoxic-ischemic encephalopathy)

Migrating focal seizures of infancy Is a devastating syndrome with early onset of almost continuous
multi-focal seizures arising independently from multiple regions of both hemispheres, and relentless psychomotor deterioration.
onset is between the first day of life and 7 months of age (mean age 1 month).
Patients are normal prior to the onset of seizures. Seizures manifest with motor and autonomic symptoms. Lateral deviation of the head and eyes, unilateral eyelid and
eye jerking, and unilateral tonic or clonic convulsions of one limb are common motor manifestations that frequently progress to secon darily GTCSs. Both sides are alternately affected.
Autonomic manifestations of apnoea, cyanosis, flushing, hiccups, sweating and hyper salivation are striking.

Contd.-
Aetiology is unknown but a functional or metabolic disorder is suspected with negative family history.
Screening of the CLCN2 gene revealed a homozygous mutation. CT and MR brain scans are normal. Ictal EEG discharges involve multiple independent sites randomly,
moving from one cortical area to another in consecutive seizurs. Anti-epileptic drug treatment and a ketogenic diet are ineffective. All children regress developmentally and develop severe
psychomotor abnormalities. They become quadriplegic with major axial hypotonia, pyramidal and
extra-pyramidal signs and athetosis. Death often occurs soon after onset of the disease, within 1 year.

79
Migrating seizures of infantileMalignant migrating partial epilepsy of infancy

80
Migrating seizures of infantile

SUMMARY (NEONATAL EPILEPTIC SYNDROME)Epileptic disorder
Age of onset Seizure type Diagnosis Remark
Neonatal epileptic seizure
1-2 days ( 1st week)
Subtle, tonic, clonic, myoclonic
CT, MRI brain, EEG ( zips)
50% normal, third develop recurrence
BFNS 2nd- 3rd days ( 1st week)
tonic, clonic, apnoea, autonomic
b/l flattening, spike & sharp wave discharge
Remit in mid-infancy, 10-14% recurrence
BNS (non-familial)
1-7 days Clonic status epilepticus, apnoea
Rhythmic spike & slow wave in rolandic area
Excellent prognosis
Early myoclonic epilepsy
Usually <10 days
Erratic myoclonia, simple focal, tonic
Burst suppression 50% mortality
Ohtahara syndrome
Usually <10 days
Tonic spasms Burst suppression 50% mortality
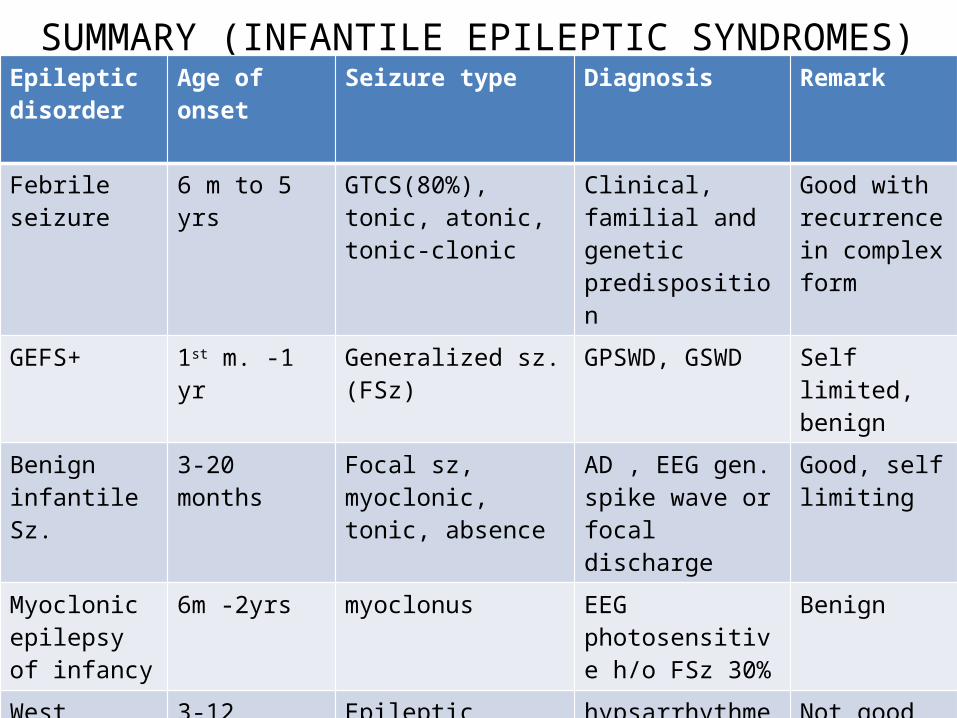
SUMMARY (INFANTILE EPILEPTIC SYNDROMES)Epileptic disorder
Age of onset Seizure type Diagnosis Remark
Febrile seizure 6 m to 5 yrs GTCS(80%), tonic, atonic, tonic-clonic
Clinical, familial and genetic predisposition
Good with recurrence in complex form
GEFS+ 1st m. -1 yr Generalized sz. (FSz) GPSWD, GSWD Self limited, benign
Benign infantile Sz.
3-20 months Focal sz, myoclonic, tonic, absence
AD , EEG gen. spike wave or focal discharge
Good, self limiting
Myoclonic epilepsy of infancy
6m -2yrs myoclonus EEG photosensitive h/o FSz 30%
Benign
West syndrome
3-12 months Epileptic spasms hypsarrhythmea Not good
Dravet syndrome
< 1 yr Febrile clonic, myoclonic, focal
Genetic SCN1A poor
MENPD Peak 12 months
Erratic myoclonus multiple Poor prognosis

TAKE HOME MESSAGEEvery shaking baby is not having epileptic seizure.Wide range of presentation.Efficiently obtain information regarding clinical, family
history, peri-partum events and neurological examination.
Establish underlying etiology and epileptic syndrome.Treatment usually successful in stopping seizures, but
risk of neuro-developmental abnormalities remains high.
Prevention of causes and education and reassurance to parents always remains a priority.

Thankyou


















