
Speaker: Yu-Ching Fang Advisors: Hsueh-Fen Juan and Hsin-Hsi Chen
Upload
ernest-oconnorCategory
view
225download
0
Protein function and classification
www.ebi.ac.uk/interpro
Hsin-Yu Chang
www.ebi.ac.uk

Greider and Balckburn discovered telomerase in 1984 and were awarded Nobel prize in 2009. Which model organism they used for this study ?
1. Tetrahymena
2. Saccharomyces cerevisiae3. Mouse
4. Human
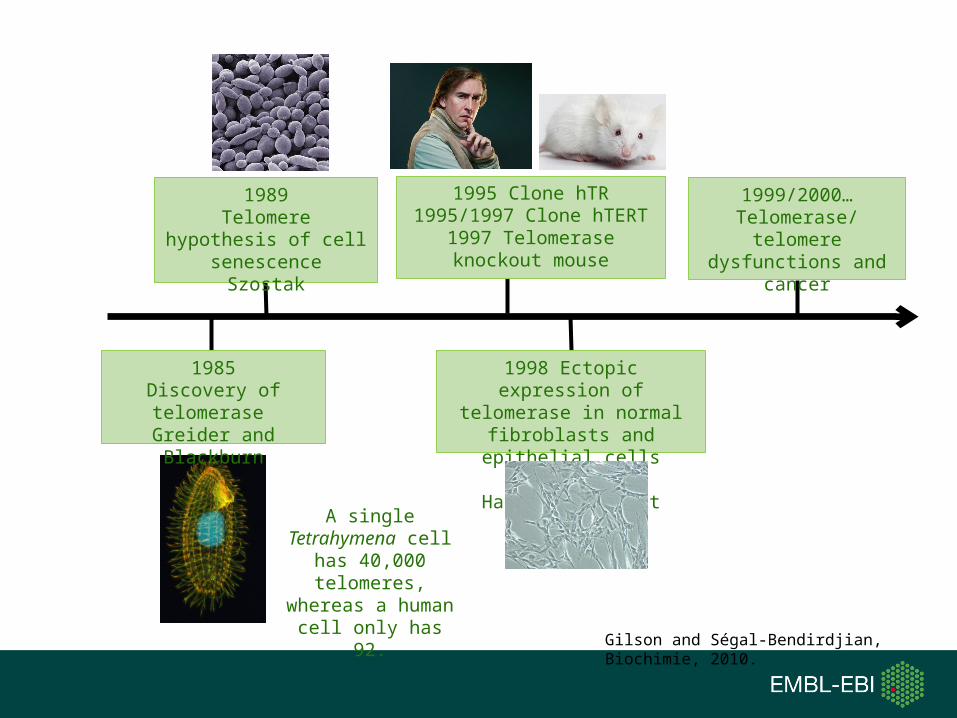
A single Tetrahymena cell has 40,000
telomeres, whereas a human cell only has
92.
1985Discovery of telomerase Greider and Blackburn
1989Telomere hypothesis of
cell senescenceSzostak
1995 Clone hTR1995/1997 Clone hTERT
1997 Telomerase knockout mouse
1998 Ectopic expression of telomerase in normal
fibroblasts and epithelial cells bypasses the Hayflick’s limit
1999/2000…Telomerase/telomere
dysfunctions and cancer
Gilson and Ségal-Bendirdjian, Biochimie, 2010.

Therefore, protein classification could help scientists to gain information about protein
functions.

In the lab, what do we usually do to analyse protein sequences and find out their functions?

• Protein BLAST
• Publications - text books or papers
• UniProt
• PDB
• Specialized protein databases such as SGD, the human
protein atlas, etc.
What I used to do:

BLAST it?
Advantages:
• Relatively fast
• User friendly
• Very good at recognising similarity between closely related sequences
Drawbacks:
• sometimes struggle with multi-domain proteins
• less useful for weakly-similar sequences (e.g., divergent homologues)

Using BLAST to find clues of protein functions-when it goes well

Pairwise alignment of two proteins: CD4 from two closely-related species
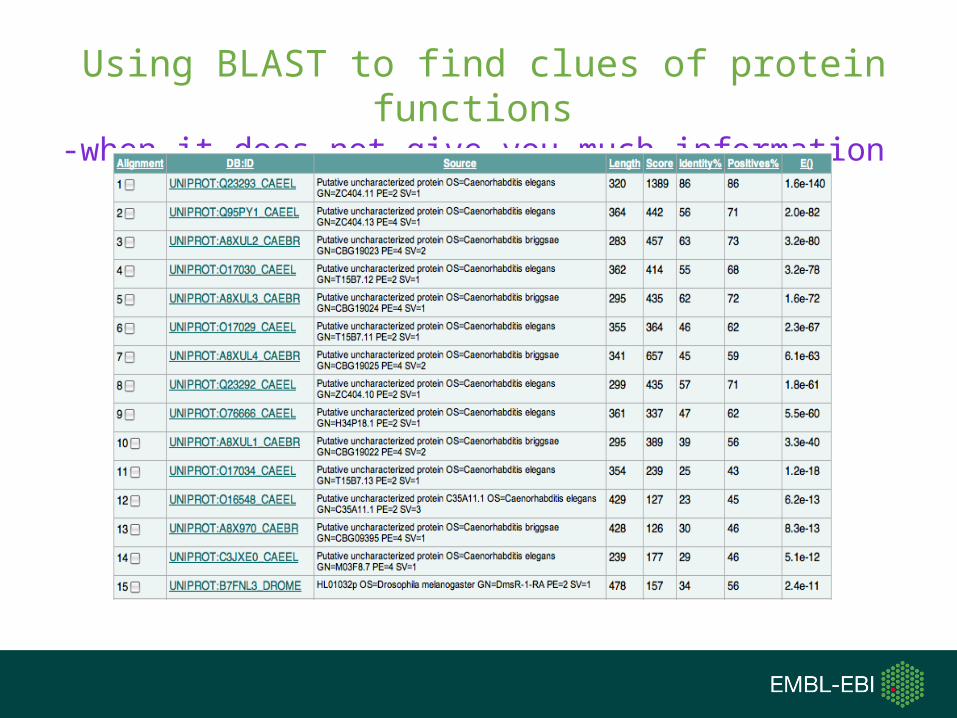
Using BLAST to find clues of protein functions-when it does not give you much information
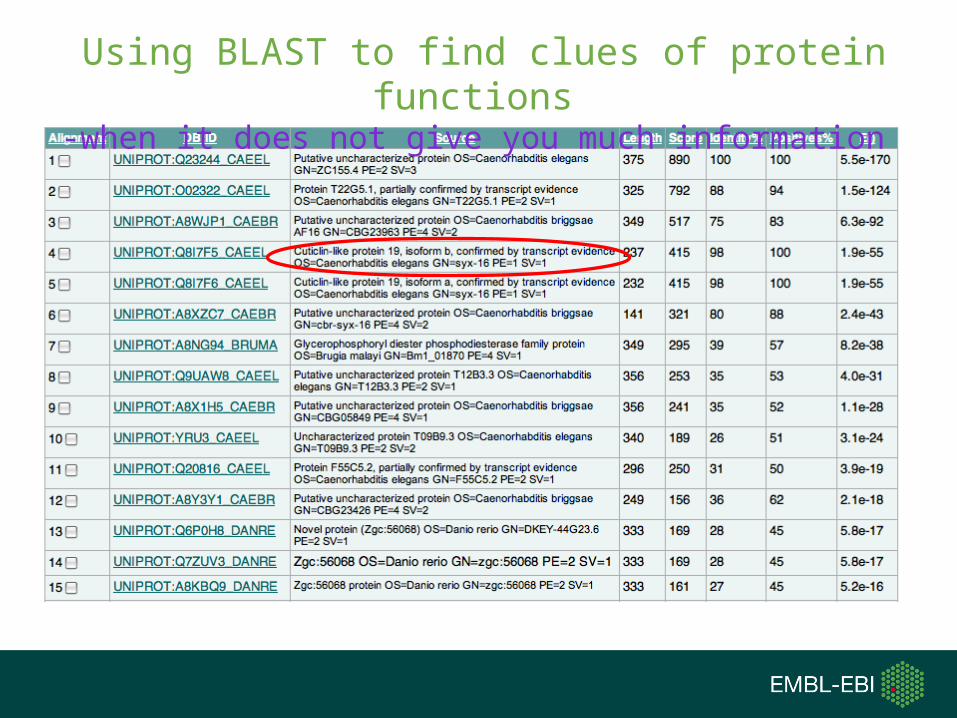
Using BLAST to find clues of protein functions-when it does not give you much information

Because BLAST performs local pairwise alignment, it:
•Cannot encode the information found in an multiple sequence alignment that show you conserved sites.
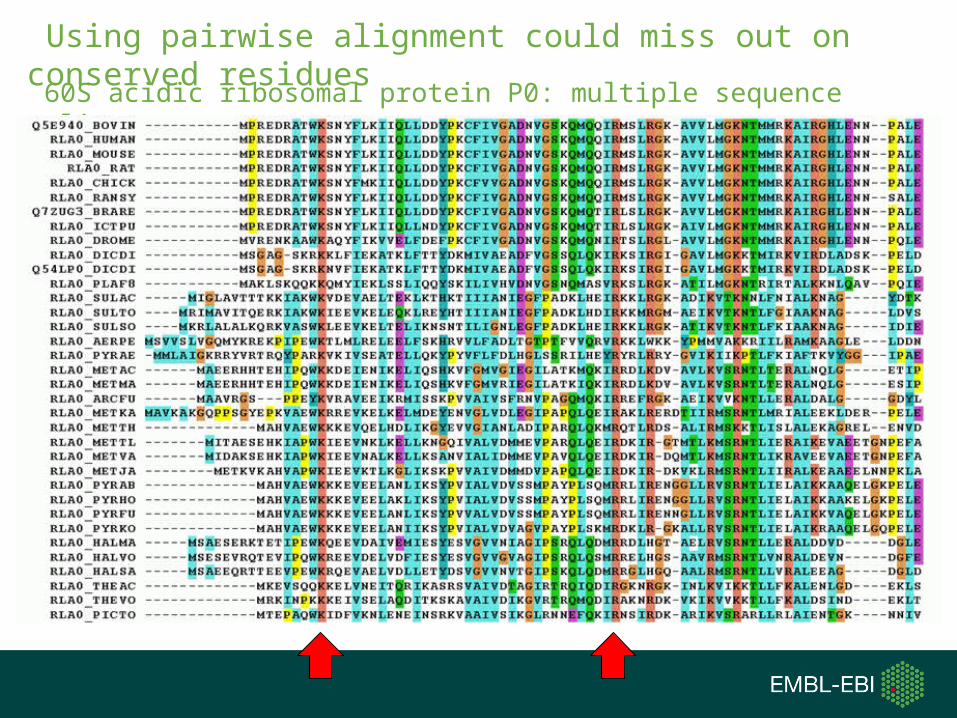
60S acidic ribosomal protein P0: multiple sequence alignment
Using pairwise alignment could miss out on conserved residues

An alternative approach: protein signature search
• Model the pattern of conserved amino acids at specific positions within a multiple sequence alignment
• Use these models to infer relationships with the characterised sequences (from which the alignment was constructed)
• This is the approach taken by protein signature databases
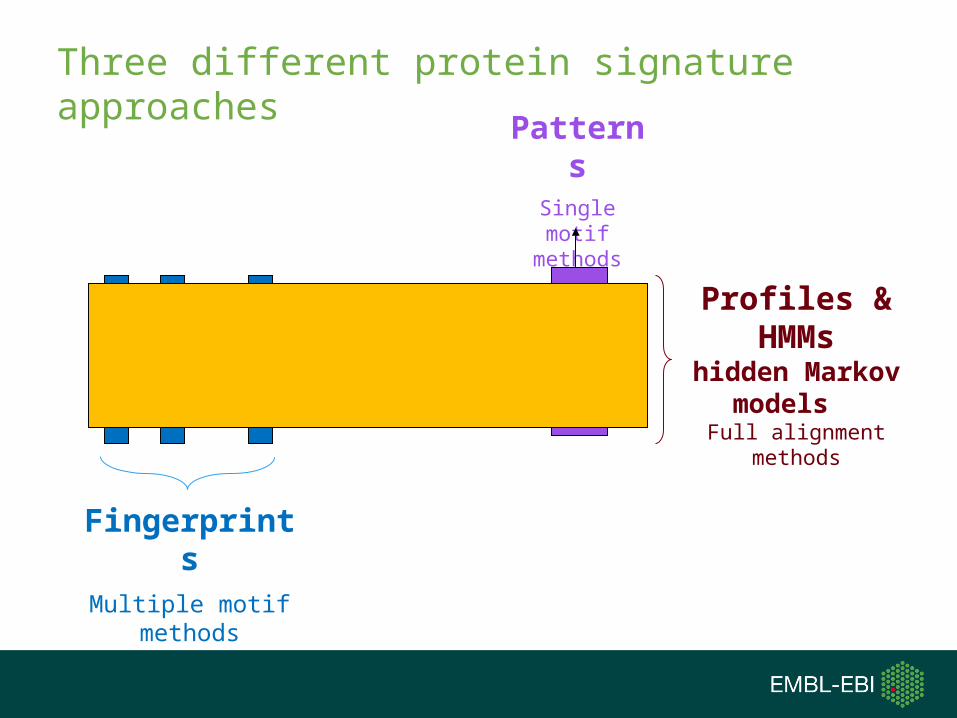
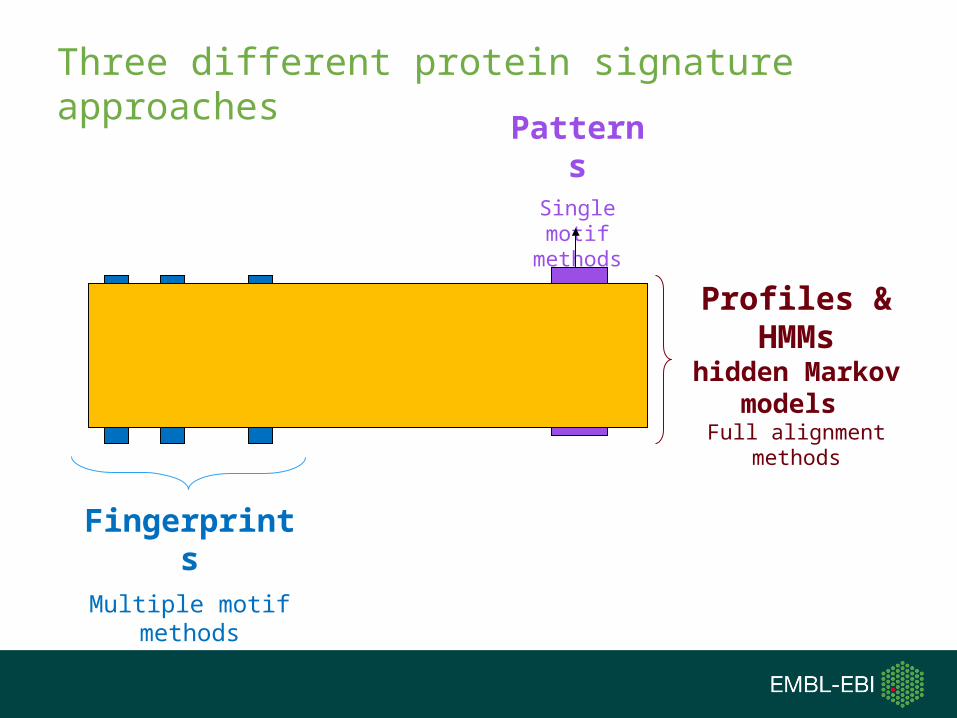
Three different protein signature approaches
PatternsSingle motif
methods
FingerprintsMultiple motif
methods
Profiles & HMMs
hidden Markov models
Full alignment methods
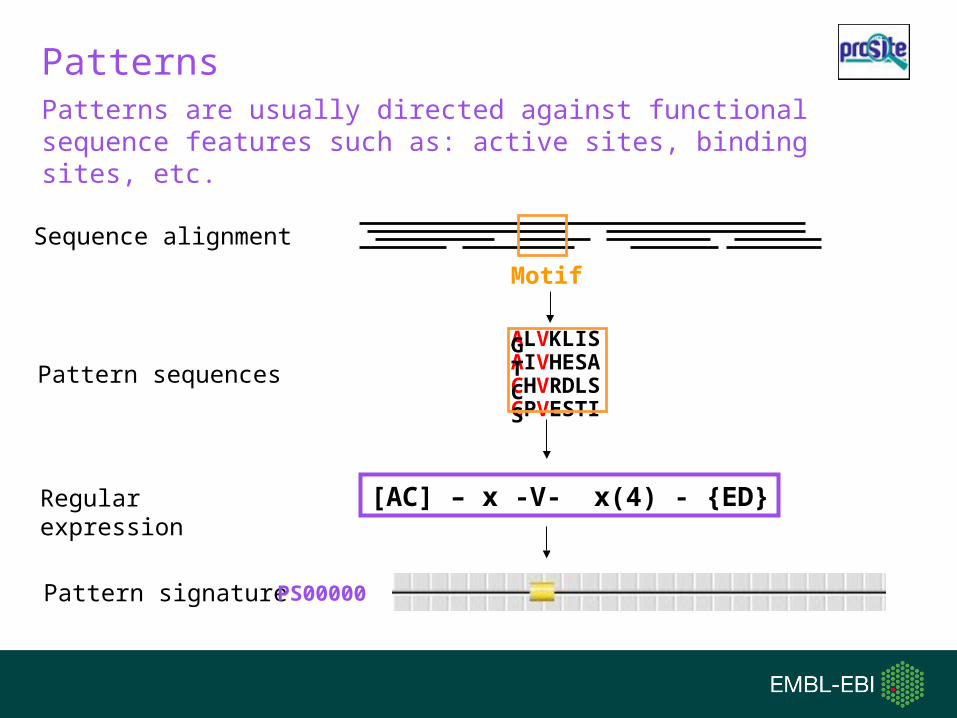
Patterns
Sequence alignment
Motif
Pattern signature
[AC] – x -V- x(4) - {ED}Regular expression
PS00000
Pattern sequences
ALVKLISGAIVHESATCHVRDLSCCPVESTIS
Patterns are usually directed against functional sequence features such as: active sites, binding sites, etc.

Patterns
Advantages:
• Can anchor the match to the extremity of a sequence
<M-R-[DE]-x(2,4)-[ALT]-{AM}
• Strict - a pattern with very little variability and forbidden residues can produce highly accurate matches
Drawbacks:
• Simple but less flexible
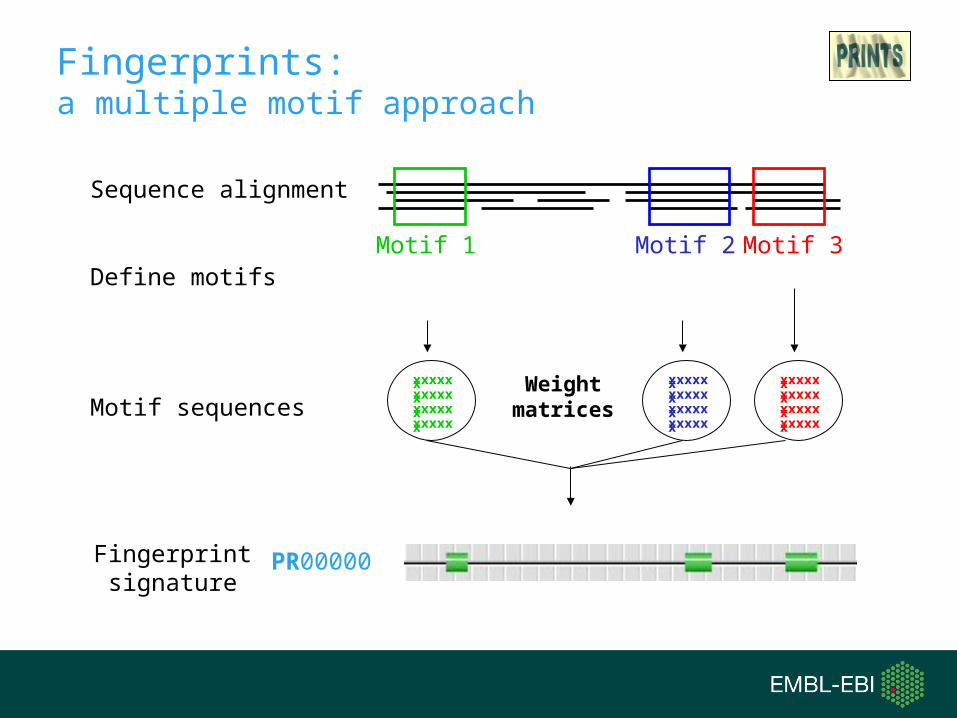
Fingerprints: a multiple motif approach
Sequence alignment
Motif 2 Motif 3Motif 1Define motifs
Fingerprint signature
PR00000
Motif sequencesxxxxxxxxxxxxxxxxxxxxxxxx
xxxxxxxxxxxxxxxxxxxxxxxx
xxxxxxxxxxxxxxxxxxxxxxxx
Weight matrices
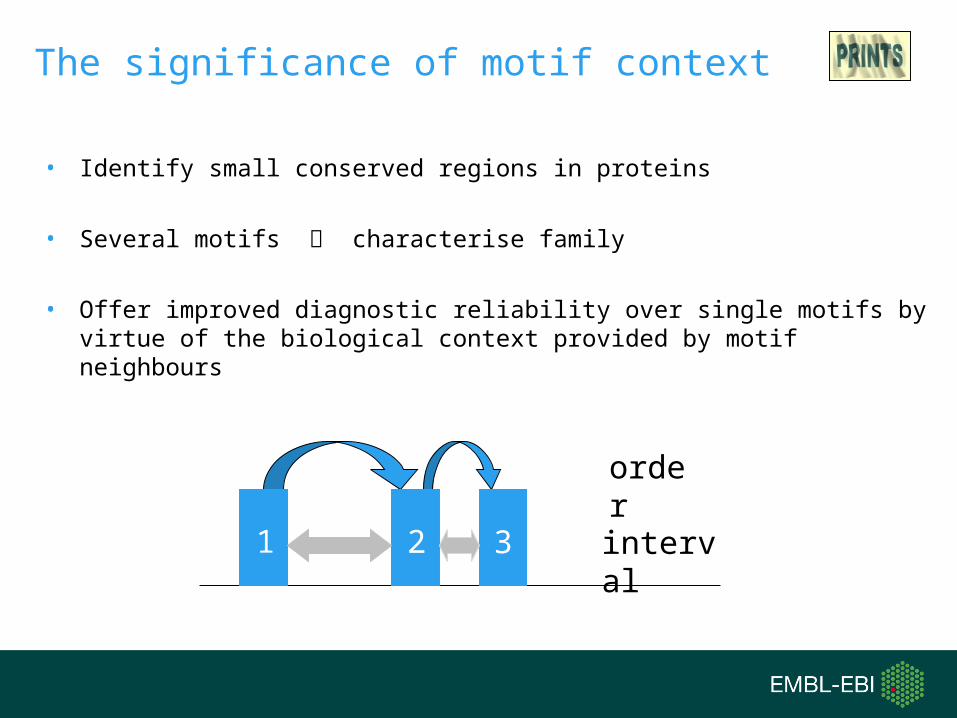
The significance of motif context
order
interval
• Identify small conserved regions in proteins
• Several motifs characterise family
• Offer improved diagnostic reliability over single motifs by virtue of the biological context provided by motif neighbours
1 2 3

• Good at modeling the often small differences between closely related proteins
• Distinguish individual subfamilies within protein families, allowing functional characterisation of sequences at a high level of specificity
Fingerprints

Sequence alignment
Entire domain Define coverage
Whole protein
Use entire alignment of domain or protein family xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
Build model
Profile or HMM signature
Profiles & HMMs

Profiles
Start with a multiple sequence alignment
Amino acids at each position in the alignment are scored according to the frequency
with which they occur
Scores are weighted according to
evolutionary distance using a BLOSUM matrix
• Good at identifying homologues

HMMs
Amino acid frequency at each position in the alignment and their transition probabilities
are encoded
Insertions and deletions are also modelled
Start with a multiple sequence alignment
• Very good at identifying evolutionarily distant homologues
• Can model very divergent regions of alignment
Three different protein signature approaches
PatternsSingle motif
methods
FingerprintsMultiple motif
methods
Profiles & HMMs
hidden Markov models
Full alignment methods

www.ebi.ac.uk/interpro
InterPro
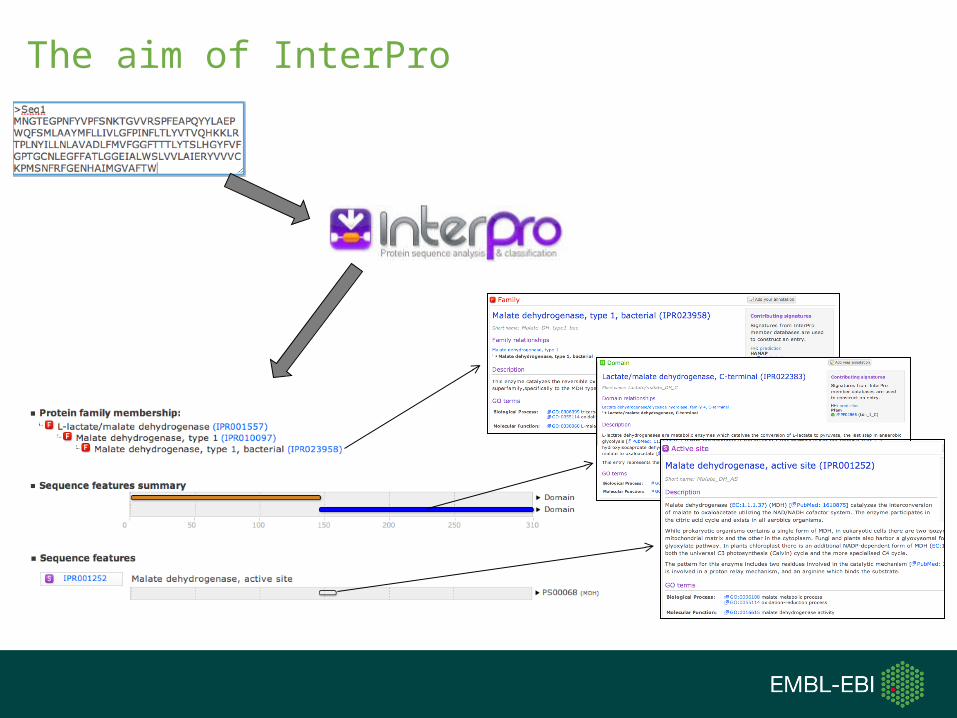
The aim of InterPro

What is InterPro?
• InterPro is an integrated sequence analysis resource
• It combines predictive models (known as signatures)
from different databases to provide functional analysis of
protein sequences by classifying them into families and
predicting domains and important sites

• First release in 1999
• 11 partner databases
• Forms part of the automated system that adds annotation to UniProtKB/TrEMBL
• Provides matches to over 80% of UniProtKB
• Source of >60 million Gene Ontology (GO) mappings to >17 million distinct UniProtKB sequences
• 50,000 unique visitors to the web site per month> 2 million sequences searched online per month. Plus offline searches with downloadable version of software
Facts about InterPro

Structuraldomains
Functional annotation of families/domains
Protein features
(sites)
Hidden Markov Models Finger prints
Profiles Patterns
HAMAP

• Signatures are provided by member databases
• They are scanned against the UniProt database to see which
sequences they match
• Curators manually inspect the matches before integrating the
signatures into InterPro
InterPro signature integration process
Signatures representing the same entity are integrated together
Relationships between entries are traced, where possible
Curators add literature referenced abstracts, cross-refs to other databases, and GO terms
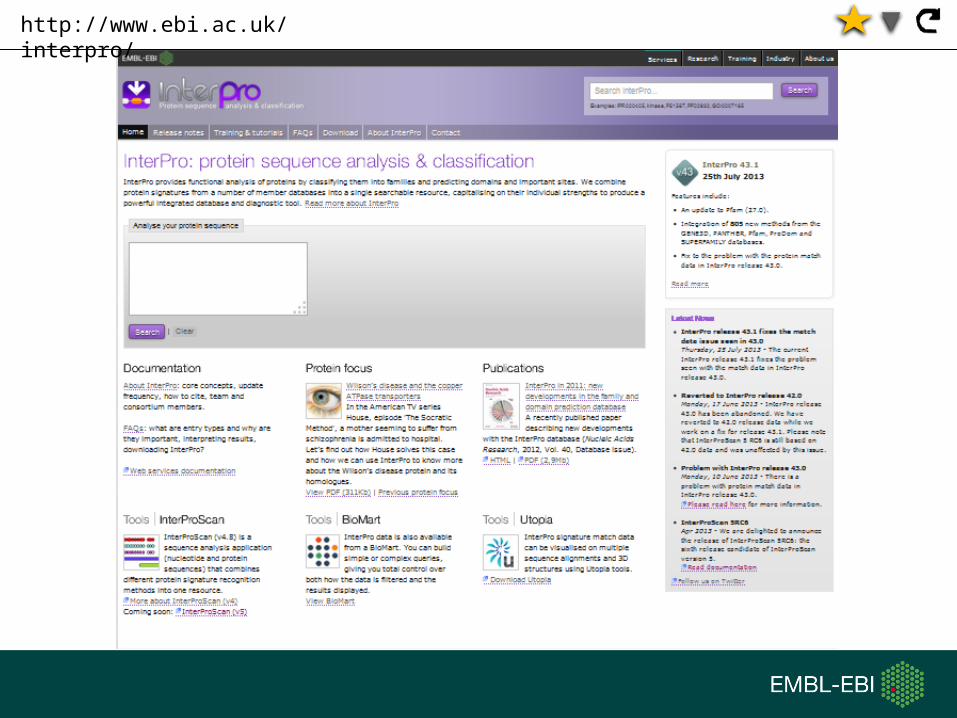
http://www.ebi.ac.uk/interpro/

Search using protein sequences
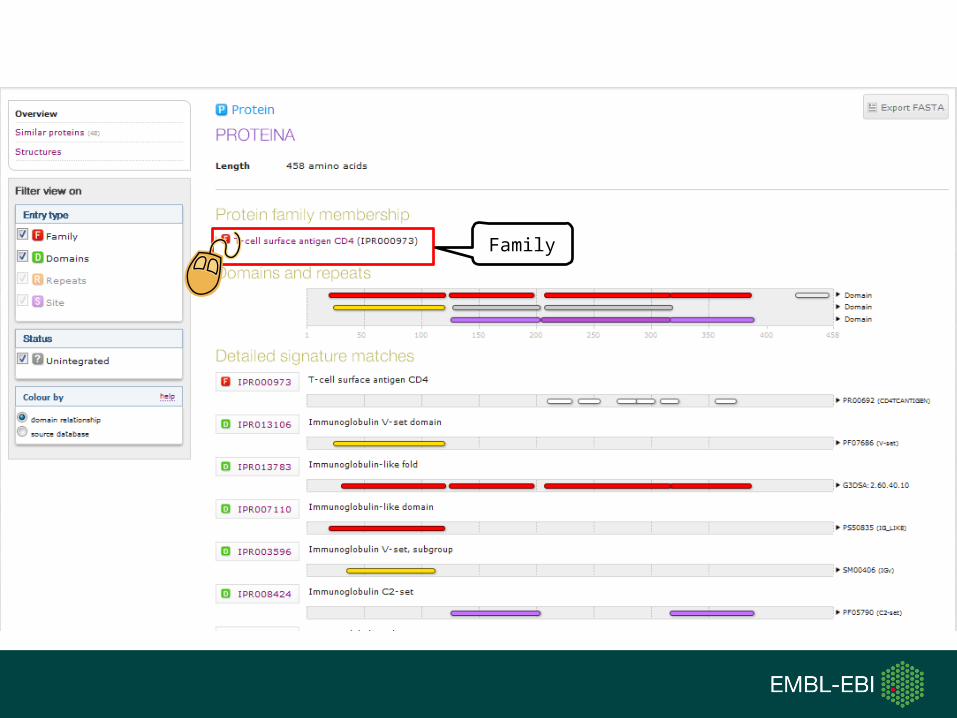
Family

Type

InterPro entry types
Proteins share a common evolutionary origin, as reflected in their related functions, sequences or structure
Family
Distinct functional, structural or sequence units that may exist in a variety of biological contextsDomain
Short sequences typically repeated within a proteinRepeats
PTM Active Site
Binding Site
Conserved Site
Sites

TypeName Identifier Contributing
signatures
Description
GO terms
References
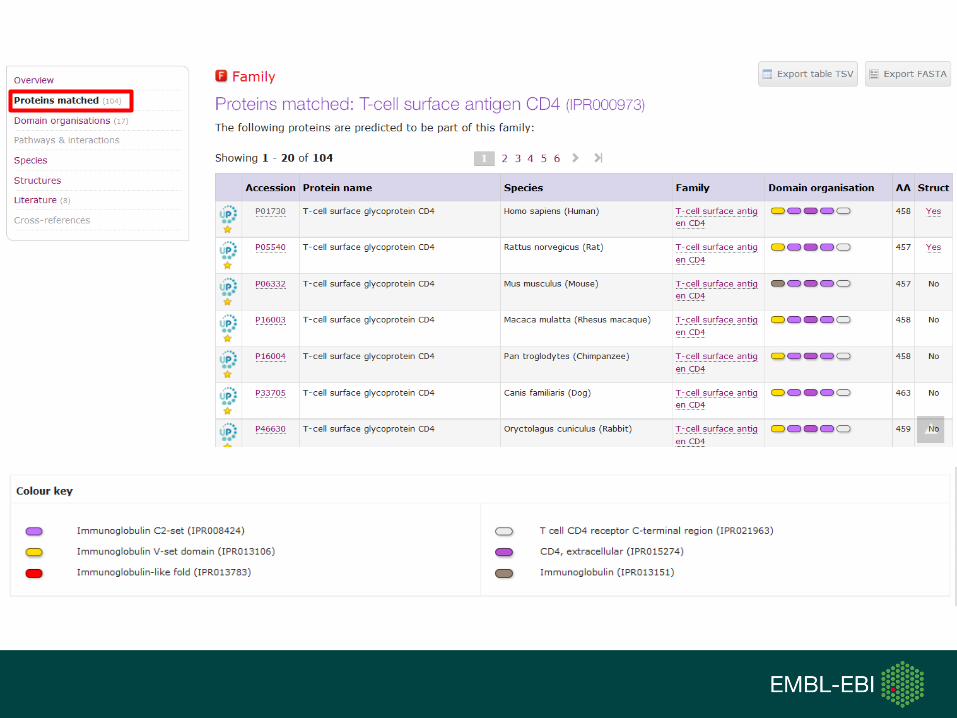
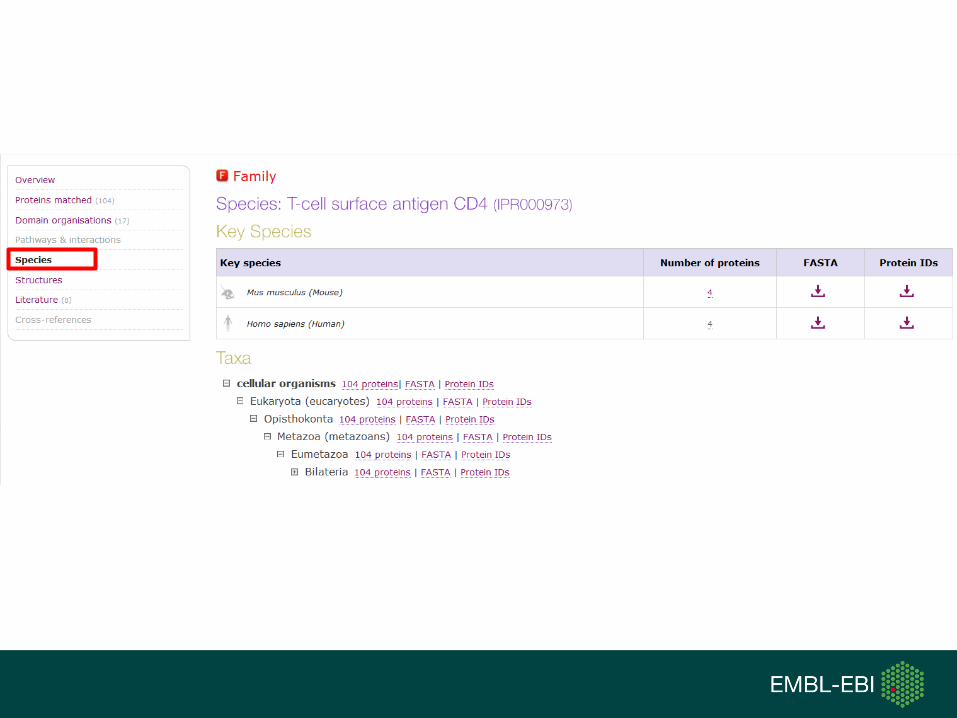


TypeName Identifier Contributing
signatures
Description
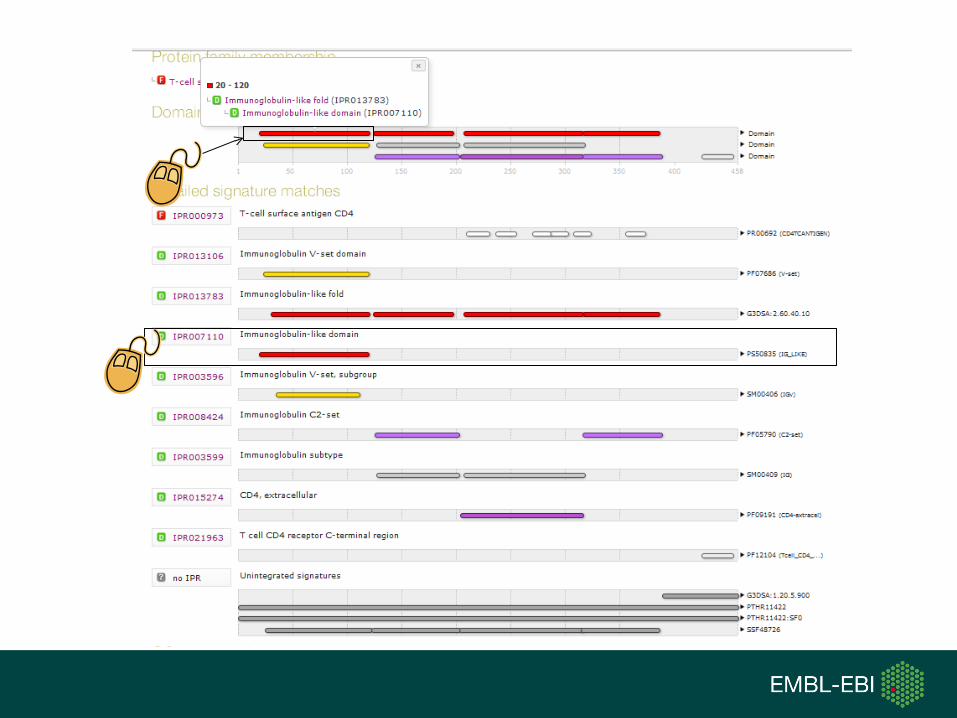
References
Relationships

InterPro family and domain relationships
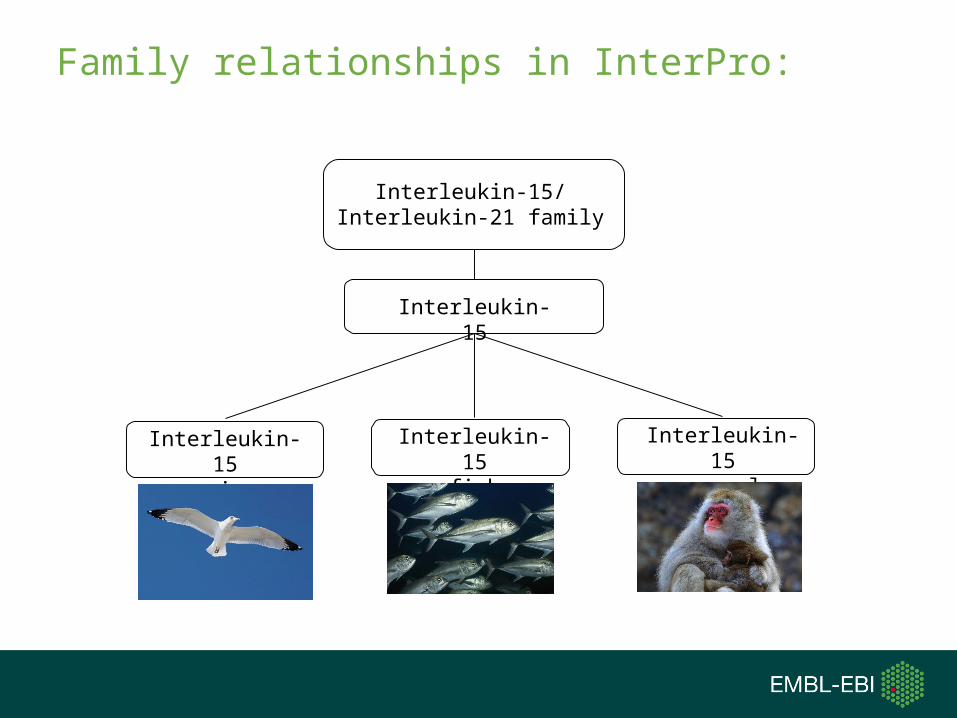
Family relationships in InterPro:
Interleukin-15/Interleukin-21 family
Interleukin-15
Interleukin-15avian
Interleukin-15fish
Interleukin-15mammal

Relationships
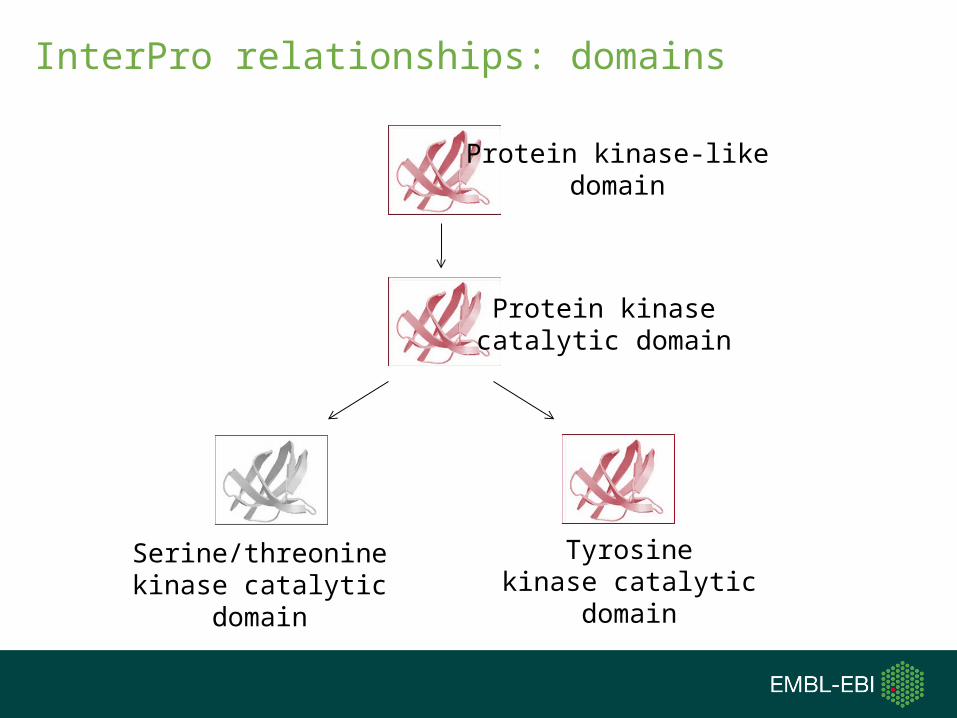
InterPro relationships: domains
Protein kinase-like domain
Protein kinase catalytic domain
Serine/threoninekinase catalytic
domain
Tyrosinekinase catalytic
domain

A brief diversion into the Gene Ontology...

Gene Ontology
• Allow cross-species and/or cross-database comparisons
• Unify the representation of gene and gene product attributes across species
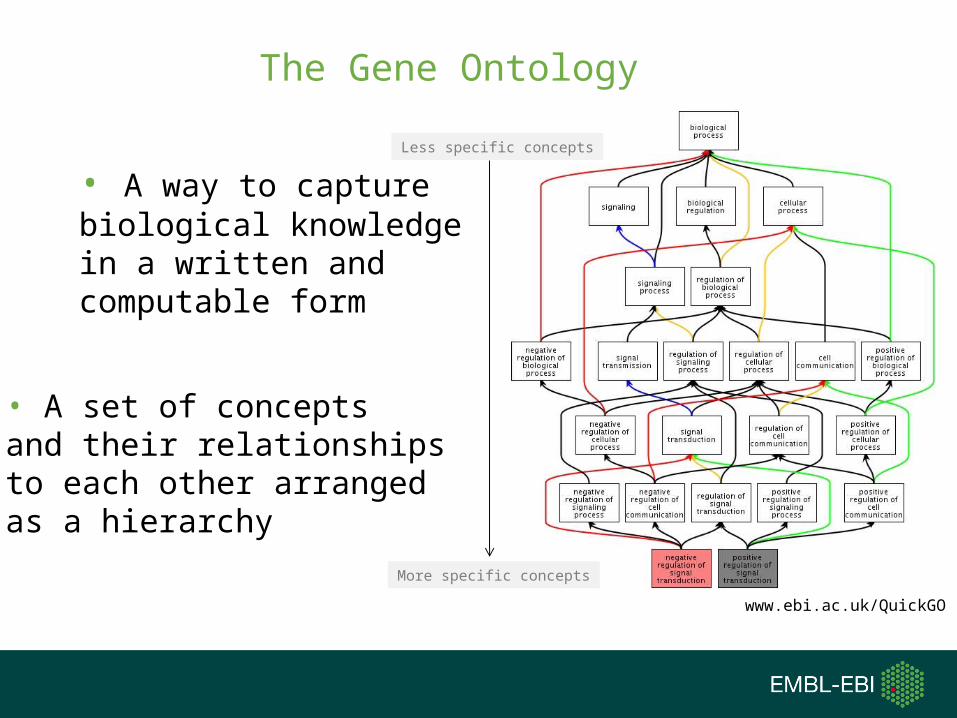
• A way to capture biological knowledge in a written and computable form
The Gene Ontology
• A set of concepts and their relationships to each other arrangedas a hierarchy
www.ebi.ac.uk/QuickGO
Less specific concepts
More specific concepts
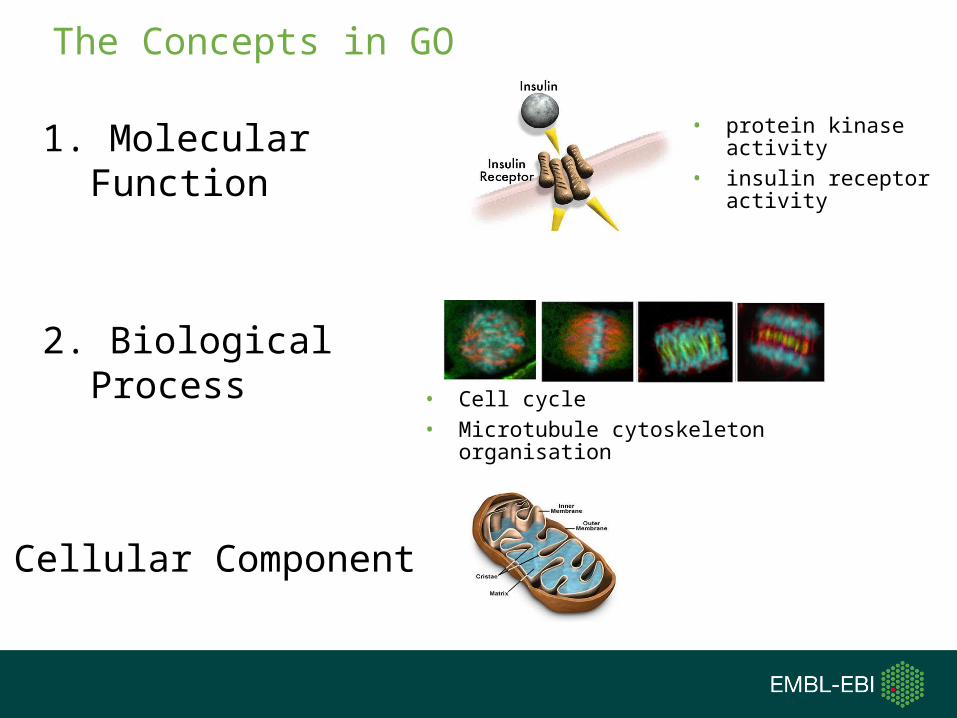
The Concepts in GO
1. Molecular Function
2. Biological Process
3. Cellular Component
• protein kinase activity• insulin receptor activity
• Cell cycle• Microtubule cytoskeleton organisation

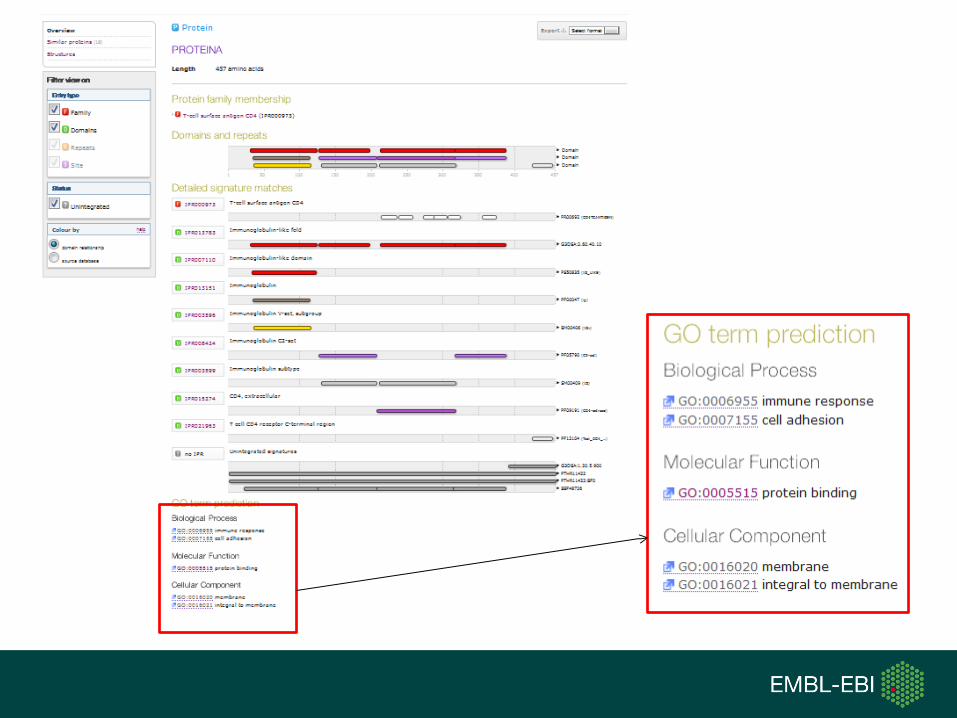
GO:0006955 Immune responseGO:0016020 membrane

Summary
Its member databases all have their particular niche or focus......but InterPro offers a combination of all their areas of expertise!
InterPro is a sequence analysis resource that classifies sequences into protein families and predicts important domains and sites
It uses protein signatures based on different methodologies from different member databases

Why use InterPro?
• Large amounts of manually curated data
• 35,634 signatures integrated into 25,214 entries
• Cites 38,877 PubMed publications
• Large coverage of protein sequence space
• Regularly updated
• ~ 8 week release schedule
• New signatures added
• Scanned against latest version of UniProtKB
Caution
We need your feedback!missing/additional referencesreporting problemsrequests
• InterPro is a predictive protein signature database - results are predictions, and should be treated as such
• InterPro entries are based on signatures supplied to us by our member databases
....this means no signature, no entry!
EBI support page.
And one more thing…..

The InterPro Team:
Amaia Sangrador
Craig McAnulla
MatthewFraser
Maxim Scheremetjew
Siew-Yit Yong
Alex Mitchell
Sebastien Pesseat
SarahHunter
GiftNuka
Hsin-YuChang
LouiseDaugherty
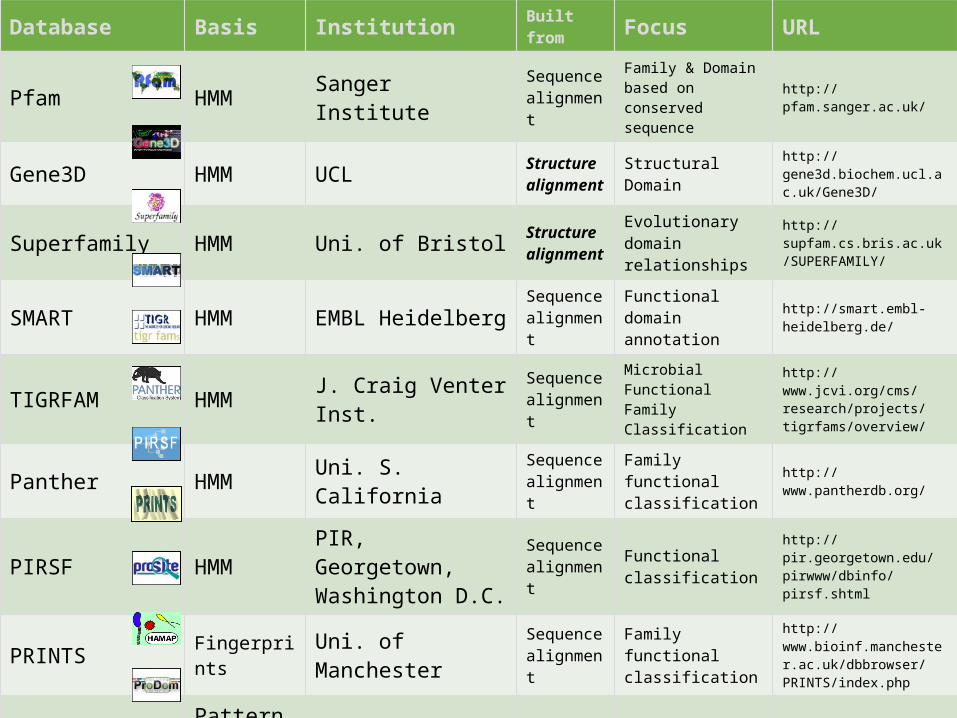
Database Basis Institution Built from Focus URL
Pfam HMM Sanger Institute Sequence alignment
Family & Domain based on conserved sequence
http://pfam.sanger.ac.uk/
Gene3D HMM UCL Structure alignment
Structural Domainhttp://gene3d.biochem.ucl.ac.uk/Gene3D/
Superfamily HMM Uni. of Bristol Structure alignment
Evolutionary domain relationships
http://supfam.cs.bris.ac.uk/SUPERFAMILY/
SMART HMM EMBL Heidelberg Sequence alignment
Functional domain annotation
http://smart.embl-heidelberg.de/
TIGRFAM HMM J. Craig Venter Inst. Sequence alignment
Microbial Functional Family Classification
http://www.jcvi.org/cms/research/projects/tigrfams/overview/
Panther HMM Uni. S. California Sequence alignment
Family functional classification
http://www.pantherdb.org/
PIRSF HMM PIR, Georgetown, Washington D.C.
Sequence alignment
Functional classification
http://pir.georgetown.edu/pirwww/dbinfo/pirsf.shtml
PRINTS Fingerprints Uni. of Manchester Sequence alignment
Family functional classification
http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/index.php
PROSITE Patterns & Profiles SIB Sequence
alignmentFunctional annotation
http://expasy.org/prosite/
HAMAP Profiles SIB Sequence alignment
Microbial protein family classification
http://expasy.org/sprot/hamap/
ProDom Sequence clustering
PRABI : Rhône-Alpes Bioinformatics Center
Sequence alignment
Conserved domain prediction
http://prodom.prabi.fr/prodom/current/html/home.php

Thank you!
www.ebi.ac.uk
Twitter: @emblebi
Facebook: EMBLEBI
YouTube: EMBLMedia


















