
Profundizando en el riesgo (epi)genético a cáncer colorrectal...
185
Profundizando en el riesgo (epi)genético a cáncer colorrectal: Nuevos genes responsables y marcadores moleculares para el cribado Eva Hernández Illán
Transcript of Profundizando en el riesgo (epi)genético a cáncer colorrectal...

Profundizando en el riesgo (epi)genético a cáncer colorrectal: Nuevos genes responsables y marcadores moleculares para el cribado
Eva Hernández Illán

Profundizando en el riesgo (epi)genético a cáncer
colorrectal: Nuevos genes responsables y
marcadores moleculares para el cribado
EVA HERNÁNDEZ ILLÁN
TESIS DOCTORAL
Marzo 2016


Departamento de Fisiología, Genética y Microbiología.
Facultad de Ciencias.
Fundación FISABIO-ISABIAL. Hospital General Universitario de Alicante.
Hospital General Universitario de Elche.
Profundizando en el riesgo (epi)genético a cáncer
colorrectal: Nuevos genes responsables y marcadores
moleculares para el cribado
EVA HERNÁNDEZ ILLÁN
Tesis presentada para aspirar al grado de DOCTORA POR LA
UNIVERSIDAD DE ALICANTE
MENCIÓN DE DOCTORA INTERNACIONAL
Programa de doctorado Biología Experimental y Aplicada.
Dirigida por:
Dra. Adela Castillejo Castillo Dr. Rodrigo Jover Martínez Dr. José Luis Soto Martínez
Marzo 2016


Departament of Physiology, Genetics and microbiology.
Faculty of Sciences.
FISABIO-ISABIAL Fundation. University General Hospital of Alicante.
University General Hospital of Elche.
Understanding further the (epi)genetic risk of colorectal
cancer: New candidate genes and molecular markers for
screening
EVA HERNÁNDEZ ILLÁN
DOCTORAL THESIS TO CONFER THE TITLE OF “INTERNATIONAL DOCTOR”
BY THE UNIVERSITY OF ALICANTE
PhD program of Experimental and Applied Biology
PhD Directors:
Dra. Adela Castillejo Castillo Dr. Rodrigo Jover Martínez Dr. José Luis Soto Martínez
Marzo 2016


Financiación
La doctoranda es beneficiaria de una Ayuda Predoctoral de Formación en
Salud (PFIS) del Instituto de Salud Carlos III en su convocatoria de 2012:
FI12/00233.
Proyectos de investigación que han financiado este trabajo de tesis doctoral:
Biomarcadores en la prevención del cáncer colorrectal (GCB13131592CAST).
Subproyecto 6: Desarrollo de biomarcadores genéticos y de metilación en
mucosa colorrectal sana.
IP. Antoni Castells; IP subproyecto 6: Rodrigo Jover Martínez.
Fundación Asociación Española Contra el Cáncer
2013-2018
1.200.000 €. Subproyecto 6: 224.880 €
Vía serrada de carcinogénesis en cáncer colorrectal: mecanismos y
marcadores moleculares. Implicaciones en el diagnóstico prevención y
tratamiento (PI11/02630).
Instituto de Salud Carlos III
IP: Rodrigo Jover Martínez.
2012-2014
171.325,11 €
Hipermetilación específica de los alelos mutados en tumores colorrectales con
inestabilidad de microsatélites (GV/2012/008).
Consellería Educación, Formación y Empleo Comunidad Valenciana.
IP: Adela Castillejo Castillo
2012-2013
12.000€
Caracterización clínica, histopatológica y molecula de los carcinomas
colorrectales con pérdida de expresión de MLH1, ausencia de hipermetilación
de su promotor y sin mutación en línea germinal (AP170/10).
Becas para el desarrollo de programas de investigación en materias sanitarias
para el año 2010. Consellería de Sanidad. Generalitat Valenciana.
IP: Artemio Payá Romá.
2010
8500€
Caracterización fenotípica y molecular de las poliposis colónicas atenuadas.
Papel de la actividad inflamatoria, resistencia insulínica y factores ambientales
(PI08/0726).
Instituto de Salud Carlos III
IP: Rodrigo Jover Martínez.
2009-2011
166.738€


A mi hermano
A mis padres


Agradecimientos
AGRADECIMIENTOS
En primer lugar, quiero agradecer haber llegado hasta aquí a mis directores de tesis,
por vuestra gran confianza depositada en mí desde el primer momento que pisé el
laboratorio. Rodrigo, gracias por dejarme formar parte de este maravilloso grupo. Por
transmitirme esa forma de ver la ciencia, en la que somos capaces de llegar mucho
más lejos de lo que creemos, y mostrarme el potencial clínico de nuestro trabajo. Por
tu capacidad de sacarnos una sonrisa en cualquier situación, tu optimismo y sosiego.
José Luis, gracias por transmitirme tu gran ilusión por la ciencia y por el permanente
aprendizaje que se consigue a tu lado. Por esa eterna disposición a ayudar y tu
asesoramiento 24h. Por abrirme la puerta a este mundo. Adela, eres mi mejor maestra
de la bancada. Gracias por tu paciencia, desde 3 columnas, 6 columnas hasta una
placa de PCR entera. Gracias por motivarme a superar los objetivos y por estar
siempre dispuesta a ayudarme.
Gracias al grupo EPICOLON, al grupo EPIPOLIP y al Programa de Cáncer Hereditario
de la Comunidad Valenciana, por su gran disposición y por permitirme sacar este
trabajo adelante. Gracias al equipo del laboratorio de Instituto Catalán de Oncología.
Sin vuestra colaboración esto no habría sido posible.
Gracias a los pacientes, porque todo esto es por y para ellos.
Porque esta aventura empezó en Elche mucho antes de mi matrícula en el doctorado,
así, tengo que agradecer en primer lugar a Isa la ayuda y paciencia desde el primer
día, te he dicho mil veces que eres incomparable profesional y personalmente. Eres el
motor del laboratorio. A Víctor, por enseñarme y ayudarme tanto en todo momento, por
ver siempre potencial en mi. A Espe, al final te adelantaste! Pero ahora volvemos a
empezar las dos, cada una una aventura diferente, en la que te deseo lo mejor! A los
demás compañeros del lab: Nati, Trini, Pilar y Miguel… A la mejor compañera de
comidas, Arantxa, por contagiarnos tu bonita sonrisa. A Vicky.
I would also like to thank all the colleagues at The Roslin Institute, during my time in
Edinburgh. Quería agradecer especialmente a Albert por acogerme y ayudarme en
todo momento. And also to Isobel, Suzanne and Amy, willing to help me whenever I
needed it. Thank you all for your patience with me. Enrique and Ricardo (English?
Spanish?) you were essential on that team too. Por otro lado, Zoe, Inga, Reyes y
Marta, fue maravilloso descubriros en aquella aventura, you are the reason why that
was one of the most exciting experiences of my life. Thank you for being part of it.

Agradecimientos
Y empieza la trayectoria en Alicante. Y tengo que empezar por ti Carla, que te conocí
mucho antes en Elche, cuando ya me impresionaste por el mero hecho de ser la
primera licenciada en Biotecnología que conocía, pero a partir de ahí has seguido
impresionándome, por todo lo que sabes, por tu gran capacidad de trabajo, tu enorme
compañerismo y como no, tu incansable espíritu de lucha. Gracias por hacerme parte
del grupo. Y las que también estáis en mi trayectoria a mitad camino entre Alicante y
Elche sois Ceci y Ana. Ceci, gracias por tu siempre desinteresada ayuda, por tu
templanza y por enseñarnos tanto, pero sobre todo a ser mejores personas. ¿Quién te
cantará que you are breaking my heart cuando nos separemos? Y Ana, desde aquel
verano de eternas sangres me llevas aguantando, y espero que muchos más, aunque
sea para ir a ver las pelis de Quim. Muchas gracias por enseñarme y apoyarme. Y
ahora sí, te toca Miri! ayyy cómo vamos a llorar cuando nos separemos, te grabaré
audios con sermones que ya te sabes de memoria. ¿Sabes que, aunque no te gusten
estas cosas, deberías compartir la autoría de la tesis conmigo no? Sin ti, esto no
podría haber sido posible. Eres una gran trabajadora y mejor persona; con un corazón
gigante que a lo mejor guardas en ese piso que no nos quieres enseñar. María,
demasiado poco tiempo coincidimos en el lab, pero el suficiente para descubrirte. Creo
que no soy la única que envidia tu determinación, tu seguridad en ti misma y tu
optimismo. Muchas gracias por tu gran ayuda y estar siempre disponible . Cristina
Alenda, muchas gracias por tu incondicional ayuda y tu capacidad de gestión y
solución de problemas, no sé qué sería del sector colon-UI sin ti. Gracias a Fany y el
resto del equipo de anatomía patológica del HGUA, imprescindibles en esta tesis. A
Lucía, que aunque casi no coincidiéramos, hiciste que “colon” pudiera ser un gran
grupo a día de hoy.
Y Araceli, aquí te ubico. Me encanta que formes parte del binomio colon-mama que
tenemos por despacho. Te deseo mucha suerte en tu futura etapa, porque te la
mereces. Laura, el diccionario de la UI con patas! Enhorabuena! El 2016 ahora sólo
puede ir a mejor, gracias por ser nuestra guía particular. También quiero agradecer al
resto de compañeros de la UI, por hacer el día a día más ameno: a Sandra, por
ayudarnos siempre; a Fer y a Reyes, que aunque os fuisteis muy pronto, sois un pilar
fundamental; a Elena y Eloy, también nos dejasteis muy pronto; a Bea y a Pura
(¿quién nos diría en el banquillo de las cadetes que aparecerías tú por aquí?); al
equipo micro –Antonio, Noemí e Inma- más vale tarde que nunca.
A los compañeros del Ciber, especialmente Paula, Alba, Bea y José Manuel, y a
Rubén, por facilitarnos siempre las cosas. A las chicas de la Fundación, especialmente

Agradecimientos
a Gema y Silvia, por su paciencia, disposición y encontrar siempre una solución a
todos nuestros problemas. Gema gracias por tus consejos.
Y como no, quiero dar las gracias a los compañeros del CEK de Barcelona por
acogerme: En primer lugar, a Jordi y a Francesc, para mi sois el cuarto y quinto
director de esta tesis. Muchas gracias por permitirme vivir esa experiencia y aceptarme
como una más. Y también especialmente a Jenny, Elena y Claudia, por su gran ayuda
y disponibilidad, y a Mini, por ser tan bonita. Pero también quiero dar las gracias a
Irene (qué penita haberte conocido tan tarde), Esther, Clara, Helena, Saray, Keyvan,
Sebas, Isa, Juanjo, y como no a Meritxell y a Sergi, por hacer de esa estancia una
etapa tan agradable.
Y siguiendo en Barcelona, en el Bar Camen and Co: Ana, Óscar, Carmen, Tania y
Gabri, sois muy bonicos, gracias por hacerme sentir como en casa. Ana, gracias por
estar ahí en tantos momentos de mi vida, las aventuras que hemos vivido son nada en
comparación con las que nos esperan! Y a Laura, el fondo norte de la calle Córcega.
Y bajando al sur tengo que parar por Valencia. Gracias a mis amores del poli y
especialmente a Eli, Carla y Mar, que con nuestros skypes a cuatro cada una en un
lado, sois parte imprescindible de esta historia.
Gracias a mis amigo/as de Ori que me hacen empezar todos los lunes con una
sonrisa. En especial a Nerea, te pongo aquí aunque eres parte de la familia. Gracias
por estar siempre a mi lado, desde antes del Gato con Botas hasta Bogotá y lo que
vendrá (RyM). A Emilio, por abrirme siempre la puerta con una sonrisa, eres un sol. A
Karlos, gracias por estar siempre, estando o sin estar, por confiar en mi más que yo
misma, por ser mi psicólogo particular y el mejor amigo. Gracias a María (por tu
felicidad, eterna comprensión y ánimo), Rocío (has llegado tarde pero pegando fuerte,
me encantas), Laura (siempre sacándonos sonrisas), y Arezu (por tu templanza, tu
alegría y tus reflexiones zen), sois imprescindibles y me encanta haberlo descubierto.
A Sito (por compartir esa forma de ver el mundo), a Marta G (por su energía y sus
martadas), a Taty y a Mari (por estar siempre ahí). Gracias a mis amores de Mar Azul,
especialmente a Paula y Andrea, por estar ahí contra viento y marea, creciendo juntas
como personas diferentes pero preciosas. Y a todos los demás, gracias.
A mi madre, a mi padre y a mi hermano (y a la otra rubia loca de la familia que me
alegra cada finde). Sin vosotros nada de esto sería posible, por creer en mí y guiarme
en esta aventura, gracias. Vicen gracias por sentarte conmigo y ayudarme a orientar
mi camino. A mi tía Nieves.


Abreviaturas
ABREVIATURAS
ACVR2A Activin A receptor type IIA
ADN Ácido desoxirribonucleico
APAF Poliposis Adenomatosa Familiar Atenuada
APC Adenomatous Polyposis Coli
ARNm Ácido ribonucleico mensajero
BAX BCL2-associated X protein
BMPR1A Bone morphogenetic protein receptor type IA
BRAF V-raf murine sarcoma viral oncogene homolog B1
BRCA2 Gen que codifica para el Breast Cancer 2
CCHNP Cáncer Colorrectal Hereditario No Polipósico
CCR Cáncer Colorrectal
CDKN2A Cyclin-dependent kinase inhibitor 2A
CE Cáncer de Endometrio
CIMP CpG Island Methylator Phenotype (Fenotipo metilador en islas CpG)
CIN Chromosome Instability (Inestabilidad cromosómica)
DCC DCC netrin 1 receptor
Dx Diagnóstico
EPCAM Epithelial cell adhesion molecule
FAN1 FANCD2/FANCI-associated nuclease 1
FBXW7 F-box and WD repeat domain containing 7
FDA Food and Drugs Administration
GALNT12 Polypeptide N-acetylgalactosaminyltransferase 12
GIST Gastrointestinal stromal tumor (Tumor gastrointestinal estromal)
GREM1 Gremlin 1, DAN family BMP antagonist
HGUA Hospital General Universitario de Alicante
HGUE Hospital General Universitario de Elche

Abreviaturas
HNRNPA0 Heterogeneous nuclear ribonucleoprotein A0
ICO Instituto Catalán de Oncología
IHQ Inmunohistoquímica
IMS Microsatellite Instability (Inestabilidad de microsatélites)
kDa kiloDalton
KRAS V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
LINE-1 Long interspersed nuclear element-1
LOH Loss of Heterozygosity (Pérdida de heterocigosidad)
LRRFIP2 Leucine rich repeat (in FLII) interacting protein 2
MAP MUTYH-associated polyposis (Poliposis asociada a MUTYH)
MGMT O-6-methylguanine-DNA methyltransferase
MLH1 MutL homolog 1, colon cancer, nonpolyposis type 2 (E. coli)
MLH1 Gen que codifica para el MutL homolog 1, colon cancer, nonpolyposis
type 2 (E. coli)
MLPA Multiplex Ligation-Dependent Probe Amplification
MMR Mismatch Repair (Sistema reparador de errores)
MSH2 MutS homolog 2, colon cancer, nonpolyposis type 1 (E. coli)
MSH2 Gen que codifica para el MutS homolog 2, colon cancer, nonpolyposis
type 1 (E. coli)
MSH6 MutS homolog 6, colon cancer, nonpolyposis type 1 (E. coli)
MSH6 Gen que codifica para el MutS homolog 6, colon cancer, nonpolyposis
type 1 (E. coli)
MS-MLPA Methylation-Specific Multiplex Ligation-Dependent Probe Amplification
MSS Microsatellite Stability (Estabilidad de microsatélites)
MUTYH MutY DNA glycosylase
NGS Next Generation Sequencing (secuenciación de nueva generación)
NRAS Neuroblastoma RAS viral (v-ras) oncogene homolog
NTHL1 Nth-like DNA glycosylase 1
OMS Organización Mundial de la Salud
PAF Poliposis Adenomatosa Familiar

Abreviaturas
pb Pares de bases
PCR Polymerase chain reaction (Reacción en cadena de la polimerasa)
PIK3CA Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
PMS2 PMS2 postmeiotic segregation increased 2 (S. cerevisiae)
PMS2 Gen que codifica para el PMS2 postmeiotic segregation increased 2 (S.
cerevisiae)
POLD1 Polymerase (DNA directed), delta 1, catalytic subunit
POLE Polymerase (DNA directed), epsilon, catalytic subunit
PPAP Polymerase proof-reading asociated polyposis (Poliposis asociada a la
actividad correctora de errores de las polimerasas)
PTEN Phosphatase and tensin homolog
qPCR PCR cuantitativa
RPS20 Ribosomal protein S20
SEMA4A Sema domain, immunoglobulin domain (Ig), transmembrane domain
(TM) and short cytoplasmic domain, (semaphorin) 4A
SIR Standarized Incidence Ratio (Tasa de Incidencia Estandarizada)
SMAD2 SMAD family member 2
SMAD4 SMAD family member 4
SNC Sistema Nervioso Central
STK11 Serine/threonine kinase 11
TCGA The Cancer Genome Atlas Network
TGBR2 Transforming Growth Factor beta receptor 2
TGF-beta Transforming Growth Factor beta
TMA Tissue Microarray
TP53 Tumor protein p53
VSI Variante de Significado Incierto
WIF1 WNT inhibitory factor 1


Índice
INDICE
Resumen de la Tesis Doctoral i
Thesis Abstract iii
1. INTRODUCCIÓN 1
1.1. El cáncer colorrectal 1
1.1.1. Epidemiología del CCR 1
1.1.2. Etiología del CCR 2
1.1.3. Estadiaje y tratamiento 5
1.1.3. Vías de carcinogénesis colorrectal 6
1.1.3.1. Vía de la Inestabilidad cromosómica (CIN) 7
1.1.3.2. Vía de la Inestabilidad de microsatélites (IMS) 8
1.1.3.3. Vía del fenotipo metilador (CIMP) o Vía Serrada 10
1.2. Cáncer colorrectal hereditario no polipósico 11
1.2.1. Síndrome de Lynch 11
1.2.1.1. Generalidades 11
1.2.1.2. Características clínicas y de vigilancia 13
1.2.1.3. Criterios diagnósticos 15
1.2.1.4. Algoritmo diagnóstico del SL 17
1.2.1.5. Tipo de mutaciones en el SL 19
1.2.1.5.1. Epimutaciones constitucionales en MLH1 20
1.2.2. CCR familiar tipo X 23
1.3. Síndromes de poliposis familiar 24
1.3.1. Poliposis adenomatosa familiar 25
1.3.2. Poliposis asociada a MUTYH 26

Indice
1.3.3. Otros síndromes polipósicos de predisposición a CCR 28
1.4. Poliposis asociada a la actividad correctora de errores de las
polimerasas (PPAP) 29
1.4.1. Función de POLE y POLD1 y su implicación en el proceso carcinogénico 30
1.4.2. Mutaciones de POLE y POLD1 descritas en cáncer 31
1.4.2.1. Mutaciones germinales en POLE y POLD1 31
1.4.2.2. Mutaciones somáticas en POLE y POLD1 33
2. HIPÓTESIS 39
3. OBJETIVOS 43
4. MATERIAL Y MÉTODOS 47
4.1. Artículo 1 47
4.1.1. Pacientes y muestras biológicas 47
4.1.2. Plan de trabajo 48
4.1.3. Cribado molecular de los tumores sospechosos de SL 49
4.1.3.1. Inmunohistoquímica de las proteínas MMR 49
4.1.3.2. Inestabilidad de microsatélites 49
4.1.3.3. Análisis de la metilación de MLH1 50
4.1.3.4. Análisis de la mutación de BRAF V600E 51
4.1.3.4.1. Genotipado por qPCR 51
4.1.3.4.2. Secuenciación Sanger 51
4.1.3.5. Análisis de mutaciones germinales en los genes MMR 52
4.1.3.6. Análisis de la secuencia de la región promotora de MLH1 53
4.1.4. Análisis estadístico 53
4.2. Artículo 2 55
4.2.1. Pacientes y muestras biológicas 55

Índice
4.2.2. Plan de trabajo 57
4.2.3. Estudio de las mutaciones germinales en POLE y POLD1 58
4.2.3.1. Serie 1 (Ensayos de genotipado KASPAR) 58
4.2.3.2. Serie 2 (Secuenciación Sanger) 59
4.2.4. Análisis de predicción in silico 59
4.2.5. Estudio de pérdida de heterocigosidad (LOH) en POLE 60
4.2.5.1. Mateo por microsatélites 60
4.2.5.2. SNAPshot para la mutación POLE_Leu424Val 60
4.2.6. Análisis de las mutaciones somáticas en BRAF, KRAS y NRAS 61
5. RESULTADOS 65
5.1. Artículo 1 67
5.2. Artículo 2 75
6. DISCUSIÓN 87
7. CONCLUSIONES 105
7. BIBLIOGRAFÍA 109
8. ANEXOS 125
ANEXO I. Algoritmo de estudio genético de SL siguiendo la Estrategia Universal 125
ANEXO II. Algoritmo de estudio genético de SL en pacientes que cumplen
criterios de Bethesda 127
ANEXO III. Algoritmo de estudio genético de poliposis adenomatosa familiar 129
ANEXO IV. Mutaciones en los dominios exonucleasa de POLE y POLD1
descritas en pacientes oncológicos previas a la publicación del Artículo 2 131
ANEXO V. Cebadores y condiciones empleados para la amplificación de
regiones de distintos genes 133

Indice
ANEXO VI. Árboles genealógicos de las cinco familias con epimutaciones
del artículo 1 135
ANEXO VII. Árboles genealógicos de las nuevas familias descritas en
nuestra serie con las mutaciones POLE_Leu424Val y POLD1_Leu474Pro
posteriores a la publicación del segundo artículo 137
ANEXO VIII. Identificación de las tres mutaciones recurrentes de POLE y POLD1 en las nuevas publicaciones posteriores al segundo artículo de esta tesis 139
ANEXO IX. Nuevas mutaciones germinales identificadas en los dominios
exonucleasa de POLE y POLD1 en pacientes con cáncer posteriores a la
publicación del segundo artículo de esta tesis. 141
ANEXO X. Propuesta de características clínicas para la sospecha de PPAP 143
ANEXO XI. Trabajo realizado por la doctoranda en esta tesis doctoral 145
ANEXO XII. Otras publicaciones en las que ha participado la doctoranda 147



Resumen de la Tesis Doctoral
i
RESUMEN DE LA TESIS DOCTORAL
El cáncer colorrectal (CCR) constituye en la actualidad la neoplasia más frecuente en
España. Hasta un 30% de los casos de CCR se incluyen dentro del CCR familiar y
alrededor de un 3-5% se deben a mutaciones en genes de elevada penetrancia que
siguen una herencia mendeliana Éstos últimos incluyen al CCR hereditario no
polipósico: principalmente el Síndrome de Lynch (SL), debido a mutaciones germinales
en los genes de sistema reparador de errores (MMR) MLH1, MSH2, MSH6 y PMS2; y
varios síndromes polipósicos, como la poliposis adenomatosa familiar, asociada al gen
APC y la poliposis asociada al gen MUTYH.
En aproximadamente un 70% de los casos analizados de pacientes con sospecha de
SL no se encuentran mutaciones germinales y en torno a un 10% de estos casos, con
pérdida de expresión de MLH1 e hipermetilación del promotor de dicho gen en su
tumor, se explican por epimutaciones constitucionales en MLH1. No obstante, la
prevalencia de este fenómeno en la población general es desconocida.
También existen casos de CCR con elevada carga familiar, pero sin alteración del
sistema MMR, de los que se desconoce su origen genético (CCR familiar tipo X). Lo
mismo ocurre para algunos casos de poliposis adenomatosa con agregación familiar,
que no se explican por mutaciones en los genes APC o MUTYH. Recientemente se
han descrito dos mutaciones recurrentes en los genes POLE y POLD1 (p.Leu424Val y
p.Ser478Asn respectivamente) como responsables de un pequeño porcentaje de estos
casos de CCR y poliposis no clasificados pero, debido a la escasez de casos descritos
hasta la fecha, fenotipo clínico exacto no se ha definido.
Así, los objetivos de este trabajo han sido: en primer lugar, determinar la prevalencia
de las epimutaciones constitucionales en MLH1 como una causa de SL en una serie
no seleccionada de CCR y compararla con la prevalencia de este fenómeno en una
serie seleccionada; y en segundo lugar, analizar la prevalencia de las mutaciones
recurrentes en los genes POLE y POLD1 en casos de CCR y poliposis familiar de
aparición temprana, sin mutación en los genes responsables de los síndromes de
sospecha, y así contribuir a definir el fenotipo clínico asociado a las mutaciones en
dichos genes.

Resumen de la Tesis Doctoral
ii
Se evaluó la prevalencia de epimutaciones constitucionales de MLH1 en una cohorte
de CCR no seleccionado, y se comparó con una cohorte de CCR familiar
seleccionado. Así, no se detectaron epimutaciones constitucionales en MLH1 en la
población no seleccionada. No obstante, el 15,6% de la serie seleccionada fueron
positivos para epimutaciones en MLH1. Por otro lado, se realizó la búsqueda de
mutaciones recurrentes en POLE y POLD1 en casos de CCR y/o poliposis familiar
mediante ensayos de genotipado KASPar y/o secuenciación Sanger. Se identificó la
mutación POLE_p.Leu424Val como mutación de novo, ningún caso presentó la
mutación POLD1_p.Ser478Asn, pero se identificó una nueva mutación en POLD1:
p.Leu474Pro, con patogenicidad sustentada mediante diversas aproximaciones, en
una familia de CCR familiar tipo X con también casos de cáncer de endometrio.
Así, estos resultados sugieren una prevalencia insignificante de epimutaciones
constitucionales en MLH1 en cohortes no seleccionadas de CCR. Así, el análisis de
epimutación constitucional en MLH1 debería dirigirse exclusivamente a pacientes que
cumplen los criterios revisados de Bethesda y cuyo tumor presente pérdida de
expresión y metilación de MLH1. Por otro lado, las mutaciones en POLE y POLD1
explican una pequeña proporción de casos de CCR y poliposis familiar. La
secuenciación de al menos estas mutaciones recurrentes debería considerarse en el
diagnóstico genético rutinario de los casos con sospecha de CCR o poliposis
hereditarios, sin mutaciones en los genes de sospecha clásicos.

Thesis Abstract
iii
THESIS ABSTRACT
Colorectal cancer (CRC) is the most common malignancy in Spain nowadays. Up to
30% of CRCs are included within the familial CRC and about 3-5% of them are caused
by mutations in high penetrance genes that follow a Mendelian inheritance The latter
include hereditary nonpolyposis CRC: mainly Lynch Syndrome (LS), caused by
germline mutations in the mismatch repair (MMR) genes: MLH1, MSH2, MSH6 and
PMS2; and various polyposis syndromes, such as familial adenomatous polyposis,
associated with the APC gene and MUTYH-associated polyposis.
in approximately 70% of the analyzed cases with suspected LS, no pathogenic
mutations are found. Around 10% of these non-informative cases that have tumor
MLH1 loss of expression and promoter hypermethylation of the gene are explained by
constitutional epimutations of MLH1. However, the prevalence of this phenomenon in
general population is unknown.
There are also cases of CRC with high family aggregation, but with proficient MMR
system, where the genetic origin is unknown (familial CRC type X). The same is shown
in some cases of familial adenomatous polyposis, that are not explained by APC either
MUTYH mutations. Recently, two recurrent mutations in the POLE and POLD1 genes
(p.Leu424Val and p.Ser478Asn respectively) has been described as responsible for a
small percentage of these unclassified cases of familiar CRC and polyposis, but due to
the scarcity of cases reported to date, its accurate clinical phenotype has not been
defined yet.
Thus, the aims of this work were: first, to determine the prevalence of constitutional
epimutations in MLH1 as a cause of SL in an unselected CRC series and compare it
with the prevalence of this phenomenon in a selected series; and secondly, to analyze
the prevalence of the two recurrent mutations in POLE and POLD1 in familial CRC and
polyposis cases with early age of onset and no mutation in the genes responsible for
the suspected syndromes, and so contribute to define the clinical phenotype
associated to the mutations in these genes.
We firstly evaluated the prevalence of MLH1 constitutional epimutations among in a
cohort unselected CRC, and compared it to a cohort of selected familial CRC. No
constitutional MLH1 epimutations were detected in the unselected population.

Thesis Abstract
iv
However, 15.6% of the selected series were positive for epimutations in MLH1.
Secondly we analyzed the recurrent mutations in POLE and POLD1 in familial CRC
cases and / or polyposis by Kaspar genotyping assays or Sanger sequencing. The
mutation POLE_p.Leu424Val was identified as de novo mutation in one patient, no
case harbored POLD1_p.Ser478Asn mutation, but a new mutation in POLD1 was
identified: p.Leu474Pro with supported pathogenicity by different approaches, in a
family of familial CRC type X with also cases of endometrial cancer.
Thus, our results suggest a negligible prevalence of MLH1 constitutional epimutations
in non-selected cohort of CRC. Therefore, the constitutional analysis in MLH1
epimutation should be exclusively directed to patients who meet revised Bethesda
criteria and whose tumors show MLH1 loss of expression and methylation. Besides,
mutations in POLE and POLD1 account for a small proportion of familial cases of
polyposis and CRC. Sequencing of at least these recurrent mutations should be
considered on the routine genetic diagnosis of cases with suspected hereditary CRC or
polyposis without mutations in the classical suspected genes.

INTRODUCCIÓN

Introducción
1
1.1. El cáncer colorrectal
1.1.1. Epidemiología del CCR
El cáncer es, junto con las enfermedades cardiovasculares, la principal causa de
morbi-mortalidad en los países desarrollados (1).
El cáncer colorrectal (CCR) representa un problema de salud pública importante y
global, tratándose del tercer cáncer más común en varones en todo el mundo y el
segundo en mujeres. A pesar de ello, es más frecuente en varones que en mujeres.
Datos de Globocan de 2012 reflejan una prevalencia global de CCR en 2012 de 1,59
millones de casos a los 5 años, con una incidencia de 1,36 millones de nuevos casos,
siendo más frecuente en los países desarrollados (2-3). Debido a la mejora en la
prevención, diagnóstico y tratamiento, la tasa de muerte por CCR ha disminuido,
siendo los datos mundiales para 2012 de 694.000 casos. La media de edad de
diagnóstico del CCR es 68 años para los hombres y 72 para las mujeres (4).
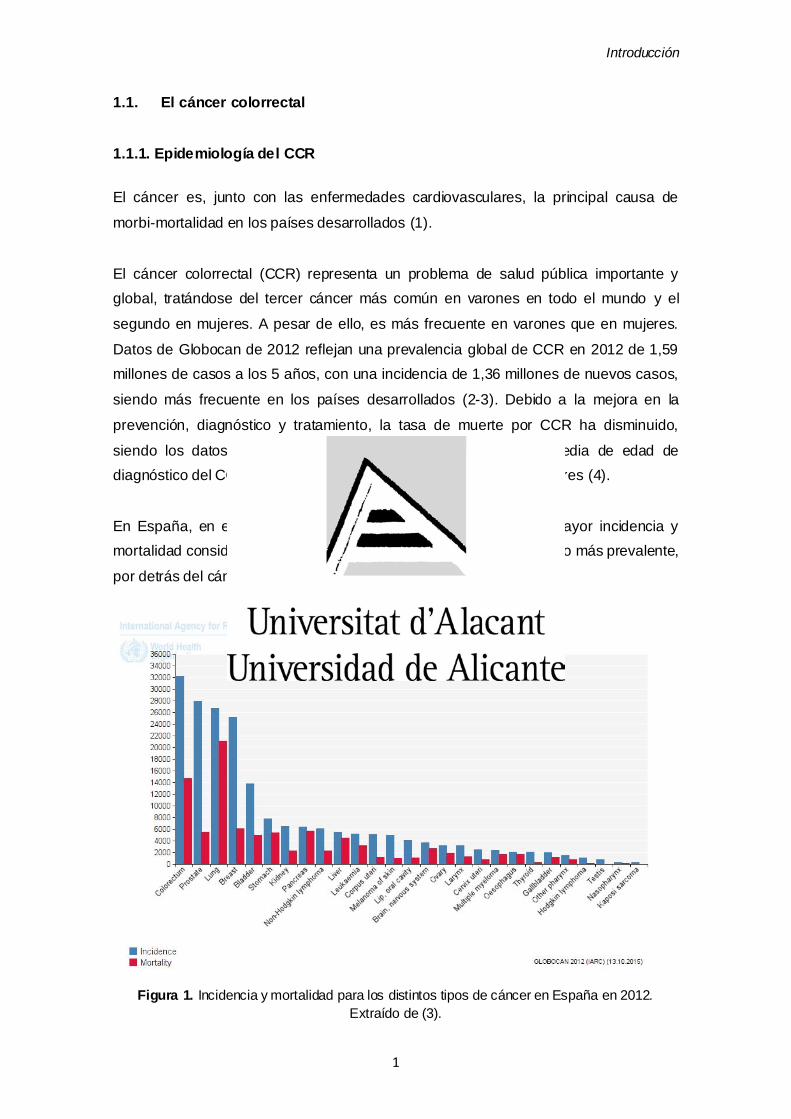
En España, en el año 2012 el CCR fue el tumor que presentó mayor incidencia y
mortalidad considerando ambos sexos (3) (Figura 1), siendo el tercero más prevalente,
por detrás del cáncer de mama y de próstata.
Figura 1. Incidencia y mortalidad para los distintos tipos de cáncer en España en 2012.
Extraído de (3).

Introducción
2
Las estimaciones de la Sociedad Española de Oncología Médica sugieren una
continuación de esta tendencia en la actualidad, diagnosticándose más de 220.000
nuevos casos en 2015, lo que supone un coste sanitario de 37.000€ por paciente
afecto. No obstante, sólo el 39% de los pacientes se diagnostican en estadios
precoces (4), por lo que la implantación de medidas de diagnóstico precoz son
requeridas para frenar el avance de estas cifras.
1.1.2. Etiología del CCR
El cáncer es un conjunto de enfermedades caracterizadas por un crecimiento celular
excesivo y una capacidad de invasión de otros tejidos u órganos. El epitelio colónico
se encuentra en renovación constante; para ello, las células madre de las criptas
migran a la superficie a medida que se van diferenciando. Durante este proceso, las
células pueden acumular alteraciones genéticas (mutaciones) en el ADN (ácido
desoxirribonucleico) y cambios epigenéticos que hacen que dejen de responder a los
mecanismos que regulan el crecimiento celular y controlan la homeostasis tisular.
Cuando este crecimiento excesivo se acompaña de la capacidad de invadir tejidos
circundantes, y diseminarse por todo el torrente sanguíneo o el sistema linfático para
colonizar otros órganos secundarios, aparece la enfermedad metastásica invasiva.
Así, estos hechos son el resultado de la sucesiva acumulación de alteraciones
genéticas y epigenéticas, que superan los umbrales de las tasas de mutación
normales, confiriéndole a la célula una mayor capacidad proliferativa, que sobrepasa
los mecanismos de control de la división celular (5). Por tanto, la inestabilidad genética
o genómica, que proporcione las herramientas celulares para superar las barreras de
homeostasis tisular y tener una ventaja en el crecimiento, es un requisito para el
desarrollo del cáncer.
De esta forma, en el caso del CCR, se pasa por varios estadios de desarrollo: las
células normales del epitelio colónico pasan a ser lesiones benignas o pólipos –
principalmente de histología adenomatosa o serrada-, éstas derivan a
adenocarcinomas totalmente desarrollados pero que no llegan a ser invasivos, y
finalmente aparece la enfermedad metastásica invasiva. No obstante, los adenomas
convencionales habitualmente no progresan, y los que progresan a una lesión maligna
suelen hacerlo de forma lenta. Por ello, las estrategias de cribado mediante
colonoscopias y resección de adenomas son de gran importancia.

Introducción
3
El origen de esta inestabilidad genética es multifactorial: puede ser adquirido por
alteraciones somáticas, como consecuencia de factores ambientales carcinogénicos; o
bien heredado por mutaciones en línea germinal, dando lugar a los cánceres de tipo
esporádico y familiar respectivamente.
La gran mayoría de casos de CCR se deben a alteraciones de tipo somático. Hasta un
30% se incluyen dentro del CCR familiar (aquellos que tienen uno o más familiares con
diagnóstico de CCR) y alrededor de un 3-5% de los casos de CCR se deben a
mutaciones en genes de elevada penetrancia que siguen una herencia mendeliana (6-
7) (Figura 2). Los casos de CCR hereditario pueden ser de tipo polipósico o no
polipósico, en función del número total de pólipos a lo largo del colon que tenga e l
paciente. Los primeros engloban los síndromes de poliposis adenomatosa (cuando el
individuo presenta más de 10 adenomas) y los síndromes hamartomatosos, donde se
incluyen la poliposis juvenil, el síndrome de Cowden y el síndrome de Peutz-Jeghers.
El síndrome de cáncer hereditario más prevalente es el Cáncer Colorrectal Hereditario
No Polipósico (CCHNP), que engloba al Síndrome de Lynch (SL) y al CCR familiar tipo
X, que representa en torno al 1-3% de todos los tumores colorrectales (8), seguido por
la poliposis adenomatosa familiar (PAF) (0,5% del total de CCR), la poliposis asociada
a MUTYH (MAP), y otros síndromes menos frecuentes. La prevalencia de estos
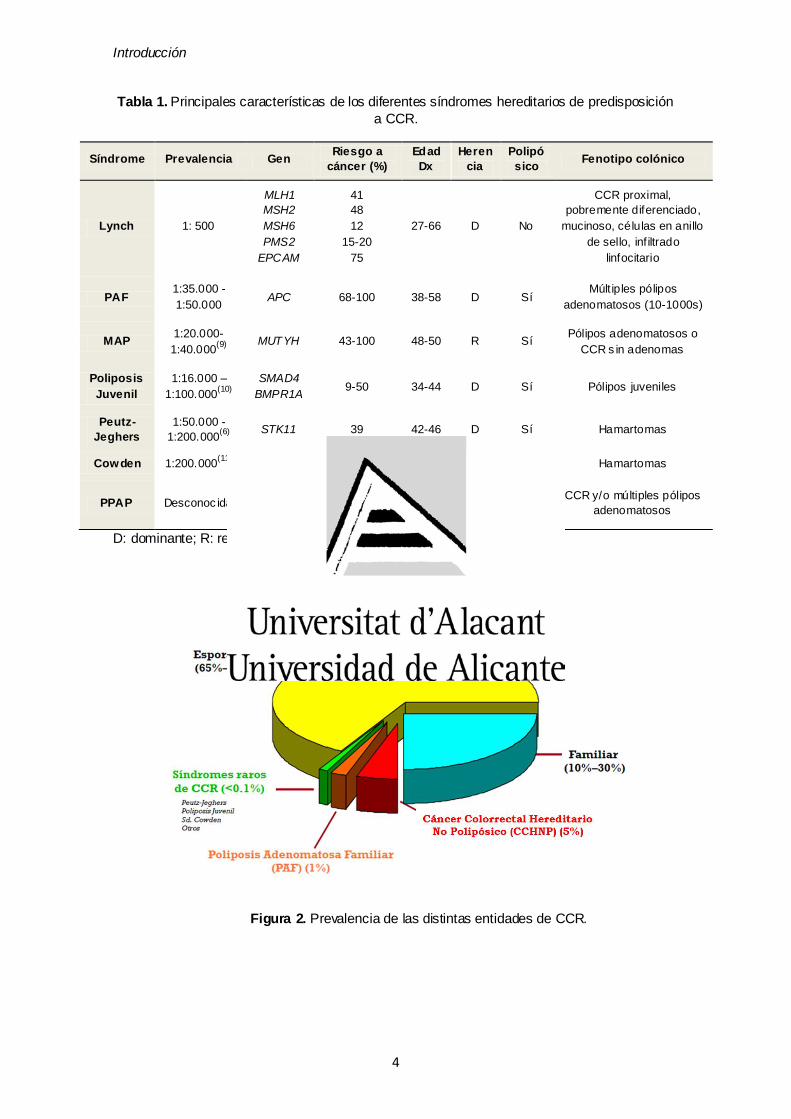
síndromes, los genes responsables, riesgo a CCR a lo largo de la vida y principales
características, se engloban en la tabla 1:
Introducción
4
Tabla 1. Principales características de los diferentes síndromes hereditarios de predisposición
a CCR.
D: dominante; R: recesivo; Dx: diagnóstico
Figura 2. Prevalencia de las distintas entidades de CCR.
Síndrome Prevalencia Gen Riesgo a
cáncer (%)
Edad
Dx
Heren
cia
Polipó
sico Fenotipo colónico
Lynch 1: 500
MLH1
MSH2
MSH6
PMS2
EPCAM
41
48
12
15-20
75
27-66 D No
CCR proximal,
pobremente diferenciado,
mucinoso, células en anillo
de sello, infiltrado
linfocitario
PAF 1:35.000 -
1:50.000 APC 68-100 38-58 D Sí
Múlt iples pólipos
adenomatosos (10-1000s)
MAP 1:20.000-
1:40.000(9)
MUTYH 43-100 48-50 R Sí
Pólipos adenomatosos o
CCR s in adenomas
Poliposis
Juvenil
1:16.000 –
1:100.000(10)
SMAD4
BMPR1A 9-50 34-44 D Sí Pólipos juveniles
Peutz-
Jeghers
1:50.000 -
1:200.000(6)
STK11 39 42-46 D Sí Hamartomas
Cowden 1:200.000(11)
PTEN 9 44-48 D Sí Hamartomas
PPAP Desconoc ida POLE
POLD1 Desconoc ido 20 D Sí
CCR y/o múltiples pólipos
adenomatosos

Introducción
5
1.1.3. Estadiaje y tratamiento
El estadio tumoral es el principal factor pronóstico del CCR, determinando la
supervivencia y el tratamiento de los pacientes.
La clasificación de los distintos estadios del CCR se basa en el sistema TNM, que se
definió en la International Union Against Cancer (12) y que considera la base
anatómica y la biología del tumor. Así, se definen los distintos estadios de CCR en
función del nivel de extensión del tumor primario en las paredes del intestino y la
invasión de los tejidos anexos (T), la presencia o no de afectación de los ganglios
linfáticos (N) y la presencia confirmada de metástasis a distancia (M). La supervivencia
de los pacientes con CCR se relaciona inversamente con estos estadios, según se
refleja en la tabla 2.
Tabla 2. Estadios del CCR y supervivencia. Adaptado de (4, 13-14).
ESTADIO DESCRIPCIÓN Correspondencia TNM Supervivencia
a 5 años (%)
0
Fase más temprana del CCR. Las
células tumorales están situadas en
la parte más superficial de la mucosa
sin traspasarla.
TisN0M0 >95%
I
El tumor afecta a la pared sin
traspasar la capa muscular. No existe
afectación de ganglios linfáticos.
T1N0M0
T2N0M0 74-93,2%
II
El tumor ha infiltrado todas las capas
de la pared. Puede invadir los
órganos de alrededor. No se aprecia
afectación ganglionar.
IIA: T3N0M0
IIB: T4aN0M0
IIC: T4bN0M0
64,5-84,7%
51,6-79,6%
32,3-58,8%
III
El tumor ha invadido los tejidos
anexos y afecta a los ganglios
linfáticos.
IIIA: T1-T2N1M0
IIIB: T3-T4N1M0
IIIC: T(cualquier)N2M0
73,1-83,4%
46,3-64,1%
8-44,3%
IV
El cáncer se ha diseminado
afectando a órganos alejados del
colon o recto como hígado, pulmón o
huesos.
T(cualquier)N(cualquier)
M1 5,7-8,1%
Tis: tumor “in situ”, confinado a la mucosa, que no traspasa las capas de la misma; T1: tumor que invade
la submucosa; T2: tumor que invade la muscularis propia; T3: tumor que llega hasta la subserosa o los
tejidos grasos perirectales; T4a: tumor que invade la superficie del peritoneo; T4b: tumor que invade
tejidos de órganos adyacentes; N0: ausencia de afectación ganglionar; N1: presencia de afectación
tumoral en 1 a 3 glánglios linfáticos perirectales; N2: afectación de 4 o más ganglios linfáticos; M0:
ausencia de metástasis; M1: presencia de metástasis a distancia.

Introducción
6
El tratamiento para los pacientes con cáncer de colon y recto varía según la
localización del tumor y el estadio al momento del diagnóstico. En estadios iniciales (I
y II) la cirugía normalmente es el único tratamiento necesario, siendo más común en
cáncer de colon (94%) que de recto (74%). Sin embargo, en estadios más avanzados
la quimioterapia sola o en combinación con radioterapia antes o después de la cirugía,
es más frecuente, especialmente en cáncer de recto (50-70%) (15).
1.1.3. Vías de carcinogénesis colorrectal
Como se ha comentado anteriormente, la carcinogénesis colorrectal implica varias
etapas secuenciales, en las que primero se forma la lesión benigna, y mediante la
acumulación de alteraciones genéticas y/o epigenéticas a nivel tisular se desarrolla
finalmente el carcinoma invasivo. Así, la inestabilidad genética y/o epigenética es
imprescindible para el desarrollo y avance del tumor; y además, el tipo de alteraciones
que se vayan acumulando va a condicionar en gran manera la historia natural del
tumor y el pronóstico del paciente.
En la actualidad se aceptan tres vías de carcinogénesis colorrectal desde el punto de
vista de las alteraciones moleculares subyacentes: vía de la inestabilidad cromosómica
(CIN), vía de la inestabilidad de microsatélites (IMS) y vía del fenotipo metilador de
islas CpG (CIMP) o vía serrada. Estas tres vías no son excluyentes y puede haber
interacciones entre ellas y tumores que presenten simultáneamente características de
más de una vía (Figura 3). Constantemente se están describiendo nuevos genes e
interacciones entre rutas, matizando la definición y características de cada una de las
vías. Además, en los últimos años se han establecido otros sistemas y vías
involucrados en la patogénesis del CCR, incluyendo aspectos relacionados con la
inflamación o los perfiles de microRNAs (16).

Introducción
7
CIN
IMS
CIMP
Célula madre colon normal
Célula madre colon normal
Célula madre colon normal
FCA displásicas
FCA displásicas
FCA displásicas
Adenoma temprano
Adenoma temprano
Lesión serrada
temprana
Adenoma avanzado
Adenoma avanzado
Adenoma serrado CCR
CCR
CCRCCR
metastásico
CCRmetastásico
CCRmetastásico
KRAS
APCVía señalización Wnt
TP53TGFBR2/SMAD4
PIK3CAKRAS
EMASTOtros factores
MLH1 BRAF
Acumulación de aneuploidía
Acumulación de mutaciones frameshift
Acumulación de genes metilados
Figura 3. Progresión de las distintas vías de carcinogénesis colorrectal e interacción entre las
mismas. Adaptado de (17).
FCA: Focos de criptas aberrantes. EMAST: Elevated microsatellite instability at selected
tetranucleotide repeats.
1.1.3.1. Vía de la Inestabilidad Cromosómica (CIN)
La gran mayoría de los CCR se originan por una inestabilidad generalizada de
tipo cromosómico. En población española representa en torno al 92% de los casos
(18). Estos tumores se desarrollan de acuerdo al modelo clásico de progresión
adenoma-carcinoma propuesto por Vogelstein y Fearon en 1990, produciéndose
numerosas alteraciones que implican la activación en determinados oncogenes
(KRAS) y la inhibición de genes supresores de tumores (APC, TP53, DCC, SMAD4)
(19). Estos tumores se vuelven aneuploides de manera temprana en la progresión,
presentando pérdidas de material genético por errores de recombinación mitótica
(pérdida de heterocigosidad o LOH), así como grandes reordenamientos
cromosómicos que llevan a la progresión tumoral.

Introducción
8
El factor iniciador de la vía es la inactivación del gen supresor de tumores APC (20), lo
que tiene lugar de manera temprana durante el desarrollo de pólipos. Posteriormente,
mutaciones en KRAS aparecen durante el estadio de adenoma, y por último, la
transición a la malignidad coincide con mutaciones en TP53, TGF-beta, PIK3CA y
deleciones del cromosoma 18q (21-24) (Figura 3). En esta vía, generalmente, las
aneuploidías y pérdida de genes supresores han sido asociadas a mal pronóstico (25).
1.1.3.2. Vía de la Inestibilidad de Microsatélites (IMS)
Los microsatélites son secuencias cortas (1-6 pares de bases) repetidas en tándem,
distribuidas por todo el genoma. Su estructura repetitiva las hace proclives a que se
produzca el deslizamiento de la polimerasa durante la replicación previa a la división
celular, produciendo errores de emparejamiento en las hembras de ADN y pequeños
bucles. En condiciones normales, estos errores serán reparados por el sistema de
reparación de ADN de los errores de la replicación (MMR). Este sistema está formado
principalmente por cuatro proteínas: MLH1, MSH2, MSH6 y PMS2. Estas proteínas
forman dos complejos heterodiméricos que, en primer lugar reconocen el error (MSH2-
MSH6) y posteriormente se le une el segundo complejo (MLH1-PMS2) que coordina la
interacción con otras proteínas necesarias para la reparación del mismo en la hebra
recién sintetizada (26).
La pérdida de función de cualquiera de los elementos que intervienen en este
mecanismo produce la acumulación de errores de tipo inserción o deleción (frameshift)
a lo largo de todo el genoma tras la replicación del ADN. Si los microsatélites se
encuentran en la región codificante de los genes, estos errores pueden ocasionar
pérdida de función por generar frameshift, codones de stop prematuros y por
consiguiente, proteínas truncadas y defectuosas. Genes que codifican para procesos
como el control del ciclo celular, apoptosis o la reparación del ADN (oncogenes y
genes supresores de tumores) presentan microsatélites en su región codificante, y por
tanto, son diana de este tipo de mutaciones. Solamente si la pérdida de funcionalidad
de estas proteínas supone una ventaja en la progresión tumoral los clones mutados
serán seleccionados y contribuirán al fenotipo oncológico (26). De esta forma surgen
los tumores con fenotipo IMS, que tienen una elevada tasa de mutaciones puntuales –
fenotipo hipermutador- y pocas anomalías cromosómicas, siendo la gran mayoría de
ellos diploides (26), al contrario que los tumores que siguen la vía CIN (27).

Introducción
9
El principal gen diana de mutación en los tumores que presentan IMS es TGFBR2, que
aparece en el 80% de los casos en lesiones avanzadas como adenomas con displasia
de alto grado (28-29). Otros genes frecuentemente mutados son SMAD2 y SMAD4
(30), ACVR2A (31) y el gen supresor de tumores BAX (32).
Actualmente, el diagnóstico de IMS se realiza de modo consensuado analizando un
panel de cinco marcadores de microsatélites casi monomórficos que muestran una alta
sensibilidad y especificidad para su detección (BAT25, BAT26, NR21, NR24 y NR27).
Patrones alterados en al menos dos de los cinco marcadores evidencian fenotipo IMS-
alta, cuando un solo marcador está alterado se considera IMS-baja y la ausencia de
alteración en los cinco marcadores define un tumor estable (MSS) (33) (Figura 4). El
estudio de la actividad de las proteínas MMR mediante inmunohistoquímica (IHQ)
también se emplea activamente para la detección indirecta de la IMS, ya que evidencia
la pérdida de función del sistema MMR y muestra una correlación prácticamente
absoluta con la IMS.
Las revisiones realizadas en población general muestran que en torno al 12-17% de
todos los CCR se desarrollan a través de esta vía de carcinogénesis (26). En
población española, no obstante, este porcentaje no supera el 10% (18).
Aproximadamente el 80-85% de estos CCR IMS son casos esporádicos, donde el
fenotipo IMS se debe a la hipermetilación somática del promotor del gen MLH1 (34).
Pero hay un pequeño porcentaje de casos (15-20%) que son de origen hereditario: los
pacientes con SL, donde la alteración del sistema MMR ocurre a nivel germinal (26).
Los CCR que se desarrollan a través de la vía de la IMS, sean esporádicos o
hereditarios, comparten determinadas características:
- Se encuentran con más frecuencia en colon proximal y muestran crecimiento
tumoral de tipo expansivo (35).
- Su presentación histológica es frecuentemente de tipo mucinoso, medular, en
anillo de sello; siendo poco diferenciados y con frecuente infiltración linfocitaria
peri e intratumoral (36-39).
- En general tienen mejor pronóstico y supervivencia que el resto de los CCR
(40-41).
- No parecen responder a tratamientos quimioterápicos basados en 5-
fluorouracilo (42-43).

Introducción
10
1.1.3.3. Vía del fenotipo metilador (CIMP) o Vía Serrada
Las islas CpG son regiones que comprenden de 0,5 a 2 Kb, ricas en dinucleótidos
citosina-guanina que están presentes en la región promotora de aproximadamente el
50% de los genes humanos (44-45). La metilación de las mísmas puede producir el
silenciamiento transcripcional de aquellos genes que las contienen.
Recientemente se ha descrito la vía del fenotipo metilador de islas CpG (CIMP) o vía
serrada como vía iniciadora del CCR, la cual se caracteriza por la hipermetilación de
islas CpG en zonas promotoras de genes supresores de tumores. En este caso la
célula requiere de una inestabilidad epigenética para garantizar la progresión tumoral.
Se puede asumir que la metilación de estas regiones en la mayoría de los tumores se
origina de forma estocástica, pudiendo afectar a la expresión de genes importantes en
el desarrollo neoplásico, tales como CDKN2A, MGMT y MLH1 (46-48).
En esta vía, los llamados pólipos serrados, que han sido tradicionalmente
considerados como lesiones benignas sin capacidad de malignización, sustituyen al
adenoma clásico como lesión precursora al cáncer. Parece que entre el 15 y el 30%
de todos los CCR podrían estar asociados a esta vía (49).
El perfil mutacional de estos tumores difiere con respecto a aquellos que tienen el
adenoma como lesión precursora, aunque existe cierto solapamiento con los
subgrupos de tumores CIN e IMS, ya que la hipermetilación puede inactivar genes
requeridos para la inestabilidad cromosómica o la reparación de errores en el ADN
(Figura 3).
El CIMP normalmente es el primer evento de la acumulación de una serie de
alteraciones, que parecen tener la mutación activadora V600E del oncogén BRAF
como precursora. Esta mutación se considera un fuerte marcador de CCR de or igen
esporádico y de lesiones serradas. Existe una fuerte asociación entre el CIMP, la
mutación de BRAF y la IMS, probablemente debida ésta última a la hipermetilación
somática de MLH1 vinculada a este fenotipo metilador (50). Así, la mayoría de los
CCR esporádicos IMS son CIMP+. Sin embargo, los adenomas clásicos estarían
asociados a tumores MSS y, donde la presencia de mutaciones en BRAF y el fenotipo
CIMP son poco frecuentes (51).

Introducción
11
De forma similar a los tumores esporádicos IMS, los CCR CIMP+ parecen asociarse a
buen pronóstico (52) y no parecen responder a la quimioterapia basada en el 5-
fluoracilo (27, 53).
1.2. Cáncer colorrectal hereditario no polipósico (CCHNP)
Se estima en un 20-30% los casos de CCR en los que hay un componente hereditario,
bien sea de alta penetrancia (los síndromes de CCR hereditario típicos citados
anteriormente) o de penetrancia moderada o baja (54). Estos últimos son menos
evidentes y su componente genético es menos conocido. Los síndromes de CCR
familiar pueden ser polipósicos o no polipósicos, en función del número total de
pólipos. Los últimos son los más frecuentes e implican normalmente un número total
de pólipos adenomatosos a lo largo del colon inferior a 10. Dentro de los síndromes de
CCHNP cabe señalar el SL y el CCR familiar tipo X, cuyo origen genético es
desconocido.
1.2.1. Síndrome de Lynch
1.2.1.1. Generalidades
El SL es el síndrome de CCR hereditario más prevalente, representando
aproximadamente el 3% de todos los casos de CCR (26). En población española no
seleccionada se ha observado una frecuencia del 0,7% del total de casos de CCR
(18), en un estudio donde se analizaron únicamente los genes MLH1 y MSH2. El SL
sigue una herencia mendeliana de tipo autosómico dominante, debido a mutaciones
germinales en los genes del sistema MMR. Principalmente MLH1 y MSH2, que
explican un 70% de los casos, seguidos de MSH6 que explicaría un 15-20% de los
casos, y en menor medida PMS2 que explica menos del 5% de los casos (55-56).
Posteriormente se describieron deleciones en EPCAM, gen localizado aguas arriba de
MSH2, como responsables de alrededor del 1% de los casos de SL (8, 57-58).
EPCAM es un gen que codifica para una molécula de adhesión presente en todos los
tejidos epiteliales. Las deleciones en línea germinal del mismo provocan la
hipermetilación somática indirecta de MSH2 en aquellos tejidos donde EPCAM se
expresa. Por consiguiente, la deleción implica la hipermetilación de MSH2 del alelo
que la contiene en todo el epitelio del tracto digestivo.

Introducción
12
El mecanismo de actuación genético implica la alteración a nivel germinal de uno de
los alelos de estos genes, y posteriormente, en el colon o tejido correspondiente se
inactivaría a nivel somático el segundo alelo no afectado, según la teoría del segundo
hit. Esta inactivación se puede dar por una mutación puntual, LOH, o por
hipermetilación del alelo no afecto (59-60). Así, se da lugar a un CCR con fenotipo
hipermutador, cuyo sello molecular característico es la IMS, presente en más del 90%
de estos tumores (61).
Los pacientes con SL tienen un riesgo elevado de desarrollar CCR, pero también otros
tumores: principalmente endometrio, y menos frecuentemente ovario, tracto urinario
superior, gástrico, intestino delgado, sistema hepatobiliar, páncreas, de piel
(adenomas sebáceos y queratoacantomas) y cerebrales (62). Típicamente los tumores
y pólipos aparecen a edades tempranas, con un riesgo estimado de desarrollo de CCR
a lo largo de la vida de entre el 22-75% y entre un 32-45% para el desarrollo de cáncer
de endometrio (CE) (63-66). No obstante, la penetrancia de la enfermedad, edad de
aparición y riesgo de tumores extracolónicos varía en función del gen mutado y el tipo
de mutación: siendo más agresivo el comportamiento para los individuos portadores
de mutaciones en MLH1 o MSH2, con más riesgo de tumores extracolónicos estos
últimos, y menor para los portadores de mutaciones en MSH6 y PMS2; aunque parece
que el riesgo de CE es más elevado en los portadores de mutaciones en MSH6 (67-
70).
Tabla 3. Características epidemiológicas de los individuos portadores de mutaciones en los
distintos genes MMR.
MLH1 MSH2 MSH6 PMS2
Frecuencia (8) 50% 40% 7-10% <5%
Edad aparición (68) 43-46 43-46 51-57 61-66*
Riesgo a desarrollar CCR (70
años)(71) 40-70% 40-70% 10-22% 15-20%
Riesgo a desarrollar CE (70 años) (71) 35-40% 35-40% 17-44% 15%
* Para CCR

Introducción
13
También existen diferencias en la penetrancia de estas mutaciones asociadas a SL en
función del sexo del individuo, siendo tradicionalmente más frecuente en varones, y en
función del origen geográfico. Así, el riesgo de CCR a los 70 años es mayor en países
como Finlandia (52-82%) (66, 72) y Escocia, con diferencias más acentuadas en
función del sexo en este último (74% varones versus 30% mujeres) (73), y más bajo
en los Países Bajos, con pocas diferencias en los dos sexos (27% en varones versus
22% en mujeres) (63).
Como se ha explicado anteriormente, las características histológicas de los CCR de
los pacientes con SL coinciden con las características de los CCR esporádicos con
fenotipo IMS. Así, estos tumores se caracterizan por ser más frecuentemente
mucinosos, pobremente diferenciados, presentar linfocitosis intraepitelial y ser más
frecuentes en colon proximal.
Existen pacientes con mutaciones germinales en estos genes MMR que muestran
otros fenotipos diferentes al convencional. En estos casos, el síndrome adopta otra
nomenclatura (8):
Síndrome de Muir-Torre: cuando aparecen neoplasias sebáceas en la piel
(adenomas sebáceos, epiteliomas sebáceos, carcinomas sebáceos y
queratoacantomas) en combinación con los tumores típicos del SL.
Síndrome de Turcot: cuando aparece un CCR o adenomas colorrectales en
combinación con tumores del sistema nervioso central (a veces también puede
estar causado por mutaciones en APC).
Pacientes con deficiencia constitucional para los genes MMR: de forma
muy poco frecuente se han observado individuos con mutaciones germinales
bialélicas patogénicas en MMR. Estos individuos presentan CCR o cáncer de
intestino delgado a edades muy tempranas (<20 años), y suelen presentar
además un mayor número de pólipos, tumores cerebrales, neoplasias
hematológicas, y un fenotipo similar al de la neurofibromatosis tipo I.
1.2.1.2. Características clínicas y de vigilancia
La identificación de familias portadoras de mutaciones en los genes reparadores
responsables del SL es de vital importancia para la prevención del cáncer y su

Introducción
14
seguimiento, habiéndose demostrado el coste-efectividad del mismo (74-76). Tan
importante es la identificación y seguimiento del caso índice como el estudio genético
predictivo en familiares a riesgo, ya que el manejo y seguimiento de alto riesgo de los
portadores de mutación es lo que aporta el componente de eficiencia en la predicción-
prevención de la enfermedad. Además, la metodología para el estudio directo de la
mutación encontrada en el familiar de primer grado del caso índice a riesgo es rápida y
económica.
Se ha observado que el seguimiento de los pacientes portadores de mutaciones
germinales previene más del 60% de los tumores y de las muertes relacionadas con
cáncer (77). La realización de una colonoscopia cada tres años conduce a una
reducción de la mortalidad en un 65% (77), ya que permite extirpar pólipos y detectar
el CCR en estadios tempranos. La reducción del riesgo a CCR y la mortalidad se ve
aumentada cuando las colonoscopias se realizan cada 1-2 años (78). Así, en la
actualidad, se recomienda la realización de colonoscopias de control cada 1 -2 años,
iniciando el cribado a los 20-25 años o 10 años antes del diagnóstico más joven en la
familia, en función de lo que ocurra primero.
La prevalencia de CE en mujeres portadoras de mutaciones en los genes del SL es
muy elevada (30-45%) (79). Aunque el pronóstico no es malo, el 20% de estas
mujeres morirán debido a esta enfermedad. Sin embargo, el método de cribado de CE
en SL es controvertido. Actualmente, los consensos de expertos recomiendan la
realización de una ecografía transvaginal y biopsia endometrial de manera anual en
mujeres portadoras a partir de los 30-35 años de edad (80). El mismo seguimiento se
recomienda para cáncer de ovario (81).
La eficacia del cribado de otras neoplasias asociadas al SL no está validada y su
realización se basa en opiniones de expertos. Para cáncer gástrico se recomienda el
cribado inicial mediante endoscopia digestiva alta y biopsia en personas a riesgo a
partir de los 30-35 años cada 2-3 años, y tratamiento de la infección Helicobacter pylori
si se detecta, aunque no existe un consenso generalizado. Para otras neoplasias, o
bien no se recomienda cribado (páncreas, intestino delgado), o bien las
recomendaciones son las mismas que las de la población general (mama, próstata)
(81).
Una vez la mutación causante del SL es detectada, además del seguimiento
exhaustivo del paciente, otras medidas para la disminución del riesgo a desarrollar

Introducción
15
cáncer deben considerarse: El consumo de aspirina como estrategia de
quimioprevención, ya que se ha visto una reducción significativa de la incidencia de
cánceres en pacientes con SL. Así como cirugías reductoras del riesgo, como la
colectomía total con anastomosis ileorrectal para CCR, que ha demostrado la
reducción en el riesgo de aparición de lesiones colónicas metacronas; o la
histerectomía y salpingooforectomía bilateral profiláctica en tumores ginecológicos,
que se recomiendan después de que la paciente haya completado sus deseos de
descendencia (81).
1.2.1.3. Criterios diagnósticos
Se han establecido unos criterios clínicos consenso para el diagnóstico de SL, que
tienen como finalidad identificar pacientes con alto riesgo de SL y establecer
estrategias de vigilancia adecuadas en ellos y sus familiares para reducir el riesgo de
aparición de cáncer.
Los criterios de Amsterdam I fueron definidos en 1991 (82). Estos criterios consideran
la historia familiar de cáncer y la edad al diagnóstico de CCR y detectan familias con
un elevado riesgo de tener SL. Posteriormente en 1999 aparecieron los cr iterios de
Amsterdam II, que tienen en cuenta, además de los casos de CCR, otros tumores
extracolónicos (83) (Tabla 4). Sin embargo, estos criterios son muy estrictos,
presentan una elevada especificidad pero baja sensibilidad para la detección de
portadores de mutaciones germinales en MMR. Por ello, en la conferencia del National
Cancer Institute de 1997, se definieron los Criterios de Bethesda, que han sido
revisados posteriormente (84). Estos últimos criterios son menos estrictos que los
anteriores, aumentando la sensibilidad mediante la identificación de los tumores
extracolónicos, y la adición de características patológicas del tumor, que tienen en
cuenta el fenotipo inestable presente en la práctica totalidad de los tumores SL (Tabla
4).

Introducción
16
Tabla 4. Criterios clínicos para el diagnóstico del SL.
CRITERIOS DE AMSTERDAM II
Tres o más individuos afectados de CCR o neoplasia asociada al SL (CE, intestino
delgado, SNC, ovario, uréter o pelvis renal), uno de ellos familiar de primer grado de los
otros dos.
Afectación de 2 generaciones consecutivas.
Como mínimo un caso diagnosticado antes de los 50 años.
Exclusión del diagnóstico de poliposis adenomatosa familiar
Los tumores deben estar confirmados histopatológicamente.
CRITERIOS REVISADOS DE BETHESDA
CCR diagnosticado en paciente antes de los 50 años.
Presencia de CCR sincrónico o metacrónico u otra neoplasia asociada al SL (CE,
estomago, ovario, páncreas, vía biliar, urinario, cerebro, intestino delgado, adenomas
sebáceos o queratoancatomas), con independencia de la edad.
CCR con infiltración linfocitaria, células en anillo de sello o crecimiento medular
diagnosticado antes de los 60 años.
Paciente con CCR y uno o más familiares de 1er grado con CCR o neoplasia asociada a
SL diagnosticada antes de los 50 años.
Paciente con CCR y dos o más familiares de 1er o 2º grado con CCR o asociada a SL, con
independencia de la edad.
SNC: Sistema nervioso central
A pesar de ello, cuando la presentación clínica y la historia familiar son indicadoras de
SL, la probabilidad de encontrar mutación es tan sólo del 50% (85) y en el mejor de los
casos, cuando la familia cumple criterios de Amsterdam y los tumores son inestables,
esta probabilidad aumenta a un 70-80% (61, 86). Globalmente en aproximadamente el
40% de las familias que cumplen criterios de Amsterdam para el diagnóstico de SL, no
se encuentra defecto en el sistema MMR (87). Estos casos, conocidos como CCR
familiar tipo X no presentan, por tanto, IMS en sus tumores, ni alteraciones germinales
en los genes MMR.
Así, a pesar de la implementación de los criterios revisados de Bethesda, más de la
tercera parte de los portadores de mutaciones en el sistema MMR no son detectados
(88) y en ocasiones, estos criterios no son bien implementados en la práctica clínica, o
son criticados por su compleja aplicación. Por lo que se ha recomendado el estudio del
sistema MMR a todos los pacientes con CCR (o todos los individuos con CCR o CE
diagnosticado antes de los 70 años de edad), independientemente de los criterios
clínicos, para la detección de casos sospechosos de SL, lo que se conoce como la

Introducción
17
estrategia de cribado universal (Anexo I). Se han publicado diversos estudios en este
sentido que sostienen una mayor sensibilidad de esta estrategia de manera coste-
efectiva (89-90) respecto a la realización del cribado únicamente en los pacientes que
cumplen los criterios de revisados Bethesda (91-93).
El estudio de la alteración del sistema MMR se puede realizar mediante el análisis de
IMS o el análisis de expresión de las proteínas MMR mediante IHQ en tejido tumoral.
Así, esta estrategia universal permite identificar individuos con sospecha de SL y
alteración del sistema MMR, en los que en muchas ocasiones la causa genética
germinal subyacente es desconocida, por lo que puede contribuir a la acumulación de
casos sospechosos de SL no resueltos.
1.2.1.4. Algoritmo diagnóstico del SL
Actualmente en la mayoría de sistemas sanitarios la selección de pacientes para la
realización de pruebas genéticas para detectar SL se basa en el cumplimiento de al
menos uno de los criterios revisados de Bethesda o en alguna estrategia del análisis
del estado del sistema de reparación del ADN (MMR). Existen algoritmos diagnósticos
que se implementan en las unidades de Consejo Genético en Cáncer para la correcta
identificación de mutaciones germinales responsables del SL (Anexo II).
Así, el estudio genético del SL consta de dos etapas principales: en la primera se
realiza un cribado en el que se estudia la pérdida de actividad de las proteínas del
sistema MMR por IHQ y/o IMS; y en los casos en los que este cribado resulte positivo,
se procede a la segunda etapa de rastreo de mutaciones germinales en los genes
MMR.
El estudio de IMS se realiza sobre el ADN del tejido tumoral. Para ello se utilizan los
cinco marcadores quasi-monomórficos de mononucleótidos repetidos descritos (94),
que se analizan mediante PCR y electroforesis capilar (Figura 4).
Por su parte, el estudio de las proteínas reparadoras mediante IHQ se basa en la
tinción histológica con anticuerpos y determina si éstas están presentes o no en el
tumor, lo que supone un buen indicador de su inactivación como consecuencia de una
mutación germinal o un silenciamiento epigenético (Figura 4).

Introducción
18
Figura 4. Ejemplos del resultado del estudio de la inactivación del sistema MMR
mediante IHQ (a) o IMS (b).
a) Ejemplo de análisis IHQ de un tumor con pérdida de expresión de MLH1/PMS2 (tumor 1) y
de MSH2/MSH6 (tumor 2). b) Resultados del análisis de los 5 marcadores de IMS por análisis
de fragmentos.
Así, el resultado del estudio IHQ permite clasificar el tejido tumoral como con
expresión conservada, pérdida de expresión o expresión no valorable para cada
proteína; y crear un patrón de expresión del sistema MMR. A partir del mismo se
puede orientar el rastreo de mutaciones según se muestra en la tabla 5.
Tabla 5. Genes diana para el rastreo mutacional en función del patrón de expresión de las
proteínas MMR.
Genes diana
de
mutaciones
Patrón de pérdida IHQ
MLH1 MSH2 MSH6 PMS2
MLH1 - + + -
MSH2, MSH6 + - - +
MSH6 + + - +
PMS2 + + + -
MLH1, MSH2,
MSH6, PMS2 - - - -

Introducción
19
La correlación de resultados entre ambas metodologías es muy alta, pero pueden
emplearse de forma complementaria, sobre todo para resolver situaciones de falsos
negativos que, aunque son poco frecuentes, existen (18). La IHQ, además de ser más
asequible en la práctica clínica habitual, tiene como valor añadido que permite orientar
el estudio genético hacia el/los genes que muestran expresión alterada. La mayoría de
mutaciones en los genes MLH1 y MSH2 dan lugar a una expresión anormal de sus
proteínas correspondientes; en cambio las mutaciones en MSH6 pueden mostrar un
patrón IHQ normal. Lo mismo ocurre con el fenotipo IMS, habiéndose descrito tumores
estables con mutación germinal en MSH6 (95).
La presencia de IMS o la pérdida de expresión de proteínas MMR por IHQ no son
exclusivas de SL; como ya se ha comentado, la IMS se asocia fuertemente a la
mutación BRAF_V600E, exclusivamente en CCR esporádico (96). Por otro lado, la
pérdida de actividad en MLH1 puede estar debida a la inactivación somática del gen
por metilación de su promotor. Por ello, en aquellos estudios genéticos de SL en los
que hay sospecha del posible origen esporádico del tumor (casos Bethesda), conviene
descartar estos eventos –mutación en BRAF o metilación en MLH1- antes de orientar
el estudio genético de los genes MMR.
El algoritmo diagnóstico para el análisis genético del SL se muestra en el anexo II.
Finalmente, se realizará el estudio guiado de las mutaciones germinales, que implica,
por un lado, el estudio de mutaciones puntuales mediante secuenciación Sanger de la
región codificante completa y las uniones intrón-exón del gen en cuestión; y por otro
lado, el estudio de grandes reordenamientos genéticos en dichos genes mediante
MLPA (Multiplex Ligation Probe Amplification).
1.2.1.5. Tipos de mutaciones en el SL
Así, siguiendo el respectivo algoritmo diagnóstico de SL (Anexo I o II) se pueden
detectar diferentes tipos de mutaciones germinales en MMR:
Mutaciones puntuales: pueden ser mutaciones sin sentido (nonsense); de
desplazamiento del patrón de lectura (frameshift), mutaciones de sentido
erróneo (missense) y mutaciones de splicing. Las dos primeras generalmente
suelen tener efectos deletéreos, y las dos últimas en ocasiones pueden dar
lugar a variantes de significado incierto (VSI), en las que se desconoce su
patogenicidad. Las mutaciones missense son mucho más frecuentes en MLH1

Introducción
20
que en MSH2, ya que en éste último el 83% del total de mutaciones son sin
sentido o frameshift (97).
Grandes reordenamientos: se trata de deleciones o duplicaciones que afectan
a grandes regiones exónicas en alguno de los genes reparadores. Se detectan
fácilmente mediante la técnica MLPA. Las grandes deleciones explican un 22%
de las mutaciones en MLH1 y PMS2, un 26% de las mutaciones en MSH2 y un
7% de las mutaciones en MSH6 (54). Además, como ya se ha descrito, se han
observado deleciones en los últimos exones de EPCAM asociadas al
silenciamiento epigenético por metilación del gen MSH2 (57-58). Estos tumores
presentan pérdida de MSH2 en su análisis IHQ. Las duplicaciones en genes
reparadores en SL son menos frecuentes.
Inversiones: Se han identificado inversiones paracéntricas en MSH2 (98-101),
así como en el cromosoma 3p22.2, lo que crea dos nuevas fusiones de
transcritos entre MLH1 y LRRFIP2 (102-103), como causantes de SL. Estas
alteraciones no son detectables mediante la metodología de análisis
mutacional clásica empleada para el diagnóstico del SL (Anexos I y II).
1.2.1.5.1. Epimutaciones constitucionales en MLH1
Aún con todo, en un porcentaje elevado de casos con sospecha de SL no se detectan
mutaciones a nivel germinal de los genes del sistema MMR.
Recientemente se ha observado que algunos casos con fenotipo de SL son causados
por inactivación epigenética a nivel constitucional de MLH1 mediante metilación de su
promotor, lo que implica la inactivación transcripcional del mismo (104). En estos
casos se produce la metilación hemialélica en las citosinas seguidas de guaninas del
promotor MLH1 a nivel germinal, en tejidos normales y, al igual que ocurre cuando hay
una mutación, es la inactivación somática del segundo alelo la que desencadenaría el
desarrollo de cáncer.
En el promotor de MLH1 hay una isla CpG constituída por varias regiones. En las
regiones C y D (posiciones -248 a -178 y -109 a +15 respectivamente) se localiza el
core del promotor, responsable de la regulación transcripcional de MLH1, y cuya
metilación se asocia directamente con la pérdida de expresión del gen (104). Estas
regiones controlan la transcripción a través de la regulación de la unión de los factores

Introducción
21
de transcripción al promotor de MLH1 (105). La metilación interfiere en la unión de los
mismos e incrementa la afinidad de las proteínas de unión a grupos metilos y
desacetilasas de histonas, que conducen a configuraciones inactivas de la cromatina,
provocando el silenciamiento genético. Por lo tanto, los estudios de metilación deben
enfocarse en esta zona (104). La técnica MS-MLPA ha demostrado ser coste-efectiva
para la realización de este análisis (106).
Figura 5. Metilación en el promotor de MLH1, sondas del MS-MLPA. Adaptado de (107)
La región enmarcada es aquella cuya metilación está relacionada con la inactivación del gen.
La revisión del tema realizada por Hitchins en 2013 (108) recopila hasta la fecha 54
casos índice con tumor con fenotipo de SL, en los que la causa de la enfermedad es la
epimutación constitucional en MLH1. La mayoría de estos casos cumplen al menos un
criterio de Bethesda. No suelen cumplir criterios de Amsterdam, posiblemente debido a
la falta de patrón de herencia mendeliano. Presentan una edad de aparición temprana
(media 39 ± 11 años) y desarrollan frecuentemente tumores metacronos relacionados
con SL (108). Además, estos tumores presentan ausencia de la mutación V600E de
BRAF.
La frecuencia de las epimutaciones en MLH1 oscila entre el 0 y el 14% de los casos
analizados, en función de los criterios de selección de los mismos: si bien se han
descartado sólo las mutaciones germinales en los genes MMR, o, en cambio se ha
filtrado por edad joven, IMS, pérdida IHQ o metilación de MLH1 (108). Se ha calculado
que las epimutaciones en MLH1 explican en torno al 10% de los casos que cumplen
los siguientes criterios: 1) cumplimiento de al menos un criterio de Bethesda; 2) tumor
con pérdida de MLH1 e IMS; 3) no mutación patogénica encontrada mediante el
rastreo genético habitual (109).

Introducción
22
La mayoría de estos casos con epimutaciones constitucionales en MLH1 descritos
hasta la fecha no tienen una fuerte agregación familiar de cáncer. En los pocos casos
en los que se ha podido estudiar a la familia, se han descrito tres patrones diferentes
de herencia intergeneracional:
1) Aparición de la epimutación constitucional de MLH1 de novo y desaparición
en la descendencia. Esto se ha observado en tres familias en las que se han
estudiado tres generaciones de individuos, y se ha observado la herencia del
alelo materno que contiene la epimutación en las tres generaciones, pero esta
metilación está ausente en la generación ancestral y en la descendencia, así
como en los espermatozoides de los individuos portadores. Estos casos, por
tanto, no contribuyen a la heredabilidad del cáncer (108, 110).
2) Transmisión no mendeliana de la epimutación. Se ha observado en una
familia en la que la madre afecta, de los tres hijos a los que transmite el alelo
portador de la epimutación, sólo en un caso transmite la metilación. Además, al
igual que antes, este caso no muestra epimutación en sus espermatozoides
(108, 111).
3) Posible transmisión autosómica dominante de la epimutación ligada a una
alteración genética en cis o en trans: cabe destacar el caso de una familia de
tres generaciones en la que se observa el ligamiento de la epimutación en
MLH1 con el cambio c.-27C>A en el mismo gen, a pesar de que en uno de los
individuos se observa de nuevo la eliminación de esta metilación en sus células
germinales (108, 110).
Con todo ello, el mecanismo por el que aparecen estas epimutaciones constitucionales
en MLH1 y se transmiten a una proporción de la descendencia actualmente es
desconocido. Puede haber una herencia de base epigenética, aunque se sabe que
esto es muy lábil, debido a la reprogramación epigenética que tiene lugar en la
gametogénesis; o bien puede resultar de una predisposición genética (caso 3), como
ocurre en el caso de EPCAM y MSH2. Este mecanismo de inactivación ha sido
descrito únicamente en un individuo por el momento (112). No obstante, esta hipótesis
está en duda, dado que si así fuera, debería observarse una herencia mendeliana de
la enfermedad como ocurre con EPCAM, y no parece ser el caso.

Introducción
23
En definitiva, el carácter hereditario de estos casos es débil en comparación con las
mutaciones germinales, por lo que las epimutaciones deben sospecharse en
individuos jóvenes, sin historia familiar, que presentan tumores inestables con pérdida
de MLH1. En cualquier caso es imprescindible considerar el posible origen germinal de
estos casos con metilación en MLH1, ya que el fenotipo de los pacientes parece
agresivo, similar al de los pacientes con SL. Por ello, se considera necesario incluir el
análisis de epimutaciones constitucionales de MLH1 dentro del algoritmo diagnóstico
del SL, a pesar del desconocimiento del mecanismo subyacente de esta alteración.
Las estrategias, por tanto, de seguimiento de los individuos y los familiares deben ser
las mismas que las consideradas para los pacientes con SL.
1.2.2. CCR familiar tipo X
Lindor y colaboradores (113) describieron por primera vez en 2005 el CCR familiar tipo
X como aquellos individuos con CCR que cumplen los criterios de Amsterdam para el
diagnóstico de SL, pero sus tumores son MSS, y no presentan pérdida de expresión
de las proteínas reparadoras ni mutación germinal en los genes MMR. Estos casos
representan aproximadamente la mitad de los casos de CCHNP que cumplen los
criterios de Amsterdam (114) y sus familias presentan una tasa de incidencia
estandarizada de CCR del doble respecto a la población general (SIR: 2,3) (113).
En comparación con los pacientes con SL, los individuos con CCR familiar tipo X
tienen una incidencia más baja de CCR, menor riesgo de desarrollar tumores
extracolónicos, y presentan una edad al diagnóstico del CCR más avanzada, en torno
a 10 años superior (7, 87). Los tumores de estos pacientes, además de diferir con los
del fenotipo del SL en mantener el sistema MMR intacto, suelen presentar CIN y
ausencia de CIMP (87) a pesar de presentar metilación aberrante en LINE-1 (115); y
no predomina la localización proximal ni los tumores mucinosos (116).
Desde el punto de vista genético, parece tratarse de un grupo heterogéneo de
individuos, que abarca varios grupos con diversas etiologías que podrían explicar el
origen de la enfermedad: genes de elevada penetrancia; mecanismos alternativos de
silenciamiento génico como epimutaciones o microRNAs; alelos de baja penetrancia
en combinación con factores ambientales,etc. (7, 87).

Introducción
24
Los estudios enfocados en la búsqueda de genes candidatos hasta la fecha han sido
escasos y sólo se han encontrado mutaciones de forma aislada. Las nuevas
tecnologías de secuenciación masiva están permitiendo un abordaje más eficiente,
mediante la búsqueda de genes de forma no dirigida, aunque pocos son los resultados
descritos en este sentido. La siguiente tabla muestra un resumen de potenciales genes
causantes de CCR familiar tipo X identificados hasta la fecha, siguiendo tanto
metodologías clásicas de biología molecular como técnicas de análisis genético de
nueva generación:
Tabla 6. Genes mutados en familias con CCR familiar tipo X.
Gen Frecuencia
n (%) Referencia
BRCA2 4/48 (8,3) (117)
BMPR1A 2/18 (11,1) (118)
POLD1 ND (119)
POLE ND (119)
FAN1 5/176 (2,8) (120)
GALNT12 4/118 (3.4)*
(0/103) (121) (122)
HNRNPA0-WIF1 ND (123)
RPS20 1/26 (3,8) (124)
SEMA4A 1/54 (1,8) (125)
*En los casos analizados en este estudio se incluyen pacientes que cumplen criterios de
Bethesda.
ND: No hay datos.
Otros eventos genéticos germinales, como la elongación de los telómeros, también
han sido asociados a las familias con este síndrome (126).
1.3. Síndromes de poliposis familiar
El diagnóstico diferencial del SL debe excluir pacientes con CCR de aparición
temprana y riesgo familiar con otros síndromes de predisposición a cáncer, que se
caracterizan principalmente por la aparición de múltiples pólipos. Estos síndromes

Introducción
25
incluyen la PAF, MAP, los síndromes de poliposis hamartomatosa (poliposis juvenil,
síndrome de Cowden y Peutz-Jeghers) y la polipolis mixta hereditaria. La prevalencia
de los síndromes de poliposis hereditarios es muy baja (Tabla 1).
Cada síndrome de poliposis colorrectal presenta un patrón de herencia , unas
características fenotípicas y manifestaciones extracolónicas propias. No obstante, en
ocasiones existen problemas de asociación fenotipo-genotipo, debido al solapamiento
de los mismos o incluso, entre los síndromes polipósicos y no polipósicos, habiéndose
encontrado casos de síndrome de poliposis familiar que debutan directamente como
CCR.
Los pólipos se pueden clasificar en distintos tipos según su histología, principalmente:
adenomas, pólipos serrados, pólipos hamartomatosos y pólipos inflamatorios. Estas
lesiones son benignas pero un pequeño porcentaje de ellas puede llegar a malignizar,
dando lugar finalmente al CCR. La capacidad de malignización difiere en función de la
histología, tamaño, displasia y características intrínsecas del individuo. Por lo que,
además del problema de los fenotipos solapantes asociados a distintos defectos
germinales, la dificultad diagnóstica de estos síndromes se debe en muchas ocasiones
al difícil reconocimiento de los pólipos más pequeños o planos, a un desarrollo
incompleto del pólipo a la hora de la cirugía y a una difícil clasificación histológica.
1.3.1. Poliposis adenomatosa familiar
La PAF (OMIM #175100) se trata de una enfermedad con herencia autosómica
dominante responsable de alrededor de un 0,5% de los CCR (127). En el fenotipo
adulto clásico, el desarrollo de los adenomas colorrectales provoca la aparición de
CCR en prácticamente el 100% de los casos si no se actúa, por lo que es necesario el
tratamiento quirúrgico profiláctico. El riesgo de CCR en pacientes con PAF aumenta de
forma significativa después de la segunda década de vida y éste suele aparecer a
partir de la 4ª-5ª década.
No obstante, se trata de una enfermedad con fenotipo variable en la que se pueden
distinguir claramente dos fenotipos: la PAF clásica, caracterizada por la presencia de
cientos a miles de pólipos adenomatosos a lo largo del intestino, con mayor tendencia
en el colon distal; y la poliposis adenomatosa familiar atenuada (APAF), cuyos

Introducción
26
individuos desarrollan menos de 100 pólipos y lo hacen más frecuentemente en colon
proximal (128). Parece que el riesgo a CCR en estos pacientes con APAF (<70%) es
menor que en PAF y el CCR es de aparición más tardía (129).
Este síndrome tiene una expresividad variable, con la presencia frecuente de
manifestaciones extracolónicas: pólipos gástricos y duodenales, tumores desmoides,
tumores cerebrales y de tiroides, osteomas, hipertrofia congénita del epitelio retinoso
pigmentario, dientes supernumerarios y quistes epidermoides (129-130); aunque éstas
son menos comunes en los individuos con el fenotipo atenuado.
Mutaciones en línea germinal en el gen APC (5q21-q22), que codifica para una
proteína con función supresora de tumores, explican el 70-90% de los casos. Además,
aproximadamente un 20-25% de los casos de PAF son debidos a mutaciones de novo
(127). Las mutaciones descritas se distribuyen de forma homogénea a lo largo del gen,
exceptuando dos hot spots (codones 1061 y 1309) que representan el 30% de los
casos. El fenotipo expresado y las manifestaciones extracolónicas estarán
determinados por la región de APC afectada. No obstante, en casi un tercio de familias
con PAF y un 85% en el caso de pacientes con APAF, el resultado del estudio
genético para APC es negativo.
1.3.2. Poliposis asociada a MUTYH (MAP)
Hasta un 7,5% de los casos con un fenotipo de PAF y un 35% adicional en el caso de
APAF, pueden ser explicados por mutaciones bialélicas germinales en MUTYH (131).
Se calcula que la incidencia de este síndrome es de 1/10.000, representando el 0,5-
1% de todos los casos de CCR, siendo tan prevalente como los casos de PAF
asociados a mutaciones en APC. Los pacientes con MAP presentan un riesgo
acumulado de desarrollar CCR a los 70 años de aproximadamente el 80% en ausencia
de tratamiento, siendo la edad al diagnóstico de CCR de 45-50 años, con una
localización predominante en colon proximal (132).
El fenotipo clínico típico de MAP es de poliposis atenuada, aunque en ocasiones
puede mimetizar a la PAF (9). También es frecuente encontrar pacientes con pólipos
serrados en este síndrome, pudiendo algunos de ellos cumplir los criterios de la OMS
para el diagnóstico del Síndrome de Poliposis Serrada (133-134). No obstante, en un
25% de los casos MAP no se observan pólipos en el momento del diagnóstico del

Introducción
27
CCR (135), mostrando en algunos casos fenotipo clásico de SL (136-137) o incluso de
Muir-Torre (138). Existe, por tanto, un solapamiento fenotípico considerable con otros
síndromes de predisposición a CCR, tanto polipósicos como no polipósicos. Esto nos
lleva a pensar que se trata de un síndrome infradiagnosticado.
Aunque en menor frecuencia que los pacientes con PAF, los pacientes con MAP
también presentan riesgo de desarrollar distintos tumores extracolónicos, tales como
cáncer de mama, tiroides, gástrico, ovario, vejiga, piel (139). Incluso se ha descrito un
caso con cáncer de testículo (140).
MAP presenta herencia mendeliana de tipo autosómico recesiva, asociada a
mutaciones bialélicas en el gen MUTYH, localizado en 1p34.1. MUTYH codifica para
una ADN-glicosilasa que funciona en el mecanismo de reparación del ADN por
escisión de bases, reparando los errores de tipo transversión G>T. Por lo tanto,
individuos deficientes para MUTYH tienden a acumular mutaciones somáticas en
genes relacionados con la carcinogénesis colorrectal, como KRAS o APC (133), que
ocurren de manera temprana en la secuencia adenoma-carcinoma.
En población europea existen tres mutaciones recurrentes en MUTYH, localizadas en
los exones 7 y 13: c.536A>G p.Tyr179Cys; c.1187G>A p.Gly396Asp y
c.1227_1228dup p.Glu410GlyfX43 (141). En casos de sospecha de MAP (>10
adenomas), generalmente se sigue una estrategia diagnóstica en tres pasos, que
implica en primer lugar el análisis de estas mutaciones recurrentes, y en el caso de
que éste sea positivo, el estudio entonces del gen completo para el rastreo de la
segunda mutación. Siguiendo esta estrategia se consiguen diagnosticar más del 95%
de los casos MAP que cumplen los criterios establecidos (134).
Recientemente, se han identificado mutaciones homocigotas en el gen NTHL1, que
también es un gen reparador por escisión de bases, como responsables de un
pequeño porcentaje (6%, 3/48 familias analizadas) de poliposis adenomatosas, en
familias con herencia recesiva que no presentaban mutaciones en los genes APC ni
MUTYH (142).

Introducción
28
1.3.3. Otros síndromes polipósicos de predisposición a CCR
Existen otros síndromes polipósicos de predisposición a CCR que son menos
frecuentes. Entre ellos están los síndromes de poliposis hamartomatosa: poliposis
juvenil, síndrome de Peutz-Jeghers, síndrome de Cowden y el síndrome de Bannayan-
Riley-Rubalcaba; y la poliposis mixta hereditaria. Todos estos síndromes presentan
herencia mendeliana de tipo autosómico dominante.
La poliposis juvenil (MIM #174900) es el síndrome hamartomatoso más común (Tabla
1). Se diagnostica por el cumplimiento de uno de los siguientes criterios: 1) múltiples
(3-10) pólipos juveniles en el colorrecto; 2) cualquier número de pólipos juveniles en un
paciente con historia familiar de poliposis juvenil; 3) presencia de pólipos juveniles
extracolónicos. Los pólipos juveniles son hamartomas con un epitelio normal y un
denso estroma y glándulas císticas en la lámina propia. La mayoría de los pólipos son
benignos, pero en un pequeño porcentaje puede tener lugar la transformación maligna,
que ocurre principalmente en el colon, aunque también puede ocurrir en otros órganos
del tracto digestivo, como el estómago. En torno al 40% de estos pacientes presentan
mutaciones germinales en SMAD4 o en BMPR1A, genes que codifican para proteínas
implicadas en la vía de señalización del TGF-beta (87).
El síndrome de Peutz-Jeghers (MIM #175200) se caracteriza por la presencia de
múltiples pólipos hamartomatosos gastrointestinales (de epitelio elongado y dilatación
glandular), acompañado de una pigmentación mucocutánea, que puede afectar a
distintas partes de la cara y dedos. Los criterios de diagnóstico clínico son similares a
los de la poliposis juvenil, basados en la presentación fenotípica y los antecedentes
familiares. Tiene un riesgo elevado a desarrollar tumores intestinales y
extraintestinales. Entre el 91 y el 100% de los casos se deben a mutaciones en el gen
STK11, que codifica para una serina-treonina kinasa (87).
Otro síndrome más raro es el síndrome de Cowden, que se caracteriza por la aparición
de hamartomas gastrointestinales, orales y cutáneos, aunque también pueden
aparecer lesiones adenomatosas e inflamatorias. Los pacientes tienen un riesgo
incrementado de padecer cáncer de mama y tiroides, mientras que el riesgo a CCR es
mucho menor (~9%). Este síndrome se asocia a mutaciones germinales en PTEN,
aunque existe una elevada heterogeneidad genética en este gen, ya que también está
relacionado con otros síndromes, como el de Bannayan-Riley-Rubalcaba (87). Este

Introducción
29
síndrome, aunque similar fenotípicamente al de Cowden, aparece en la infancia y se
caracteriza por una pigmentación en forma de mancha en el pene.
Finalmente, la poliposis mixta hereditaria está caracterizada por la presencia de
múltiples lesiones colónicas de diferentes tipos (pólipos hamartomatosos, serrados,
adenomas, CCR) y la ausencia de manifestaciones extracolónicas. El gen GREM1 se
ha visto asociado a este síndrome, y duplicaciones de la región 15q13.3 que lo
contiene, que llegan hasta la región reguladora aguas arriba del gen GREM1,
cosegregan con la enfermedad (87).
1.4. Poliposis asociada a la actividad correctora de errores de las polimerasas
(PPAP)
Aproximadamente el 30% del total de CCR son casos familiares. Los síndromes con
herencia mendeliana descritos (SL, PAF, MAP, síndromes de poliposis
hamartomatosa, poliposis mixta hereditaria) explican un porcentaje de estos casos. No
obstante, la heredabilidad perdida de estos casos no es despreciable. El marco común
de estos síndromes es la predisposición al desarrollo de CCR y pólipos colónicos, con
riesgo familiar que normalmente sigue una herencia autosómica dominante (con
excepción de MUTYH). La aparición en los últimos años de las técnicas de
secuenciación de nueva generación está permitiendo avanzar en el conocimiento de la
etiología de los mismos.
En el año 2013, el equipo de Palles y colaboradores (119) mediante la combinación de
secuenciación del genoma completo y análisis de ligamiento, descubrieron mutaciones
germinales en los dominios de corrección de errores (proofreading) de las polimerasas
δ y ε (genes POLD1 y POLE) como causantes del cáncer en familias con elevada
agregación familiar de CCR y poliposis adenomatosas. Poco después, Briggs y
Tomlinson denominaron este síndrome como: poliposis asociada a la actividad
correctora de errores de las polimerasas (PPAP) (143).
Este síndrome, recientemente acuñado, parece presentar una herencia de tipo
autosómico dominante, una penetrancia elevada y una edad de aparición temprana
(119). Cuando las mutaciones germinales tienen lugar en POLD1 se asocia también

Introducción
30
fuertemente a CE (119). Otras manifestaciones extracolónicas se han observado de
manera menos frecuente.
La replicación del ADN previa a la división celular tiene una alta tasa de fidelidad, con
sólo 1 x 10-10 errores por base (144). Distintos mecanismos moleculares colaboran
secuencialmente durante la replicación para mantener este número mínimo de errores.
El alto grado de conservación de estos mecanismos entre especies subraya su
importancia biológica. En la corrección de errores en la replicación del ADN se
observan tres etapas (144): 1) correcta incorporación del nucleótido complementario
por las ADN polimerasas, 2) eliminación de los nuevos nucleótidos incorporados
incorrectamente previo a la extensión, por la actividad 3’ exonucleasa (proofreading)
asociada a algunas ADN polimerasas, 3) control post-replicativo de las bases mal
incorporadas, mediante el sistema reparador de errores (MMR) y la reparación del
ADN por escisión de bases. Como ya se ha explicado, está demostrado que la
inactivación a nivel germinal de cualquiera de estos dos últimos mecanismos se asocia
a síndromes de CCR hereditario o poliposis (SL o MAP respectivamente), que tienen
un perfil hipermutador a lo largo del genoma (145). El estudio realizado por Palles y
colaboradores (119) demuestra que mutaciones inactivantes en los dominios
exonucleasa de las polimerasas POLE y POLD1 provocan la acumulación de multitud
de mutaciones a lo largo del genoma, causando finalmente el desarrollo del cáncer.
1.4.1. Función de POLE y POLD1 y su implicación en el proceso carcinogénico
Pol ε y Pol δ son las únicas polimerasas en eucariotas con capacidad intrínseca
exonucleasa 3’-5’ de corrección de errores, mediante la cual se elimina el nucleótido
erróneamente incorporado, previo a la extensión de la hebra de ADN. Ambas
polimerasas son heterotetrámetros que actúan conjuntamente con Pol α en la horquilla
de replicación para garantizar la replicación del ADN previa a la división. Así, Pol α
prepara la síntesis de ambas hebras y hace pequeñas copias del ADN, mientras que
Pol ε y δ son las responsables de la elongación del ADN en la hebra principal y
complementaria, respectivamente (143, 146).
En humanos, las subunidades catalíticas de estas enzimas (Pol ε y Pol δ), están
codificadas por los genes POLE y POLD1, respectivamente, que pertenecen a la
familia B de las polimerasas (143). POLE es un gen localizado en 12q24.3 que
contiene 54 exones y codifica para la mayor de las cuatro subunidades de Pol ε, de

Introducción
31
140 kDa. Por otro lado, POLD1, localizado en el cromosoma 19 (19q13.3), contiene 29
exones, y codifica para una subunidad de 125 kDa. Ambos genes son parálogos,
presentando estructura y funciones homólogas, mostrando su mayor conservación y
homología (23% identidad, 37% similaridad) en los dominios exonucleasa (residuos
268–471 de POLE y 304–517 de POLD1) (143).
Estudios in vitro en levadura y Escherichia coli, que inducen mutaciones en los
dominios exonucleasa de Pol ε y Pol δ, causan un incremento del ratio de mutaciones
espontáneas de entre 10 y 1.000-10.000 veces para cada modelo, demostrando así la
implicación de estos dominios en la fidelidad en la replicación (144, 147-149). Al
parecer, la inactivación de Pol ε provocaría más frecuentemente mutaciones
puntuales, con respecto a Pol δ, que provocaría tanto mutaciones puntuales como
frameshift (150).
Las primeras evidencias de la implicación de estas polimerasas en cáncer vienen de
estudios en ratones. La pérdida de la funcionalidad del dominio exonucleasa en Pol ε
en ratones provoca la aparición de tumores intestinales, con respecto a la alteración
en el respectivo dominio en Pol δ, que provoca más frecuentemente linfomas y
neoplasia de células escamosas de la piel, con una peor tasa de supervivencia (146).
Estos fenotipos sólo se observaban cuando las mutaciones eran homocigotas, lo que
apoya la necesidad de un segundo hit. Las diferencias fenotípicas parecen sugerir que
ambas polimerasas participan en vías independientes de progresión tumoral. Así,
parece que POLD1 también participa en el sistema reparador de errores (MMR) y en la
reparación por escisión de bases.
1.4.2. Mutaciones de POLE y POLD1 descritas en cáncer
1.4.2.1. Mutaciones germinales en POLE y POLD1
Las mutaciones descritas por Palles y colaboradores (119) como causantes CCR y
adenomas en familias seleccionadas con elevada carga fueron dos cambios de
sentido erróneo (missense): p.Leu424Val (c.1270C>G) en el exón 13 de POLE y
p.Ser478Asn (c.1433G>A) en el exón 11 de POLD1.
En este estudio, la variante p.Leu424Val en POLE fue encontrada en una familia y
posteriormente en 12 familias independientes en una cohorte de validación. Todas las

Introducción
32
familias excepto una presentaban múltiples adenomas y CCR, sin claras
manifestaciones extracolónicas. Algunos de los tumores presentaban LOH en dicha
región de POLE como segundo hit. Todos los tumores eran MSS y dos de ellos
presentaban CIN. El rastreo de las mutaciones somáticas clásicas en la
carcinogénesis colorrectal mostró en algunos tumores la presencia de mutaciones en
APC (R1114X, Q1338X), mutaciones en KRAS (codones 12 y 146) en torno al 18% de
los casos y la ausencia de la mutación BRAF_V600E (119).
Al ser POLE y POLD1 genes parálogos, sus secuencias tienen un alto grado de
homología. De este modo, la variante p.Ser478Asn en POLD1, es prácticamente
equivalente a la encontrada en POLE, ya que el residuo 478 en POLD1 corresponde
con el residuo 428 en POLE, muy cercano a la alteración encontrada. La alteración
p.Ser478Asn fue encontrada en dos familias del estudio que posteriormente se
demostró que estaban relacionadas, y más tarde en otra familia aparentemente no
relacionada en la cohorte de validación. El fenotipo tumoral de los portadores de esta
mutación fue muy similar al encontrado para los portadores de la mutación en POLE.
No obstante, había una elevada prevalencia de CE de aparición temprana en las
mujeres de estas familias. El perfil mutacional de los tumores de los individuos
portadores de mutaciones en POLD1 resultó similar al de los portadores de la
mutación en POLE, con la excepción de que las mutaciones en KRAS aparecieron en
los codones 13 y 61, y algunos tumores presentaron mutación en BRAF (119).
En definitiva, los resultados obtenidos en el estudio de Palles y colaboradores,
muestran que el fenotipo del síndrome PPAP puede solapar con el de otros síndromes
de predisposición a CCR, como el SL o la MAP (119).
Las evidencias descritas por el equipo de Palles y colaboradores para considerar estas
mutaciones como patogénicas y causantes del síndrome en estas familias eran
considerables:
- Ninguna de estas dos mutaciones estaban presentes en una cohorte de
10.755 controles.
- Ambos residuos están altamente conservados entre especies.
- Se encuentran en el sitio activo del dominio exonucleasa (Leu424 en POLE) o
muy cercano al mismo (Ser474 en POLD1).
- Ambos residuos en la conformación de la T4 ADN polimerasa/exonucleasa se
encuentran localizados muy cercanos al ADN de cadena simple, por lo que

Introducción
33
estas mutaciones pueden alterar el empaquetado de las hélices, pudiendo
afectar la actividad exonucleasa (ver Figura 6).
- La alteración de dichos residuos en levaduras confiere un fenotipo mutador.
- Los programas de predicción bioinformáticos muestran severas
consecuencias funcionales en la actividad de la proteína.
- El alelo mutado se expresa de forma estable a nivel de ARNm.
En este estudio, se identificó además una tercera mutación probablemente patogénica
en POLD1: p.Pro327Leu (c.981C>G). Esta mutación, identificada en un individuo de
70 años con 10 adenomas, está muy conservada evolutivamente y está localizada en
la zona de interacción del dominio exonucleasa de POLD1 con el ADN (119).
Además de las tres variantes encontradas en este estudio, sólo se han identificado dos
mutaciones germinales más en los dominios exonucleasa de estos genes en pacientes
con fenotipos similares de cáncer (Anexo IV):
En el estudio realizado por Church y colaboradores (151) en pacientes con CE
identificaron una mutación germinal en POLE (c.1337G>A; p.Arg446Gln;
rs151273553) y otra en POLD1 (c.931C>T; p.Arg311Cys; rs201010746), previamente
descritas, que parece que por el momento su significado clínico es desconocido. La
primera está conservada en Saccharomyces cerevisiae pero no en Saccharomyces
pombe, las predicciones muestran que parece que la función proteica se ve afectada,
pero está bastante distante al dominio exonuclesasa. La segunda, p.Arg311Cys en
POLD1, no ha sido descrita en cáncer, pero flanquea la región exonucleasa y las
predicciones in silico muestran una afectación a nivel de proteína.
Por otro lado, en el estudio realizado por Smith y colaboradores identificaron una
mutación germinal tipo frameshift en POLE (c.5621_5622delGT) en un paciente con
diagnóstico de CCR a los 26 años, sin antecedentes familiares de cáncer (152). No
obstante, esta mutación, localizada en el exón 39, está muy alejada del dominio
exonucleasa del gen, por lo que su relación con cáncer está en duda.
1.4.2.2. Mutaciones somáticas en POLE y POLD1
Mutaciones somáticas en distintas regiones del gen POLE principalmente, y en menor
medida POLD1, también han sido descritas en pacientes con CCR o CE. La potencial
patogenicidad de las mismas ha sido especialmente considerada cuando las

Introducción
34
mutaciones tienen lugar en los dominios exonucleasa de estos genes, ya que afectan
a residuos altamente conservados y la predicción de la alteración de la proteína suele
ser patogénica (Anexo IV); en comparación con la predicción de las mutaciones que
tienen lugar en otras regiones, que es más variable y su patogenicidad está puesta en
duda.
En primer lugar, en el proyecto de secuenciación del exoma completo del TCGA (The
Genome Cancer Atlas Network ), se subrayó la presencia de mutaciones tipo missense
en POLE de forma muy representativa en tumores colorrectales con fenotipo
hipermutador (tasa de mutación >12/106 pb), en ausencia de IMS (153). En este
estudio se encontraron 18 mutaciones diferentes en POLE, 5 de ellas localizadas en el
dominio exonucleasa: p.Pro286His, p.Phe367Ser, p.Val411Leu, p.Pro436Arg y
p.Ser459Phe (119, 153). Las tres primeras han sido descritas a su vez en otros
trabajos: p.Pro286His y p.Phe367Ser, en tres individuos con CCR (144, 154); y la
tercera (p.Val411Leu) en CE en el estudio realizado por el TCGA para este tumor y por
Church y colaboradores (151, 155). Estos estudios realizados en CE detectaron
además otras cuatro mutaciones diferentes más en el dominio exonucleasa de POLE:
p.Asp275Val, p.Pro286Arg, p.Ser297Phe, p.Ala456Pro. Al igual que para CCR, casi
todas estas mutaciones en POLE en CE estaban presentes en tumores estables y
permitían definir un subgrupo de tumores hipermutados con características
moleculares concretas y mejor pronóstico, en comparación con el resto de subgrupos
(155).
Globalmente, los resultados de estas investigaciones en CE muestran una prevalencia
de mutaciones somáticas en POLE del 6,8-8,5%, siendo más recurrentes las
mutaciones p.Pro286Arg y p.Val411Leu (151, 155). Esta prevalencia es más baja en
el caso del CCR, de un 3,1% (153).
Según estos estudios, estos tumores con mutaciones en el dominio exonucleasa de
POLE muestran un fenotipo hipermutador, con ausencia de IMS (aunque no fue
determinada en la cohorte del TCGA de CE), un patrón atípico de mutaciones en los
genes típicos de cáncer, y una tendencia incrementada a acumular substituciones de
tipo G:C>T:A; en comparación con aquellos tumores sin mutación en estos dominios
(151, 153, 155).
En contraposición con estos resultados, las mutaciones somáticas en el dominio
exonucleasa de POLD1 son muy poco prevalentes, habiéndose encontrado

Introducción
35
únicamente en los tumores de dos pacientes hasta la fecha: un CCR, que mostraba
además IMS, y un CE; lo que equivale a una prevalencia de estas mutaciones de en
torno al 0,4% (151, 153).
Las razones de estas diferencias en la frecuencia de mutaciones somáticas en las
regiones exonucleasas de estos dos genes son desconocidas hasta la fecha.
Figura 6. Imagen generada por Pymol de las mutaciones en los dominios exonucleasa de
POLE y POLD1 en la estructura de Pol δ en levadura en el complejo ADN polimerasa T4.
Extraída de (143).
El ADN de simple cadena se muestra en amarillo. Los residuos mutados se muestran en rojo
(POLE) o azul (POLD1).


HIPÓTESIS


Hipótesis
39
Hay una proporción considerable de las familias, que cumpliendo criterios para el
estudio genético de síndromes hereditarios de CCR y/o poliposis colónica, no
presentan mutación germinal en los genes conocidos como responsables de dichos
síndromes.
Al menos una parte de estos casos, no explicados desde el punto de vista genético,
pueden ser consecuencia de otras alteraciones de tipo genético o epigenético que no
son analizados de rutina en los algoritmos diagnósticos establecidos. La identificación
y caracterización de las mismas puede contribuir a una mejora de la eficacia en el
diagnóstico genético en individuos y familias con alto riesgo de CCR.


OBJETIVOS


Objetivos
43
Objetivo general
Mejorar la eficacia del diagnóstico genético en individuos y familias con alto riesgo de
CCR.
Objetivos específicos
Artículo 1
1.1. Analizar la prevalencia de las epimutaciones constitucionales en MLH1 como una
causa de SL en casos no seleccionados de CCR.
1.2. Comparar los datos obtenidos en población no seleccionada con los datos de una
población seleccionada atendida en Unidades de Consejo Genético en Cáncer y que
cumplían los criterios revisados de Bethesda.
1.3. Proponer la adaptación del algoritmo diagnóstico del SL al cribado universal de los
CCR por inmunohistoquímica, en lo referente al estudio de epimutaciones en MLH1.
Artículo 2
2.1. Analizar la prevalencia de las mutaciones recurrentes en los genes POLE y
POLD1 en casos con sospecha de CCR familiar sin mutación en los genes
responsables de los síndromes de sospecha.
2.2. Contribuir a definir el fenotipo clínico asociado a las mutaciones en dichos genes.


MATERIAL Y MÉTODOS


Material y métodos
47
4.1.1. Pacientes y muestras biológicas
En este estudio se incluyeron un total de 2970 pacientes con diagnóstico de CCR que
se dividieron en dos grupos: el grupo no seleccionado y el grupo seleccionado.
En el grupo no seleccionado se incluyeron un total de 2123 pacientes diagnosticados
de CCR procedentes de dos cohortes: 671 pacientes del estudio multicéntrico nacional
EPICOLON II y 1452 pacientes que fueron diagnosticados y tratados de CCR en el
Hospital General Universitario de Alicante (HGUA) entre los años 1999-2011.
Por otra parte, el grupo seleccionado procedía del programa de Cáncer Hereditario de
la Comunidad Valenciana, y por lo tanto, son pacientes con agregación familiar de
cáncer y sospecha de síndrome hereditario, por lo que cumplen al menos alguno de
los criterios de Bethesda de riesgo familiar de CCR (Tabla 4).
Los tumores de todos los pacientes se encontraban fijados en formol e incluidos en
parafina. El tejido germinal correspondía a linfocitos de sangre periférica en el grupo
seleccionado y en la cohorte EPICOLON II del grupo no seleccionado; y a mucosa
colónica sana fijada en formol e incluida en parafina en la cohorte del HGUA.
Las muestras de tejido tumoral (cortes histológicos o punch en zona seleccionada del
tumor) fueron desparafinadas con aceite mineral o xilol. El ADN tumoral se extrajo
mediante un sistema automatizado (QiaCube, QIAGEN, Alemania) usando el kit
QIAamp DNA Mini (QIAGEN, Alemania). Esta misma metodología se empleó para la
extracción de ADN de las muestras de mucosa intestinal sana y mucosa bucal, de los
casos del HGUA, de la cohorte no seleccionada y de los casos con epimutación
constitucional. De la misma manera se extrajo el ADN de sangre periférica de los
casos de la cohorte seleccionada previa lisis de eritrocitos. Finalmente, el ADN de
saliva de los pacientes con epimutación constitucional se extrajo usando el Oragene-
DNA kit (DNA Genotek, Canadá).
4.1. Artículo 1. Castillejo A et al. “Prevalence of MLH1 constitutional
epimutations as a cause of Lynch syndrome in unselected versus selected
consecutive series of patients with colorectal cancer”. J Med Genet. (2015)

Material y métodos
48
4.1.2. Plan de trabajo
Para la realización de este estudio se siguió el siguiente diagrama de flujo de trabajo
para las dos series. El diagrama está basado en al algoritmo diagnóstico del SL
explicado en el apartado 1.2.1.4 (Anexo II):
Figura 7. Plan de trabajo para la realización del estudio reflejado en el artículo 1.
*El análisis de IMS no se realizó en todos los casos. El resultado se empleó para apoyar el
resultado IHQ, según lo explicado en el siguiente apartado.

Material y métodos
49
4.1.3. Cribado molecular de los tumores sospechosos de SL
4.1.3.1. IHQ de las proteínas MMR
En primer lugar se realizó el análisis IHQ de las cuatro proteínas reparadoras: MLH1,
MSH2, MSH6 y PMS2 en tumores parafinados de los casos de CCR. Esto se hizo
disponiendo los tumores en TMAs (Tissue Microarrays), estudiando cada muestra por
duplicado, según lo descrito por Pérez-Carbonell y colaboradores (92).
El análisis de las muestras de la cohorte de Cáncer Hereditario (cohorte seleccionada)
se realizó en los respectivos hospitales de referencia según sectorización de la
Comunidad Valenciana (DOGV– Núm. 7559/29.06.2015). Por otro lado, el análisis de
las dos cohortes no seleccionadas, EPICOLON y HGUA, se realizó íntegramente en el
HGUA.
Se consideró que el tumor presentaba expresión normal para MLH1, MSH2, MSH6 y
PMS2 cuando se observaba tinción nuclear inequívoca en células neoplásicas
epiteliales. En los casos con pérdida de expresión se observó falta de tinción nuclear
en las células tumorales, en presencia de células normales del estroma con tinción
nuclear intacta (control positivo interno). Todos los casos donde los datos de IMS e
IHQ fueron discordantes fueron reevaluados.
4.1.3.2. Inestabilidad de microsatélites
El estudio de IMS se realizó en el ADN tumoral mediante análisis de microsatélites
(homopolímeros quasi-monomórficos) por PCR fluorescente, electroforesis capilar y
análisis de fragmentos. Para la cohorte de EPICOLON II se estudiaron los marcadores
BAT26 y NR24 como se ha descrito previamente (115), considerándose el caso IMS-
positivo cuando al menos uno de los dos marcadores presentó un patrón inestable.
Para el resto de cohortes la IMS se estudió mediante una única PCR pentaplex de los
cinco marcadores quasi-monomórficos considerados estándar (33): BAT25, BAT26,
NR21, NR24 y NR27 según se ha descrito (156). Se consideraron los tumores como
IMS-positivos cuando al menos dos de los cinco marcadores resultaron inestables. El
análisis e interpretación de los resultados se hizo según lo reflejado en la figura 4.

Material y métodos
50
4.1.3.3. Análisis de la metilación de MLH1
El análisis de la metilación somática de MLH1 se realizó en ADN de tejido parafinado
de todos los tumores que presentaban pérdida de la expresión proteica de MLH1 por
IHQ y había material tumoral disponible. En los casos que presentaban metilación en
el tumor, se realizó el estudio de la epimutación constitucional de MLH1 en sangre
(grupo seleccionado y cohorte EPICOLON II) o tejido sano (cohorte HGUA del grupo
no seleccionado) usando la misma metodología. En los casos con metilación MLH1 en
sangre, se analizaron otros tejidos sanos disponibles del mismo paciente (ver Tabla 1
del Artículo 1).
El análisis de la metilación en MLH1 se realizó mediante la técnica MS-MLPA
(Methylation-Sensitive Multiplex Ligation-dependent Probe Amplification; MRC-Holland,
Amsterdam, Paises Bajos) y posterior análisis de fragmentos en el secuenciador 3500
Genetic Analyzer (Life Technologies), según se ha descrito en (106).
Se empleó el kit SALSA MS-MLPA ME011 Mismatch Repair Genes (MMR) que
interroga la metilación en las islas CpG de los promotores de los genes reparadores de
errores MLH1, MSH2, MLH3, PMS2, MSH3, MSH6 y MGMT. Para ello, este kit
contiene 22 sondas que hibridan en regiones de estos genes que contienen un sitio de
restricción para HhaI (enzima de restricción sensible a metilación), además de otras 16
sondas de referencia que hibridan en regiones que no contienen sitios de restricción
para esta enzima. Cuatro de estas sondas hibridan en las regiones A, B, C y D del
promotor de MLH1 respectivamente. Un ADN se considera que presenta metilación en
el promotor de MLH1 cuando tiene un patrón positivo para las sondas que interrogan
las regiones C (-246) y D (-13) (ver figura 5). Solamente la metilación en estas
regiones C y D se correlaciona con la pérdida de expresión de MLH1 (104).
Brevemente, la técnica de MS-MLPA consiste en lo siguiente: en primer lugar,
desnaturalización de la muestra de ADN tumoral, hibridación y ligación de las sondas
de forma específica. Posteriormente se hacen dos alícuotas de la muestra tratada, una
de ellas se digiere con HhaI (New Englands Biolabs) y luego ambas se amplifican por
PCR de forma paralela. Los fragmentos resultantes de esta PCR se separan y se
cuantifican mediante electroforesis capilar en un secuenciador 3500 Genetic Analyzer
(Life Technologies) y se analizan mediante el software Genemapper v 4.0 (Life
Technologies). Finalmente, estos resultados se procesan y se normalizan con los

Material y métodos
51
controles internos de referencia a través de unos algoritmos proporcionados por MRC-
Holland. Así, se obtiene un ratio de metilación de la muestra digerida con respecto a la
no digerida, considerándose como punto de corte para considerar que la región de
estudio está metilada un 25%.
4.1.3.4. Análisis de la mutación de BRAF_V600E
El estudio de la mutación somática BRAF_V600E (exón 15) se hizo de todos los
tumores que tenían pérdida de MLH1 por inmunohistoquímica y de los cuales había
ADN tumoral disponible. Esta determinación se realizó por genotipado por qPCR para
el grupo no seleccionado y por secuenciación Sanger en el grupo seleccionado.
Cuando el tumor resultaba mutado, el caso se consideraba esporádico.
4.1.3.4.1. Genotipado por qPCR
La determinación de la mutación BRAF_V600E mediante PCR en tiempo real (qPCR)
se llevó a cabo a partir de ADN de cada tumor parafinado empleando sondas TaqMan
MGB (Minor Grove Binding) para discriminación alélica. Los cebadores y sondas
fueron diseñados y descritos por Benlloch y colaboradores (157). Los resultados
fueron analizados en el sistema de detección ABI Prism 7500 (Life Technologies).
4.1.3.4.2. Secuenciación Sanger
La determinación de la mutación BRAF_V600E mediante secuenciación Sanger se
hizo amplificando por PCR el exón 15, que contiene dicho codón, con cebadores
específicos y posterior secuenciación y análisis en un secuenciador automático.
Para ello, se amplificó por PCR la región, utilizando AmpliTaq Gold MM 2X (Life
Technologies), usando una pareja de cebadores específicos extraídos de Nagasaka y
colaboradores (158) y las condiciones explicadas en el Anexo V. Posteriormente se
purificó el producto de amplificación mediante una reacción enzimática usando el kit
ExoSAp IT (USB Corporation), y seguidamente se llevó a cabo la reacción de
secuencia utilizando el kit BigDye® Terminator v3.1 Cycle Sequencing (Life
Technologies). La purificación del producto de esta amplificación se realizó con las
columnas Performa DTR Gel Filtration Cartridges (Edge Biosystems). Finalmente, el

Material y métodos
52
producto de esta amplificación fue secuenciado en el secuenciador 3500 Genetic
Analyzer (Life Technologies) y analizado usando los programas Sequencing Analysis
v.5.1 y Variant Reporter v.1.1 (Life Technologies).
4.1.3.5. Análisis de mutaciones germinales en los genes MMR
Análisis de mutaciones puntuales por secuenciación Sanger
El análisis de mutaciones germinales se realizó en ADN genómico procedente de
leucocitos de sangre periférica (grupo seleccionado completo y cohorte EPICOLON II
del grupo no seleccionado) o de tejido no tumoral (cohorte HGUA del grupo no
seleccionado).
El estudio de mutaciones en los genes MMR se realizó basándose en los resultados
de IHQ, IMS, metilación de MLH1 y mutación BRAF_V600E, según lo descrito en
(106), utilizando el algoritmo diagnóstico descrito en el Anexo II.
La detección de mutaciones puntuales se analizó mediante amplificación por PCR y
secuenciación directa de la región codificante completa, así como de las zonas de
unión intrón-exón de cada gen, según lo que se ha explicado en el apartado anterior.
Las condiciones de PCR y los cebadores para cada gen y región han sido descritos
previamente (159-161).
Interpretación de resultados
Se utilizó la nomenclatura recomendada por Human Genome Variation Society para
la descripción de las variantes: http://www.hgvs.org/mutnomen/
Clasificación de las variantes genéticas: se utilizan los criterios establecidos en las
recomendaciones del Colegio Americano de Genética Médica (162) e InSiGHT
(163). Las variantes se clasifican en cinco clases: 1: No patogénicas; 2:
probablemente no patogénicas; 3: variantes de significado clínico
desconocido; 4: probablemente patogénicas y 5: patogénicas. Desde el punto
de vista clínico, las variantes de clase 1 y 2 son consideradas neutrales y no
confieren riesgo. Las variantes 4 y 5 son consideradas patogénicas y de alto riesgo,
mientras que las de clase 3 son de significado clínico desconocido y por

Material y métodos
53
consiguiente, en el momento actual, no se puede establecer su implicación con la
enfermedad.
Análisis de grandes reordenamientos por MLPA
Se estudiaron los grandes reordenamientos (deleciones/inserciones) en los genes
MMR mediante la técnica MLPA, usando los siguientes kits: P003 (MLH1-MSH2);
P248 (MLH1-MSH2 confirmación); P008 (PMS2-MSH6) y P072 (EPCAM), de acuerdo
con las instrucciones del fabricante (MRC-Holland).
4.1.3.6. Análisis de la secuencia de la región promotora de MLH1
En aquellos casos con epimutación constitucional en MLH1 se secuenció la región
promotora de dicho gen, con el fin de identificar variantes asociadas con dicho
fenotipo. La secuenciación se hizo según lo descrito en el apartado 4.1.3.4.2., usando
los cebadores y condiciones de PCR que figuran en el Anexo V.
4.1.4. Análisis estadístico
Para el análisis de los datos generados se empleó el paquete estadístico IBM SPSS
v.21.0. (Chicago, IL, Estados Unidos). Las variables categóricas se representaron
como frecuencias o porcentajes. Las variables cuantitativas (edad) se representaron
como mediana e intervalo intercuartílico, tras comprobar su distribución no normal
mediante el test de Kolmogorov-Smirnov. Para la comparación de las variables
categóricas se usó el test chi-cuadrado, y para las variables cuantitativas
independientes con distribución asimétrica se usó el test U de Mann-Whitney. Todos
los p-valores tienen dos colas y se consideraron como estadísticamente significativos
aquellos valores de p<0,05.


Material y métodos
55
En este artículo se recogen los resultados de dos estudios similares, realizados de
forma paralela, en dos series de casos seleccionados. La primera serie fue analizada
en el laboratorio del programa de Cáncer Hereditario del Instituto Catalán de
Oncología (ICO); y la segunda serie (Comunidad Valenciana) fue analizada por el
grupo de investigación al que pertenece la doctoranda (Unidad de Investigación del
HGUA y Genética Molecular del HGUE).
4.2.1. Pacientes y muestras biológicas
En este estudio se incluyeron un total de 858 pacientes diagnosticados de CCR y/o
poliposis provenientes de 840 familias de dos series diferentes (Figura 8). Se
seleccionaron casos con sospecha de origen hereditario en los que no se había
encontrado mutación. Estos casos no explicados desde el punto de vista genético
fueron analizados para los genes responsables, siguiendo los algoritmos diagnósticos
de los síndromes de sospecha en cada caso (SL (Anexos II) o síndromes de poliposis
colónica (MAP o PAF, Anexo III)).
En la serie 1 se incluyeron un total de 612 pacientes de CCR y/o poliposis familiar de
aparición temprana, correspondientes a 594 familias provenientes de las unidades de
consejo genético del ICO. Se seleccionaron 524 individuos de 506 familias de CCR no
polipósico que cumplían, al menos, los criterios revisados de Bethesda. Estos
pacientes habían sido previamente testados por IHQ de las proteínas MMR y/o IMS en
sus tumores, y no presentaban mutación germinal en estos genes MMR, ni metilación
en MLH1. Para el estudio de mutaciones germinales en los genes MMR de estos
casos, se siguió el algoritmo explicado en el apartado 1.2.1.4. (Anexo II).
Así mismo, se incluyeron 88 casos de poliposis que habían sido estudiados para
MUTYH siguiendo el esquema habitual, así como para APC cuando el número de
adenomas era superior a 20 (Anexo III). Para MUTYH se siguió la estrategia propuesta
Artículo 2. Valle L et al. “New insights into POLE and POLD1 germline
mutations in familial colorectal cancer and polyposis”. Hum Mol Genet. (2014)

Material y métodos
56
en (134): se estudiaron las tres variantes recurrentes en población española, mediante
secuenciación Sanger de los exones 7 y 13. En caso de detectar la presencia de
alguna variante en heterocigosis, se secuenciaba la región codificante completa y las
uniones intrón-exón correspondientes (ver apartado 1.3.2.). Además se realizaba
MLPA para el análisis de grandes reordenamientos en MUTYH (P378, MRC Holland).
Para APC se estudió toda la región codificante y las uniones intrón-exón por
secuenciación Sanger y se realizó el estudio de grandes reordenamientos por MLPA
(P043, MRC Holland).
El estudio de mutaciones germinales se realizó a partir de ADN extraído de sangre
periférica mediante el kit Flexigene DNA (QIAGEN, Alemania). Para la extracción de
ADN de muestras tumorales fijadas en formol e incluidas en parafina se utilizó el kit
DNA Mini (QIAGEN, Alemania).
Figura 8. Casos de CCR y poliposis estudiados.
En la serie 2 se incluyeron un total de 246 casos índice diagnosticados de CCR no
polipósico y/o poliposis. Los casos de CCR no polipósico procedían, al igual que el
grupo seccionado del primer artículo, del programa de Cáncer Hereditario de la
Comunidad Valenciana. Todos los casos presentaban agregación familiar de cáncer:
63 casos cumplían criterios de Amsterdam I o II y el resto (n=80) presentaban 2 o más
familiares de primer o segundo grado diagnosticados con un tumor relacionado con el
858 pacientes
Serie 1 (ICO)
n=612
CCR
n=524
MMR+
n=438
MMR–
n=86
Poliposis
n=88
Serie 2
(C. Valenciana)
n=246
CCR
n=143
MMR+
n=143
Poliposis
n=103

Material y métodos
57
SL (criterio 5 de Bethesda) (Tabla 4). Todos los casos eran MMR-positivos, lo que se
comprobó por IHQ y/o IMS y se siguió el algoritmo diagnóstico de mutaciones
germinales en MMR según lo explicado en el apartado 1.2.1.4.
Se incluyeron además 103 casos de poliposis atenuada (10-100 pólipos) provenientes
del proyecto multicéntrico nacional EPIPOLIP (164). Todos estos individuos tenían al
menos un familiar de primer grado afectado de CCR y todos habían sido interrogados
para MUTYH, y para APC cuando tenían más de 10 adenomas, según lo descrito para
la serie 1.
En ambos grupos de pacientes, tanto el ADN de tejido tumoral como el de sangre
periférica ya habían sido extraídos previamente en el proceso para el estudio de
mutaciones germinales en sus respectivas unidades. En el caso de la serie 2 esto se
hizo siguiendo la metodología previamente indicada en el artículo 1.
4.2.2. Plan de trabajo
El plan de trabajo para la realización del estudio reflejado en el segundo artículo se
muestra a continuación en la Figura 9:
Material y métodos
58
Figura 9. Plan de trabajo para la realización del estudio reflejado en el artículo 2.
*El resultado del análisis de IMS se empleó para apoyar el resultado IHQ, priorizando el
resultado de éste último.
4.2.3. Estudio de las mutaciones germinales en POLE y POLD1
El estudio de las mutaciones recurrentes en POLE y POLD1 se hizo por metodologías
diferentes en cada serie: en la serie 1 se estudiaron específicamente las mutaciones
POLE_Leu424Val y POLD1_Ser478Asn mediante ensayos KASPar, validando los
resultados encontrados por secuenciación directa; en la serie 2 se estudiaron por
secuenciación Sanger los exones 13 y 11 de POLE y POLD1 respectivamente, donde
se encuentran dichas mutaciones.
4.2.3.1. Serie 1 (Ensayos de genotipado KASPAR)
Ambas mutaciones se genotiparon mediante ensayos KASPar (KASP-By-Design
genotyping assays. LGC group, Teddington, Reino Unido), según se ha descrito (165),
usando cebadores específicos para la detección de estas variantes (Tabla

Material y métodos
59
Suplementaria 1 del Artículo 2). Las reacciones se llevaron a cabo en un sistema de
detección de PCR a tiempo real LightCycler 480 (Roche DiagnosticsGmbH, Alemania).
En cada carrera se introdujo un control positivo para cada una de las mutaciones
testadas.
La validación de estos resultados de genotipado, junto con el análisis de las muestras
que fallaron, así como los estudios de cosegregación se realizaron mediante
secuenciación directa, como se ha descrito en el apartado 4.1.3.4.2. Los cebadores y
condiciones empleados figuran en el anexo V.
4.2.3.2. Serie 2 (Secuenciación Sanger)
Se estudió el exón 13 de POLE y el exón 11 de POLD1, así como los sitios de unión
intrón-exón a partir de ADN de sangre periférica, mediante PCR y secuenciación,
según lo descrito en el apartado 4.1.3.4.2; usando los cebadores y condiciones citados
en el Anexo V.
4.2.4. Análisis de predicción in silico
Se usaron diferentes programas bioinformáticos para evaluar el potencial dañino de la
variante germinal de cambio de sentido (missense) p.Leu474Pro encontrada en
POLD1 a nivel de la afectación en la estructura y función de la proteína: SNPs3D,
PolyPhen-2, SIFT, CONDEL (166).
Estos programas se utilizan como método para predecir el posible impacto del cambio
de nucleótido en la funcionalidad de la proteína mutada que se genera: la
conservación del residuo en la evolución, la severidad de la substitución desde el
punto de vista bioquímico (polaridad, carga y tamaño), la localización en dominios
estructurales y funcionales y el potencial impacto estructural entre las proteínas salvaje
y mutada.

Material y métodos
60
Tabla 7. Programas de predicción in silico empleados.
Programa de predicción Página web
SNPs3D http://www.snps3d.org/
PolyPhen-2 http://genetics.bwh.harvard.edu/pph2/
SIFT http://sift.jcvi.org/
CONDEL(166) http://bg.upf.edu/fannsdb/
4.2.5. Estudio de pérdida de heterocigosidad (LOH) en POLE
En el tumor de colon del individuo de la serie 1 portador de la mutación germinal
POLE_Leu424Val se realizó el estudio de LOH mediante dos metodologías distintas:
4.2.5.1. Mapeo por microsatélites
Se estudiaron dos microsatélites informativos (D12S1628 y D12S357) localizados en
una región cromosómica de 1.356 Mb circundante al codón POLE_Leu424. Esto se
realizó mediante PCR fluorescente y posterior análisis de fragmentos, de forma similar
al estudio de la IMS (apartado 4.1.3.2.), usando los cebadores e indicaciones
proporcionadas por el equipo de Palles y colaboradores (119).
4.2.5.2. SNaPshot para la mutación POLE_Leu424Val
Esta técnica permite estudiar la abundancia de cada alelo (mutado versus salvaje) del
codón Leu424. Consiste en la amplificación por PCR de la región de interés y posterior
extensión del cebador en un solo nucleótido marcado por fluorescencia, lo que se
detecta también por electroforesis capilar y análisis de fragmentos. Los cebadores y las
condiciones de PCR figuran en el anexo V.
Para ambas metodologías, se considera que hay LOH cuando la intensidad relativa de
cualquier alelo se reduce ≥50% con respecto al otro alelo, considerando la intensidad
alélica relativa en el ADN germinal.

Material y métodos
61
4.2.6. Análisis de las mutaciones somáticas en BRAF, KRAS y NRAS
Con el fin de definir la vía de carcinogénesis que siguen los tumores de los pacientes
portadores de la mutación germinal POLD1_Leu474Pro encontrados en la serie 1, se
realizó el análisis de las mutaciones somáticas en el ADN de los tumores disponibles
de los individuos portadores de la mutación: en el CCR del caso índice y en el CE de
la tía materna de éste. La siguiente tabla muestra las mutaciones estudiadas y la
metodología y condiciones empleadas para ello:
Tabla 8. Mutaciones somáticas analizadas en BRAF, KRAS y NRAS y metodología empleada.
GEN MUTACIÓN METODOLOGÍA CONDICIONES
BRAF V600E Secuenciación Sanger
(ver apartado 4.1.3.4.2)
Cebadores y
condiciones en Anexo V
KRAS
Codones 12 y 13 KRAS StripAssay
(ver siguiente párrafo)
Según instrucciones del
fabricante
Exón 3 (codones 59 y 61) Secuenciación Sanger
(ver apartado 4.1.3.4.2)
Cebadores y
condiciones en Anexo V Exón 4 (codones 117 y 146)
NRAS
Exón 2 (codones 12 y 13)
Secuenciación Sanger
(ver apartado 4.1.3.4.2)
Cebadores y
condiciones en Anexo V Exón 3 (codones 117)
Exón 4 (codón 146)
La determinación de las mutaciones en los codones 12 y 13 de KRAS mediante KRAS
StripAssay (VienaLab Diagnostics GmbH, Vienna Austria) permite la detección de las
10 mutaciones más frecuentes en esta localización. El método utilizado se ajusta a las
instrucciones del fabricante e implica las siguientes etapas: 1) amplificación por PCR
del ADN tumoral con cebadores específicos marcados con biotina; 2) hibridación y
lavado del producto de la amplificación en una tira de nitrocelulosa que contiene
sondas de oligonucleótido alelo-específico, fijadas en líneas paralelas de forma
automatizada en Auto-LiPA 48; 3) detección de las secuencias de cada codón
mediante revelado de color por estreptavidina-fosfatasa-alcalina y sustrato de color.


RESULTADOS


Resultados
65
En esta sección de resultados de esta Tesis Doctoral se presentan los dos artículos
que constituyen el trabajo desarrollado para la defensa de la misma. A continuación se
muestra el resumen de cada uno de ellos.
En el Anexo XII se incluyen otros trabajos publicados de los cuales la doctoranda es
coautora.


Resultados
67
Adela Castillejo*, Eva Hernández-Illán*, María Rodriguez-Soler, Lucía Pérez-
Carbonell, Cecilia Egoavil, Victor M Barberá, María–Isabel Castillejo, Carla Guarinos,
Eduardo Martínez-de-Dueñas, María-Jose Juan, Ana-Beatriz Sánchez-Heras, Zaida
García-Casado, Clara Ruiz-Ponte, Alejandro Brea-Fernández, Miriam Juárez, Luis
Bujanda, Juan Clofent, Xavier Llor, Montserrat Andreu, Antoni Castells, Angel
Carracedo, Cristina Alenda, Artemio Payá, Rodrigo Jover**, José-Luis Soto**.
*Adela Castillejo y Eva Hernández-Illán han contribuido igualmente a este trabajo y
comparten la primera autoría.
**Jose-Luis Soto y Rodrigo Jover han contribuido igualmente a este trabajo y
comparten la autoría sénior.
Journal of Medical Genetics. 2015 Jul;52(7):498-502.
Factor de impacto (2014): 6.335
Primer Cuartil (Genetics and Heredity 18/167; Q1)
Artículo 1. Castillejo A et al. “Prevalence of MLH1 constitutional epimutations
as a cause of Lynch syndrome in unselected versus selected consecutive
series of patients with colorectal cancer”. J Med Genet. (2015)

Resultados
68
RESUMEN
Introducción: La prevalencia de las epimutaciones constitucionales en MLH1 en la
población general es desconocida. Nuestro objetivo fue determinar la prevalencia de
las epimutaciones constitucionales en MLH1 en una serie no seleccionada de
pacientes con cáncer colorrectal, y compararla con la prevalencia de este mismo
fenómeno en una serie seleccionada.
Métodos: En el grupo no seleccionado se incluyeron pacientes con diagnóstico de
cáncer colorrectal (n=2123). Para comparar, se incluyó además un grupo seleccionado
de 847 pacientes con cáncer colorrectal que cumplían los criterios revisados de
Bethesda. En casos con ausencia de expresión de MLH1, se evaluó la metilación
somática y constitucional de MLH1 mediante amplificación multiplex de sondas
dependientes de ligación y específicas de metilación. Se evaluaron alteraciones
germinales en los genes reparadores de errores por secuenciación Sanger y
amplificación multiplex de sondas dependientes de ligación.
Resultados: El 5,5% de los casos de la serie no seleccionada y el 12,5% de la serie
selecccionada presentaban pérdida de expresión de MLH1 (p<0,0001). No se
detectaron epimutaciones constitucionales en MLH1 en la población no seleccionada
(0/62); cinco casos de la serie seleccionada fueron positivos para epimutaciones en
MLH1 (15,6%, 5/32; p=0,004).
Conclusiones: Nuestros resultados sugieren una prevalencia insignificante de
epimutaciones constitucionales en MLH1 en cohortes no seleccionadas de cáncer
colorrectal. Por tanto, el análisis de epimutación constitucional en MLH1 debería
dirigirse exclusivamente a pacientes que cumplen los criterios revisados de Bethesda y
cuyo tumor presente pérdida de expresión y metilación de MLH1.

Fd





Supplementary Table 1. Clinical and pathological characteristics of CRC patients.
UNSELECTED COHORT*
n (%)
SELECTED COHORT
n (%) Age, median (IQI) 71.5 (62.3-78) 49 (43-62)
Sex, males 1194 / 2073 (57.6) 285/574 (49.7) Amsterdam criteria 20/764 (2.6) 152/847 (17.9) Bethesda critera 377/2121 (17.8) 847/847 (100) Tumour pathologic features Location - Proximal - Distal (esplenic flexure included)
542/1696 (32) 1154/1696 (68)
NDA
Stage - I and II - III and IV
576/1141 (50.5) 565/1141 (49.5)
NDA
Grade - Low - High
1426/1596 (89.3) 170/1596 (10.7)
NDA
Histology (WHO) - Adenocarcinoma - Medullary carcinoma - Signet-ring cell carcinoma - Mucinous adenocarcinoma - Neuroendocrine carcinoma - Undifferentiated carcinoma - Other
1292/1449 (89.2) 25/1449 (1.7) 14/1449 (1) 90/1449 (6.2) 8/1449 (0.5) 6/1449 (0.4) 14/1449 (1)
NDA
Lymphocytic infiltration 416/2086 (19.9) NDA Molecular features MLH1 loss of expression 117/2123 (5.5) 106/847 (12.5) MSI 92/1179 (7.8) 183/840 (21.8) BRAF mutation 54/691 (7.8) 9/197 (4.6)
NDA: no data available.
* Frequency of patients fulfilling Bethesda criteria was 21.6% (158/669) for the Epicolon II
sub-cohort, versus 15.1% (219/1452) for the Hospital General Universitario de Alicante sub-
cohort (p<0.0001). This difference might be explained because only the Bethesda criterion
#1 (age at diagnosis of CRC <50y) was considered in the retrospective series from Hospital
General Universitario de Alicante, while the five canonical criteria for Bethesda classification
was used in Epicolon II sub-cohort.
No significant differences were found among the two sub-cohorts of the unselected series of
cases for the rest of variables.

Resultados
75
Laura Valle†, Eva Hernández-Illán†, Fernando Bellido, Gemma Aiza, Adela Castillejo,
María-Isabel Castillejo, Matilde Navarro, Nuria Seguí, Gardenia Vargas, Carla Guarinos,
Miriam Juarez, Xavier Sanjuán, Silvia Iglesias, Cristina Alenda, Cecilia Egoavil, Ángel
Segura, María-José Juan, María Rodriguez-Soler, Joan Brunet, Sara González, Rodrigo
Jover, Conxi Lázaro, Gabriel Capellá, Marta Pineda, José Luís Soto, Ignacio Blanco.
†Laura Valle y Eva Hernández-Illán han contribuido equitativamente a este trabajo.
Human Molecular Genetics. 2014 Jul 1;23(13):3506-12.
Factor de impacto (2014): 6.393
Primer Cuartil (Genetics and Heredity 17/167; Bioquemistry and Molecular Biology 32/290;
Q1)
Artículo 2. Valle L et al. “New insights into POLE and POLD1 germline
mutations in familial colorectal cancer and polyposis”. Hum Mol Genet. (2014)

Resultados
76
RESUMEN
Introducción: Se han identificado recientemente mutaciones germinales en las ADN
polimerasas ε (POLE) y δ (POLD1), en familias con múltiples adenomas colorrectales y con
CCR. Todos los casos descritos eran portadores de la mutación POLE c.1270C>G
(p.Leu424Val) o POLD1 c.1433G>A (p.Ser478Asn). Debido a la escasez de casos descritos
hasta la fecha, no se ha descrito un fenotipo clínico exacto. Nuestro objetivo fue evaluar la
presencia de estas mutaciones recurrentes en casos no explicados de CCR y poliposis
familiar de aparición temprana; y proporcionar información adicional para definir las
características clínicas de los casos con mutación.
Métodos: Se estudiaron un total de 858 pacientes de CCR y poliposis familiar de aparición
temprana: 581 casos de CCR familiar y de aparición temprana con el sistema reparador de
errores intacto (MMR), 86 casos con deficiencia en MMR, y 191 casos de poliposis. La
búsqueda de mutaciones se hizo mediante ensayos de genotipado KASPar y/o
secuenciación Sanger del respectivo exón.
Resultados: Se identificó la mutación POLE p.Leu424Val como mutación de novo en una
paciente de 28 años con CCR y poliposis. Ninguno de los 858 casos estudiados presentó la
mutación POLD1 p.Ser478Asn. Se identificó una nueva mutación POLD1 c.1421T>C
(p.Leu474Pro) en una familia Amsterdam II sin alteración del sistema reparador de errores.
La patogenicidad de esta variante se ve sustentada por la cosegregación genotipo-fenotipo
en la familia, las predicciones in silico, y ensayos funcionales en levaduras previamente
publicados.
Conclusiones: Las mutaciones en POLE y POLD1 explican un porcentaje de los casos de
CCR familiar y de poliposis. La secuenciación de los dominios de corrección de errores de
POLE y POLD1 se debería considerar en el diagnóstico genético rutinario. Hasta que no se
proporcione mayor evidencia, el estudio genético de POLE y POLD1 no se debería restringir
a los casos con poliposis y debería considerarse la presencia de mutaciones de novo.






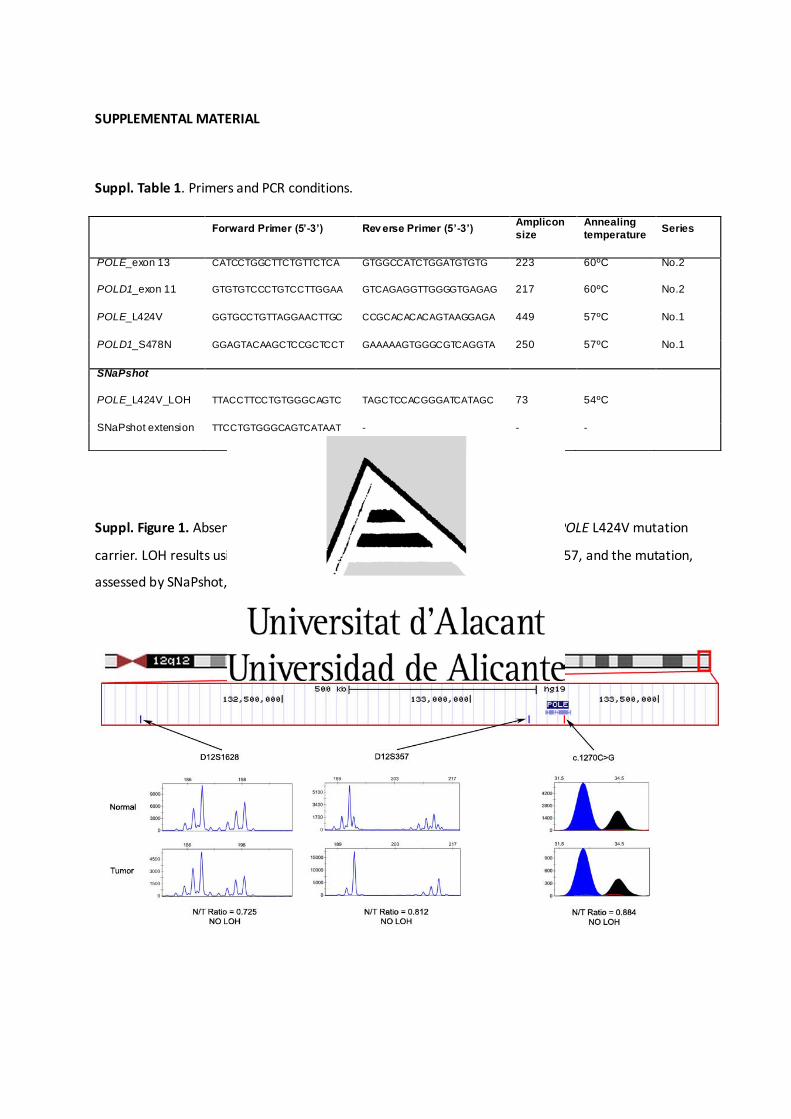
SUPPLEMENTAL MATERIAL
Suppl. Table 1. Primers and PCR conditions.
Suppl. Figure 1. Absence of LOH at POLE in the colon tumor developed by the POLE L424V mutation
carrier. LOH results using two informative microsatelites, D12S1628 and D12S357, and the mutation,
assessed by SNaPshot, are shown.
Forward Primer (5’-3’) Rev erse Primer (5’-3’) Amplicon
size
Annealing
temperature Series
POLE_exon 13 CATCCTGGCTTCTGTTCTCA GTGGCCATCTGGATGTGTG 223 60ºC No.2
POLD1_exon 11 GTGTGTCCCTGTCCTTGGAA GTCAGAGGTTGGGGTGAGAG 217 60ºC No.2
POLE_L424V GGTGCCTGTTAGGAACTTGC CCGCACACACAGTAAGGAGA 449 57ºC No.1
POLD1_S478N GGAGTACAAGCTCCGCTCCT GAAAAAGTGGGCGTCAGGTA 250 57ºC No.1
SNaPshot
POLE_L424V_LOH TTACCTTCCTGTGGGCAGTC TAGCTCCACGGGATCATAGC 73 54ºC
SNaPshot extension TTCCTGTGGGCAGTCATAAT - - -


DISCUSIÓN


Discusión
87
En la oncogénesis y desarrollo del cáncer colorrectal intervienen una combinación de
factores fundamentalmente de tipo ambiental. No obstante, se estima que el
componente hereditario puede estar presente hasta en un 30% de los casos. En la
mayor parte de éstos se trata de variantes genéticas de penetrancia moderada o baja;
mientras que los casos donde se evidencia una alta penetrancia con herencia
mendeliana, y que configuran entidades clínicas sindrómicas identificables, suponen
entre un 3-5% del total de los CCR. Los principales genes implicados en cada uno de
los síndromes hereditarios de CCR son conocidos (a excepción de la Poliposis Mixta
Hereditaria) y se utilizan en el diagnóstico genético y predictivo de riesgo en las
familias que cumplen determinados criterios clínicos y patológicos.
En los últimos 25 años el avance en el conocimiento de estas patologías ha sido
meteórico y ha permitido una mejora sustancial en el manejo de las familias. No
obstante, la mayoría de estos síndromes hereditarios muestran un alto grado de
complejidad desde el punto de vista genético. La mayoría de estos síndromes tienen
una herencia autosómica dominante (excepto MAP con herencia recesiva), con
penetrancia incompleta y expresividad variable. Algunos de ellos presentan una
notable heterogeneidad de loci o de alelo, además de patrones de herencia atípicos,
con mutaciones de novo o mosaicismo somático. El rendimiento del diagnóstico
genético sigue siendo muy mejorable, ya que en la mayoría de los casos no se llega a
identificar la alteración genética responsable. Las causas pueden ser múltiples, desde
aspectos que tienen que ver con la estrategia o algoritmos diagnósticos, a los
puramente técnicos y de interpretación de resultados, como a la existencia de genes y
mecanismos moleculares responsables de los síndromes que son desconocidos en el
momento actual.
En este escenario, en los últimos años se están acumulando evidencias que muestran
un solapamiento significativo en el fenotipo clínico de diferentes síndromes, lo que
complica el diagnóstico genético y en consecuencia, el manejo de los pacientes y sus
familiares a riesgo. Se están describiendo un número creciente de casos en los que el
síndrome de sospecha inicial es diferente del que termina resultando tras un análisis
genético masivo, fundamentalmente utilizando amplios paneles de genes. Así pues,
casos con sospecha clínica de síndrome de mama-ovario hereditario terminan siendo
genéticamente un SL (167), o casos que cumpliendo criterios clínicos, patológicos y
moleculares somáticos de SL presentan mutación en MUTYH y son realmente MAP
(136-137) o incluso Li-Fraumeni (168).

Discusión
88
El SL es el paradigma de complejidad genética: es el síndrome de CCR hereditario
más prevalente y presenta una herencia autosómica dominante, heterogeneidad de
loci, penetrancia incompleta y variable según las poblaciones (18, 63, 66, 72-73) y con
expresividad también muy variable. Se han descrito casos con mutaciones de novo
(169) y fenómenos de anticipación (170), así como mutaciones en zonas intrónicas
(171), profundas traslocaciones e inversiones (98-101). Las mutaciones en
heterocigosis en PMS2 presentan una moderada penetrancia (15-20%) (71), pudiendo
pasar desapercibidas en la mayoría de los casos. Existen formas con mutación
bialélica y un fenotipo muy agresivo con cánceres en la infancia (8). Algunas familias
con SL presentan alteraciones epigenéticas (hipermetilación en el promotor de MSH2)
como causa directa, siendo ésta el resultado de una deleción que afecta a un gen
próximo (EPCAM) no relacionado funcionalmente con la reparación de errores de tipo
mismatch. Finalmente, en hasta un 10-15% de los casos con sospecha de SL (criterios
de Bethesda), que presentan pérdidas de expresión inmunohistoquímica de MLH1 y
metilación de su promotor en el tejido tumoral, la causa de la enfermedad es una
epimutación constitucional en dicho gen.
En el presente trabajo de Tesis Doctoral se ha pretendido aportar nuevo conocimiento
que ayude a mejorar la eficacia del diagnóstico genético en individuos y familias con
alto riesgo de CCR. El primero de los dos estudios que conforman la Tesis Doctoral se
enmarca precisamente en este último aspecto de las epimutaciones constitucionales
de MLH1. Se trata de un estudio colaborativo multidisciplinar con la participación del
grupo EPICOLON II y el Programa de Cáncer Hereditario de la Comunidad
Valenciana.
Hasta la fecha, y utilizando el algoritmo de análisis establecido para pacientes con
sospecha de SL (pacientes que cumplen criterios de Bethesda) , el estudio de la
metilación de MLH1 en sangre está indicado en todos aquellos casos con pérdida de
expresión de MLH1 y metilación de su promotor en tejido tumoral. El nuevo escenario
que se nos presenta con la incorporación del cribado universal, mediante el estudio
inmunohistoquímico de las proteínas MMR de todos los CCR y CE, o aquellos
diagnosticados antes de los 71 años, conlleva ineludiblemente la identificación de un
elevado número de casos con pérdida de expresión de MLH1 y metilación de su
promotor en el tumor. Se hacía necesario por tanto, establecer cuál es la prevalencia
real de epimutaciones constitucionales de MLH1 en población no seleccionada, con
objeto de precisar cuándo hacer el estudio de metilación en sangre en el algoritmo del
cribado universal.

Discusión
89
Para abordar esta cuestión se incluyeron dos cohortes de pacientes con CCR: una
serie de casos de CCR no seleccionados (n=2.123) y una serie seleccionada de casos
que cumplían al menos los criterios revisados de Bethesda (n=847). Ambas series
mostraron diferencias en variables demográficas y moleculares, relacionadas con los
criterios de selección. Así, la serie no seleccionada presentó una mayor proporción de
varones, de acuerdo con lo descrito para CCR no seleccionado (3) y mayor edad al
diagnóstico, tanto cuando se consideran las series completas, como entre los casos
que presentan metilación de MLH1 en el tumor. Esta metilación puede estar
relacionada con el fenotipo CIMP típico en estos tumores con IMS, que se caracteriza
por la hipermetilación somática de las regiones promotoras de los genes supresores
de tumores y que típicamente se asocia al envejecimiento, lo que podría explicar estas
diferencias de edad encontradas en los casos con metilación de MLH1 entre ambas
cohortes. Además, la serie no seleccionada presentó un menor porcentaje de tumores
con pérdida de expresión de MLH1; pero cuando se considera el subgrupo de estos
casos con pérdida de expresión de MLH1 se observa una mayor frecuencia de
metilación somática de MLH1 en estos casos no seleccionados. Esto puede responder
a una mayor representación de casos hereditarios en la serie seleccionada: mayor
frecuencia de mutaciones germinales en los genes MMR y epimutaciones
constitucionales en MLH1.
No se observaron epimutaciones constitucionales en el grupo no seleccionado. Sin
embargo, un 15,6% de los pacientes que acudían a las consultas de consejo genético
por sospecha clínica de SL (cumplimiento de los criterios revisados de Bethesda) que
presentaban pérdida de expresión de MLH1 y metilación somática de este gen en el
tumor, presentaron epimutación contitucional de MLH1. Ninguno de los casos
identificados presentaba mutaciones en línea germinal de MLH1. Estas epimutaciones
se han considerado como una causa poco frecuente de CCHNP. Nuestros resultados
confirman la frecuencia de las epimutaciones en MLH1 previamente descrita, en torno
al 10-15% de estos casos (109, 172-174), avalando así la incorporación de su estudio
en el algoritmo diagnóstico de SL.
El estudio de la epimutación de MLH1 hasta la fecha se ha restringido a pacientes
seleccionados de CCR con fenotipo de riesgo; ningún estudio había abordado el
análisis de la prevalencia de esta alteración en una gran serie de pacientes con CCR
de base poblacional. Los resultados de este estudio sugieren que la prevalencia de
esta alteración epigenética en población de CCR no seleccionada es insignificante.
Por el contrario, su prevalencia no es despreciable, como hemos podido confirmar, en

Discusión
90
pacientes que cumplen los criterios de Bethesda y presentan pérdida de expresión de
MLH1.
La historia familiar de cáncer en los cinco casos con epimutación constitucional es
ausente o escasa, mostrando únicamente uno de los cinco pacientes un familiar de
primer grado afecto de un cáncer relacionado con el SL (Anexo VI). Además, se
analizaron once familiares de tres casos índice con epimutación (Anexo VI) y ninguno
mostró metilación de MLH1 en sangre, lo que sugiere que este evento en nuestra serie
puede ser el resultado de una alteración de novo. No obstante, en dos casos índices
no se pudo estudiar la presencia de metilación constitucional de MLH1 en sus
familiares, por lo que no se puede descartar algún tipo de herencia de este evento en
los mismos. Las epimutaciones constitucionales son reversibles en la meiosis, por lo
que presentan una herencia no mendeliana atípica, a diferencia de la herencia
autosómica dominante asociada a las mutaciones en los genes del SL (109, 111). Se
han propuesto distintos mecanismos de herencia transgeneracional de estas
epimutaciones, aunque realmente han sido identificados en un escaso número de
familias (175). En los casos con epimutación de nuestra serie se secuenció el
promotor de MLH1 en busca de alteraciones genéticas en cis que pudieran explicar la
epimutación en estos casos, pero no se observó ninguna alteración significativa. El
único hallazgo fue en el caso #5, que resultó heterocigoto para el polimorfismo -
93G>A. Esta variante tiene una frecuencia del alelo menos frecuente de 31,5% (1000
genomas) y se ha asociado a CCR con IMS y CIMP (176), habiendo sido también
descrita anteriormente en otro caso con epimutación (172).
Atendiendo al ratio de metilación encontrado en la región activa del promotor de MLH1
(regiones C y D, Tabla 1 del Artículo 1) en estos cinco casos con epimutación,
podemos considerar que todos ellos presentan metilación constitucional monoalélica.
Hasta la fecha, sólo se ha descrito un caso con metilación constitucional bialélica
(174). En los tumores de tres de estos cinco casos la metilación en MLH1 resultó
bialélica (ratio de metilación >80%), mientras que en los tumores y demás tejidos
analizados de los otros dos casos, el ratio de metilación sugería un carácter
monoalélico, por lo que en estos casos la inactivación somática del segundo alelo
tendría lugar presumiblemente por otro tipo de alteraciones genéticas (mutaciones
puntuales, grandes reordenamientos, LOH…).
La mutación de BRAF_V600E se asocia fuertemente a la inactivación de MLH1 por
hipermetilación de su promotor (177-178) y este marcador ha sido ampliamente

Discusión
91
utilizado para la discriminación entre CCR del SL y CCR esporádico con IMS. Como
era previsible, en nuestro estudio las mutaciones de BRAF en los casos con metilación
en MLH1 fueron significativamente más frecuentes en la serie no seleccionada que en
la serie seleccionada. No obstante, ninguno de los cinco casos con metilación
constitucional en MLH1 presentó la mutación en BRAF, lo que podría sugerir que la
presencia de la mutación de BRAF también podría comportarse como un marcador
negativo para la presencia de epimutación en MLH1, como ya lo hace en los casos
con mutación de MLH1 en línea germinal. En los estudios realizados hasta la fecha,
sólo se han descrito tres casos con epimutación en MLH1 y mutación en BRAF (175,
179-180). Se precisaría disponer de la información referida a un tamaño muestral
suficientemente representativo para poder establecer la existencia de tal asociación y
su valor como marcador excluyente de epimutación constitucional de MLH1.
La inclusión del análisis de las epimutaciones constitucionales de MLH1 en el
algoritmo diagnóstico del SL, en casos que cumplen criterios de sospecha, está
ampliamente aceptada. No obstante, la realidad en nuestro entorno es que su análisis
rutinario no está implementado de forma generalizada en los laboratorios diagnósticos.
Aspectos como limitaciones de disponibilidad tecnológica, la baja prevalencia de estas
alteraciones o la incertidumbre respecto a su penetrancia y modo de herencia pueden
influir en su utilización. En cualquier caso, estos pacientes tienen un riesgo
incrementado de desarrollar tumores metacronos relacionados con el SL y un riesgo
desconocido de cáncer en sus familiares, por lo que consideramos que el análisis de
epimutación constitucional no se puede obviar, aunque solo sea para no interpretar
erróneamente que la metilación de MLH1 presente en el tumor del caso índice implica
un origen esporádico, sino que se trata realmente de una condición hereditaria de SL
atípica.
En conclusión, nuestros resultados confirmaron que alrededor de un 10-15% de los
casos con sospecha de SL, con pérdida de expresión e hipermetilación de MLH1 en el
tumor, presentan epimutación constitucional en dicho gen. En casos de CCR no
seleccionados no se detectó ningún caso con epimutación constitucional. En
consecuencia, dicho análisis se recomienda únicamente en casos con pérdida de
expresión e hipermetilación de MLH1 en tumor que cumplen criterios de Bethesda, ya
sean remitidos como tal (Anexo II) o que provengan de un cribado universal (Anexo I).
Los criterios de Bethesda aplicables en este último caso van a corresponder
fundamentalmente a lo concerniente a su historia personal de cáncer y sus
características clínicas y patológicas, más que a los criterios relacionados con los

Discusión
92
antecedentes familiares, que generalmente están ausentes o no recogidos con rigor en
la historia clínica del paciente.
Con el estudio presentado se añade una pequeña pieza al puzle que está permitiendo
gestionar adecuadamente el manejo de los casos con metilación de MLH1 en tumor
provenientes del cribado universal; que a su vez, aporta un incremento en la
sensibilidad en la detección del SL y una mejora en su diagnóstico. Como
consecuencia, se ofrece la posibilidad de un mejor manejo de la población a riesgo , de
una forma proactiva y eficiente.
Existen, no obstante, muchos otros ámbitos de mejora dentro del complejo contexto de
la identificación de formas de predisposición hereditaria a CCR e individuos a riesgo.
Con el conocimiento actual, todavía son muchas las familias con elevada carga de
cáncer y que cumplen criterios clínicos de cualquiera de los síndromes relacionados
con CCR hereditario, en las que no se llega a identificar la causa genética responsable
del alto riesgo patente en la familia. Las nuevas tecnologías de secuenciación masiva
aportan una herramienta muy eficaz en el descubrimiento de nuevos genes
responsables de formas hereditarias no explicadas. Un ejemplo reciente y notable de
esto mismo, ha sido la identificación de dos mutaciones recurrentes en dos genes que
codifican para las subunidades catalíticas de las polimerasas ε y δ (POLE y POLD1,
respectivamente) como responsables de casos de CCR y poliposis adenomatosas, y
CE con agregación familiar de cáncer (119). Este nuevo conocimiento nos ofreció la
posibilidad de reanalizar aquellos casos de familias con elevada carga de CCR o
poliposis en los que no se encontró mutación en los genes responsables de los
síndromes de sospecha, para así establecer la prevalencia de las mutaciones
recurrentes en POLE y POLD1 en nuestra población, y poder contribuir a una mejor
definición del fenotipo clínico asociado a estas mutaciones.
En este contexto se planteó el segundo estudio del presente trabajo de Tesis Doctoral.
El artículo publicado es fruto de una colaboración multicéntrica y multidisciplinar donde
se recogen los resultados obtenidos de tres series independientes de pacientes con
CCR y/o poliposis, con carga familiar de cáncer o poliposis provenientes de: i)
pacientes del Instituto Catalán de Oncología (pacientes con CCR y/o poliposis), ii)
Programa de Cáncer Hereditario de la Comunidad Valenciana (pacientes con CCR) y
iii) estudio multicéntrico nacional EPIPOLIP (pacientes con poliposis atenuadas: 10-
100 pólipos). Se realizó el cribado de las dos mutaciones recurrentes descritas
(POLE_p.Leu424Val y POLD1_p.Ser478Asn) en un total de 858 pacientes.

Discusión
93
Globalmente, en el estudio se identificó un individuo portador de la mutación
POLE_p.Leu424Val, que resultó ser de novo, y ningún caso portador de la mutación
POLD1_p.Ser478Asn; lo que pone de manifiesto la escasa prevalencia de esta
mutación en población española.
No obstante, se identificó una nueva mutación en POLD1 no descrita hasta la fecha:
p.Leu474Pro en una familia que cumple criterios de Amsterdam II, sin alteración del
sistema MMR detectada por IHQ ni IMS (CCR familiar tipo X). La Leu474 de POLD1
es el residuo parálogo de Leu424 en POLE, que es claramente patogénico; además,
está muy conservado en la evolución, los diferentes programas de predicción in silico
utilizados sugieren patogenicidad y los estudios funcionales in vitro (levaduras) indican
una pérdida de funcionalidad de la polimerasa con esta alteración, produciendo un
fenotipo mutador. La mutación encontrada cosegrega con la enfermedad en la familia,
estando presente en los individuos afectos. La sumatoria de todas estas evidencias
nos permite clasificarla como patogénica y responsable de la predisposición a cáncer
en los individuos portadores.
La mutación en esta familia se identificó en dos mujeres afectas de múltiples tumores:
el caso índice con un CCR y un tumor gastrointestinal estromal (GIST) sincronos
diagnosticados a los 36 años, y la tía materna con CCR y CE diagnosticados a los 33 y
56 años respectivamente. La madre del caso índice, con CE a los 52 años, era por
tanto portadora obligada de la mutación. Ninguna de estas tres mujeres ni ningún otro
miembro de la familia presentó pólipos en el colorrecto. Previamente ya se ha visto
una prevalencia elevada de CE en mujeres portadoras de mutaciones en POLD1,
concretamente de la mutación p.Ser478Asn (119). Esto pone de manifiesto la
importancia de las medidas de prevención para este tipo de tumor en las pacientes
portadoras de mutaciones en este gen.
Esta nueva mutación identificada en nuestra serie ha sido descrita posteriormente en
una familia en un estudio publicado por nuestros colaboradores del ICO en Cataluña
(181). Esta familia, aparentemente no relacionada con la anterior procede también del
Programa de Cáncer Hereditario de la Comunidad Valenciana. Se trata de una familia
que cumple criterios de Amsterdam I, con casos tanto MSS como IMS (pérdida de
MSH6), en la que se ha identificado la mutación en 7 individuos: dos sanos y 5
diagnosticados de CCR, pólipos colónicos y pólipos gástricos (Anexos: VII-A.1., VIII,
XII-C.1.). Más recientemente, nuestro grupo ha identificado también esta mutación en
una tercera familia del Programa de Cáncer Hereditario de la Comunidad Valenciana,

Discusión
94
aparentemente no relacionada con las dos anteriores. Se trata de una mujer que
desarrolló un CCR a los 51 años, sin antecedentes familiares de cáncer y que
provenía del cribado universal. Presentaba pérdida IHQ de MLH1 y PMS2, IMS, con
ausencia de metilación de MLH1 y mutación de BRAF en tumor y ausencia de
mutación de MLH1 en línea germinal (Anexos VII-A.2.; Anexo VIII) (datos no
publicados).
Por tanto, las tres familias identificadas con la mutación p.Leu474Pro en POLD1
proceden de la Comunidad Valenciana y aparentemente no están relacionadas.
Estudios posteriores de otros grupos que han analizado, o bien el exoma completo, o
bien el dominio exonucleasa de POLD1 en casos de CCR o poliposis familiar en busca
de mutaciones germinales, no han vuelto a identificar esta alteración (182-185). Esta
situación sugiere un posible efecto fundador de esta mutación en esta región. El
estudio de haplotipos en las tres familias para establecer el carácter fundador de esta
mutación está en desarrollo.
Algo similar podría suceder con la mutación POLD1_p.Ser478Asn, ya que el único
estudio realizado hasta la fecha en el que ésta se vuelve a encontrar, es en el llevado
a cabo por Chubb y colaboradores (185), en el que se observa esta mutación en una
familia con CCR y poliposis de una cohorte del Reino Unido (Anexo VIII), mismo origen
que el de las familias donde se describió esta mutación por primera vez (119).
Otras cinco mutaciones germinales nuevas en el dominio exonucleasa de POLD1 se
han descrito posteriormente a la publicación del segundo artículo de esta tesis y hasta
la fecha, en familias con CCR principalmente, pero también cáncer de mama y otros
tumores, en ausencia de pólipos y de IMS (Anexo IX) (181, 185). En estas cinco
familias descritas no se han encontrado casos con adenomas u otro tipo de pólipos
colónicos. No obstante, es necesario esperar a que se identifiquen más familias
portadoras de mutaciones para redefinir con mayor evidencia el espectro fenotípico
completo asociado a la pérdida de funcionalidad de estos genes.
Por otro lado, la mutación recurrente descrita en POLE (p.Leu424Val) en nuestro
estudio se identificó en un único individuo diagnosticado con poliposis adenomatosa y
CCR a los 28 años. Este individuo fue además diagnosticado de un oligodendroglioma
dos años más tarde (Dx: 30 años), lo que está descrito en el estudio realizado por
Bellido y colaboradores (181). El análisis realizado en los progenitores concluyó que la
mutación en esta familia tuvo lugar de novo, lo que supone el primer caso de mutación

Discusión
95
germinal de novo en los genes POLE/POLD1, y hasta la fecha sólo se ha descrito otro
caso, con mutación en el dominio exonucleasa de POLE (Anexo IX) (182). Este evento
sí que se produce de manera relativamente frecuente en otros genes de
predisposición a cáncer, como es el caso de APC en el 20-25% de casos (127). No
obstante, no se puede descartar que la aparición de novo pueda ser consecuencia de
un mosaicismo somático en alguno de los progenitores. La existencia de mutaciones
de novo en el codón Leu424 de POLE y su mayor prevalencia con respecto a POLD1
sugiere que se trate de un hotspot (punto caliente) que pueda generar casos mutados
de forma independiente y recurrente.
Esta mutación se ha identificado en estudios posteriores en 22 individuos
correspondientes a 10 familias aparentemente no relacionadas (182, 185-186), más
una familia extra en nuestra cohorte de Cáncer Hereditario de la Comunidad
Valenciana (Anexos VII-B; VIII; XII-C.1.). Esto, junto con los datos publicados
previamente y la familia identificada en el segundo artículo de esta tesis doctoral
hacen un total de 51 individuos portadores de la mutación POLE_p.Leu424Val,
pertenecientes a 24 familias aparentemente no relacionadas. El fenotípico típico de los
pacientes portadores es de CCR y/o poliposis colónicas, pero también se ha
observado la presencia de múltiples tipos de cáncer: carcinomas en otras partes del
tracto digestivo, pólipos gástricos y duodenales, otro tipo de cánceres como mama o
gliomas, etc; todos normalmente MSS (119, 182, 185-186) (Anexos IV y VIII).
Posteriormente a la publicación de nuestro artículo, se han descrito doce mutaciones
nuevas en el dominio exonucleasa de POLE, cada una detectada en una única familia,
con excepción de la mutación p.Lys425Arg, descrita en dos familias aparentemente no
relacionadas (Anexo IX). Estas trece familias presentan CCR, pólipos colorrectales y
una amplia gama de otros tipos tumorales, destacando la presencia de melanoma y
gliomas (182-185, 187).
En definitiva, los resultados obtenidos en nuestro estudio, junto con los obtenidos por
el resto de estudios realizados hasta la fecha, ponen de manifiesto la existencia de
mutaciones patogénicas en el dominio exonucleasa de las polimerasas en familias con
CCR, poliposis, CE y otra gran variedad de tumores diferentes que no presentaban
mutaciones en los genes responsables de los síndromes de sospecha.

Discusión
96
A modo de recapitulación, podemos concluir que las características de las familias con
mutaciones germinales en los genes POLE y POLD1 y por tanto, la propuesta de
criterios clínicos para la sospecha de PPAP serían las que se muestran en el Anexo X.
Globalmente, parece que el perfil fenotípico de las familias con mutaciones germinales
en POLE se asemeja más a aquellas familias con PAF o MAP y, por su parte, el perfil
de las familias con mutación en POLD1 es más similar al del SL, lo que vuelve a poner
de manifiesto el solapamiento fenotípico entre los distintos síndromes de
predisposición a CCR. No obstante, se requieren estudios adicionales que analicen
familias con mutaciones germinales en los dominios exonucleasa de estas
polimerasas para definir mejor los criterios clínicos, características y fenotipo de las
mismas.
Como ya se ha demostrado (153), los tumores con mutaciones en los dominios
exonucleasa de las polimerasas, ya sean éstas somáticas o germinales, muestran un
fenotipo hipermutador, que en ocasiones lleva a una tasa de mutaciones del orden de
106 sustituciones por tumor. En estos tumores, el espectro mutacional es diferente, con
una proporción incrementada de transversiones G:C a T:A y A:T a C:G. Por ello, se
esperaría encontrar una elevada cantidad de mutaciones en los genes comúnmente
implicados con CCR. Sin embargo, en nuestro estudio, en dos tumores de dos mujeres
portadoras de la variante POLD1_p.Leu474Pro (un CCR y un CE) de los que había
disponibilidad de material, no se encontraron mutaciones en los codones típicos de
KRAS, NRAS y BRAF, aun cuando muchas de estas mutaciones son transversiones
de este tipo. Además, estos tumores de nuestro estudio presentaban el sistema MMR
funcional e intacto. Esto concuerda con los resultados obtenidos en estudios previos
(119, 143), donde la tasa de mutaciones de genes típicamente implicados de forma
temprana en la carcinogénesis (incluyendo APC) es particularmente baja. En el
estudio realizado por el TGCA en CE se muestra un porcentaje elevado de mutaciones
en los genes PIK3CA y FBXW7 en los casos con mutaciones tipo missense en POLE
(155). No obstante, estos estudios están realizados en tumores con mutaciones
somáticas en POLE y no POLD1, ya que éstas últimas son mucho menos prevalentes.
En este estudio del TCGA se observó además que los tumores con mutaciones en
POLE parecían tener mejor pronóstico que el resto de tumores con otros perfiles
somáticos. No obstante, estudios posteriores no mostraron asociación de la presencia
de estas mutaciones con la supervivencia o variables patológicas (188-189).

Discusión
97
Incluso, en el estudio realizado por Billingsley y colaboradores se vio que la
distribución de las mutaciones de POLE era prácticamente equivalente entre tumores
estables e inestables (188). Otros estudios también han mostrado la presencia de
estas mutaciones a nivel somático en tumores con alteración de la vía reparadora
(155, 190). Estos casos serían los llamados “SL-like” (191), ya que muestran alteración
somática del sistema MMR, pero sin embargo no se encuentra mutación germinal en
estos genes, ni hipermetilación tumoral en MLH1 que la explique. Una posibilidad
sobre el origen del tumor en estos pacientes, ya postulada por algunos autores (167,
186), es que mutaciones germinales en otros genes distintos a los del sistema MMR
causen indirectamente la inactivación del mismo, posiblemente por alterar a nivel
somático los genes que lo codifican. Esto ya se ha visto en algunos pacientes con
deficiencia en MUTYH (136-137). Lo mismo podría suceder en casos con mutaciones
germinales en POLE/POLD1 que, por tener estos casos una tasa tan elevada de
mutaciones en sus tumores, podrían alterar indirectamente el sistema MMR, creando
un tumor con un fenotipo Lynch, lo que explicaría aquellos casos PPAP con alteración
del sistema MMR (186). De hecho, es probable que la paciente de la tercera familia
descrita anteriormente con la mutación p.Leu474Pro de POLD1 y CCR con pérdida de
MLH1, sin metilación ni BRAF mutado, ni mutación de MLH1 en línea germinal sea un
ejemplo de esto. Además, también podría ocurrir este efecto de deficiencia de MMR
indirecta a nivel somático, si se alteran los genes POLE y POLD1 en determinados
tejidos y estocásticamente alteran los genes MMR. En cualquier caso, se espera que
estudios futuros evalúen el espectro mutacional de los tumores con mutaciones
somáticas en POLE y POLD1, y proporcionen una imagen más clara del perfil
patológico molecular de los tumores de este síndrome.
En definitiva, los resultados descritos en el segundo artículo de esta tesis, junto con la
información publicada hasta la fecha por otros autores, avalan el estudio de la región
que codifica para el dominio exonucleasa de los genes POLE y POLD1 en todos los
casos de CCR y/o poliposis; y otros espectros tumorales resumidos en el Anexo X, con
agregación familiar de cáncer y sin mutación en los genes de predisposición
tradicionales. Dependiendo de la metodología de análisis, esto se puede realizar, bien
incluyendo dichos genes en los paneles diseñados para el análisis por secuenciación
masiva, o bien incorporando su estudio en el algoritmo secuencial de análisis, una vez
se hayan descartado alteraciones en otros genes que sean más prevalentes.
Los resultados derivados de los dos trabajos incluidos en el presente trabajo de Tesis
Doctoral ofrecen dos pequeñas mejoras en los algoritmos que siguen siendo utilizados

Discusión
98
de manera generalizada en nuestro entorno para el abordaje del diagnóstico genético
tanto de CCHNP, como de las poliposis atenuadas mediante secuenciación Sanger.
1. Cuando se observe alteración de la expresión de MLH1 en el tumor e
hipermetilación del promotor de MLH1 se debe analizar la presencia/ausencia de
epimutación constitucional en MLH1 en una muestra de sangre del paciente como
causa de SL, solamente si la familia cumple criterios de Bethesda.
2. Cuando no se encuentran alteraciones en el análisis de mutaciones germinales en
los genes de sospecha, según se trate de SL o poliposis, se deberá proceder al
cribado mutacional en los dominios exonucleasa de los genes POLE y POLD1,
independientemente de si el tumor tiene el sistema MMR alterado o no.
En cualquier caso, para ambas situaciones de CCHNP y poliposis debería
considerarse en estas familias la posibilidad de aparición de múltiples fenotipos
tumorales y de eventos mutacionales de novo. De hecho, el estudio de estos genes no
debería restringirse a casos índice con CCR o CE, sino que debería considerarse
también en presencia de otros tumores del tracto digestivo distintos y de tumores
extracolónicos fuera del espectro Lynch (como cáncer de mama, glioma, melanoma,
etc). Por ello, según los resultados del segundo trabajo de esta tesis y de otros
estudios publicados hasta la fecha, no basta con aplicar los criterios clínicos de
Bethesda para filtrar a las familias, sino que habría que estandarizar unos criterios en
base a los aquí propuestos en el Anexo X, y a los que se proponen en el estudio
realizado por el grupo de Bellido y colaboradores (181) para cribar a las familias
PPAP.
Los individuos de las familias de cáncer familiar en las que no se encuentran
alteraciones germinales en los genes de sospecha, reciben un seguimiento de alto
riesgo, por el desconocimiento del riesgo específico a desarrollar cáncer de cada uno
de los miembros. Esto conlleva una importante repercusión psicológica en la familia y
económica en el sistema sanitario. La identificación de alteraciones germinales permite
personalizar el riesgo de cáncer en las familias portadoras de las mismas,
identificando los individuos portadores y no portadores, y pudiendo así definir un
seguimiento específico para los individuos de alto riesgo, lo que supone una mejora
sustancial de la eficiencia del manejo clínico de estas familias.

Discusión
99
Basado en las evidencias proporcionadas hasta la fecha, se recomienda que tanto los
pacientes y familias con epimutaciones como con mutaciones germinales en los
dominios exonucleasa de las polimerasas, tengan un seguimiento equivalente al de los
pacientes con SL y poliposis; teniendo en cuenta la herencia no mendeliana de las
epimutaciones y el riesgo a desarrollar tumores fuera del espectro Lynch para los
pacientes PPAP. Para estos últimos, se han propuesto recientemente medidas de
seguimiento (181-182, 185), como son la realización de colonoscopias cada 1-2 años y
gastroduodenoscopias cada tres, empezando a los 20-25 años (181-182). Añadiendo
el cribado de CE a partir de los 40 años para las mujeres portadoras de mutaciones en
POLD1 (181), y la consideración de la cirugía profiláctica (185). Lógicamente, el nivel
de evidencia de estas recomendaciones es bajo (recomendaciones de expertos), pero
de momento es todo de lo que disponemos en este ámbito específico.
La implementación de estos análisis en el algoritmo genético para el diagnóstico de
síndromes de cáncer y poliposis hereditarios, contribuiría a reducir la heredabilidad
perdida que hay en estos síndromes que, a día de hoy, sigue siendo bastante elevada.
No obstante, la prevalencia de las epimutaciones en MLH1 y de las mutaciones en
POLE y POLD1 es baja, en comparación con los genes clásicos de CCR hereditario.
En nuestro estudio hemos visto que en casos que cumplen criterios de Bethesda para
el cribado del SL, la prevalencia de las epimutaciones constitucionales de MLH1 es del
0,6%; y para POLE y POLD1 del 0,23%, aunque nuestro estudio sólo interroga las dos
mutaciones recurrentes. En el estudio realizado por Chubb y colaboradores en 646
individuos con CCR de aparición temprana (<56 años) con al menos un familiar de
primer grado afecto de CCR, en el que analizan los genes de elevada penetrancia en
la susceptibilidad a CCR, observan una prevalencia de estas mutaciones del 0,5%
(0,3% para POLE y 0,2% para POLD1 respectivamente), en comparación con la
prevalencia más elevada de las mutaciones en los genes de Lynch, del 10,9%, y de
los genes de poliposis adenomatosas, que explican un 2,7% de los casos (185).
Globalmente, este estudio muestra que la heredabilidad perdida de los casos de CCR
familiar está en torno al 76%.
En familias de CCHNP, con elevada agregación de cáncer y con el sistema MMR
intacto (CCR familiar tipo X), estudios recientes han descrito mutaciones en otros
genes distintos a los clásicos de predisposición a CCR (como BMPR1A, FAN1,
GALNT12, HNRNPA0-WIF1, RPS20, SEMA4A; Tabla 6) de forma puntual (117-118,
120-121, 123-125). Lo mismo ha ocurrido para una serie de casos de poliposis
adenomatosa, donde se identificaron mutaciones en el gen NTHL1 (142).

Discusión
100
La mayoría de estos estudios han podido identificar estos nuevos genes candidatos
mediante el empleo de las nuevas tecnologías de secuenciación masiva (NGS)
(análisis del exoma o genoma completos). El uso de NGS en la investigación en este
campo está permitiendo generar nueva evidencia científica para la creación de
paneles amplios de genes de cáncer para el diagnóstico en el ámbito asistencial, que
evalúen simultáneamente los genes candidatos clásicos de predisposición a cáncer; y
vayan incorporando aquellos genes nuevos que se van descubriendo. El uso de estos
paneles de genes por NGS a día de hoy ya es coste-efectivo, en comparación con los
algoritmos de diagnóstico genético clásicos, que dirigen el estudio genético en función
del fenotipo clínico de los pacientes (192). Su utilización permite además, solventar el
problema del solapamiento genotípico-fenotípico que empezamos a ver en un número
creciente de casos.
En estos momentos nos encontramos en un punto de inflexión, donde se está
produciendo precisamente la incorporación de la NGS al ámbito asistencial de una
forma generalizada. Y es importante tener en cuenta que la secuenciación Sanger
continúa siendo el gold standard para la secuenciación, y que, dado que son muy
pocos los paneles de genes de NGS que tienen aprobación para diagnóstico por la
FDA o “marcado CE” (de Conformidad Europea), éstos deben de ser validados frente
a Sanger antes de su uso; o bien, se debe realizar un cribado con NGS y confirmar los
resultados con Sanger.
El uso del estudio de paneles de genes por NGS en el ámbito clínico presenta algunas
limitaciones y/o complicaciones, tanto desde el punto de vista del asesoramiento
genético pre-test, como a nivel analítico y postanalítico que deben ser considerados:
- El abordaje de genes que tienen pseudogenes, como es el caso de PMS2, debe
seguir realizándose mediante el uso previo de PCR largas.
- Las alteraciones de tipo gran reordenamiento y otras alteraciones estructurales
como inversiones o traslocaciones no son, generalmente, abordables por NGS.
- Las mutaciones patogénicas en regiones reguladoras o en zonas intrónicas
profundas no están cubiertas.
- Las alteraciones epigenéticas constitucionales quedan fuera del alcance de los
paneles de genes.

Discusión
101
- Se genera un incremento en el número de variantes genéticas de significado
clínico desconocido directamente proporcional al número y tamaño de los genes
analizados.
- Detección de hallazgos incidentales: mutaciones patogénicas en genes
responsables de síndromes no sospechados que exceden del interés diagnóstico
motivo del estudio. Esto es especialmente problemático cuando se utilizan amplios
paneles que contienen genes responsables de diferentes tipos de patologías, más
allá del cáncer hereditario, o con el estudio de exomas.
- En el asesoramiento pre-test se debe informar al paciente sobre el alcance del
estudio genético a realizar, sus limitaciones y la posibilidad de encontrar hallazgos
inesperados. Así mismo, se debe informar adecuadamente sobre la alta
probabilidad de detección de variantes de significado clínico desconocido y cómo
se van a gestionar.
Como conclusión de todo lo expuesto en relación a la NGS, y teniendo en cuenta sus
beneficios y limitaciones, se considera que la implementación de estas metodologías
en la práctica clínica supone un gran avance, y contribuye a una mayor eficacia en el
rendimiento diagnóstico, con un ahorro de tiempo y costes económicos, lo que
repercute muy positivamente en el sistema sanitario y en la salud de las personas.
Los avances tecnológicos y la información acumulada en el ámbito del CCR
hereditario están favoreciendo una rápida evolución del conocimiento, que pone de
manifiesto, una vez más, la gran complejidad a nivel molecular de la biología y
genética del cáncer, y el gran reto que supone ofrecer a los pacientes y familias
mejoras en la predicción, la prevención y la personalización de sus tratamientos.


CONCLUSIONES


Conclusiones
105
1. La prevalencia de las epimutaciones constitucionales de MLH1 en casos no
seleccionados de CCR es insignificante. El análisis de las epimutaciones
constitucionales de MLH1 es innecesario en casos de CCR poblacional. Este
análisis sólo debería realizarse en aquellos pacientes que cumplan los criterios
revisados de Bethesda, presenten pérdida de expresión de MLH1 y tengan
hipermetilación de MLH1 en sus tumores.
2. Las mutaciones de POLE y POLD1 explican una pequeña proporción de casos
de CCR y poliposis familiar. La secuenciación de al menos las mutaciones
recurrentes de estos genes debería considerarse en el análisis genético
rutinario de los casos con sospecha de CCR o poliposis hereditarios sin
mutaciones en los genes de sospecha clásicos.


BIBLIOGRAFÍA


Bibliografía
109
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011 Mar-Apr;61(2):69-90.
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015 Mar 1;136(5):E359-86.
3. Ferlay J SI, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 Lyon, France: International Agency for Research on Cancer: http://globocan.iarc.fr; 2013 [cited 2015 October].
4. Howlader N NA, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, Edwards BK (eds). SEER Cancer Statistics Review, 1975-2008, National Cancer Institute. Bethesda, MD: http://seer.cancer.gov/csr/1975_2008/; [updated 2011]; Available from: http://seer.cancer.gov/csr/1975_2008/.
5. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74.
6. Peters U, Bien S, Zubair N. Genetic architecture of colorectal cancer. Gut. 2015 Oct;64(10):1623-36.
7. Esteban-Jurado C, Garre P, Vila M, Lozano JJ, Pristoupilova A, Beltran S, et al. New genes emerging for colorectal cancer predisposition. World J Gastroenterol. 2014 Feb 28;20(8):1961-71.
8. Kohlmann W, Gruber SB. Lynch Syndrome. 2004 Feb 5 [Updated 2014 May 22]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1211/(Kohlmann W 2004 Feb 5 [Updated 2014 May 22])
9. Nielsen M, Lynch H, Infante E, et al. MUTYH-Associated Polyposis. 2012 Oct 4 [Updated 2015 Sep 24]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015.Available from: http://www.ncbi.nlm.nih.gov/books/NBK107219/
10. Larsen Haidle J, Howe JR. Juvenile Polyposis Syndrome. 2003 May 13 [Updated 2014 May 22]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1469/
11. Eng C. PTEN Hamartoma Tumor Syndrome (PHTS) 2001 Nov 29 [Updated 2014 Jan 23]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1488/
12. Sobin LH, Fleming ID. TNM Classification of Malignant Tumors, fifth edition (1997). Union Internationale Contre le Cancer and the American Joint Committee on Cancer. Cancer. 1997 Nov 1;80(9):1803-4.
13. Arena EA, Bilchik AJ. What is the optimal means of staging colon cancer? Adv Surg. 2013;47:199-211.

Bibliografía
110
14. Edge SB BD, Compton CC, Fritz AG, Green FL,Trotti A. Colon and rectal cancer. American Joint Committee on Cancer (AJCC) Staging Manual. 7 ed. New York: Springer; 2010. p. 143–63.
15. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012 Jul-Aug;62(4):220-41.
16. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013;14(8):16365-85.
17. Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015 Oct;149(5):1177-90 e3.
18. Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005 Apr 27;293(16):1986-94.
19. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990 Jun 1;61(5):759-67.
20. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001 Apr;3(4):433-8.
21. Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995 Jun 2;268(5215):1336-8.
22. Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996 Jul;13(3):343-6.
23. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004 Oct;3(10):1221-4.
24. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989 Apr 14;244(4901):217-21.
25. Markman B, Rodriguez-Freixinos V, Tabernero J. Biomarkers in colorectal cancer. Clin Transl Oncol. 2010 Apr;12(4):261-70.
26. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010 Jun;138(6):2073-87 e3.
27. Schulz W. Molecular Biology of Human Cancers: An Advanced Student's Textbook: Springer Netherlands; 2007.
28. Grady WM, Rajput A, Myeroff L, Liu DF, Kwon K, Willis J, et al. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998 Jul 15;58(14):3101-4.

Bibliografía
111
29. Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. J Gastroenterol. 2006 Mar;41(3):185-92.
30. Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997 Jul 1;57(13):2578-80.
31. Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004 Mar;126(3):654-9.
32. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997 Feb 14;275(5302):967-9.
33. Buhard O, Cattaneo F, Wong YF, Yim SF, Friedman E, Flejou JF, et al. Multipopulation analysis of polymorphisms in five mononucleotide repeats used to determine the microsatellite instability status of human tumors. J Clin Oncol. 2006 Jan 10;24(2):241-51.
34. Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998 Aug 1;58(15):3455-60.
35. Jass JR, Do KA, Simms LA, Iino H, Wynter C, Pillay SP, et al. Morphology of sporadic colorectal cancer with DNA replication errors. Gut. 1998 May;42(5):673-9.
36. Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001 Jun 15;91(12):2417-22.
37. Jover R, Paya A, Alenda C, Poveda MJ, Peiro G, Aranda FI, et al. Defective mismatch-repair colorectal cancer: clinicopathologic characteristics and usefulness of immunohistochemical analysis for diagnosis. Am J Clin Pathol. 2004 Sep;122(3):389-94.
38. Wright CL, Stewart ID. Histopathology and mismatch repair status of 458 consecutive colorectal carcinomas. Am J Surg Pathol. 2003 Nov;27(11):1393-406.
39. Ogino S, Brahmandam M, Cantor M, Namgyal C, Kawasaki T, Kirkner G, et al. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod Pathol. 2006 Jan;19(1):59-68.
40. Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000 Jan 13;342(2):69-77.
41. Wright CM, Dent OF, Barker M, Newland RC, Chapuis PH, Bokey EL, et al. Prognostic significance of extensive microsatellite instability in sporadic clinicopathological stage C colorectal cancer. Br J Surg. 2000 Sep;87(9):1197-202.
42. Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur J Cancer. 2009 Feb;45(3):365-73.

Bibliografía
112
43. Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012 Mar 15;18(6):1506-12.
44. Chan AO, Issa JP, Morris JS, Hamilton SR, Rashid A. Concordant CpG island methylation in hyperplastic polyposis. Am J Pathol. 2002 Feb;160(2):529-36.
45. Kambara T, Simms LA, Whitehall VL, Spring KJ, Wynter CV, Walsh MD, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004 Aug;53(8):1137-44.
46. Wynter CV, Walsh MD, Higuchi T, Leggett BA, Young J, Jass JR. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut. 2004 Apr;53(4):573-80.
47. Young J, Jass JR. The case for a genetic predisposition to serrated neoplasia in the colorectum: hypothesis and review of the literature. Cancer Epidemiol Biomarkers Prev. 2006 Oct;15(10):1778-84.
48. Hesson LB, Hitchins MP, Ward RL. Epimutations and cancer predisposition: importance and mechanisms. Curr Opin Genet Dev. 2010 Jun;20(3):290-8.
49. Anderson JC. Pathogenesis and management of serrated polyps: current sta tus and future directions. Gut Liver. 2014 Nov;8(6):582-9.
50. Minoo P, Moyer MP, Jass JR. Role of BRAF-V600E in the serrated pathway of colorectal tumourigenesis. J Pathol. 2007 Jun;212(2):124-33.
51. Jass JR. Colorectal polyposes: from phenotype to diagnosis. Pathol Res Pract. 2008;204(7):431-47.
52. Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009 Jan;58(1):90-6.
53. Jover R, Nguyen TP, Perez-Carbonell L, Zapater P, Paya A, Alenda C, et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology. 2011 Apr;140(4):1174-81.
54. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008 Dec 10;26(35):5783-8.
55. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010 Jun;138(6):2044-58.
56. Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4-5):269-76.
57. Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet. 2009 Jan;41(1):112-7.

Bibliografía
113
58. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009 Feb;30(2):197-203.
59. Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971 Apr;68(4):820-3.
60. Hemminki A, Peltomaki P, Mecklin JP, Jarvinen H, Salovaara R, Nystrom-Lahti M, et al. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet. 1994 Dec;8(4):405-10.
61. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003 Mar 6;348(10):919-32.
62. Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Jarvinen HJ, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008 Jul 15;123(2):444-9.
63. Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005 Jun;42(6):491-6.
64. Jenkins MA, Baglietto L, Dowty JG, Van Vliet CM, Smith L, Mead LJ, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol. 2006 Apr;4(4):489-98.
65. Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009 Nov;137(5):1621-7.
66. Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999 Apr 12;81(2):214-8.
67. Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011 Jun 8;305(22):2304-10.
68. Peltomaki P, Gao X, Mecklin JP. Genotype and phenotype in hereditary nonpolyposis colon cancer: a study of families with different vs. shared predisposing mutations. Fam Cancer. 2001;1(1):9-15.
69. Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004 Jul;127(1):17-25.
70. Kastrinos F, Stoffel EM. History, genetics, and strategies for cancer prevention in Lynch syndrome. Clin Gastroenterol Hepatol. 2014 May;12(5):715-27; quiz e41-3.
71. Cohen SA, Leininger A. The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014;7:147-58.
72. Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005 Aug;129(2):415-21.

Bibliografía
114
73. Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L, Burn J, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997 Jan;6(1):105-10.
74. Jarvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 1995 May;108(5):1405-11.
75. Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med. 2010 Feb;12(2):93-104.
76. Vasen HF, van Ballegooijen M, Buskens E, Kleibeuker JK, Taal BG, Griffioen G, et al. A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer. 1998 May 1;82(9):1632-7.
77. Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000 May;118(5):829-34.
78. Vasen HF, Abdirahman M, Brohet R, Langers AM, Kleibeuker JH, van Kouwen M, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology. 2010 Jun;138(7):2300-6.
79. Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer. 2013 Jun;12(2):229-40.
80. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006 Sep 27;296(12):1507-17.
81. Pascual Bolufer Gilabert, Isabel Chirivella González, José Antonio López
Guerrero, Eduardo Martínez Dueñas, Juan Silvestre Oltra, Dolores Salas Trejo, Ángel
Segura Huerta, José Luis Soto Martínez. Guía de práctica clínica en Cáncer
Hereditario. 3ª Edición. Generalitat Valenciana, Consellería de Sanitat. 2015; En
prensa.
82. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991 May;34(5):424-5.
83. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999 Jun;116(6):1453-6.
84. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004 Feb 18;96(4):261-8.
85. Salovaara R, Loukola A, Kristo P, Kaariainen H, Ahtola H, Eskelinen M, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2000 Jun;18(11):2193-200.

Bibliografía
115
86. Lynch HT, Boland CR, Rodriguez-Bigas MA, Amos C, Lynch JF, Lynch PM. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J Clin Oncol. 2007 Aug 10;25(23):3534-42.
87. Valle L. Genetic predisposition to colorectal cancer: where we stand and future perspectives. World J Gastroenterol. 2014 Aug 7;20(29):9828-49.
88. Hampel H. Point: justification for Lynch syndrome screening among all patients with newly diagnosed colorectal cancer. J Natl Compr Canc Netw. 2010 May;8(5):597-601.
89. Konishi K, Yamochi T, Makino R, Kaneko K, Yamamoto T, Nozawa H, et al. Molecular differences between sporadic serrated and conventional colorectal adenomas. Clin Cancer Res. 2004 May 1;10(9):3082-90.
90. Jass JR. Serrated adenoma and colorectal cancer. J Pathol. 1999 Apr;187(5):499-502.
91. Jass JR, Iino H, Ruszkiewicz A, Painter D, Solomon MJ, Koorey DJ, et al. Neoplastic progression occurs through mutator pathways in hyperplastic polyposis of the colorectum. Gut. 2000 Jul;47(1):43-9.
92. Perez-Carbonell L, Ruiz-Ponte C, Guarinos C, Alenda C, Paya A, Brea A, et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut. 2012 Jun;61(6):865-72.
93. Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012 Oct 17;308(15):1555-65.
94. Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002 Dec;123(6):1804-11.
95. Zhao YS, Hu FL, Wang F, Han B, Li DD, Li XW, et al. Meta-analysis of MSH6 gene mutation frequency in colorectal and endometrial cancers. J Toxicol Environ Health A. 2009;72(11-12):690-7.
96. Loughrey MB, Waring PM, Tan A, Trivett M, Kovalenko S, Beshay V, et al. Incorporation of somatic BRAF mutation testing into an algorithm for the investigation of hereditary non-polyposis colorectal cancer. Fam Cancer. 2007;6(3):301-10.
97. Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000 Jul;47(1):148-53.
98. Chen JM. The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: re-annotation and mutational mechanisms. Genes Chromosomes Cancer. 2008 Jun;47(6):543-5.
99. Wagner A, van der Klift H, Franken P, Wijnen J, Breukel C, Bezrookove V, et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer. 2002 Sep;35(1):49-57.

Bibliografía
116
100. Liu Q, Hesson LB, Nunez AC, Packham D, Williams R, Ward RL, et al. A cryptic paracentric inversion of MSH2 exons 2-6 causes Lynch syndrome. Carcinogenesis. 2015 Oct 24.
101. Rhees J, Arnold M, Boland CR. Inversion of exons 1-7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam Cancer. 2014 Jun;13(2):219-25.
102. Morak M, Koehler U, Schackert HK, Steinke V, Royer-Pokora B, Schulmann K, et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet. 2011 Aug;48(8):513-9.
103. Pinheiro M, Pinto C, Peixoto A, Veiga I, Mesquita B, Henrique R, et al. A novel exonic rearrangement affecting MLH1 and the contiguous LRRFIP2 is a founder mutation in Portuguese Lynch syndrome families. Genet Med. 2011 Oct;13(10):895-902.
104. Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999 May 1;59(9):2029-33.
105. Deng G, Chen A, Pong E, Kim YS. Methylation in hMLH1 promoter interferes with its binding to transcription factor CBF and inhibits gene expression. Oncogene. 2001 Oct 25;20(48):7120-7.
106. Perez-Carbonell L, Alenda C, Paya A, Castillejo A, Barbera VM, Guillen C, et al. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn. 2010 Jul;12(4):498-504.
107. Gausachs M, Mur P, Corral J, Pineda M, Gonzalez S, Benito L, et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Hum Genet. 2012 Jul;20(7):762-8.
108. Hitchins MP. The role of epigenetics in Lynch syndrome. Fam Cancer. 2013 Jun;12(2):189-205.
109. Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009 Dec;46(12):793-802.
110. Hitchins MP. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat Rev Cancer. 2015 Oct;15(10):625-34.
111. Hitchins MP, Wong JJ, Suthers G, Suter CM, Martin DI, Hawkins NJ, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007 Feb 15;356(7):697-705.
112. Gylling A, Ridanpaa M, Vierimaa O, Aittomaki K, Avela K, Kaariainen H, et al. Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer. 2009 May 15;124(10):2333-40.
113. Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005 Apr 27;293(16):1979-85.

Bibliografía
117
114. Lindor NM. Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome. Surg Oncol Clin N Am. 2009 Oct;18(4):637-45.
115. Goel A, Xicola RM, Nguyen TP, Doyle BJ, Sohn VR, Bandipalliam P, et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010 May;138(5):1854-62.
116. Shiovitz S, Copeland WK, Passarelli MN, Burnett-Hartman AN, Grady WM, Potter JD, et al. Characterisation of familial colorectal cancer Type X, Lynch syndrome, and non-familial colorectal cancer. Br J Cancer. 2014 Jul 29;111(3):598-602.
117. Garre P, Martin L, Sanz J, Romero A, Tosar A, Bando I, et al. BRCA2 gene: a candidate for clinical testing in familial colorectal cancer type X. Clin Genet. 2015 Jun;87(6):582-7.
118. Nieminen TT, Abdel-Rahman WM, Ristimaki A, Lappalainen M, Lahermo P, Mecklin JP, et al. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2011 Jul;141(1):e23-6.
119. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013 Feb;45(2):136-44.
120. Segui N, Mina LB, Lazaro C, Sanz-Pamplona R, Pons T, Navarro M, et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology. 2015 Sep;149(3):563-6.
121. Clarke E, Green RC, Green JS, Mahoney K, Parfrey PS, Younghusband HB, et al. Inherited deleterious variants in GALNT12 are associated with CRC susceptibility. Hum Mutat. 2012 Jul;33(7):1056-8.
122. Segui N, Pineda M, Navarro M, Lazaro C, Brunet J, Infante M, et al. GALNT12 is not a major contributor of familial colorectal cancer type X. Hum Mutat. 2014 Jan;35(1):50-2.
123. Wei C, Peng B, Han Y, Chen WV, Rother J, Tomlinson GE, et al. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam Cancer. 2015 Jun;14(2):297-306.
124. Nieminen TT, O'Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014 Sep;147(3):595-8 e5.
125. Schulz E, Klampfl P, Holzapfel S, Janecke AR, Ulz P, Renner W, et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat Commun. 2014;5:5191.
126. Segui N, Guino E, Pineda M, Navarro M, Bellido F, Lazaro C, et al. Longer telomeres are associated with cancer risk in MMR-proficient hereditary non-polyposis colorectal cancer. PLoS One. 2014;9(2):e86063.
127. Jasperson KW, Burt RW. APC-Associated Polyposis Conditions. 1998 Dec 18 [Updated 2014 Mar 27]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors.

Bibliografía
118
GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1345/
128. Umar A, Risinger JI, Hawk ET, Barrett JC. Testing guidelines for hereditary non-polyposis colorectal cancer. Nat Rev Cancer. 2004 Feb;4(2):153-8.
129. Leoz ML, Carballal S, Moreira L, Ocana T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet. 2015;8:95-107.
130. Ficari F, Cama A, Valanzano R, Curia MC, Palmirotta R, Aceto G, et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br J Cancer. 2000 Jan;82(2):348-53.
131. Thirlwell C, Howarth KM, Segditsas S, Guerra G, Thomas HJ, Phillips RK, et al. Investigation of pathogenic mechanisms in multiple colorectal adenoma patients without germline APC or MYH/MUTYH mutations. Br J Cancer. 2007 Jun 4;96(11):1729-34.
132. Goodenberger M, Lindor NM. Lynch syndrome and MYH-associated polyposis: review and testing strategy. J Clin Gastroenterol. 2011 Jul;45(6):488-500.
133. Boparai KS, Dekker E, Van Eeden S, Polak MM, Bartelsman JF, Mathus-Vliegen EM, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008 Dec;135(6):2014-8.
134. Guarinos C, Juarez M, Egoavil C, Rodriguez-Soler M, Perez-Carbonell L, Salas R, et al. Prevalence and characteristics of MUTYH-associated polyposis in patients with multiple adenomatous and serrated polyps. Clin Cancer Res. 2014 Mar 1;20(5):1158-68.
135. Kastrinos F, Syngal S. Recently identified colon cancer predispositions: MYH and MSH6 mutations. Semin Oncol. 2007 Oct;34(5):418-24.
136. Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014 Nov;22(11):1334-7.
137. Castillejo A, Vargas G, Castillejo MI, Navarro M, Barbera VM, Gonzalez S, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer. 2014 Sep;50(13):2241-50.
138. Guillen-Ponce C, Castillejo A, Barbera VM, Pascual-Ramirez JC, Andrada E, Castillejo MI, et al. Biallelic MYH germline mutations as cause of Muir-Torre syndrome. Fam Cancer. 2010 Jun;9(2):151-4.
139. Vogt S, Jones N, Christian D, Engel C, Nielsen M, Kaufmann A, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009 Dec;137(6):1976-85 e1-10.
140. Castillejo A, Sanchez-Heras AB, Jover R, Castillejo MI, Guarinos C, Oltra S, et al. Recurrent testicular germ cell tumors in a family with MYH-associated polyposis. J Clin Oncol. 2012 Aug 10;30(23):e216-7.

Bibliografía
119
141. Balaguer F, Castellvi-Bel S, Castells A, Andreu M, Munoz J, Gisbert JP, et al. Identification of MYH mutation carriers in colorectal cancer: a multicenter, case-control, population-based study. Clin Gastroenterol Hepatol. 2007 Mar;5(3):379-87.
142. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015 Jun;47(6):668-71.
143. Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013 Jun;230(2):148-53.
144. Yoshida R, Miyashita K, Inoue M, Shimamoto A, Yan Z, Egashira A, et al. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur J Hum Genet. 2011 Mar;19(3):320-5.
145. Church JM. Polymerase proofreading-associated polyposis: a new, dominantly inherited syndrome of hereditary colorectal cancer predisposition. Dis Colon Rectum. 2014 Mar;57(3):396-7.
146. Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009 Oct 6;106(40):17101-4.
147. Scheuermann R, Tam S, Burgers PM, Lu C, Echols H. Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: a fidelity subunit for DNA replication. Proc Natl Acad Sci U S A. 1983 Dec;80(23):7085-9.
148. Maki H, Horiuchi T, Sekiguchi M. Isolation of conditional lethal mutator mutants of Escherichia coli by localized mutagenesis. J Bacteriol. 1983 Mar;153(3):1361-7.
149. Morrison A, Sugino A. The 3'-->5' exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet. 1994 Feb;242(3):289-96.
150. Henninger EE, Pursell ZF. DNA polymerase epsilon and its roles in genome stability. IUBMB Life. 2014 May;66(5):339-51.
151. Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013 Jul 15;22(14):2820-8.
152. Smith CG, Naven M, Harris R, Colley J, West H, Li N, et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat. 2013 Jul;34(7):1026-34.
153. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012 Jul 19;487(7407):330-7.
154. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012 Aug 30;488(7413):660-4.
155. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013 May 2;497(7447) :67-73.

Bibliografía
120
156. Xicola RM, Llor X, Pons E, Castells A, Alenda C, Pinol V, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007 Feb 7;99(3):244-52.
157. Benlloch S, Paya A, Alenda C, Bessa X, Andreu M, Jover R, et al. Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn. 2006 Nov;8(5):540-3.
158. Nagasaka T, Koi M, Kloor M, Gebert J, Vilkin A, Nishida N, et al. Mutations in both KRAS and BRAF may contribute to the methylator phenotype in colon cancer. Gastroenterology. 2008 Jun;134(7):1950-60, 60 e1.
159. Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006 Aug 1;66(15):7810-7.
160. Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, et al. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002 Jun 15;62(12):3485-92.
161. Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999 Oct 15;59(20):5068-74.
162. Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008 Apr;10(4):294-300.
163. Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014 Feb;46(2):107-15.
164. Guarinos C, Sanchez-Fortun C, Rodriguez-Soler M, Perez-Carbonell L, Egoavil C, Juarez M, et al. Clinical subtypes and molecular characteristics of serrated polyposis syndrome. Clin Gastroenterol Hepatol. 2013 Jun;11(6):705-11; quiz e46.
165. Cuppen E. Genotyping by Allele-Specific Amplification (KASPar). CSH Protoc. 2007;2007:pdb prot4841.
166. Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011 Apr 8;88(4):440-9.
167. Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J Gastroenterol. 2015 Aug 21;21(31):9253-61.
168. Akashi M, Koeffler HP. Li-Fraumeni syndrome and the role of the p53 tumor suppressor gene in cancer susceptibility. Clin Obstet Gynecol. 1998 Mar;41(1):172-99.
169. Win AK, Jenkins MA, Buchanan DD, Clendenning M, Young JP, Giles GG, et al. Determining the frequency of de novo germline mutations in DNA mismatch repair genes. J Med Genet. 2011 Aug;48(8):530-4.

Bibliografía
121
170. Nilbert M, Timshel S, Bernstein I, Larsen K. Role for genetic anticipation in Lynch syndrome. J Clin Oncol. 2009 Jan 20;27(3):360-4.
171. Clendenning M, Buchanan DD, Walsh MD, Nagler B, Rosty C, Thompson B, et al. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam Cancer. 2011 Jun;10(2):297-301.
172. Pineda M, Mur P, Iniesta MD, Borras E, Campos O, Vargas G, et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur J Hum Genet. 2012 Dec;20(12):1256-64.
173. Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013 Jan;15(1):25-35.
174. Auclair J, Vaissiere T, Desseigne F, Lasset C, Bonadona V, Giraud S, et al. Intensity-dependent constitutional MLH1 promoter methylation leads to early onset of colorectal cancer by affecting both alleles. Genes Chromosomes Cancer. 2011 Mar;50(3):178-85.
175. Crepin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, et al. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2012 Jan;33(1):180-8.
176. Raptis S, Mrkonjic M, Green RC, Pethe VV, Monga N, Chan YM, et al. MLH1 -93G>A promoter polymorphism and the risk of microsatellite-unstable colorectal cancer. J Natl Cancer Inst. 2007 Mar 21;99(6):463-74.
177. Toon CW, Walsh MD, Chou A, Capper D, Clarkson A, Sioson L, et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am J Surg Pathol. 2013 Oct;37(10):1592-602.
178. Capper D, Voigt A, Bozukova G, Ahadova A, Kickingereder P, von Deimling A, et al. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int J Cancer. 2013 Oct 1;133(7):1624-30.
179. Goel A, Nguyen TP, Leung HC, Nagasaka T, Rhees J, Hotchkiss E, et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011 Feb 15;128(4):869-78.
180. van Roon EH, van Puijenbroek M, Middeldorp A, van Eijk R, de Meijer EJ, Erasmus D, et al. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer. 2010;10:180.
181. Bellido F, Pineda M, Aiza G, Valdes-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2015 Jul 2.
182. Spier I, Holzapfel S, Altmuller J, Zhao B, Horpaopan S, Vogt S, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015 Jul 15;137(2):320-31.

Bibliografía
122
183. Aoude LG, Heitzer E, Johansson P, Gartside M, Wadt K, Pritchard AL, et al. POLE mutations in families predisposed to cutaneous melanoma. Fam Cancer. 2015 Dec;14(4):621-8.
184. Rohlin A, Eiengard F, Lundstam U, Zagoras T, Nilsson S, Edsjo A, et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes Chromosomes Cancer. 2015 Oct 23.
185. Chubb D, Broderick P, Frampton M, Kinnersley B, Sherborne A, Penegar S, et al. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J Clin Oncol. 2015 Feb 10;33(5):426-32.
186. Elsayed FA, Kets CM, Ruano D, van den Akker B, Mensenkamp AR, Schrumpf M, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet. 2015 Aug;23(8):1080-4.
187. Hansen MF, Johansen J, Bjornevoll I, Sylvander AE, Steinsbekk KS, Saetrom P, et al. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam Cancer. 2015 Sep;14(3):437-48.
188. Billingsley CC, Cohn DE, Mutch DG, Stephens JA, Suarez AA, Goodfellow PJ. Polymerase varepsilon (POLE) mutations in endometrial cancer: clinical outcomes and implications for Lynch syndrome testing. Cancer. 2015 Feb 1;121(3):386-94.
189. Stenzinger A, Pfarr N, Endris V, Penzel R, Jansen L, Wolf T, et al. Mutations in POLE and survival of colorectal cancer patients--link to disease stage and treatment. Cancer Med. 2014 Dec;3(6):1527-38.
190. Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014 Dec;147(6):1308-16 e1.
191. Rodriguez-Soler M, Perez-Carbonell L, Guarinos C, Zapater P, Castillejo A, Barbera VM, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013 May;144(5):926-32 e1; quiz e13-4.
192. Lu JT, Campeau PM, Lee BH. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N Engl J Med. 2014 Aug 14;371(7):593-6.
193. Rohlin A, Zagoras T, Nilsson S, Lundstam U, Wahlstrom J, Hulten L, et al. A mutation in POLE predisposing to a multi-tumour phenotype. Int J Oncol. 2014 Jul;45(1):77-81.

ANEXOS


Anexos
125
ANEXO I: Algoritmo de estudio genético de SL siguiendo la Estrategia Universal
(pacientes con CCR o CE diagnosticado antes de los 70 años) (81).

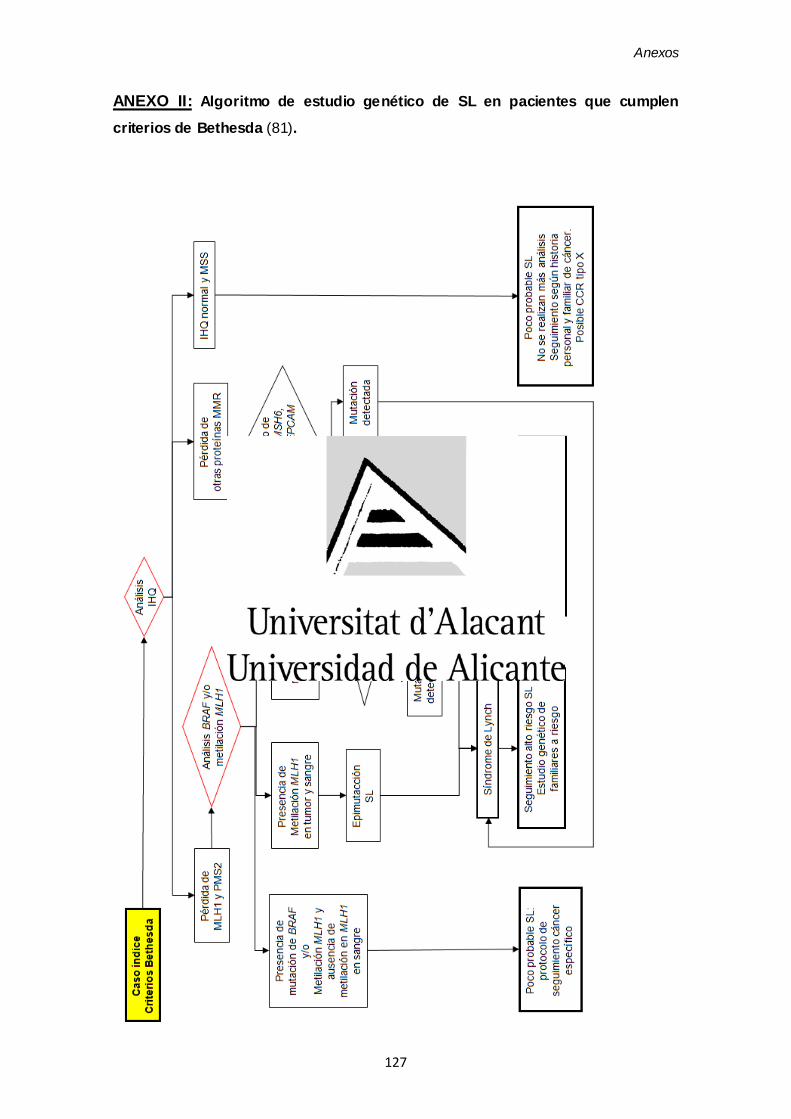
Anexos
127
ANEXO II: Algoritmo de estudio genético de SL en pacientes que cumplen
criterios de Bethesda (81).


Anexos
129
ANEXO III: Algoritmo de estudio genético de poliposis adenomatosa familiar

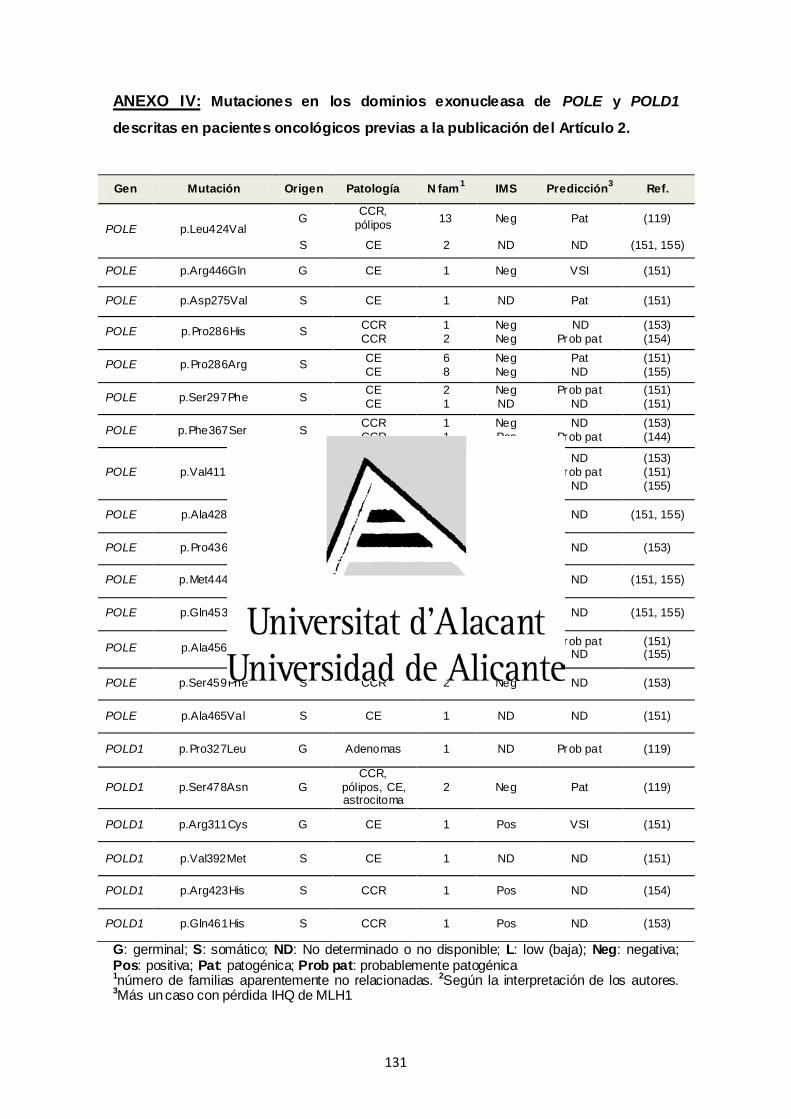
131
ANEXO IV: Mutaciones en los dominios exonucleasa de POLE y POLD1
descritas en pacientes oncológicos previas a la publicación del Artículo 2.
G: germinal; S: somático; ND: No determinado o no disponible; L: low (baja); Neg: negativa; Pos: positiva; Pat: patogénica; Prob pat: probablemente patogénica 1número de familias aparentemente no relacionadas. 2Según la interpretación de los autores. 3Más un caso con pérdida IHQ de MLH1
Gen Mutación Origen Patología N fam1 IMS Predicción
3 Ref.
POLE p.Leu424Val G
CCR,
pólipos 13 Neg Pat (119)
S CE 2 ND ND (151, 155)
POLE p.Arg446Gln G CE 1 Neg VSI (151)
POLE p.Asp275Val S CE 1 ND Pat (151)
POLE p.Pro286His S CCR
CCR
1
2
Neg
Neg
ND
Prob pat
(153)
(154)
POLE p.Pro286Arg S CE
CE
6
8
Neg
Neg
Pat
ND
(151)
(155)
POLE p.Ser297Phe S CE
CE
2
1
Neg
ND
Prob pat
ND
(151)
(151)
POLE p.Phe367Ser S CCR
CCR
1
1
Neg
Pos
ND
Prob pat
(153)
(144)
POLE p.Val411Leu S
CCR
CE
CE
2
2
5
Neg; L
Neg
2: L3
ND
Prob pat
ND
(153)
(151)
(155)
POLE p.Ala428Thr S CE 1 ND ND (151, 155)
POLE p.Pro436Arg S CCR 1 L ND (153)
POLE p.Met444Lys S CE 1 ND ND (151, 155)
POLE p.Gln453Arg S CE 1 ND ND (151, 155)
POLE p.Ala456Pro S CE CE
1 1
Neg ND
Prob pat ND
(151) (155)
POLE p.Ser459Phe S CCR 2 Neg ND (153)
POLE p.Ala465Val S CE 1 ND ND (151)
POLD1 p.Pro327Leu G Adenomas 1 ND Prob pat (119)
POLD1 p.Ser478Asn G
CCR,
pólipos, CE, astrocitoma
2 Neg Pat (119)
POLD1 p.Arg311Cys G CE 1 Pos VSI (151)
POLD1 p.Val392Met S CE 1 ND ND (151)
POLD1 p.Arg423His S CCR 1 Pos ND (154)
POLD1 p.Gln461His S CCR 1 Pos ND (153)

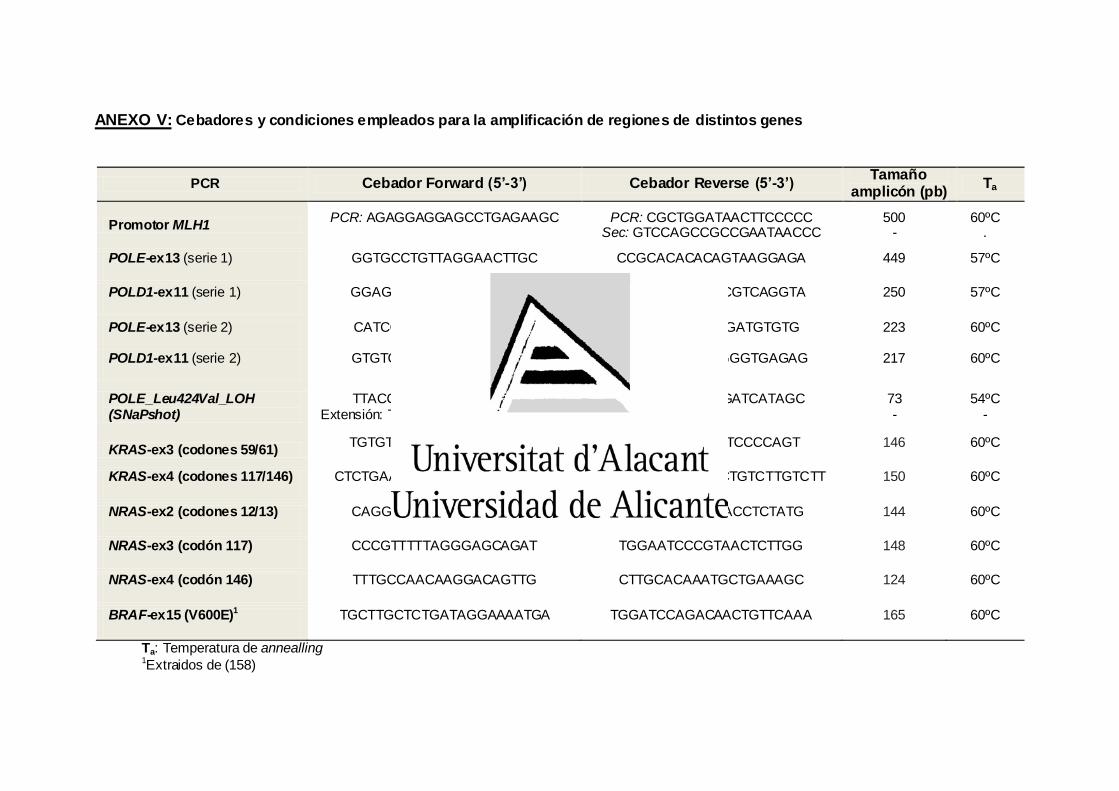
ANEXO V: Cebadores y condiciones empleados para la amplificación de regiones de distintos genes
PCR Cebador Forward (5’-3’) Cebador Reverse (5’-3’) Tamaño
amplicón (pb) Ta
Promotor MLH1 PCR: AGAGGAGGAGCCTGAGAAGC PCR: CGCTGGATAACTTCCCCC
Sec: GTCCAGCCGCCGAATAACCC 500
- 60ºC
.
POLE-ex13 (serie 1) GGTGCCTGTTAGGAACTTGC CCGCACACACAGTAAGGAGA 449 57ºC
POLD1-ex11 (serie 1) GGAGTACAAGCTCCGCTCCT GAAAAAGTGGGCGTCAGGTA 250 57ºC
POLE-ex13 (serie 2) CATCCTGGCTTCTGTTCTCA GTGGCCATCTGGATGTGTG 223 60ºC
POLD1-ex11 (serie 2) GTGTGTCCCTGTCCTTGGAA GTCAGAGGTTGGGGTGAGAG 217 60ºC
POLE_Leu424Val_LOH (SNaPshot)
TTACCTTCCTGTGGGCAGTC Extensión: TTCCTGTGGGCAGTCATAAT
TAGCTCCACGGGATCATAGC -
73 -
54ºC -
KRAS-ex3 (codones 59/61) TGTGTTTCTCCCTTCTCAGGA AAAGAAAGCCCTCCCCAGT 146 60ºC
KRAS-ex4 (codones 117/146) CTCTGAAGATGTACCTATGGTCCT TCAGTGTTACTTACCTGTCTTGTCTT 150 60ºC
NRAS-ex2 (codones 12/13) CAGGTTCTTGCTGGTGTGAA CACTGGGCCTCACCTCTATG 144 60ºC
NRAS-ex3 (codón 117) CCCGTTTTTAGGGAGCAGAT TGGAATCCCGTAACTCTTGG 148 60ºC
NRAS-ex4 (codón 146) TTTGCCAACAAGGACAGTTG CTTGCACAAATGCTGAAAGC 124 60ºC
BRAF-ex15 (V600E)1 TGCTTGCTCTGATAGGAAAATGA TGGATCCAGACAACTGTTCAAA 165 60ºC
Ta: Temperatura de annealling 1Extraidos de (158)


Anexos
135
ANEXO VI: Árboles genealógicos de las cinco familias con epimutaciones del
artículo 1.
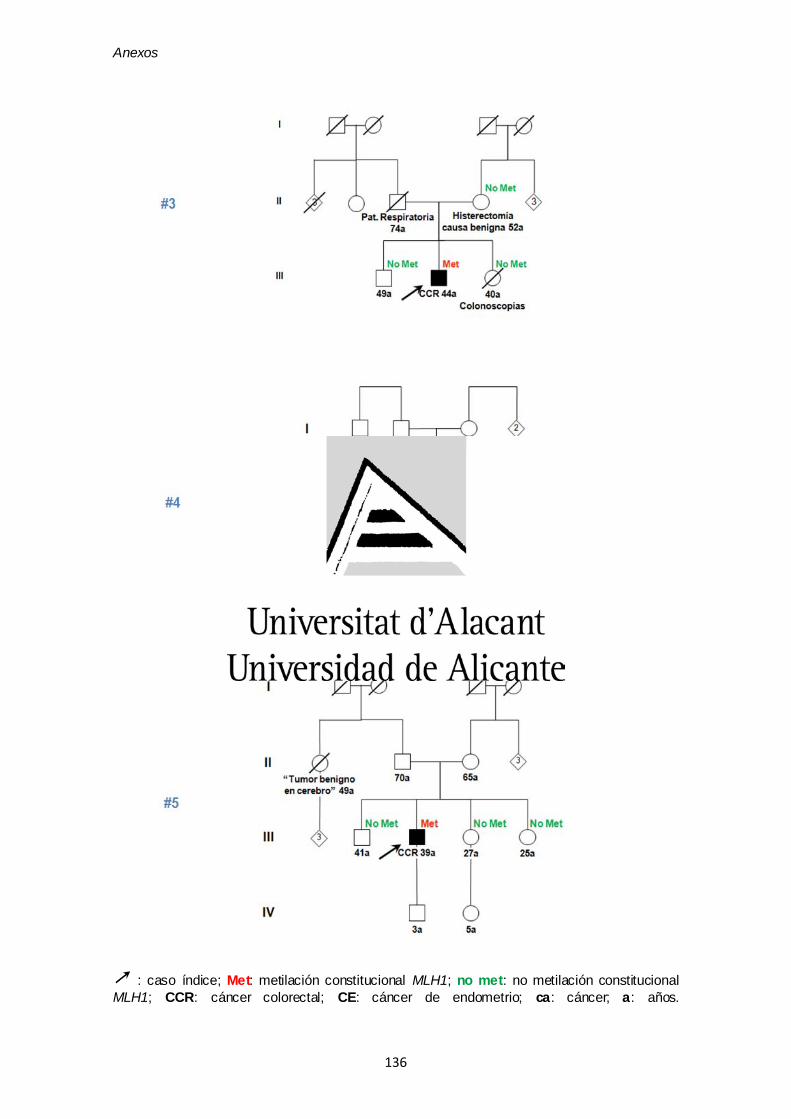
Anexos
136
: caso índice; Met: metilación constitucional MLH1; no met: no metilación constitucional
MLH1; CCR: cáncer colorectal; CE: cáncer de endometrio; ca: cáncer; a: años.

Anexos
137
ANEXO VII: Árboles genealógicos de las nuevas familias descritas en nuestra
serie con las mutaciones POLE_Leu424Val y POLD1_Leu474Pro posteriores a la
publicación del segundo artículo.

Anexos
138
: caso índice; +: portador de la mutación; (+): portador obligado de la mutación; -: no portador de la mutación; CCR: cáncer colorectal; CE: cáncer de endometrio; TSNC: tumor del sistema nervioso central; ca: cáncer; a: años.
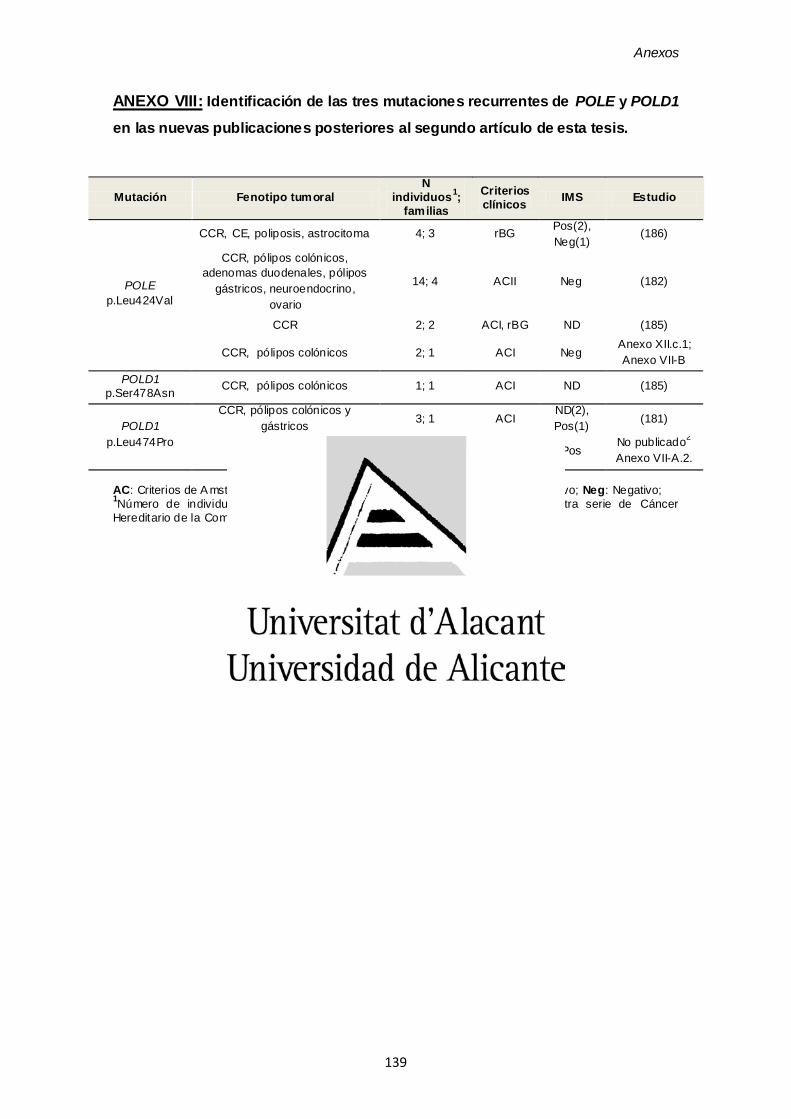
Anexos
139
ANEXO VIII: Identificación de las tres mutaciones recurrentes de POLE y POLD1
en las nuevas publicaciones posteriores al segundo artículo de esta tesis.
Mutación Fenotipo tumoral
N
individuos1;
familias
Criterios
clínicos IMS Estudio
POLE
p.Leu424Val
CCR, CE, poliposis, astrocitoma 4; 3 rBG Pos(2),
Neg(1) (186)
CCR, pólipos colónicos,
adenomas duodenales, pólipos
gástricos, neuroendocrino,
ovario
14; 4 ACII Neg (182)
CCR 2; 2 ACI, rBG ND (185)
CCR, pólipos colónicos 2; 1 ACI Neg Anexo XII.c.1;
Anexo VII-B
POLD1
p.Ser478Asn CCR, pólipos colónicos 1; 1 ACI ND (185)
POLD1
p.Leu474Pro
CCR, pólipos colónicos y
gástricos 3; 1 ACI
ND(2),
Pos(1) (181)
CCR 1; 1 rBG Pos No publicado
2
Anexo VII-A.2.
AC: Criterios de A msterdam ( I o II); r BG: criterios revisados de Bethesda; Pos : positivo; Neg: Negativo; 1Número de individuos portadores de la mutación enfermos.
2Familia de nuestra serie de Cáncer
Hereditario de la Comunidad Valenciana.

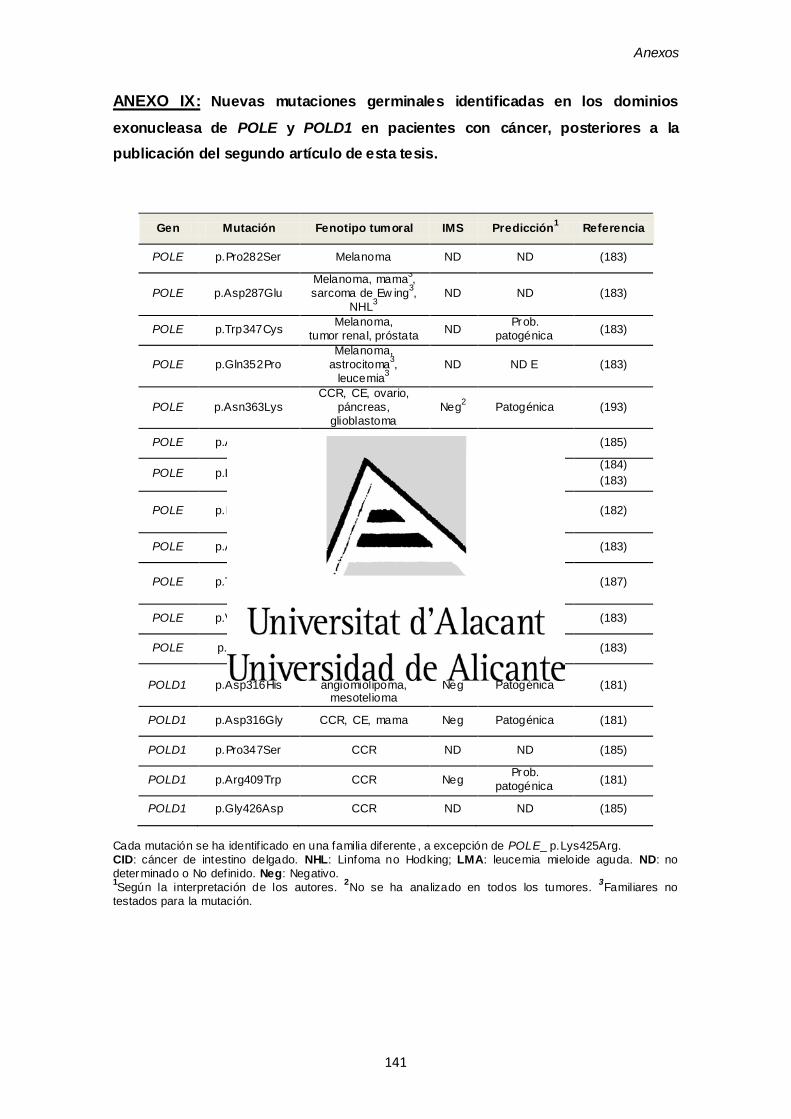
Anexos
141
ANEXO IX: Nuevas mutaciones germinales identificadas en los dominios
exonucleasa de POLE y POLD1 en pacientes con cáncer, posteriores a la
publicación del segundo artículo de esta tesis.
Gen Mutación Fenotipo tumoral IMS Predicción1 Referencia
POLE p.Pro282Ser Melanoma ND ND (183)
POLE p.Asp287Glu
Melanoma, mama3,
sarcoma de Ew ing3,
NHL3
ND ND (183)
POLE p.Trp347Cys Melanoma,
tumor renal, próstata ND
Prob.
patogénica (183)
POLE p.Gln352Pro
Melanoma,
astrocitoma3,
leucemia3
ND ND E (183)
POLE p.Asn363Lys
CCR, CE, ovario,
páncreas,
glioblastoma
Neg2 Patogénica (193)
POLE p.Asp368Val CCR Neg VSI (185)
POLE p.Lys425Arg CCR
Melanoma
Neg
ND
Prob. Patog
ND
(184)
(183)
POLE p.Pro436Ser
Poliposis atenuada,
CCR, adenomas
duodenales
Neg Patogénica (182)
POLE p.Arg446Gln Melanoma, pulmón3 ND ND (183)
POLE p.Tyr458Phe
CCR, adenomas
colorrectales, ovario,
páncreas, CID
ND Patogénica (187)
POLE p.Val460Met Melanoma, LMA
3,
esófago3, próstata
3
ND ND (183)
POLE p.Ile515Met Melanoma ND ND (183)
POLD1 p.Asp316His
CCR, mama,
angiomiolipoma, mesotelioma
Neg Patogénica (181)
POLD1 p.Asp316Gly CCR, CE, mama Neg Patogénica (181)
POLD1 p.Pro347Ser CCR ND ND (185)
POLD1 p.Arg409Trp CCR Neg Prob.
patogénica (181)
POLD1 p.Gly426Asp CCR ND ND (185)
Cada mutación se ha identif icado en una familia diferente , a excepción de POLE_ p.Lys425Arg.
CID: cáncer de intestino delgado. NHL: Linfoma no Hodking; LMA: leucemia mieloide aguda. ND: no
determinado o No definido. Neg: Negativo. 1Según la interpretación de los autores.
2No se ha analizado en todos los tumores.
3Familiares no
testados para la mutación.


Anexos
143
ANEXO X: Propuesta de características clínicas para la sospecha de PPAP
Propuesta de características clínicas para la sospecha de PPAP
Edad diagnóstico Diagnóstico del cáncer a edad temprana (<50 años).
Espectro tumoral
Pr incipales: CCR, poliposis colónicas, CE (POLD1).
Secundar ios: Pólipos duodenales (POLE), gliomas, melanoma (POLE),
cáncer de mama (POLD1), cáncer del tracto digestivo distinto al CCR, otros.
Agregación familiar Alta (criterios de A msterdam II principalmente, pero no excluir Bethesda).
Herencia dominante.
Estado sistema MM R Generalmente intacto, tumores MSS (no excluir casos IMS).


Anexos
145
ANEXO XI: Trabajo realizado por la doctoranda en esta tesis doctoral
La doctoranda ha participado en los siguientes aspectos de la concepción y diseño,
desarrollo de la metodología, adquisición de datos, análisis e interpretación de
resultados y elaboración y revisión de la comunicación de resultados del presente
trabajo de tesis doctoral:
Artículo 1:
- Realización del análisis de la metilación de MLH1 somática y germinal.
Interpretación de resultados.
- Análisis de la mutación de BRAF_V600E. Interpretación de resultados.
- Recopilación de la información clinicopatológica y la elaboración de la base de
datos.
- Análisis estadístico de los datos.
- Redacción y revisión del manuscrito.
Artículo 2:
- Selección de pacientes y muestras.
- Puesta a punto de la metodología y análisis de las mutaciones germinales en
POLE y POLD1. Interpretación de resultados.
- Análisis de predicción in silico.
- Análisis de las mutaciones somáticas en BRAF, KRAS y NRAS. Interpretación
de resultados.
- Recopilación de la información clinicopatológica y la elaboración de la base de
datos.
- Redacción y revisión del manuscrito.


Anexos
147
ANEXO XII: Otras publicaciones en las que ha participado la doctoranda.
A) Artículos científicos
A.1. Serrated colorectal cancer: molecular classification, prognosis, and
response to chemotherapy.
Óscar Murcia, Miriam Juárez, Eva Hernández-Illán, Cecilia Egoavil, Mar Giner, María
Rodriguez-Soler, Rodrigo Jover.
World J of Gastroenterol. Aceptado 2016.
Factor de Impacto: 2,369 (2014) (Q2).
ABSTRACT:
Molecular advances support the existence of an alternative pathway of colorectal
carcinogenesis that is based on the hypermethylation of specific DNA regions that
silences tumor suppressor genes. This alternative pathway has been called the
serrated pathway due to the serrated appearance of tumors in histological analysis.
New classifications for colorectal cancer (CRC) were proposed recently based on
genetic profiles that show four types of molecular alterations: BRAF gene mutations,
KRAS gene mutations, microsatellite instability, and hypermethylation of CpG islands.
This review summarizes what is known about the serrated pathway of CRC, including
CRC molecular and clinical features, prognosis, and response to chemotherapy.
A.2. Prevalence of germline MUTYH mutations among Lynch-like syndrome
patients.
Castillejo A, Vargas G, Castillejo MI, Navarro M, Barberá VM, González S, Hernández-
Illán E, Brunet J, Ramón y Cajal T, Balmaña J, Oltra S,Iglesias S, Velasco A, Solanes
A, Campos O, Sánchez Heras AB, Gallego J, Carrasco E, González Juan D, Segura
A, Chirivella I, Juan MJ,Tena I, Lázaro C, Blanco I, Pineda M, Capellá G, Soto JL.
Eur J Cancer. 2014 Sep;50(13):2241-50. doi: 10.1016/j.ejca.2014.05.022. Epub 2014
Jun 18.

Anexos
148
Factor de Impacto: 5,417 (Q1).
ABSTRACT:
Background and aims
Individuals with tumours showing mismatch repair (MMR) deficiency not linked to
germline mutations or somatic methylation of MMR genes have been recently referred
as having 'Lynch-like syndrome' (LLS). The genetic basis of these LLS cases is
unknown. MUTYH-associated polyposis patients show some phenotypic similarities to
Lynch syndrome patients. The aim of this study was to investigate the prevalence of
germline MUTYH mutations in a large series of LLS patients.
Methods
Two hundred and twenty-five probands fulfilling LLS criteria were included in this study.
Screening of MUTYH recurrent mutations, whole coding sequencing and a large
rearrangement analysis were undertaken. Age, sex, clinical, pathological and molecular
characteristics of tumours including KRAS mutations were assessed.
Results
We found a prevalence of 3.1% of MAP syndrome in the whole series of LLS (7/225)
and 3.9% when only cases fulfilling clinical criteria were considered (7/178). Patients
with MUTYH biallelic mutations had more adenomas than monoallelic (P=0.02) and
wildtype patients (P<0.0001). Six out of nine analysed tumours from six biallelic
MUTYH carriers harboured KRAS-p.G12C mutation. This mutation was found to be
associated with biallelic MUTYH germline mutation when compared with reported
series of unselected colorectal cancer cohorts (P<0.0001).
Conclusions
A proportion of unexplained LLS cases is caused by biallelic MUTYH mutations. The
obtained results further justify the inclusion of MUTYH in the diagnostic strategy for
Lynch syndrome-suspected patients.
A.3. TGFBR1 intralocus epistatic interaction as a risk factor for colorectal
cancer.
Martinez-Canto A, Castillejo A, Mata-Balaguer T, Castillejo MI, Hernandez-Illan E, Irles
E, Barbera VM, Egoavil C, Guarinos C, Alenda C, Ochoa E, Lazaro R,Fajardo
S, Lacueva J, Calpena R, Soto JL.

Anexos
149
PLoS One . 2012;7(1):e30812. doi: 10.1371/journal.pone.0030812. Epub 2012 Jan 23.
Factor de Impacto: 3,73 (Q1).
ABSTRACT:
In colorectal cancer (CRC), an inherited susceptibility risk affects about 35% of
patients, whereas high-penetrance germline mutations account for <6% of cases. A
considerable proportion of sporadic tumors could be explained by the coinheritance of
multiple low-penetrance variants, some of which are common. We assessed the
susceptibility to CRC conferred by genetic variants at the TGFBR1 locus. We analyzed
14 polymorphisms and the allele-specific expression (ASE) of TGFBR1 in 1025
individuals from the Spanish population. A case-control study was undertaken with 504
controls and 521 patients with sporadic CRC. Fourteen polymorphisms located at the
TGFBR1 locus were genotyped with the iPLEX Gold (MassARRAY-Sequenom)
technology. Descriptive analyses of the polymorphisms and haplotypes and association
studies were performed with the SNPator workpackage. No relevant associations were
detected between individual polymorphisms or haplotypes and the risk of CRC. The
TGFBR1*9A/6A polymorphism was used for the ASE analysis. Heterozygous
individuals were analyzed for ASE by fragment analysis using cDNA from normal
tissue. The relative level of allelic expression was extrapolated from a standard curve.
The cutoff value was calculated with Youden's index. ASE was found in 25.4% of
patients and 16.4% of controls. Considering both bimodal and continuous types of
distribution, no significant differences between the ASE values of patients and controls
were identified. Interestingly, a combined analysis of the polymorphisms and ASE for
the association with CRC occurrence revealed that ASE-positive individuals carrying
one of the most common haplotypes (H2: 20.7%) showed remarkable susceptibility to
CRC (RR: 5.25; 95% CI: 2.547-5.250; p<0.001) with a synergy factor of 3.7. In our
study, 54.1% of sporadic CRC cases were attributable to the coinheritance of the H2
haplotype and TGFBR1 ASE. These results support the hypothesis that the allelic
architecture of cancer genes, rather than individual polymorphisms, more accurately
defines the CRC risk.

Anexos
150
B) Capítulos de libro
B.1. Serrated Polyposis Syndrome.
Miriam Juárez, Eva Hernández-Illán, Oscar Murcia, María Rodriguez-Soler, Rodrigo
Jover.
LIBRO: Intestinal Polyposis Syndromes. Editorial Springer . 2016 En prensa
ABSTRACT:
Serrated polyposis syndrome (SPS), formerly known as Hyperplastic polyposis
syndrome, is an uncommon disease characterized by the presence of multiple serrated
polyps throughout the colorectum. Until now, its genetics, molecular and clinical
characteristics continue to be unknown. This syndrome could be considered as the
paradigm of the serrated pathway of carcinogenesis. Diagnosis is made by the
fulfilment of any of the WHO criteria, recently re-defined: 1) At least 5 serrated polyps
proximal to the sigmoid colon, with 2 or more of these being larger than 10mm; 2) Any
number of serrated polyps proximal to the sigmoid colon in an individual who has a first
degree relative with SPS and 3) More than 20 serrated polyps of any size, distributed
throughout the colon.
In this chapter we will review the clinical and epidemiological characteristics of the
syndrome, the main hallmarks of the serrated pathway of carcinogenesis and the risk
of cancer and recommendations for surveillance for patients and their relatives.
C) Comunicaciones a congresos relacionadas con la tesis doctoral
C.1. Prevalencia de mutaciones POLE/POLD1 en Cáncer Colorectal Familiar
tipo X
Castillejo A, Hernández-Illán E, Castillejo MI, Pena L, Urioste M, Soto JL en nombre
del Programa de Cáncer Hereditario de la Comunidad Valenciana.
XXVIII Congreso Nacional de Genética Humana – AEGH. Palma de Mallorca, 13-15
Mayo 2015.
Comunicación oral. C0029.

Anexos
151
ABSTRACT:
El Cancer Colorectal Familiar tipo X (fCRC-X) es un subtipo de cáncer colorrectal
hereditario no polipósico que no presenta deficiencia en el mecanismo de reparación
de DNA de tipo mismatch. Las bases genéticas del fCRC-X son desconocidas.
Recientemente se han descrito mutaciones en línea germinal en el dominio de
corrección de pruebas las subunidades de la DNA polimerasa ε (POLE) y δ (POLD1)
en familias con múltiples adenomas y cánceres colorrectales.
El objetivo del presente trabajo fue determinar la prevalencia de las mutaciones de
POLE y POLD1 en fCRC-X.
Se incluyeron 72 probandos de familias que cumplían criterios de Amsterdam II con
expresión normal de las proteínas reparadoras MLH1, MSH2, MSH6 y PMS2 y
ausencia de inestabilidad de microsatélites. Los pacientes fueron atendidos en las
Unidades de Consejo Genético en Cáncer de la Comunidad Valenciana (2005-2014).
Se analizaron mediante secuenciación Sanger los exones 13 de POLE y 11 de
POLD1, donde se localizan los puntos calientes de mutación descritos.
Se detectaron dos casos con una nueva mutación en POLD1: c.1421T>C;
p.(Leu474Pro) y un caso con mutación en POLE: c.1270C>G; p.(Leu424Val). La
prevalencia en nuestra serie es de un 4.2% (3/72). La mutación de POLD1
p.(Leu474Pro) ha sido recientemente descrita como patogénica en una de nuestras
familias (Valle et al, 2014). La descripción de la misma mutación en otra familia
aparentemente no relacionada indica el posible carácter fundador de dicha mutación.
Se han diagnosticado siete individuos portadores de mutación entre ambas familias
POLD1 con cinco CCR y dos cánceres de endometrio diagnosticados entre los 23 y 56
años. La familia con mutación en POLE presenta seis casos diagnosticados de CCR
con edades comprendidas entre los 11 y 46 años.
Nuestros resultados avalan la inclusión del estudio genético de POLE y POLD1 en
familias con Cáncer Colorectal Familiar tipo X.

Anexos
152
C.2. A new POLD1 germline mutation as cause of Familial Colorectal Cancer
Type X
Hernández-Illán E, Castillejo A, Castillejo MI, Segura A, Juan MJ, Jover R, Soto JL.
European Conference of Human Genetics – ESHG. Milán, May 31- June 3, 2014.
European Journal of Human Genetics. Vol 22. Suppl. 1. Mayo 2014. P12.116-M
ABSTRACT:
The genetic basis for Familial Colorectal Cancer Type-X (fCRC-X) is unknown.
Mutations at the proof-reading domains of DNA polymerase ε (POLE) and δ (POLD1)
have been recently identified in families with multiple colorectal adenomas and CRC.
We aimed to assess the prevalence of POLE and POLD1 mutations in fCRC-X.
A total of 63 index cases fulfilling Amsterdam II criteria with normal expression of
mismatch repair proteins and microsatellite stable tumors were included. Sanger
sequencing of POLE-exon13 and POLD1-exon11 was performed.
None of the cases carried mutations in POLE. We found one case with a new POLD1
variant in heterozygosis: c.1421T>C (p.Leu474Pro). The patient was a woman who
underwent a synchronous CRC and large bowel gastrointestinal stromal tumor at age
36. Her maternal aunt had metachronous CRC (dx33y) and endometrial cancer (dx56y)
and was a carrier of this variant. Therefore, the index case’s mother, who underwent
endometrial cancer (dx52y), was an obligate variant carrier. It has been described that
the homologous residue of POLD1 p.Leu479Pro mutation in S. cerevisiae causes a
mutator phenotype. Furthermore, this is the paralogous residue of the hot spot at POLE
(p.Leu424). In silico prediction analysis strongly suggests a pathogenic nature for this
variant. Integration of all these evidences drove us to classify this new variant as
probably damaging.
POLD1 mutations might explain about 1.6% of fCRC-X. POLD1 testing should be
included in the diagnostic strategy for unexplained familial CRC.




















