
Prediction and refinement of complexes using coarse ... · Prediction and refinement of complexes...
38
Prediction and refinement of complexes using coarse-grained and atomistic approaches in ATTRACT Martin Zacharias Theoretical Biophysics T38 Physics Department Technical University of Munich
Transcript of Prediction and refinement of complexes using coarse ... · Prediction and refinement of complexes...

Prediction and refinement of complexes using coarse-grained and atomistic approaches in ATTRACT
Martin ZachariasTheoretical Biophysics T38Physics DepartmentTechnical University of Munich

The ATTRACT approach
• 31 LJ-atom types• Real charges
Multi-start systematic search by Energy Minimization
Multi-start systematic search by Energy Minimization
Score
distanceZacharias, Protein Sci. 2003,12;1271;Saladin et al., BMCStruct Biol 2009, 9:27; de Vries, Schindler, de Beauchene, Zacharias, Biophys. J. 2015,108:462.
min
68
min
min
68
68
2
:
rrifr
r
R
r
ReV
rrifr
r
R
r
RV
pairrepulsive
pairattractiveofcaseinr
r
R
r
RV
ijij
ji
ij
AB
ij
AB
ijij
ji
ij
AB
ij
AB
ij
ji
ij
AB
ij
AB

Multi-start systematic search by Energy Minimization
Multi-start systematic search by Energy Minimization
The ATTRACT approach

Multi-start systematic search by Energy Minimization
Multi-start systematic search by Energy Minimization
The ATTRACT approach

Multi-start systematic search by Energy Minimization
Multi-start systematic search by Energy Minimization
The ATTRACT approach

Multi-start systematic search by Energy Minimization
Multi-start systematic search by Energy Minimization
The ATTRACT approach

Systematic improvement of the scoring function
receptor
Score
distance
AimScoring optimization of near-native vs. alternative docking minima for a large set of training complexes
Target functionTop ranking of native solution
(large gap to incorrect solutions)
Step 1Generation of „high-ranked“
incorrect solutions
Step 2Optimization of pairwise
interactions with respect to target function
Step 3Test of scoring on separate
set of test complexes

Efficient inclusion of flexibility
Local flexibility:• Side chains and small loops
represented by several conformational copies
– Mean field representation
– Simultaneous optimization of docking geometry and side chain and loop structure
Global flexibility:• Inclusion of global soft collective
degrees of freedom from normal mode analysis– Accounting for most important
global motion using very few new variables (1-10)
Computationally very fast
May & Zacharias, BBA 2005; May & Zacharias, Proteins 2009;Bastard, Prevost & Zacharias, Proteins 2006
Docking with multiple loop copies
Softest global mode of Xylanase

Induced fit Docking using normal mode coordinates
• Illustration of a docking process including minimization in normal mode variables for an enzyme-inhibitor complex

New methods implemented in ATTRACT
de Beauchene, Plos Comput Biol, 2016; deVries et al., Biophys. J. 2015, 2016; Schindler et al., Proteins, 2014; Schindler et al., Structure, 2015; de Vries & Zacharias, Proteins, 2013; Setny et al., BMC Bioinfo., 2012; De Vries & Zacharias, PLosOne, 2012; Setny & Zacharias, NAR 2011
Multisubunit-Docking into CryoEM density starting from millions of start arrangements
ATTRACTProtein-Protein
Docking
Docking using GPU accleration
Web-Interfacewww.attract.ph.tum.de
Protein-DNA and Protein-RNA
docking
Protein-Peptide docking using pepATTRACT
Protein-Protein docking including
SAXS during docking
Single stranded RNA-Protein docking using combination of
RNA fragments
Refinement using iATTRACT

Refinement of predicted complexes
• Current procedure– Superposition of atomic resolution structures onto
coarse-grained models
– Energy minimization + Refinement at atomic resolution using Molecular Dynamic Simulations
• Problem:– Atomistic simulations are time consuming and often
don‘t move far from the start structure.

Refinement with iATTRACT• iATTRACT: combined energy minimization in rigid-body degrees of
freedom and atomistic motion at interface.
• Use of the normal mode concept implemented in ATTRACT: the motion of each interface atom is assigned one „mode“

• Motion of atoms at the interface is controlled by a structure-based force field with the unbound structure as reference.
• Atomistic interactions between proteins (OPLS force field)
Total energy
Refinement with iATTRACT
Schindler, de Vries, Zacharias Proteins, 2014, 83:248.

• iATTRACT allows relatively large translational and rotational displacements triggered by local adjustments at the interface.
• Test on 166 protein-protein complexes from benchmark 4.0
• Start: best 200 (in terms of iRMSD) rigid body docking solutions obtained using unbound partner structures
• Evaluation by interface root mean square deviation (iRMSD) and fraction of native contacts (fnat)
Refinement with iATTRACT
pdb1WDW
pdb2OUL
pdb2MTA





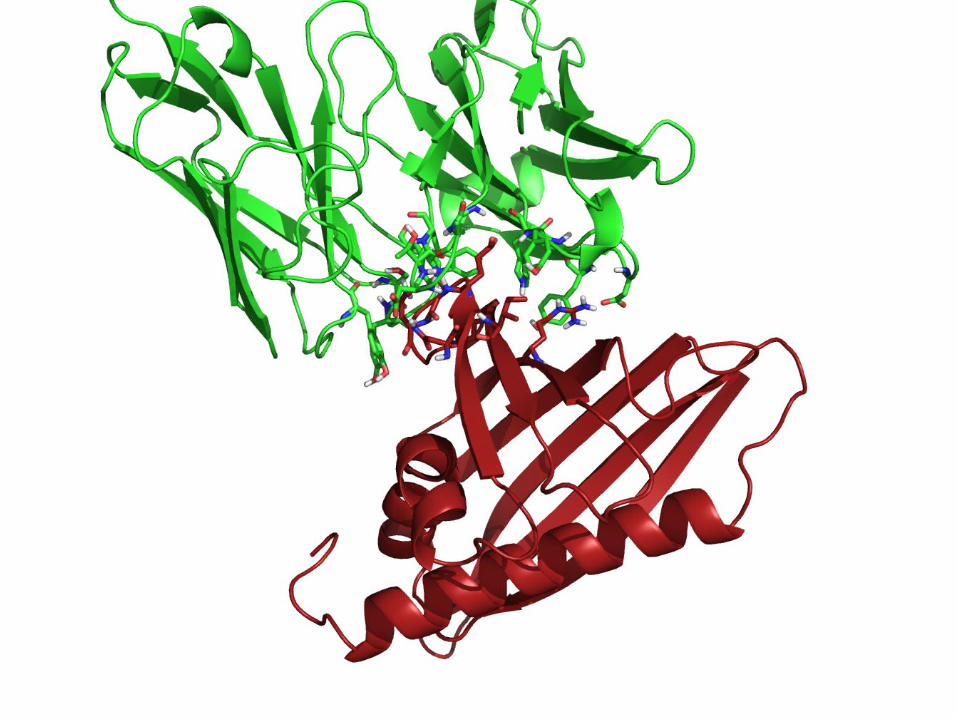





Docking refinement using iATTRACT
• Significant improvement of iRMSD and fnat after refinement – In some cases from an initial iRMSD of 6-8 Å to a final iRMSD of
~1 Å
• High probability to find structurally improved models for start structures with initial iRMSD < 5 Å.

Ranking after refinement using iATTRACT
• For a fraction of complexes a „funnel-like“ behavior of the scoring vs. iRMSD was observed.
• Only modest improvement in scoring near-native docking solutions

Prediction of Protein-Ligand complexes using Biasing-Replica-Exchange
• MD-simulations allow in principle for full flexibility.
• Limited timescale of search and trapping at the surface caused by non-optimal binding
• Trapping can be avoid using a special ambiguity restraint:
• The ambiguity restraint includes all heavy atoms on the surface of the two partners.
2ambiguityambiguity )()V(d refddk=
d
reference replica R0
d
replica R10

Illustration of the method

Test on simple system in vacuum• Receptor: two-Helix bundle
• Ligand: single helix• Partners kept semi-rigid (distance restraints between CA atoms in each partner)
~40 ps ~200 ps ~800 ps ~1500 ps

Test on simple system in vacuum• Receptor: two-Helix bundle
• Ligand: single helix• Partners kept semi-rigid (distance restraints between CA atoms in each partner)
~40 ps ~200 ps ~800 ps ~1500 ps

Refinement of unbound docking cases
• 20 start structures with 4 Å <RMSDlig < ~15 Å
• (a) Perform energy minimization
• (b) 12 MD simulations (short-range CA distance restraints in each partner and a center-of-mass restraint between partners)
• (c) H-REMD with repulsive potential and 12 replicas (otherwise same as MD)
Ligand RMSD (Å)
Ligand RMSD (Å)Ligand RMSD (Å)
Inte
ract
ion
energ
y (
kca
l/m
ol)
)
Inte
ract
ion
energ
y (
kca
l/m
ol)
)In
tera
ctio
n e
nerg
y (
kcal/m
ol)
)
pdb1GCQ
pdb1PPEpdb2OOB

Results in CAPRI rounds 28-36Target best model fnat IRMSD[Å] category
59 5 0.18 3.8 acceptable quality (2*)60 5/6 1.0/0.94 0.44 high quality(2***,4**)
61 1 0.76 0.5 medium quality (5**)62 1 0.92 0.39 high quality (2***, 3**)63 1 0.81 0.49 high quality (2***, 3*)64 2 0.87 0.42 high quality (3***, 2**)65 2 0.27 3.7 incorrect66 1/1 0.5/0.75 1.3/2.1 acceptable (1*)67 5 0.88 0.8 medium quality (2**, 8*)95 3 0.5 3.3 acceptable quality (5*)96 3 0.15 2.96 acceptable quality (1*)97 8 0.05 12.2 incorrect98-99 - - - incorrect103 10 0.27 12.2 incorrect104 4 0.68 0.92 high quality (1***,9**) water (5++)105 3 0.66 1.2 medium quality (10**) water (7+++)107 - - - incorrect

Target 60-64: importin alpha in complex with peptides
experimental complex, target 63 predicted(***)
Distinctive Conformation of Minor Site-Specific Nuclear Localization Signals Bound to Importin-Alpha.Chang, C.-W., Counago, R.M., Williams, S.J., Boden, M., Kobe, B.(2013) Traffic 14: 1144
• Docking strategy:
• Docking using information from available complexes
• Atomistic refinement with restraint MD simulations
• (***) solutions for all except target61

Target 66: pria-helicase bound to ssb C-terminal tail peptide
Structural mechanisms of PriA-mediated DNA replication restart.Bhattacharyya et al. Proc Natl Acad Sci U S A. 111,1373 (2014).
• Systematic peptide-protein surface using pepATTRACT
• Refinement with iATTRACT and atomistic MD simulations
• More extended binding surface in predicted complexes.
experimental complex: yellow/bluepredicted(*) : orange/atom-coded

Target 67:Peptide in complex with WW3 domain/NEDD4
Structural and Biochemical Basis for Ubiquitin Ligase Recruitment by Arrestin-Related Domain-Containing Protein-3 (ARRDC3).
Qi et al. J Biol. Chem. 289, 4743 (2014)
• Docking including exprimental data on binding site
• Molecular dynamics based refinement

Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome.McGinty, Henrici, Tan. Nature 514, 591 (2014)
• Docking using standard approach– Use restraint to keep active site in
enzyme near H3 tail ubiquination site
– Acidic patch at H2A/H2B interface
– differs from previous published model
Target 95:Ubiquitin-conjugating enzyme in complex with
nucleosome

Summary
• Several new features in ATTRACT docking package
• Rapid refinement using iATTRACT and/or Biasing potential replica exchange MD simulations
• Several successes in CAPRI challenge
• Extension of refinement to more realistic conditions

Acknowledgements
Group membersRainer BombliesIsaure de BeaucheneUwe EhrmannChristina FrostFlorian KandziaAlexander KnipsManuel LuitzKatja OstermeirGuiseppe La RosaAlexander SasseChristina SchindlerNadine SchwierzSjoerd de VriesFabian Zeller
Current Funding:DFG (RNA-Protein-Interaction)CIPSM, SFB-749 ;SFB1035,
SFB863
Collaboration of Protein-Protein docking
Chantal Prevost, Benjamin Boyer, Adrien Saladin, IBPC, Paris
Sebastien Fiorucci, University of Nice
CAPRI-DOCKING-TEAM:Christina Schindler, Sjoerd de Vries, Isaure de
Beauchene


















