
Nano-magnesium oxide reinforced polylactic acid biofilms ...

527
16Polylactic Acid Technology
David E. Henton, Patrick Gruber, Jim Lunt, and Jed Randall
CONTENTS16.1 Introduction ..............................................................................................52816.2 Lactic Acid ................................................................................................53116.3 Life Cycle Analysis ..................................................................................53216.4 Polymerization of Lactide ......................................................................53616.5 PLA Physical Properties ..........................................................................538
16.5.1 Linear Optical Copolymer Structures and Blends ..................53816.5.1.1 Density ..........................................................................54016.5.1.2 Glass Transition and the Amorphous Phase ............540
16.5.2 Rheology ........................................................................................54216.5.2.1 Melt Rheology of Linear PLA ....................................54216.5.2.2 Melt Stability ................................................................54416.5.2.3 Solution Properties ......................................................54416.5.2.4 Branching ......................................................................54516.5.2.5 Solid-State Viscoelastic Properties ............................547
16.5.3 Crystallinity ..................................................................................54716.5.3.1 Morphology ..................................................................54716.5.3.2 Degree of Crystallinity ................................................55016.5.3.3 Crystallization Kinetics ..............................................55216.5.3.4 Stereocomplex ..............................................................55316.5.3.5 Stress-Induced Crystallization ..................................554
16.5.4 Degradation and Hydrolysis ......................................................55516.5.4.1 Hydrolysis ....................................................................55516.5.4.2 Hydrolysis of Solid Samples Suspended in
Aqueous Media ............................................................55816.5.4.3 Solution Hydrolysis ....................................................56016.5.4.4 Hydrolysis of Samples Exposed to Humidity ........561
16.5.5 Enzymatic Degradation ..............................................................56316.6 Applications and Performance ..............................................................56316.7 Summary ....................................................................................................568
1741_C16.qxd 2/11/2005 9:57 AM Page 527

Acknowledgments ............................................................................................569Reference ..............................................................................................................569
ABSTRACT Poly(lactic acid) (PLA) is the first commodity polymer pro-duced from annually renewable resources. Some of the environmental ben-efits of PLA, such as low energy to produce and reduced green house gasproduction, are presented and opportunities for the future are given. Thischapter on PLA reviews the chemistry and material properties of the poly-mer and its copolymers. The current technology for the production of lacticacid, lactide, and polymer are presented. Applications for fabricated articlessuch as films and fibers are also discussed.
16.1 Introduction
Polylactic acid (PLA) is a rigid thermoplastic polymer that can be semi-crystalline or totally amorphous, depending on the stereopurity of the poly-mer backbone. L(�)-lactic acid (2-hydroxy propionic acid) is the natural andmost common form of the acid, but D(�)-lactic acid can also be produced bymicroorganisms or through racemization and this “impurity” acts much likecomonomers in other polymers such as polyethylene terephthalate (PET) orpolyethylene (PE). In PET, diethylene glycol or isophthalic acid is copoly-merized into the backbone at low levels (1–10%) to control the rate of crys-tallization. In the same way, D-lactic acid units are incorporated into L-PLAto optimize the crystallization kinetics for specific fabrication processes andapplications.
PLA is a unique polymer that in many ways behaves like PET, but alsoperforms a lot like polypropylene (PP), a polyolefin. Ultimately it may be thepolymer with the broadest range of applications because of its ability to bestress crystallized, thermally crystallized, impact modified, filled, copoly-merized, and processed in most polymer processing equipment. It can beformed into transparent films, fibers, or injection molded into blow-moldable preforms for bottles, like PET. PLA also has excellent organolepticcharacteristics and is excellent for food contact and related packaging appli-cations.
In spite of this unique combination of characteristics, the commercial via-bility has historically been limited by high production costs (greater than$2/lb). Until now PLA has enjoyed little success in replacing petroleum-based plastics in commodity applications, with most initial uses limited tobiomedical applications such as sutures.1
PLA is not new to the world of polymers. Carothers2 investigated the pro-duction of PLA from the cyclic dimer (lactide) of lactic acid as early as 1932.Even before that, low molecular weight dimers and oligomers were detected
528 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:57 AM Page 528

when water was removed from an aqueous solution of lactic acid.3 Theannouncement of the formation of a new company, Cargill Dow LLC, in 1997brought two large companies together to focus on the production and mar-keting of PLA with the intention of significantly reducing the cost of pro-duction and making PLA a large-volume plastic.4
In today’s world of green chemistry and concern for the environment,PLA has additional drivers that make it unique in the marketplace. The start-ing material for the final polymer, lactic acid, is made by a fermentationprocess using 100% annually renewable resources. The polymer will alsorapidly degrade in the environment and the by-products are of very low tox-icity, eventually being converted to carbon dioxide and water.
PLA can be prepared by both direct condensation of lactic acid and by the ring-opening polymerization of the cyclic lactide dimer, as shown inFigure 16.1. Because the direct condensation route is an equilibrium reaction,difficulties of removing trace amounts of water in the late stages of polymer-ization generally limit the ultimate molecular weight achievable by thisapproach. Most work has focused on the ring-opening polymerization of lac-tide, although other approaches, such as azeotropic distillation to drive theremoval of water in the direct esterification process, have been evaluated.5
Cargill Dow LLC has developed a patented, low-cost continuous processfor the production of lactic acid-based polymers.6 The process combines thesubstantial environmental and economic benefits of synthesizing both lactideand PLA in the melt rather than in solution and, for the first time, provides acommercially viable biodegradable commodity polymer made from renew-able resources. The process starts with lactic acid produced by fermentation ofdextrose, followed by a continuous condensation reaction of aqueous lactic acid to produce low molecular weight PLA prepolymer (Figure 16.2).Next, the low molecular weight oligomers are converted into a mixture of lactide stereoisomers using a catalyst to enhance the rate and selectivity of the
Polylactic Acid Technology 529
OH
O
H CH3
HO
L-Lactic acid
H2O
H2O
O
O
O
O
H
H
CH3CH3
L-Lactide
O
O
H CH3n
L-PLA
FIGURE 16.1Polymerization routes to polylactic acid.
1741_C16.qxd 2/11/2005 9:57 AM Page 529
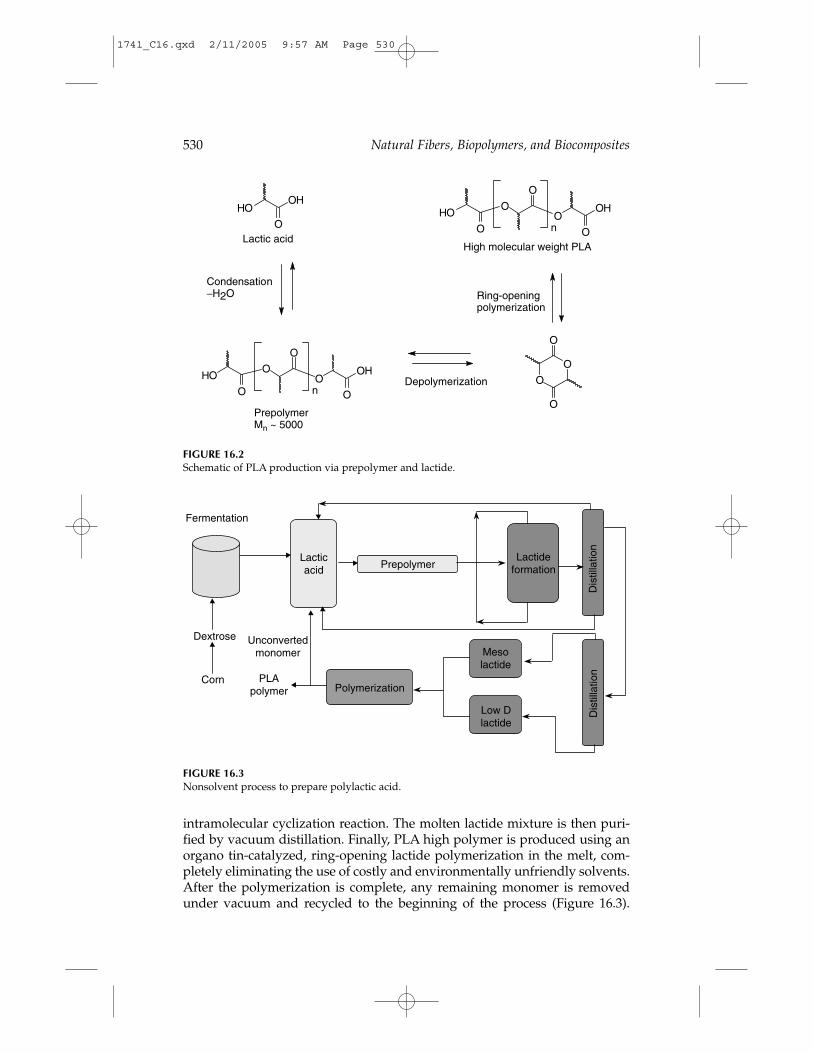
intramolecular cyclization reaction. The molten lactide mixture is then puri-fied by vacuum distillation. Finally, PLA high polymer is produced using anorgano tin-catalyzed, ring-opening lactide polymerization in the melt, com-pletely eliminating the use of costly and environmentally unfriendly solvents.After the polymerization is complete, any remaining monomer is removedunder vacuum and recycled to the beginning of the process (Figure 16.3).
530 Natural Fibers, Biopolymers, and Biocomposites
HOOH
O
O
O
O
O
O
O
HO
OO
OH
On
O
O
HO
OO
OH
On
Condensation−H2O
Lactic acid
Depolymerization
Ring-openingpolymerization
PrepolymerMn ~ 5000
High molecular weight PLA
FIGURE 16.2Schematic of PLA production via prepolymer and lactide.
Fermentation
Lacticacid Prepolymer
Lactideformation
Polymerization
Mesolactide
Low Dlactide
Dis
tilla
tion
PLApolymer
Unconvertedmonomer
Corn
Dextrose
Dis
tilla
tion
FIGURE 16.3Nonsolvent process to prepare polylactic acid.
1741_C16.qxd 2/11/2005 9:57 AM Page 530

This process is currently in operation in a recently constructed 300 million lb/yrcommercial-scale PLA plant in Blair, Nebraska, USA. Cargill Dow LLC hasalso announced the construction of an additional plant in Europe in the nearfuture.7
PLA has been extensively studied by many researchers and reviewed indetail in several recent publications.8 This chapter will not review all of thegrowing volume of information on PLA, but will focus on the key elementsof technology that will change this polymer from a specialty material to alarge-volume commodity plastic.
16.2 Lactic Acid
Lactic acid (2-hydroxypropionic acid) is the basic building block for PLA. Itis a highly water-soluble, three-carbon chiral acid that is naturally occurringand is most commonly found in the L(�) form. It is used as an acidulant infoods, as a building block for biodegradable polymers (PLA), and is con-verted to esters and used as a green solvent for metal cleaning, paints, andcoatings. PURAC has been the world’s largest producer of lactic acid, but therecent start-up of the Cargill Dow lactic acid plant, with a capacity of400,000,000 lb, exceeds that of all producers combined. There are differentgrades of lactic acid, each varying in purity depending on their end useapplication. The lowest to highest purity is technical grade, edible grade,pharmaceutical grade, and analytical grade. The lower grades contain sul-fates, metals, amino acids, and various carbohydrates. With the need forpolymerization-grade lactic acid it has become clear that even the highestpurity grade, analytical grade, is not pure enough. The slight traces of car-bohydrates and amino acids remaining in analytical grade cause color in theprocess and even parts per million traces of cations such as Na� lead toracemization of the acid, resulting in the less desired D(�) lactic acid.
A good summary of lactic acid chemistry is found in the book by Holten,9
Lactic Acid, Properties and Chemistry of Lactic Acid and Derivatives. An older,but still informative review of lactic acid production via fermentation isfound in a chapter on lactic acid by H. Schopmeyer.10 The major issues of fer-mentation, purification, and a history of lactic acid growth in the UnitedStates are reviewed. It also contains a good literature review up to that timeon lactic acid.
A more recent review11 (1976) of lactic acid processes by Cox and Macbeanlooked closely at the details of distillation, liquid extraction, esterification,salt processes, electrodialysis, thermal methods, and ion exchangeapproaches to providing lactic acid from fermentation of carbohydrates.There have been a large number of proposed routes and patents issuedclaiming to have improved processes to isolate lactic acid from fermentationbroth, but most are combinations or improvements on these basic concepts.
Polylactic Acid Technology 531
1741_C16.qxd 2/11/2005 9:57 AM Page 531

The main stages of lactic acid production consist of (1) fermentation, (2) cellmass and protein removal, (3) recovery and purification of lactic acid, (4) concentration of lactic acid, and (5) color removal.
Research and improvements in lactic acid production have been centeredaround each of these steps, with continued improvement in the cost and efficiency of each. Better microorganisms for the fermentation that are morerobust to temperature, pH, and the organic products they produce have evolvedin recent years. Engineering improvements have increased the size and effi-ciency of the plants, while improved technology for water removal (multipleeffect evaporators and membranes) has lowered the cost for production. By farthe greatest developments and diversity of approaches have been in the area ofseparation and purification. Some of the separation and purification processesand their proposed key features are listed in Table 16.1.
Most processes to produce lactic acid rely on bacterial fermentation of dex-trose in aqueous slurry with a continual addition of a base, such as calciumhydroxide, to maintain a neutral pH. Bacteria are generally quite sensitive topH and are only productive in a narrow pH range. This sensitivity to pH isdemonstrated in Table 16.2 for the production of acetic acid by Clostridiumthermoaceticum.12 Generally, deviation of pH by more than a couple of pHunits totally inhibits bacterial fermentation. For the production of neutralspecies such as ethanol, this is not an issue. However, the production oforganic acids such as acetic and lactic acid shifts the pH as the acids are pro-duced. To maintain the optimum pH, bases are continuously added.
At a pH of about 7 (neutral pH), essentially all of the lactic acid is ionized.Isolation of the lactic acid and removal of water requires that a strong min-eral acid, such as sulfuric acid, be added to protonate the lactate salt, whileat the same time producing a mole of salt for every two moles of acid pro-duced. On a weight basis, about a pound of salt per pound of acid is pro-duced. This significant amount of waste salt contributes to the cost of thelactic acid and is undesirable from a sustainability point of view.
The process of the near future will contain an organism that is tolerant oftemperature and acidic conditions and be combined with lactic acid removaltechnology that does not result in the generation of waste salts. This processwill be cost- advantaged and beneficial to the environment.
16.3 Life Cycle Analysis
In recent years, products, processes, and technology have been judged for theiroverall sustainability. Those that use large quantities of energy, deplete naturalresources, pollute the environment, or emit greenhouse gases are viewed as lesssustainable. Green chemistry is the use of chemistry for pollution prevention orthe production of materials that fulfill the definition of sustainability. Morespecifically, green chemistry is the design of chemical products and processes
532 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:57 AM Page 532

Polylactic Acid Technology 533
TAB
LE 1
6.1
Lac
tic
Aci
d P
urif
icat
ion
Uni
t Ope
rati
ons
Feat
ure
Ad
van
tage
/Dis
adva
nta
ge
Ele
ctro
dia
lysi
sC
an b
e us
ed to
con
tinu
ousl
y re
mov
e la
ctic
aci
d (
lact
ate
ions
) 1.
Doe
s no
t req
uire
aci
dif
icat
ion
of f
erm
enta
tion
.th
roug
h a
mem
bran
e d
rive
n by
ele
ctri
cal p
oten
tial
.2.
Ene
rgy
cost
and
cap
ital
.R
ever
se o
smos
isL
acti
c ac
id is
con
tinu
ousl
y re
mov
ed
1. H
ighe
r pr
oduc
tivi
ty b
ecau
se o
ne c
an
thro
ugh
a m
embr
ane.
mai
ntai
n lo
w a
cid
leve
l in
ferm
ente
r.2.
Fou
ling
of th
e m
embr
ane.
3. R
equi
res
acid
ic p
H s
tabl
e or
gani
sm.
Liq
uid
ext
ract
ion
Lac
tic
acid
is c
onti
nuou
sly
rem
oved
fro
m th
e fe
rmen
tati
on1.
Sui
tabl
e fo
r co
ntin
uous
pro
cess
and
pro
vid
es e
ffic
ient
or
aci
dif
ied
bro
th b
y pr
efer
enti
al p
arti
tion
ing
into
a s
olve
nt.
rem
oval
fro
m m
any
non-
acid
ic im
puri
ties
.2.
Hig
h co
st o
f ca
pita
l.3.
Sol
vent
loss
cos
ts.
Ion
exch
ange
(ac
idif
icat
ion)
The
lact
ate
salt
is a
cid
ifie
d b
y a
str o
ng
1. E
limin
ates
the
need
to a
dd
a s
tron
g ac
id to
the
acid
ion
exch
ange
res
in.
ferm
enta
tion
.2.
Cos
t of
resi
n an
d is
sues
of
resi
n re
gene
rati
on.
Ion
exch
ange
(pu
rifi
cati
on)
Lac
tic
acid
is r
emov
ed f
rom
the
aque
ous
solu
tion
1. T
his
is th
e so
lid e
quiv
alen
t of t
-am
ine
extr
acti
on
by c
ompl
exin
g w
ith
an a
min
o co
ntai
ning
res
in.
tech
nolo
gy w
itho
ut th
e so
lven
t los
s is
sues
.2.
Reg
ener
atio
n of
the
resi
n.3.
Cos
t and
ava
ilabi
lity
of th
e re
sin.
Dis
tilla
tion
Lac
tic
acid
is s
epar
ated
fro
m le
ss v
olat
ile
1. L
acti
c ac
id c
an b
e st
eam
dis
tille
d.
com
pone
nts
by v
acuu
m s
team
dis
tilla
tion
.2.
Sig
nifi
cant
pur
ific
atio
n m
ust b
e d
one
prio
r to
dis
tilla
tion
.3.
Dep
end
ing
on c
ond
itio
ns, s
ome
deg
rad
atio
nan
d o
ligom
eriz
atio
n ca
n oc
cur.
Inso
lubl
e sa
lt p
roce
sses
The
fer
men
tati
on o
r pu
rifi
cati
on p
roce
ss is
run
1.
Sim
ple
proc
ess
that
uti
lizes
low
-cos
t cap
ital
.at
a c
once
ntra
tion
that
exc
eed
s th
e so
lubi
lity
2. T
he c
ryst
alliz
atio
n of
CaS
O4
occl
udes
impu
riti
es a
nd
of th
e la
ctat
e sa
lt (
e.g.
, CaS
O4)
, whi
ch is
isol
ated
re
sult
s in
rel
ativ
ely
impu
re a
cid
.an
d la
ter
acid
ifie
d.
Est
erif
icat
ion
Lac
tate
est
ers
are
prep
ared
and
the
1. D
isti
llati
on a
nd s
epar
atio
n of
est
ers
vola
tile
est
ers
are
dis
tille
d.
give
s hi
gh-q
ualit
y pr
oduc
t.2.
Req
uire
s re
conv
ersi
on to
aci
d.
1741_C16.qxd 2/11/2005 9:57 AM Page 533

that reduce or eliminate the use and generation of hazardous substances. In aproduction supply chain, green products and processes integrate engineering,chemistry, and environmental issues. The supply chain evaluation incorporatesthe energy, wastes, and emissions for operations during the production andtransportation of all raw materials throughout the entire process, from produc-tion of all raw materials to disposal of the final articles.
An excellent method to quantify the sustainability of a product or processis to do a life cycle analysis (LCA). LCA looks at all factors from raw materi-als, transportation, product manufacture, end use, to disposal. The energy,air and water pollution, as well as the hazards of the chemicals are assessedand judged against alternate products or solutions. Each step from cradle tograve is quantified and the total impact on the environment in each of theareas is thus known.
Cargill Dow’s process embodies the well-known Principles of GreenChemistry by (1) preventing pollution at the source through the use of a nat-ural fermentation process to produce lactic acid, (2) substituting annuallyrenewable materials for petroleum-based feedstock, (3) eliminating the useof solvents or other hazardous materials, (4) completely recycling productand by-product streams, and (5) efficiently using catalysts to reduce energyconsumption and improve yield. In addition, NatureWorksTM PLA productscan be either recycled or composted after use.
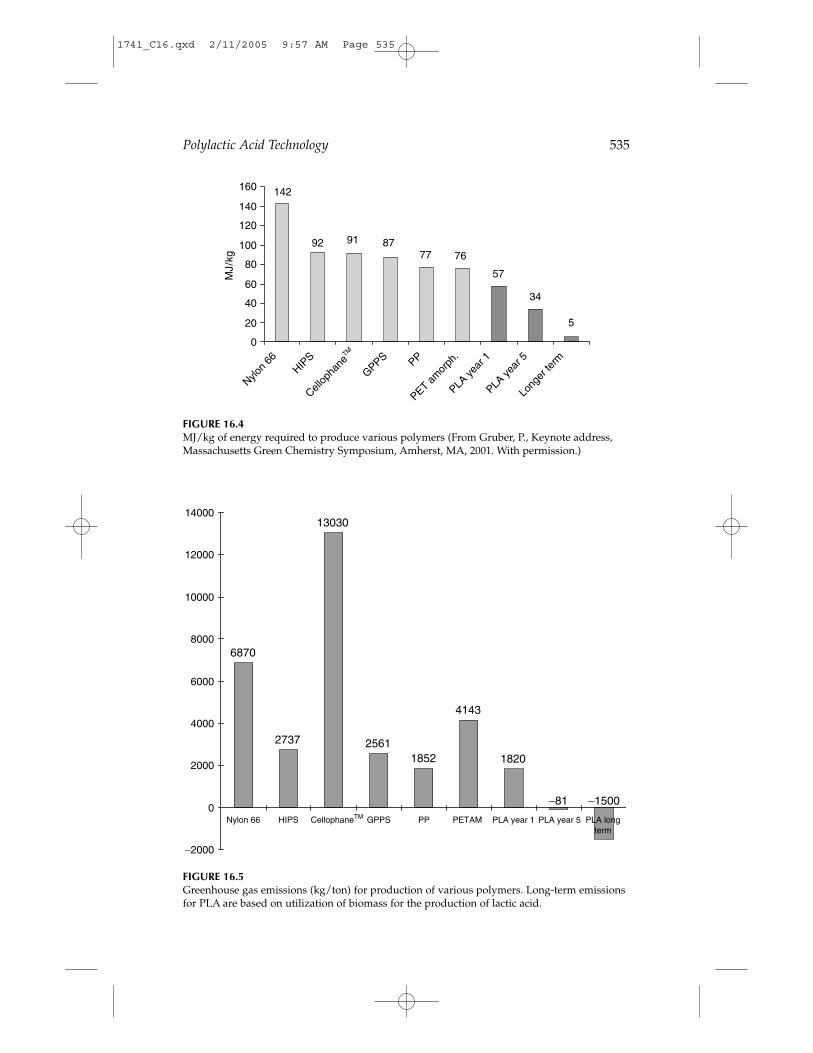
PLA, as produced by Cargill Dow LLC, uses from 20 to 50% less fossilfuel than conventional plastic resins (Figure 16.4), meaning that up to 2 to 5times more PLA can be produced with a given amount of fossil fuel thanpetrochemical-derived plastics. The ability to produce the polymer on afavorable cost/performance basis is critical to realizing these human healthand environmental benefits. This has been achieved through efficient use ofraw materials, reduced energy utilization, and elimination of solvents.
PLA is also a low-impact, greenhouse gas polymer because the CO2 gen-erated during PLA biodegradation is balanced by an equal amount takenfrom the atmosphere during the growth of plant feedstocks. In contrast,petrochemical-based polymers contribute to volatile organic compound(VOC) emissions and CO2 generation when incinerated. LCA calculationsindicate that PLA has a greenhouse gas emission rate of about 1600 kgCO2/metric ton, while polypropylene, polystyrene, PET, and nylon havegreenhouse gas values of 1850, 2740, 4140, and 7150 kg CO2/metric ton,respectively.13 Figure 16.5 shows the relative comparison of greenhouse gas
534 Natural Fibers, Biopolymers, and Biocomposites
TABLE 16.2
Fermentation Performance of ClostridiumThermoaceticum vs. pH in Acetic Acid Production
pH 6.8 5.5Productivity (g/hr) 0.8 0.6Acetate –g/l (as acid) 45 16Yield (%) 85–90 77
1741_C16.qxd 2/11/2005 9:57 AM Page 534
Polylactic Acid Technology 535
142
9192 8777 76
34
5
57
0
20
40
60
80
100
120
140
160
Nylon
66HIP
S
Cellop
hane
TM
GPPS PP
PET am
orph
.
PLA ye
ar 1
PLA ye
ar 5
Long
er te
rm
MJ/
kg
FIGURE 16.4MJ/kg of energy required to produce various polymers (From Gruber, P., Keynote address,Massachusetts Green Chemistry Symposium, Amherst, MA, 2001. With permission.)
6870
2737
13030
25611852
4143
1820
−81 −1500
−2000
0
2000
4000
6000
8000
10000
12000
14000
Nylon 66 HIPS CellophaneTM GPPS PP PETAM PLA year 1 PLA year 5 PLA long term
FIGURE 16.5Greenhouse gas emissions (kg/ton) for production of various polymers. Long-term emissionsfor PLA are based on utilization of biomass for the production of lactic acid.
1741_C16.qxd 2/11/2005 9:57 AM Page 535

contribution to the environment for common plastic products. Longer term,as PLA is produced from field wastes or other biomass, PLA can become aCO2 sink and actually contribute to a net reduction in greenhouse gases.
Although problematic in the past, recycling can offer an environmentallyattractive disposal option for plastics. However, recycled plastics are typicallycontaminated with other incompatible materials and thus have reduced properties. Consequently, recycled plastics are often relegated to low-performance/low-value applications. In contrast, PLA articles can be readilyhydrolyzed with water to form lactic acid, which is then purified and polymer-ized to remake prime polymer. PLA can also be naturally composted intohumus, carbon dioxide, and water, with complete degradation occurring withinonly a few weeks under typical compost conditions. However, polymer hydrol-ysis and monomer recovery offer the preferred method for PLA recycling.
16.4 Polymerization of Lactide
Many catalyst systems have been evaluated for the polymerization of lactideincluding complexes of aluminum, zinc, tin, and lanthanides. Even strongbases such as metal alkoxides have been used with some success. Dependingon the catalyst system and reaction conditions, almost all conceivable mecha-nisms (cationic,14 anionic,15 coordination,16 etc.) have been proposed to explainthe kinetics, side reactions, and nature of the end groups observed in lactidepolymerization. Tin compounds, especially tin(II) bis-2-ethylhexanoic acid (tinoctoate), are preferred for the bulk polymerization of lactide due to their solu-bility in molten lactide, high catalytic activity, and low rate of racemization ofthe polymer. Conversions of � 90% and less than 1% racemization can beobtained while providing polymer with high molecular weight.
The polymerization of lactide using tin octoate is generally thought to occurvia a coordination-insertion mechanism16 with ring opening of the lactide toadd two lactyl units (a single lactide unit) to the growing end of the polymerchain shown schematically in Figure 16.6. The tin catalyst facilitates the
536 Natural Fibers, Biopolymers, and Biocomposites
O
O
O
O
R-OH
Sn(Oct)2
O
O
O
OSn
(Oct)2
O
H
R
O
O
O
O O
Sn R
(Oct)2
H
OO
O
O
H
RO
Sn(Oct)2
ROO
OH
O
O Sn(Oct)2
FIGURE 16.6Generalized coordination-insertion chain growth mechanism of lactide to PLA: R, growingpolymer chain.
1741_C16.qxd 2/11/2005 9:57 AM Page 536

polymerization, but hydroxyl or other nucleophilic species are the actual ini-tiators. There is generally several hundred parts per million of hydroxyl impu-rities in the lactide from water, lactic acid, and linear dimers and trimers.
High molecular weight polymer, good reaction rate, and low levels ofracemization are obtained with tin octoate-catalyzed polymerization of lac-tide. Typical conditions for polymerization are 180°–210°C, tin octoate con-centrations of 100–1000 ppm, and 2–5 h to reach 95% conversion. Thepolymerization is first order in both catalyst and lactide. Frequentlyhydroxyl-containing initiators such as 1-octanol are used to both controlmolecular weight and accelerate the reaction.
Copolymers of lactide with other cyclic monomers such as caprolactone17
can be prepared using similar reaction conditions (Figure 16.7). Thesemonomers can be used to prepare random copolymers or block polymersbecause of the end growth polymerization mechanism. Cyclic carbonates,epoxides, and morpoline diones have also been copolymerized with lactide.
In addition to the work done on catalysts and comonomers, a significantamount of work has been done designing the optimum polymerizationprocess from a cost and versatility perspective. Most of this work has usedlactide dimer as the starting point. Batch polymerization as well as continu-ous processes can be used. Continuous processes (Figure 16.3) have a costand productivity advantage and thus are the focus of most work. Stirredtank or pipe reactors have been evaluated alone as well as in combination.Because of the low energy of the ring-opening polymerization and thepotential to obtain high rates of polymerization, melt extruders have beenextensively evaluated as reactors to produce PLA. A number of groups havereported or patented specific aspects of the use of extruders or combinationsof several reactor concepts to produce PLA.18–31
DuPont19 recognized the importance of high rates of lactide polymeriza-tion and the economic viability of using extruders. They investigated theissues surrounding hydroxyl initiators, catalysts, and acidic impurities.DuPont’s patent19 (US 5,310,599) provides examples showing the dramatic(negative) effect of acidic impurities, such as the linear dimer (DP2) on therate of lactide polymerization. The acid impurities coordinate with the tincatalyst and render it ineffective as a ring-opening polymerization site. As anexample, 389,000 MW PLA can be made with a 5-min residence time (6000:1
Polylactic Acid Technology 537
O
O
O
O
+Sn(Oct)2
OO
O
O
O
O
n m
Lactide ε-Caprolactone
FIGURE 16.7Copolymerization of lactide and caprolactone.
1741_C16.qxd 2/11/2005 9:57 AM Page 537

monomer: Sn octoate) at 180oC in the absence of acidic impurities, and 98%conversion is obtained. The conversion in 5 min drops to 50% at an impu-rity:catalyst ratio of 2:1 and to 12% in 5 min at a 6:1 ratio.
The rate of polymerization of lactide can be accelerated by the use ofhydroxyl initiators such as butanol or by higher catalyst loading. However,increased initiator levels will reduce the molecular weight while more cata-lyst will reduce the thermal stability and increase the color of the PLA.
16.5 PLA Physical Properties
Several excellent reviews have been written which include details on theproperties and characteristics of PLA.32–36 A discussion of the variety ofstructures and their thermal and mechanical behavior follows.
The physical characteristics of high molecular weight PLA are to a greatextent dependent on its transition temperatures for common qualities suchas density, heat capacity, and mechanical and rheological properties. In thesolid state, PLA can be either amorphous or semicrystalline, depending onthe stereochemistry and thermal history. For amorphous PLAs, the glasstransition (Tg) determines the upper use temperature for most commercialapplications. For semicrystalline PLAs, both the Tg (~58°C) and meltingpoint (Tm), 130°–230°C (depending on structure) are important for determin-ing the use temperatures across various applications. Both of these transitions, Tg and Tm, are strongly affected by overall optical composition,primary structure, thermal history, and molecular weight.
Above Tg amorphous PLAs transition from glassy to rubbery and willbehave as a viscous fluid upon further heating. Below Tg, PLA behaves as aglass with the ability to creep until cooled to its β transition temperature ofapproximately �45°C. Below this temperature PLA will only behave as abrittle polymer.
16.5.1 Linear Optical Copolymer Structures and Blends
Having two optical arrangements for lactic acid (L-lactic acid, D-lactic acid),and three optical arrangements for lactide (L-lactide, D-lactide, meso-lactide),the variety of primary structures available for PLA is substantial. In addi-tion, many other monomers have been copolymerized with lactic acidand/or lactide, the subject of which will not be covered in this chapter.Techniques for measuring PLA stereosequences by NMR have been pub-lished by Thakur et al.37
Containing the discussion to strictly PLA linear optical copolymers, there areseveral notable primary structures which lead to a variety of highly orderedcrystalline structures. The simplest is that of the isotactic homopolymer
538 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:57 AM Page 538

Polylactic Acid Technology 539
poly(L-lactide) (PLLA) or its opposite enantiomer poly(D-lactide) (PDLA)where all of the monomers in the chain are of the same optical composition. Agreater amount of research has been done on PLLA due to the fact that the com-mercial production of lactic acid has been predominantly L-lactic acid. The crys-tal structure of PLLA homopolymer exists in α and β forms. A more detaileddescription of the crystal structures and unit cell parameters follows.
The most common commercial polymers of PLA are optical copolymers ofpredominantly L-lactide, with small amounts of D- and meso-lactides, madethrough bulk polymerization with tin octoate catalyst via ring-opening polymerization (ROP). While these copolymers are generally described asrandom, there is evidence of some degree of stereoselectivity. Selectivity oftin octoate is discussed by Thakur et al.38 Condensation polymerization ofmostly L-lactic acid with small amounts of D-lactic acid, polymerized in solu-tion, has also been used. The optical comonomers introduce “kinks” inPLA’s natural helical conformation and “defects” in the crystal arrangement,which results in depression of the melting point, reduction in the level ofattainable crystallinity, and reduction in the rate of crystallization. Opticalpurity (OP) is a common nomenclature to describe polymers of this variety.As OP decreases, crystallization eventually becomes impossible and thepolymer is amorphous (occurring about when OP � 0.78). For the purposesof this chapter, the designation a-PLA will be used for PLAs made withinsufficient optical purity to crystallize, and c-PLA will be used to designatePLAs that are crystallizable. Stereoselective catalysis has been used todevelop optical copolymers having a highly tapered concentration of opticalpurity (OP) along the chain.39
Random copolymers made from meso-lactide result in an atactic primarystructure referred to as poly(meso-lactide) and are amorphous. Random opti-cal copolymers made from equimolar amounts of D-lactide and L-lactide arecommonly referred to as PDLLA or poly(rac-lactide). PDLLA is also essen-tially atactic, but the primary structure is segregated into optical doublets ofthe lactyl group, and it is also amorphous.
Ovitt and Coates40 have made two novel stereoregular primary structuresusing chiral catalysts. The first was syndiotactic PLA, made by ROP of meso-lactide with a chiral aluminum alkoxide catalyst, which selectively openedthe L-lactyl side of the meso-lactide molecule and resulted in alternating stere-ocenters along the growing chain. This can be visualized as an optical head-to-tail addition and results in an –LDLDLD- arrangement of lactylstereocenters along the chain. In this case, optical purity (OP) will refer to thefraction of stereoregular syndiotactic sequences. The material was shown tocrystallize with a melting point of about 152°C with an OP of 0.96. The sec-ond structure, referred to as di-syndiotactic PLA or heterotactic PLA, wasmade by ROP of meso-lactide with a racemic catalyst that would add meso-lactide. However, the reacted lactide stereocenter would add in an alternatingfashion, in what can be visualized as an optical head-to-head addition result-ing in sequences of –LLDDLLDD- along the chain. The heterotactic PLAs pro-duced did not crystallize, and had a Tg of about 40°C. Ovitt also showed that
1741_C16.qxd 2/11/2005 9:57 AM Page 539

540 Natural Fibers, Biopolymers, and Biocomposites
racemic catalysts can be used to polymerize rac-lactide into stereoblockcopolymers that have long sequences of poly(L-lactide) and poly(D-lactide)blocks, referred to as isotactic stereoblock PLA.41 Similar work had been per-formed earlier by Yui et al.42 With sufficiently long isotactic run lengths, thesepolymers are able to crystallize into the stereocomplex crystal arrangement.
Blends of PLA optical copolymers have also offered a variety of new prop-erties. The most notable is that of the stereocomplex PLA, which is made bysolvent or melt blending PLLA and PDLA where the polymers undergo stere-ocomplexation or racemic crystallization. This phenomenon has been an areaof intense study since first described by Ikada et al.43 A more detailed descrip-tion of stereocomplex will follow. A second blend receiving attention is theblend of crystallizable PLA (c-PLA) with noncrystallizable PLA (a-PLA).
Key melting point and glass transition data for PLA structures and blendsare listed in Table 16.3.
16.5.1.1 DensityVolumetric measurements show that PLA and lactide isomers in the liquidstate have approximately the same density as a function of temperature,with a thermal coefficient of expansion α1 � 7.4 � 10�4°C�1. The melt densitycan be approximated using Equation (16.1) by Witzke:32
ρ(g/cm3) � (16.1)
where ρ150 � 1.1452.The solid amorphous PLA density is ~1.25 g/cm3. The purely crystalline
PLLA density32 is estimated to be 1.37–1.49 g/cm3.
16.5.1.2 Glass Transition and the Amorphous PhaseThe PLA glass transition (Tg) increases with Mn and OP. Orientation andphysical aging also affect the Tg. The infinite molecular weight Tg for PLLA,poly(meso-lactide) and PDLLA were estimated to be 61°, 46°, and 53°C,respectively, by Witzke,32 who developed an equation describing the Tg ofunoriented poly(L-lactide-co-meso-lactide), Equation (16.2)
Tg � 45 � � 16wL-mer�7wmeso (16.2)
where wL-mer and wmeso are the starting mole fractions of L-lactide and meso-lactide, respectively.
Physical aging affects the glass transition of PLA. It can occur betweenabout �45°C and Tg.32 The free volume reduction, characteristic of physicalaging, is observed by DSC as an endothermic volume expansion immediatelyafter Tg and is often referred to as the enthalpy of relaxation. Crystallizationand orientation reduce the rate and extent of PLA aging. The rate of agingincreases with temperature up to the Tg.
180,000�
Mn
ρ150°C��1�α1(T(°C)�150)
1741_C16.qxd 2/11/2005 9:57 AM Page 540

Polylactic Acid Technology 541
TAB
LE 1
6.3
Mel
ting
Poi
nt a
nd G
lass
Tra
nsit
ion
Dat
a fo
r PL
ASt
ruct
ures
and
Ble
nds
PL
AS
tru
ctu
reD
escr
ipti
onT
m(°
C)
Tm°
(°C
)T
g(°C
)
Isot
acti
c po
ly( L
-lac
tid
e) o
r L
LL
LL
LL
LL
or D
DD
DD
DD
DD
D17
0–19
020
5 Ts
uji a
nd I
kad
a (1
995)
poly
(D-l
acti
de)
211
Run
t (20
01)
215
Kal
b an
d P
enni
ngs
55–6
5R
and
om o
ptic
al
Ran
dom
leve
l of
mes
o or
(198
0)co
poly
mer
sD
-lac
tid
e in
L-l
acti
de
165–
210
or D
-lac
tic
acid
in L
-lac
tic
acid
130–
170
Run
t (20
01)
45–6
5
PLL
A/
PDL
AL
LL
LL
LL
Lm
ixed
wit
h 22
0–23
0 27
9 65
–72
ster
eoco
mpl
exD
DD
DD
DD
DIk
ada
et a
l. (1
987)
Tsuj
i and
Ika
da
(199
6)Ts
uji a
nd I
kad
a (1
999)
PLL
A/
PDL
Ast
ereo
bloc
k L
LL
LL
LL
LL
LL
DD
DD
DD
DD
205
Yui e
t al.
(199
0)co
mpl
exes
179
Ovi
tt a
nd C
oate
s (2
000)
Synd
iota
ctic
pol
y(m
eso-
) D
LD
LD
LD
LD
LD
L15
2 40
PL
AA
l-ce
nter
ed R
- ch
iral
cat
alys
tO
vitt
and
Coa
tes
(199
9)O
vitt
and
Coa
tes
(199
9)
Het
erot
acti
c L
LD
DL
LD
DL
LD
DL
LD
D40
(d
isyn
dio
tact
ic)
poly
( mes
o-A
l-ce
nter
ed r
ac-
chir
al c
atal
yst
Ovi
tt a
nd C
oate
s (1
999)
lact
ide)
Ata
ctic
pol
y(m
eso-
lact
ide)
No
ster
eoco
ntro
l46 W
itzk
e (1
997)
Ata
ctic
PD
LL
AN
o st
ereo
cont
rol
53 Wit
zke
(199
7)
1741_C16.qxd 2/11/2005 9:57 AM Page 541

542 Natural Fibers, Biopolymers, and Biocomposites
A significant reduction in the Tg is found for PLLA crystallized under par-tially constrained conditions, where the polymer is prevented from shrink-ing.44 It was proposed that the constrained crystallization process increasesthe net free volume in the amorphous phase. Tg depression increased withincreasing crystallinity. In contrast, when samples are allowed to shrink orcrystallize quiescently, Tg increases with crystallinity. It was shown that theTg could vary by as much as 30°C between the two methods, while the melt-ing point (Tm), heat of fusion (∆Hm), and overall degree of crystallinity (xc)were not influenced. It is possible that the reduction in Tg under partiallystrained conditions may be related to suppressing physical aging.
Blends of PLAs of different optical compositions have been studied for mis-cibility and compatibility. The most notable blend is that of the PLA stereo-complex, which occurs when PLLA is mixed with PDLA and thehomopolymers cocrystallize into a new arrangement with a markedly highermelting point. The interesting features of the stereocomplex crystal will be dis-cussed later. Tsuji and Ikada45 reported that the Tg of stereocomplex PLA is 5°Chigher than that of the nonblended components. This was attributed to thestrong interaction between the L-lactyl and D-lactyl unit sequences in the amor-phous region of the blends, resulting in a more dense chain packing. A secondvery interesting blend is to blend a crystallizable polymer (c-PLA) of high opti-cal purity (OP) with a polymer of low OP (a-PLA). Urayama et al.46 investi-gated blends of PLLA with PDLLA by dynamic mechanical analysis (DMA). Tg behavior supported compatibility of both polymers. The modulus remainedconstant regardless of blend ratio. Modulus changes were consistent withhomopolymers, where the E� of the blends dropped from 2–3 � 109 Pa to a rub-bery plateau of 1–3 � 106 Pa across the Tg for amorphous specimens. The rub-bery plateau increased to a different level depending on the crystallinity of theblend. Blends of this type provide a route to modify flexibility of the polymerabove the glass transition by controlling the overall level of crystallinity (xc).
16.5.2 Rheology
Rheological properties are useful in evaluating thermoplastics for their per-formance during processing operations. The literature reports a wide variety ofprocesses and products using PLA, spanning from high orientation processessuch as fiber spinning and oriented films to lower orientation processes such asthermoforming and foams. The time scales of these processes are very differ-ent. Considerations for material flow characteristics, orientation, and crystal-lization must be made to gauge and predict the resulting properties of theproducts made from these processes. Intrinsic factors affecting the flow charac-teristics of PLA are molecular weight distribution, degree and type of branch-ing, optical composition, optical block length distributions, and melt stability.
16.5.2.1 Melt Rheology of Linear PLAPLA, like many thermoplastics, is a pseudoplastic, non-Newtonian fluid.Above the melting point, PLA behaves as a classic flexible-chain polymer
1741_C16.qxd 2/11/2005 9:57 AM Page 542
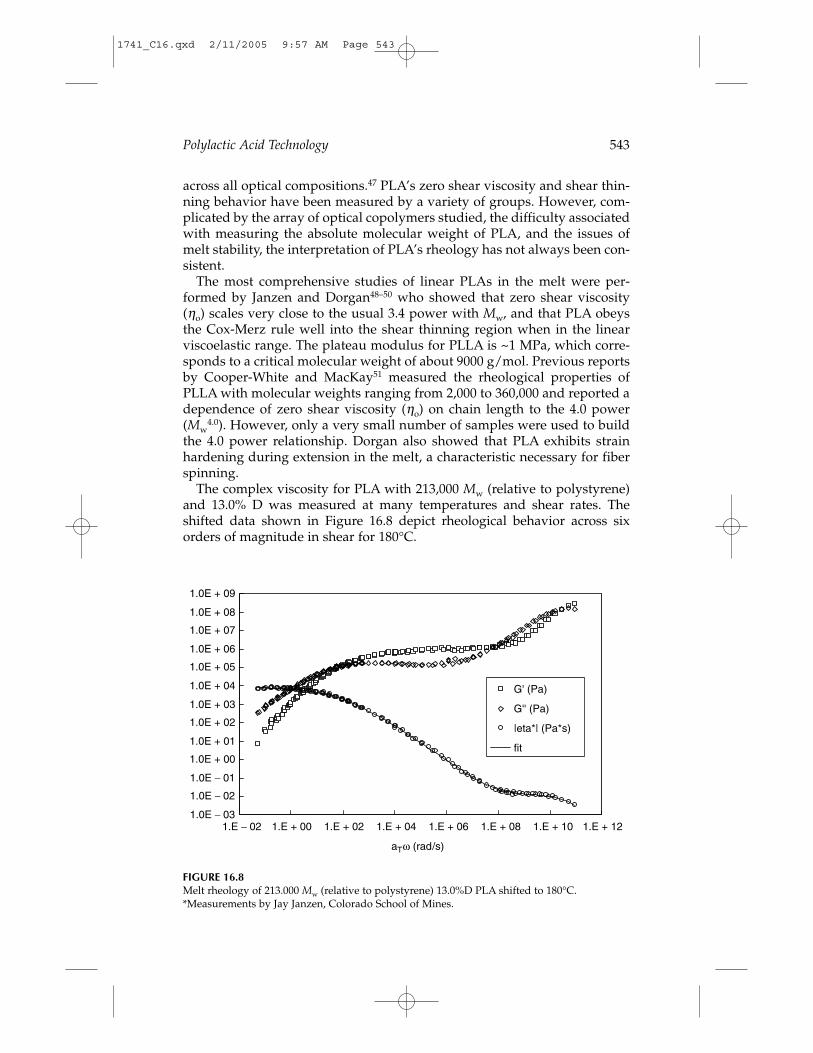
Polylactic Acid Technology 543
across all optical compositions.47 PLA’s zero shear viscosity and shear thin-ning behavior have been measured by a variety of groups. However, com-plicated by the array of optical copolymers studied, the difficulty associatedwith measuring the absolute molecular weight of PLA, and the issues ofmelt stability, the interpretation of PLA’s rheology has not always been con-sistent.
The most comprehensive studies of linear PLAs in the melt were per-formed by Janzen and Dorgan48–50 who showed that zero shear viscosity(ηo) scales very close to the usual 3.4 power with Mw, and that PLA obeysthe Cox-Merz rule well into the shear thinning region when in the linearviscoelastic range. The plateau modulus for PLLA is ~1 MPa, which corre-sponds to a critical molecular weight of about 9000 g/mol. Previous reportsby Cooper-White and MacKay51 measured the rheological properties ofPLLA with molecular weights ranging from 2,000 to 360,000 and reported adependence of zero shear viscosity (ηo) on chain length to the 4.0 power(Mw
4.0). However, only a very small number of samples were used to buildthe 4.0 power relationship. Dorgan also showed that PLA exhibits strainhardening during extension in the melt, a characteristic necessary for fiber spinning.
The complex viscosity for PLA with 213,000 Mw (relative to polystyrene)and 13.0% D was measured at many temperatures and shear rates. Theshifted data shown in Figure 16.8 depict rheological behavior across sixorders of magnitude in shear for 180°C.
1.0E − 03
1.0E − 02
1.0E − 01
1.0E + 00
1.0E + 01
1.0E + 02
1.0E + 03
1.0E + 04
1.0E + 05
1.0E + 06
1.0E + 07
1.0E + 08
1.0E + 09
1.E − 02 1.E + 00 1.E + 02 1.E + 04 1.E + 06 1.E + 08 1.E + 10 1.E + 12
aTω (rad/s)
G' (Pa)
G'' (Pa)
|eta*| (Pa*s)
fit
FIGURE 16.8Melt rheology of 213.000 Mw (relative to polystyrene) 13.0%D PLA shifted to 180°C.*Measurements by Jay Janzen, Colorado School of Mines.
1741_C16.qxd 2/11/2005 9:57 AM Page 543

544 Natural Fibers, Biopolymers, and Biocomposites
16.5.2.2 Melt StabilityIssues with PLA melt stability during processing and rheological testinghave been cited by Witzke,32 Dorgan et al.,48 Ramkumar and Bhattacharya,52
and Feng and Hanna.53 Reactions leading to poor melt stability include chainscission due to hydrolysis, chain scission due to thermal instability, and lac-tide formation by the well-documented “back-biting” mechanism.Hydrolysis can be reduced or essentially eliminated with conventional dry-ing operations where water is removed prior to melting. Recommended dry-ing conditions are available for commercial PLAs. When PLA is exposed toa humid environment following drying, moisture is quickly absorbed intothe polymer up to its equilibrium level of swelling. Kinetics for melt hydrol-ysis depend on water concentration, acid or base catalysis, and polymeriza-tion catalyst concentration.
The proton in the CH group of the main chain of PLA is labile. It has beensuggested that the proximity of this labile proton to the ester group affects thethermal sensitivity of the polymer.52 The proton may be abstracted resultingin the breakdown of the molecular chain. In practical extrusion processes,thermal stability becomes important at temperatures greater than 250°C.
Using PLA polymerized with tin octoate catalyst, Cicero et al.54 showed thatthe melt stability of PLA can be improved by the addition of small amounts oftris(nonylphenyl)phosphite (TNPP), likely due to balancing chain extensionwith degradation reactions. Adding TNPP during rheological testing greatlystabilized the polymer and lengthened the time available for testing.
The thermodynamic equilibrium between polylactide and lactidemonomer varies with temperature and is about 4.3% lactide at 210°C.32 Sincelactide imparts undesirable characteristics during processing, such as fum-ing, it is desirable to depress the lactide formation kinetics and stabilize thepolymer. Gruber et al.55 showed that lactide formation could be substantiallyreduced under normal processing by complexing the catalyst with additives,resulting in a melt-stable PLA polymer.
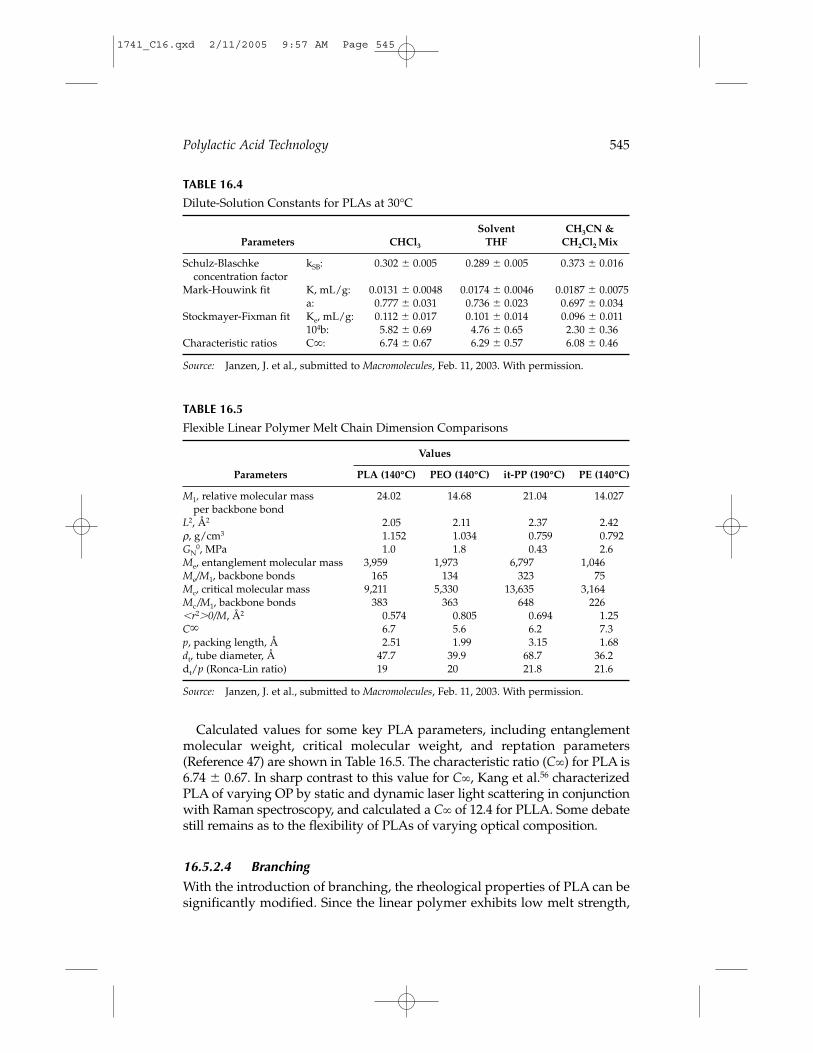
16.5.2.3 Solution PropertiesThe dilute-solution properties of PLA are critical for characterizing molecu-lar weight, chain flexibility, and defining solvent quality. Factors reported toaffect PLA’s intrinsic viscosity ([η]) are molecular weight and distribution,optical composition, and branching. A recent publication submission byJanzen et al.47 systematically measured the dilute-solution properties ofPLAs of wide OP in three solvent systems. Mark-Houwink parameters weredetermined by comparing [η] to a broad range of molecular weights deter-mined by light scattering in a mixed solvent system first used on PLA byKang et al.56 Tables 16.4 and 16.5 are taken from Janzen’s publication sub-mission. Surprisingly, no effect of optical composition was found to affect[η], within the error of the experiments. Earlier work by Schindler andHarper57 had shown a significant difference between the Mark-Houwinkparameters of PLLA and PDLLA.
1741_C16.qxd 2/11/2005 9:57 AM Page 544
Polylactic Acid Technology 545
Calculated values for some key PLA parameters, including entanglementmolecular weight, critical molecular weight, and reptation parameters(Reference 47) are shown in Table 16.5. The characteristic ratio (C) for PLA is6.74 0.67. In sharp contrast to this value for C, Kang et al.56 characterizedPLA of varying OP by static and dynamic laser light scattering in conjunctionwith Raman spectroscopy, and calculated a C of 12.4 for PLLA. Some debatestill remains as to the flexibility of PLAs of varying optical composition.
16.5.2.4 BranchingWith the introduction of branching, the rheological properties of PLA can besignificantly modified. Since the linear polymer exhibits low melt strength,
TABLE 16.4
Dilute-Solution Constants for PLAs at 30°C
Solvent CH3CN & Parameters CHCl3 THF CH2Cl2 Mix
Schulz-Blaschke kSB: 0.302 0.005 0.289 0.005 0.373 0.016concentration factor
Mark-Houwink fit K, mL/g: 0.0131 0.0048 0.0174 0.0046 0.0187 0.0075a: 0.777 0.031 0.736 0.023 0.697 0.034
Stockmayer-Fixman fit Ke, mL/g: 0.112 0.017 0.101 0.014 0.096 0.011 104b: 5.82 0.69 4.76 0.65 2.30 0.36
Characteristic ratios C: 6.74 0.67 6.29 0.57 6.08 0.46
Source: Janzen, J. et al., submitted to Macromolecules, Feb. 11, 2003. With permission.
TABLE 16.5
Flexible Linear Polymer Melt Chain Dimension Comparisons
Values
Parameters PLA (140°C) PEO (140°C) it-PP (190°C) PE (140°C)
M1, relative molecular mass 24.02 14.68 21.04 14.027per backbone bond
L2, Å2 2.05 2.11 2.37 2.42ρ, g/cm3 1.152 1.034 0.759 0.792GN
0, MPa 1.0 1.8 0.43 2.6Me, entanglement molecular mass 3,959 1,973 6,797 1,046Me/M1, backbone bonds 165 134 323 75Mc, critical molecular mass 9,211 5,330 13,635 3,164Mc /M1, backbone bonds 383 363 648 226�r2�0/M, Å2 0.574 0.805 0.694 1.25C 6.7 5.6 6.2 7.3p, packing length, Å 2.51 1.99 3.15 1.68dt, tube diameter, Å 47.7 39.9 68.7 36.2dt/p (Ronca-Lin ratio) 19 20 21.8 21.6
Source: Janzen, J. et al., submitted to Macromolecules, Feb. 11, 2003. With permission.
1741_C16.qxd 2/11/2005 9:57 AM Page 545
546 Natural Fibers, Biopolymers, and Biocomposites
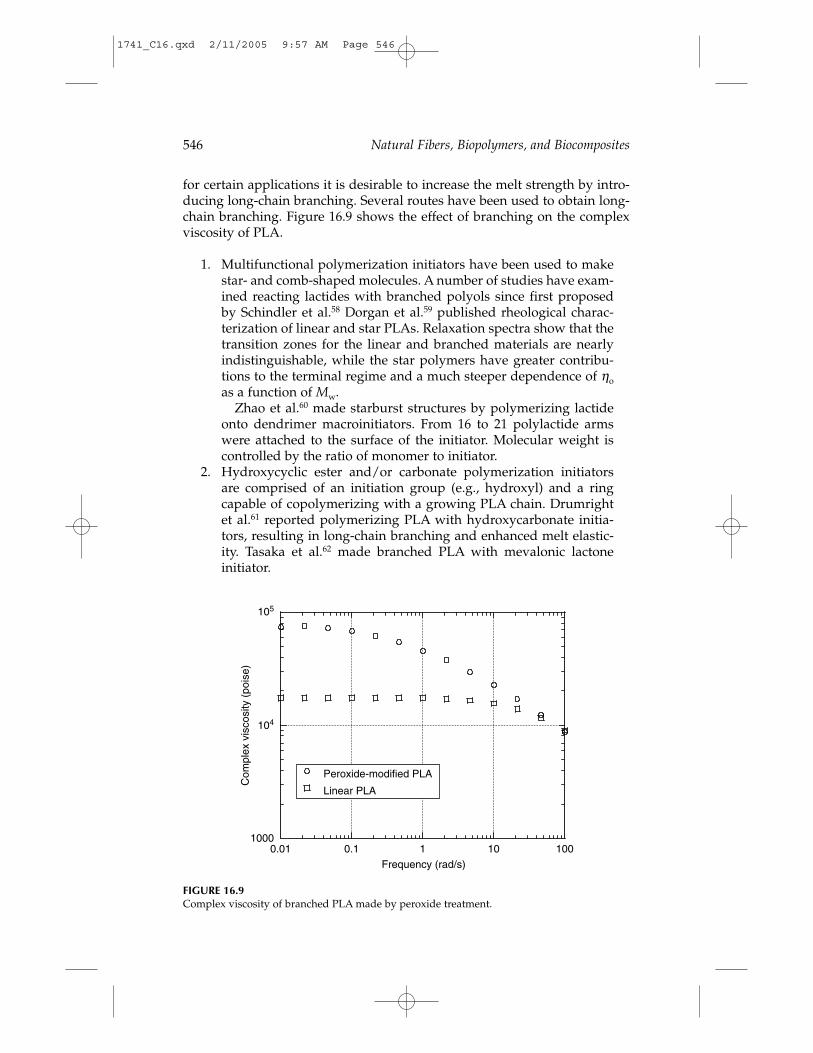
for certain applications it is desirable to increase the melt strength by intro-ducing long-chain branching. Several routes have been used to obtain long-chain branching. Figure 16.9 shows the effect of branching on the complexviscosity of PLA.
1. Multifunctional polymerization initiators have been used to makestar- and comb-shaped molecules. A number of studies have exam-ined reacting lactides with branched polyols since first proposedby Schindler et al.58 Dorgan et al.59 published rheological charac-terization of linear and star PLAs. Relaxation spectra show that thetransition zones for the linear and branched materials are nearlyindistinguishable, while the star polymers have greater contribu-tions to the terminal regime and a much steeper dependence of ηoas a function of Mw.
Zhao et al.60 made starburst structures by polymerizing lactideonto dendrimer macroinitiators. From 16 to 21 polylactide armswere attached to the surface of the initiator. Molecular weight iscontrolled by the ratio of monomer to initiator.
2. Hydroxycyclic ester and/or carbonate polymerization initiatorsare comprised of an initiation group (e.g., hydroxyl) and a ringcapable of copolymerizing with a growing PLA chain. Drumrightet al.61 reported polymerizing PLA with hydroxycarbonate initia-tors, resulting in long-chain branching and enhanced melt elastic-ity. Tasaka et al.62 made branched PLA with mevalonic lactoneinitiator.
1000
104
Com
plex
vis
cosi
ty (
pois
e)
105
0.01 0.1 1 10 100
Peroxide-modified PLA
Linear PLA
Frequency (rad/s)
FIGURE 16.9Complex viscosity of branched PLA made by peroxide treatment.
1741_C16.qxd 2/11/2005 9:57 AM Page 546

Polylactic Acid Technology 547
3. Multicyclic ester, multicyclic carbonate and/or multicyclic epoxycomonomers are comprised of multiple rings (most commonlytwo) capable of copolymerizing with a growing PLA chain.Drumright et al.63 also reported on polymerizing PLA with bicyclicdiesters and/or dicarbonates to control rheological properties. The bicyclic diesters and dicarbonates copolymerize easily withlactide to form copolymers that can have tailored levels of branch-ing. The copolymers showed excellent rheological properties,including increased melt tensions and improved shear thinning,compared to the analogous linear polymers. A typical polymer wasmanufactured by polymerization of L-lactide at 130°–185°C with0.1% α,α-2,5-dioxabicyclo[2.2.2]octane-3,6-dione. Since multicycliclactones do not contain an initiator, the chemistry can theoreticallybe used to cross-link PLA to infinite molecular weight. A patent byGruber et al.64 describes rheological modification of PLA by copoly-merizing with multifunctional expoxidized oil.
4. Cross-linking via free radical has been accomplished in the melt bytreating PLA with peroxides.65 Small amounts of peroxide result inlong-chain branches. The polymer remains melt processable and isstable. Large improvements in melt elasticity have been measuredusing die swell, melt tension, and parallel plate rheometry. Blendsof linear and branched PLAs showed good compatibility and pre-dictable melt rheology by the mixing rule.66
16.5.2.5 Solid-State Viscoelastic PropertiesSolid-state rheological properties of amorphous PLA typically show a stor-age modulus (E’) of about 2–3 � 109 Pa below the glass transition tempera-ture. As the sample is heated, a sharp drop in modulus occurs between 55°and 80°C, and E’ decreases to ~1–3 � 106 Pa as the glass transition (Tg) isexceeded.67 Semicrystalline samples show less of a drop in E’ across the Tg,and also a higher Tg with increasing crystallinity. Peak height of the tan δcurve will decrease, but the width of the tan δ will increase as the percentageof crystallinity increases. Highly crystalline samples will typically show atan δ peak at 70°–80°C. Optical composition and crystallization conditionswill strongly influence the onset of melting, and the corresponding drop inE’. The β transition of PLA is very small and is scarcely detectable by DMA.
16.5.3 Crystallinity
16.5.3.1 MorphologyThe morphology of PLA crystals is influenced by composition and thermalhistory. Abe et al.68 showed that PLLA crystals grown at different tempera-tures go through three regime changes associated with nucleation and crys-tal growth rates. A morphology change was observed between regimes I andII. For PLLA crystallized isothermally from the melt, spherulitic morphology
1741_C16.qxd 2/11/2005 9:57 AM Page 547

548 Natural Fibers, Biopolymers, and Biocomposites
is observed at crystallization temperatures below 145°C (regime II). Duringisothermal growth, the radius of the spherulites increases linearly withtime, and the slope of the curve is the lineal crystal growth rate (LCR). Athigher temperatures, a smaller number of spherulites are formed due to adecrease in the nucleation density. This leads to larger spherulites withincreasing crystallization temperature. Crystallizing PLLA at temperaturesgreater than 150°C (regime I) results in hexagonal lamellar stacking crystalmorphology.
Spherulites have been shown to contain stacked lamellar morphology,with the long axis of the lamellar crystal running parallel to the spheruliteradius. Within the crystal, PLA exists as a polymeric helix, with anorthorhombic unit cell with dimensions a � 1.070 nm and b � 0.614 nm.Amorphous polymer resides between the lamellae. The lamellar thickness ofPLA varies with the temperature of crystallization (Tc), crystallization time(tc), optical purity (OP), and molecular weight. PLLA grown at 120°, 140°,and 160°C had lamellar periodicities measured at 14–16, 16–18, and18–22 nm, respectively, measured by AFM.68
For L-rich polymers with small amounts of D- and meso-lactides, there is anobserved decrease in the equilibrium melting point with decreasing OP. Atleast two possible explanations for this have been considered. In one extremecase it is assumed that the optically impure units are incorporated into thecrystal structure as defects. This results in a decreasing melting point (Tm)due to the decrease in the enthalpy of fusion. In the other extreme case, it isassumed that the optically impure units are rejected from the crystal struc-ture, causing a reduction in Tm due to entropy effects. Baratian et al.69 stud-ied the degree of crystallinity, spherulitic growth rate, lamellar thickness,and melting temperature of a series of optical copolymers of this variety,comparing the effects of D-lactide and meso-lactide optical impurities. Small-angle x-ray scattering (SAXS) results showed that lamellar thicknessdecreases with decreasing OP at a given degree of supercooling. It was pro-posed that a critical sequence length that is free of stereo defects is necessaryfor crystallization and that the stereo defects are excluded from the crys-talline regions. In a different study by Zell et al.,70 it was shown that, at leastunder long crystallization times, a certain fraction of optical impurity (in thiscase, a small percentage of labeled L-lactide copolymerized into a polymer ofpredominantly D-lactide) is incorporated into the crystal lamellae. Zell et al.70 went on to develop a model for determining the fraction of crystal-lizable PLA based upon a critical chain length necessary for crystallizationthat allows stereo defects to be included in the crystalline region if they aresurrounded by a sufficient isotactic run length.
The spherulite structure for L-lactide-rich PLA was studied by Pyda et al.71
Figure 16.10 shows the crystal imaging using AFM and polarized lightmicroscopy.
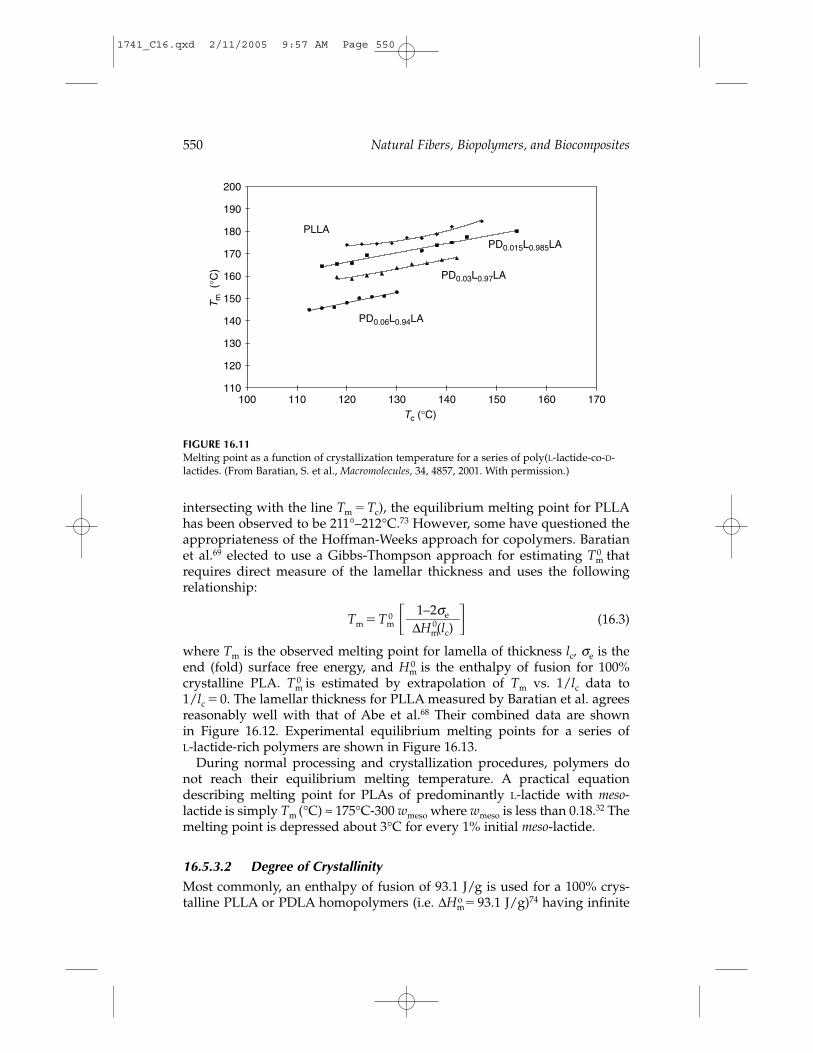
A graph showing the observed melting point by DSC after isothermaltreatments at crystallization temperature Tc is shown in Figure 16.11 for aseries of poly(L-lactide-co-D-lactide)s that are rich in L-lactide content.69
Polymers were made by ROP using tin octoate catalyst.
1741_C16.qxd 2/11/2005 9:57 AM Page 548

Polylactic Acid Technology 549
Using the Hoffman-Weeks72 method for determining equilibrium melt-ing temperature (Tm
0 ) of a polymer [linear extrapolation of experimen-tally observed melting points (Tm) vs. crystallization temperature (Tc)
FIGURE 16.10Morphology of PLA by optical microscopy and AFM. (a) Molecular formula of PLA. (b) Pictureof the central region of a spherulite by optical microscopy. (c) Copy of ab AFM picture of asection of the spherulite shown in (b). (d) Schematic drawing of the lamellae within aspherulite. (From Pyda et al., Proceedings of the 30th NATAS Annual Conference on ThermalAnalysis and Applications, 463, 2002. With permission.)
1741_C16.qxd 2/11/2005 9:57 AM Page 549
550 Natural Fibers, Biopolymers, and Biocomposites
intersecting with the line Tm � Tc), the equilibrium melting point for PLLAhas been observed to be 211°–212°C.73 However, some have questioned theappropriateness of the Hoffman-Weeks approach for copolymers. Baratianet al.69 elected to use a Gibbs-Thompson approach for estimating Tm
0 thatrequires direct measure of the lamellar thickness and uses the followingrelationship:
Tm � Tm0 � � (16.3)
where Tm is the observed melting point for lamella of thickness lc, σe is theend (fold) surface free energy, and Hm
0 is the enthalpy of fusion for 100% crystalline PLA. Tm
0 is estimated by extrapolation of Tm vs. 1/lc data to1/lc � 0. The lamellar thickness for PLLA measured by Baratian et al. agreesreasonably well with that of Abe et al.68 Their combined data are shown in Figure 16.12. Experimental equilibrium melting points for a series of L-lactide-rich polymers are shown in Figure 16.13.
During normal processing and crystallization procedures, polymers donot reach their equilibrium melting temperature. A practical equationdescribing melting point for PLAs of predominantly L-lactide with meso-lactide is simply Tm (°C) ≈ 175°C-300 wmeso where wmeso is less than 0.18.32 Themelting point is depressed about 3°C for every 1% initial meso-lactide.
16.5.3.2 Degree of CrystallinityMost commonly, an enthalpy of fusion of 93.1 J/g is used for a 100% crys-talline PLLA or PDLA homopolymers (i.e. ∆Hm
o � 93.1 J/g)74 having infinite
1–2σe��∆Hm
0(lc)
110
120
130
140
150
160
170
180
190
200
100 110 120 130 140 150 160 170Tc (°C)
Tm
(°C
)
PLLAPD0.015L0.985LA
PD0.03L0.97LA
PD0.06L0.94LA
FIGURE 16.11Melting point as a function of crystallization temperature for a series of poly(L-lactide-co-D-lactides. (From Baratian, S. et al., Macromolecules, 34, 4857, 2001. With permission.)
1741_C16.qxd 2/11/2005 9:57 AM Page 550
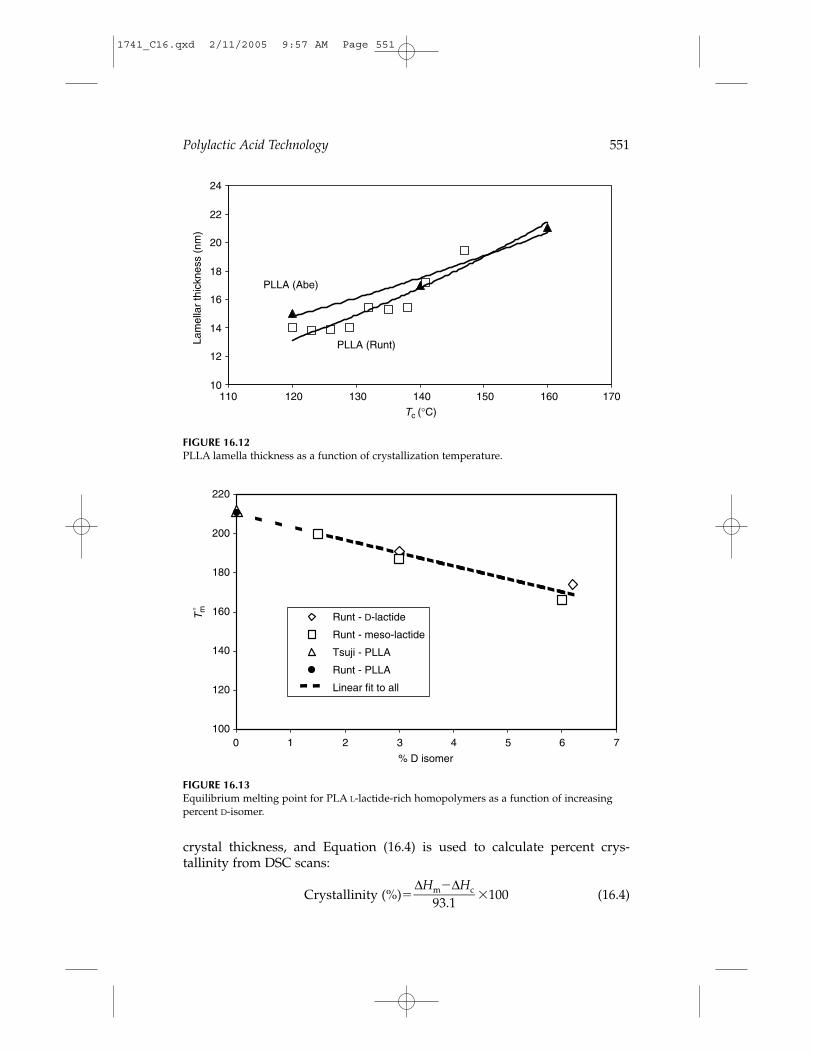
Polylactic Acid Technology 551
crystal thickness, and Equation (16.4) is used to calculate percent crys-tallinity from DSC scans:
Crystallinity (%)� �100 (16.4)∆Hm�∆Hc��93.1
10
12
14
16
18
20
22
24
110 120 130 140 150 160 170
Tc (°C)
Lam
ella
r th
ickn
ess
(nm
)
PLLA (Runt)
PLLA (Abe)
FIGURE 16.12PLLA lamella thickness as a function of crystallization temperature.
100
120
140
160
180
200
220
0 2 4 61 3 5 7
% D isomer
Tm
Runt - D-lactide
Runt - meso-lactide
Tsuji - PLLA
Runt - PLLA
Linear fit to all
°
FIGURE 16.13Equilibrium melting point for PLA L-lactide-rich homopolymers as a function of increasingpercent D-isomer.
1741_C16.qxd 2/11/2005 9:57 AM Page 551

552 Natural Fibers, Biopolymers, and Biocomposites
where ∆Hm is the measured heat of fusion and ∆Hc is the heat of crystalliza-tion. This value of 93.1 J/g is used throughout the PLA literature. However,since Tm
0 decreases with decreasing OP, it is expected that using a constantvalue for ∆Hm
o across all optical compositions will introduce error. No refer-ence was found that systematically calculated ∆Hm
o for homopolymers ofreduced OP. It is well known that for PLAs of essentially random opticalcopolymers of predominantly L-lactide, with small amounts of D- and meso-lactides, the attainable percentage of crystallinity (xc) decreases with decreas-ing optical purity (OP), and crystallization is essentially nonexistent aboutwhen OP � 0.78.
16.5.3.3 Crystallization KineticsThe crystallization kinetics of PLA are strongly dependent on the opticalcopolymer composition. The degree of crystallinity, nucleation rate, andspherulite growth rate decrease substantially with decreasing optical purity(OP). Under quiescent crystallization conditions, Kolstad showed that thebulk crystallization half time increases by roughly 40% for every 1 wt%increase in the meso-lactide.75 PLA has the highest rate of crystallizationbetween 100° and 130°C.
Two phenomena contribute to the bulk crystallization rate, nucleation andcrystal growth. For instantaneous nucleation and spherulitic growth(Avrami n � 3), the crystallization rate constant is given by
k� πNG3 (16.5)
For the rate equation,
x�1�e�ktn
(16.6)
where x � ∆Hm(t)/∆Hm(∞), N � the density of primary nucleation sites toinitiate spherulitic growth, and G is the lineal growth rate of a spherulite.
The rate of PLA lineal crystal growth (G) of a spherulite is dependent onTc, OP, and molecular weight. Nulceation density (N) depends on Tc, poly-mer characteristics, thermal history, and impurities.
When PLA is isothermally crystallized from the melt or from a quenchedstate, the size of the spherulites changes dramatically. Pluta and Galeski76
showed that spherulites were larger in samples crystallized via cooling fromthe melt than those in samples crystallized via heating from the glassy amor-phous state. On the lamellar level, the structures were similar for samplesprepared by the two methods under similar time and temperature condi-tions. The thermal properties, Tm and Hm, were similar.
Technology to increase the rate of crystallization of PLA has been investi-gated because even the fastest crystallizing PLAs are considered to be slowwhen compared to many conventional thermoplastics. Kolstad75 showedthat talc is an extremely effective nucleating agent for PLA homopolymers.
4�3
1741_C16.qxd 2/11/2005 9:57 AM Page 552

Polylactic Acid Technology 553
Schmidt and Hillmyer77 showed that a small level of stereocomplex crystalis effective for seeding the crystallization of PLA homopolymers.
Liao et al.78 studied the crystallization characteristics of blends of PLLAand PDLLA. Crystallization behavior of PLLA in the blends was similar tothat of the homopolymer when PDLLA � 30%.
16.5.3.4 StereocomplexStereocomplexation has been reported in the literature for a number ofpolymers including polylactide. The PLA racemic crystal structure consistsof alternating L- and D-PLA chains packed side by side with a 1:1 ratio of L:Dmonomer units. The chain conformation in the stereocrystal is a 31 helix vs.the 103 for the unstrained homopolymer crystal. The 31 helix winds a littletighter, going from 108° to 120º rotation per residue, resulting in a significantchange in the molecular shape. The 31 configuration allows a chain-to-chainseparation difference of 0.4 Å smaller.79 This tighter packing results in morefavorable nonbonding interactions that contribute to the higher crystallinemelting point. The equilibrium melting temperature of PLA stereocomplexhas been reported by Tsuji and Ikada to be 279°C,80 a very marked improve-ment over homocrystals, with the minimum tacticity sequencing needed toproduce stereocomplex for the L-rich and D-rich PLAs to be 15 lactyl (or 7.5lactide) units.81 The heat of fusion for pure stereocomplex has been deter-mined to be 142 J/g by Loomis and Murdoch.82
Melting points for stereocomplex are dictated by the optical purity (OP) ofboth the L-lactide-rich and D-lactide-rich polymers (in terms of optically purerun lengths) as well as molecular weight and blend ratio. Maximum regu-larity is attained with a blend of PLLA and PDLA. The greater the deviationin OP of the L-rich and/or D-rich chains, the greater the decrease in stereo-complex melting peak. This follows the equivalent behaviors of PLAhomocrystals, but is shifted to a higher temperature. A practical upperattainable melting temperature that would be achievable for commercialresins approaches 230ºC.
Melting point (Tm) and enthalpy of melting (∆Hm) of the stereocomplex isdependent on the ratio of L-lactide-rich polymer and D-lactide-rich polymer.For a constant optical purity of racemic polymers, the Tm varies only by afew degrees across the possible blend ratios, whereas the stereocomplex ∆Hmvaries directly with the blend ratio with maximum at 1:1. Tsuji et al. pub-lished an excellent series of papers on the crystallization, mechanical behav-ior, and hydrolysis properties of PLA stereocomplexes.83–94
Kang et al.95 have examined the PLA stereocomplex by Raman spectra anddetermined that C�O stretching bands are related to the packing formationof the polymer chain. By combining Raman spectroscopy and DSC tech-niques, they deduced the enthalpy of fusion for stereocomplex to be 126 J/g.During crystallization, packing order led the conformation order duringearly stages. The C�O dipole interaction is believed to be the driving forcefor crystallization.
1741_C16.qxd 2/11/2005 9:57 AM Page 553

554 Natural Fibers, Biopolymers, and Biocomposites
16.5.3.5 Stress-Induced CrystallizationWhile the slow quiescent crystallization kinetics of PLA may limit its speedof processing for some applications, opportunities utilizing stress-inducedcrystallization (SIC) with PLA have been enjoying success commerciallybecause of the naturally wide processing window. It is possible to highly ori-ent PLA in the rubbery or molten state during operations such as filmstretching or fiber spinning. Fabrication processes such as this take fulladvantage of the semicrystalline nature of PLA to develop different proper-ties. Kokturk et al.96 showed that PLA exhibited almost ideal stress-strainbehavior in the temperature range 65°–80°C. As films are stretched, crystal-lization and strain hardening leads to uniform thickness at high deforma-tion. This effect is referred to as self-leveling, and it is a highly desirableproperty for orientation processes. Crystallization of homopolymers, duringorientation processes, results in β crystals in a 31 helix conformation.
Puiggali et al.97 characterized PLA crystals made through SIC, using elec-tron diffraction and conformational energy analysis. Chains were shown tobe arranged as a frustrated packing of 31 helixes in a trigonal unit-cell ofparameters a � b � 1.052 nm, c � 0.88 nm. The frustrated packing is of thetype described as North-South-South (NSS).
The effects of uniaxial drawing conditions on the orientation of variouspoly(L-lactide) (PLLA) samples have been investigated using Fourier trans-form IR dichroism and thermal analysis.98 Orientation in uniaxially stretchedPLLA was influenced by the drawing rate, drawing temperature, and drawratio. It was shown that spherulites could be deformed at high temperatureunder drawing conditions into new crystal morphologies.
Kister et al.99 first examined the Raman and IR spectra of poly(L-lacticacid) in the amorphous and semicrystalline states from 3600 to 100 cm�1.Vibrational assignments were proposed, and Kister et al. indicated thatRaman data supported the 103 helical conformation for PLA. Raman spec-troscopy has been further developed for PLA more recently. Smith et al. andKang et al. have reported on a technique for characterizing orientation inboth the amorphous and crystalline phases for films in multiple axes.100–102
Correlations to level of crystallinity were also developed. Film shrinkagewas correlated to crystallinity level and orientation of the amorphous phase.
Randall et al.103 studied the melting points of PLAs made by SIC fabrica-tion processes of very different time scales using DSC. At a given OP, PLAhomopolymers tended to develop the same melting point during film orien-tation, bottle inflation, and fiber spinning.
Cicero et al.104,105 characterized the crystal morphology of fibers made withlinear and branched architectures. The level of crystallinity increased withdraw ratio. The morphology was fibrillar, with microfibril diameters rangingfrom 20 to 30 nm for both architectures. Cicero et al. noted that the orienta-tion of the crystal blocks was similar to nylon. Two-step drawn fibersundergo a morphology transition from class 2 to class 1 within the classifi-cation proposed by Keller et al.106 at higher draw conditions.
1741_C16.qxd 2/11/2005 9:57 AM Page 554

Polylactic Acid Technology 555
16.5.4 Degradation and Hydrolysis
Under conditions of high temperature and high humidity, as in active com-post, for example, PLA will degrade quickly and disintegrate within weeksto months. The primary mechanism of degradation is hydrolysis, followedby bacterial attack on the fragmented residues. The environmental degrada-tion of PLA occurs by a two-step process.107 During the initial phases ofdegradation, the high molecular weight polyester chains hydrolyze to lowermolecular weight oligomers. The rate of hydrolysis is accelerated by acids orbases and is dependent on moisture content and temperature. Article dimen-sions, crystallinity, and blends will affect the rate of degradation. PLA prod-ucts rapidly degrade in both aerobic and anaerobic composting conditions:
Molecular Rate of MW Weight Hydrolysis DegradationStage Weight Decrease Loss Reaction Mechanism
First stage High Slow (rate None Nonenzymatic Bulkdetermined)
Critical (Mn: 10,000 – 20,000) Onset
Second stage Low Rapid Rapid Nonenzymatic Bulk andand enzymatic surface
Under typical use conditions, PLA is very stable and will retain its molec-ular weight and physical properties for years. This is typified by its growinguse in clothing and durable applications. High molecular weight PLA is alsonaturally resistant to supporting bacterial and fungal growth, which allowsit to be safely used for applications such as food packaging and sanitation.
A recent review by Tsuji108 gives an excellent account of PLA degradationstudies.
16.5.4.1 HydrolysisDegradation of PLA is primarily due to hydrolysis of the ester linkages,which occurs more or less randomly along the backbone of the polymer. Itrequires the presence of water according to the following reaction:
CH
CH3
PLA C
O
O CH
CH3
C
O
OPLA + H2O
PLA CH
CH3
C
O
OH + HO CH
CH3
C
O
OPLA
1741_C16.qxd 2/11/2005 9:57 AM Page 555

556 Natural Fibers, Biopolymers, and Biocomposites
The rate of hydrolysis is determined by its intrinsic rate constant, waterconcentration, acid or base catalyst, temperature, and morphology. Twomajor challenges to the stabilization of PLA with regard to hydrolysis are thefact that it is quite permeable to water and that the hydrolysis reaction isautocatalytic. Polyethylene terephthalate, for example, is also a polyesterthat hydrolyzes, but the inherent rate of hydrolysis is slower than PLA andit is not autocatalytic.
The autocatalytic hydrolysis reaction is the following:
An equation describing decrease in ester concentration [E] over time is thefollowing:
�k[�COOH][H2O]� (16.7)
for random chain scission where [�COOH] ∝ 1/Mn and the product[H2O][E] is constant. Rearranging to
Mnd(1/Mn)�kdt (16.8)
the integrated form becomes
ln Mn,t � lnMn,o�kt (16.9)
where Mn,t � number average molecular weight at time t, Mn,o � number aver-age molecular weight at time zero, and k is the hydrolysis rate constant. Thesekinetics were derived by Pitt et al.,109 and were again supported by Tsuji.108
Siparsky et al.110 proposed a slightly different equation using an autocat-alytic model:
�kh[�COOH][H2O][E] (16.10)
They found that this equation did not fit their solution kinetics so theyderived a second equation using quadratic autocatalysis kinetics. This model
d[�COOH]��
dt
d(1/Mn)��
dtd[E]�
dt
R-COOH R-COO¯ + H+
CH
CH3
PLA C
O
O CH
CH3
C
O
OPLA + H2O + H+
kh
CH
CH3
PLA C
O
OH + HO CH
CH3
C
O
OPLA + H+
ka
1741_C16.qxd 2/11/2005 9:57 AM Page 556

Polylactic Acid Technology 557
fit the data much better. It is of the following form:
�kh[E][H2O][x]
where x � Ka[�COOH]1/2, and Ka is the acid dissociation constant. This formaccounts for the dissociation of the acid.
Stated differently,
�kh[H2O][E]([�COOH]ka)1/2 (16.11)
The expression makes it possible to take into account all the acid species inthe polymer. It also removes the water dependence of the rate constantexpression. A comprehensive predictive hydrolysis model for PLA auto-catalysis also requires temperature dependence for kh above and below Tg,and accurate prediction of water concentration. Combining this expressionwith mass transfer terms for [H2O] and [�COOH] as a function of positionin an article may be useful for some applications.
Strategies for stabilizing PLA to hydrolysis include reducing the level ofresidual monomer to as low a level as possible and lowering the water con-centration in PLA and preventing autocatalysis. The equilibrium moisturecontent can be decreased by controlling morphology (highly crystalline, ori-ented, fibular structure). Reduction in the rate of autocatalysis has beenaccomplished by incorporation of basic, buffering salts such as CaCO3.Certain impurities and classes of additives increase the rate of PLA hydroly-sis, including lactide and oligomers and some acidic and basic additives. Asecond approach to preventing autocatalysis was reported by Lee et al.111 byfunctionalizing the PLA end group chemistry. End groups studied includedOH-, COOH-, Cl-, and NH2-. NH2- and Cl-terminated PLAs were moreresistant to thermal and hydrolytic degradation. The thermal stability of theOH-terminated PLA was poor (likely due to lactide formation), and the hydrolytic stability of the COOH-terminated PLA was also poor.
The hydrolysis of PLA has been studied using at least four experimentaldesigns: melt hydrolysis (discussed in rheology section above) to simulateextrusion conditions, solution hydrolysis of PLA dissolved in various sol-vents, and two types of solid-state hydrolysis methods. The first type ofsolid-state hydrolysis experiment utilizes solid samples suspended in aque-ous media. The second type utilizes exposure of solid samples to humidity.Since the product of the hydrolysis reaction is also a catalyst for further reac-tion, the hydrolysis of PLA is autocatalytic. Melt hydrolysis, solution hydrol-ysis, and solid exposure to humidity experimental designs generally giverise to homogeneous hydrolysis. In contrast, exposure of solid samples sus-pended in water or aqueous buffers gives rise to very inhomogeneoushydrolysis. This is due to differences in hydrolysis rate as a function of depthwithin the sample and it is caused by extraction of PLA oligomers from thesurface of the sample into the aqueous medium. It is the chain end groups ofthe PLA oligomers that catalyze the rate of hydrolysis, and therefore the
d[E]�
dt
d[x]�
dt
1741_C16.qxd 2/11/2005 9:57 AM Page 557

558 Natural Fibers, Biopolymers, and Biocomposites
inside of the sample actually hydrolyzes faster than the surface for thicksamples submersed in water.
It is important that the data used for the determination of any type of sta-bility analysis be consistent with the type of hydrolysis conditions of theintended application. In vivo degradation of PLA is outside the scope of thischapter.
Four key features mark the hydrolytic degradation of PLA:
1. The pKa of PLA’s carboxylic acid end group and its oligomers isunusually low (~3) compared to most carboxylic acid groups (4.5to 5). PLA’s carboxylic acid end groups catalyze the ester hydroly-sis, which generally leads to a faster rate of degradation as thepolymer degrades (autocatalysis).
2. Two reaction mechanisms apparently occur. The first is a randomchain scission and the second a chain-end hydrolysis. The chain-end scission is about 10 times faster than the random scission reac-tion at low pH values but is negligible at neutral and basic pHvalues. The number of ester groups along the chain is much greaterthan that of the chain ends and is statistically much more impor-tant.
3. The crystalline regions hydrolyze much more slowly than theamorphous regions.
4. The rate of hydrolysis (slope) is much greater above the glass tran-sition temperature (Tg) than below.
In the following, we summarize each of the four hydrolysis experimentaldesign conditions independently. Melt hydrolysis is covered in the rheologysection pertaining to melt stability.
16.5.4.2 Hydrolysis of Solid Samples Suspended in Aqueous MediaHydrolytic degradation of PLA is autocatalytic due to the production of car-boxylic acid end groups that catalyze further hydrolysis.112 An effect of thisis that for a thick sample immersed in a 7.4 pH buffer at 37°C, bulk hydrol-ysis occurs faster than surface hydrolysis.113,114 The pH of the surface remainsthat of the 7.4 pH buffer whereas the pH of the interior of the sample isreduced due to the accumulation of PLA acid end groups and the inabilityof the oligomers to quickly diffuse into the buffer medium.
Vert et al.115 summarized the literature on the hydrolytic stability of solid,suspended PLA in 1997 and captured several key points including auto-catalysis and leaching effects. In addition, morphology and structure wereaddressed. The rate of degradation is strongly influenced by the numberaverage molecular weight.116 Lower Mn PLA degraded faster as did PLAwith higher levels of oligomers. The presence of lactide also increased therate of hydrolytic degradation.117 Effects of Mn, oligomers, and lactide can allbe attributed to autocatalysis. PLA’s amorphous domains are more suscepti-ble to hydrolysis than the crystalline domains. This is due to the ability of
1741_C16.qxd 2/11/2005 9:57 AM Page 558

Polylactic Acid Technology 559
water to permeate within the amorphous phase but not the crystalline phase,and leads to differences in water concentration throughout the article on themicroscale. Hydrolysis of the crystalline domain occurs mainly by a surfaceerosion mechanism.
Grizzi et al. used the fact that surface hydrolysis is slow compared to bulkhydrolysis to stabilize PLA to hydrolysis simply by using small dimen-sions.113 By suspending PLA samples of varying size in water and determin-ing the rates of hydrolysis, they showed that leaching could be used to slowdown the rate of solid-state hydrolysis. They estimated that the skin, whichwas formed due to the leaching of catalytic oligomers, was about 200 to300 µm in thickness. Samples smaller than this size were found to degrademuch more slowly than large samples.
The heterogeneity of the process makes it difficult to determine hydrolysiskinetics constants for solids suspended in aqueous media. Translation of datafrom these kinds of studies to other environments is suspect for this reason.
In alkaline media, Iwata and Doi118 studied the change in morphology andcrystalline state of solution-grown single crystals of PLLA. Samples werecharacterized by transmission electron microscopy (TEM), atomic forcemicroscopy (AFM), and gel permeation chromatography (GPC). As hydrol-ysis proceeded, the molecular weight of the degraded crystals correspondedto the value calculated from lamellar thickness measured by AFM. Theexplanation for this is that the PLA first degrades in the more loosely packedchain folding regions. In the later stages of hydrolysis, the more persistentcrystalline region’s molecular weight approaches that of the lamellar thick-ness, and the degradation mechanism will change to that of surface erosion.
Tsuji and Nakahara119 tested PLLA films at pH 2.0 (1. HCl solution, 2. D,L-lactic acid solution), 7.4 (phosphate buffered), and 12 (NaOH soln).While the alkaline media degraded faster than the neutral pH control, thesamples in acid media degraded at about the same rate as the neutral pH con-trol. Using microcapsules suspended in buffer solutions, Makino et al.120
showed that PLA degrades slowest at pH 5.0 and increases in acidic and alka-line solutions. The activation energy of hydrolysis at pH 7.4 and ionic strength0.15 was 19.9 kcal/mol for PDLLA microcapsules and 20.0 kcal/mol for PLLAmicrocapsules when the initial Mw was about 100,000 for both polymers.
The degradation characteristics of PLA in seawater are important for cer-tain applications and potential disposal routes. Yuan et al.121 tested PLLAfibers in saline buffer at 37°C for molecular weight loss and mechanicalproperties. The results suggested that the tensile properties of the PLLAfibers were stable for 35 weeks, even though their molecular weightsdecreased remarkably and obvious spotty defects appeared on their sur-faces. Tsuji and Suzuyoshi122 showed that PLA degraded faster in naturalseawater than controlled static seawater, although the rate of molecularweight loss is slow. Makino et al.120 showed that AlCl3, CaCl2, and KCl hadno or little effect on the degradation rate.
At temperatures above the glass transition, PLA’s degradation rate increasessubstantially. Tsuji et al.123 tested PLLA films in buffers at 97°C and showed
1741_C16.qxd 2/11/2005 9:57 AM Page 559

560 Natural Fibers, Biopolymers, and Biocomposites
that the hydrolysis takes place predominantly and randomly at the chains inthe amorphous region. Amorphous samples crystallized while degrading andhigher initial crystallinity slightly slowed the rate of hydrolysis.
In a very extreme case showing the effect of morphology, fibers (drawn toa draw ratio of about 8� using a solution spinning approach) were exam-ined. These fibers possessed few defects (chain folds, microvoids) togetherwith high crystallinity, low fractional free volume, and small cross-sectionaldimension.112 The PLLA fiber was very stable over a 5-year period immersedin water at room temperature under a static load and lost 10% of its diame-ter and less than 25% of its tensile strength. The perfection of morphologywas thought to reduce the penetration and diffusion of water into the fiber.
Effects of processing conditions were also examined by Iannace et al.124
Samples of PLLA crystallized under isothermal and nonisothermal condi-tions were prepared. Initial morphology developed during the isothermalcrystallization affected the rate of hydrolysis in buffer solutions. Larger crys-tals developed at higher Tc were more resistant to erosion compared to lessperfect and smaller crystals developed at higher degrees of supercooling.
Tsuji and Suzuki94 also studied stereocomplex degradation over the courseof 30 months, in 7.4 pH buffer solution using 1:1 blends and nonblendedfilms prepared from PLLA and PDLA. Properties and molecular weightwere monitored with GPC, tensile tests, DSC, SEM, optical polarizingmicroscopy, x-ray diffractometry, and gravimetry. The rate of molecularweight loss, tensile strength, Young’s modulus, melting temperature, andmass remaining of the films in the course of hydrolysis was more stable forthe stereocomplex film than for the nonblended films.
16.5.4.3 Solution HydrolysisSolution hydrolysis is generally thought to occur by random scission of theester linkages in PLA.125 It has been reported, however, that base and neutralhydrolysis occur by random chain scission but acid hydrolysis occurs by amixture of two mechanisms, random and chain-end scission, with chain-endscission being the faster.126 This was determined in dioxane/water solutionwith 1H NMR and GPC using a statistical method where the level of lacticacid produced by hydrolysis is compared to the degree of degradationacross the entire molecular weight distribution. The chain-end scission issometimes referred to as “unzipping.”
PLA solution hydrolysis was also studied in acetone/water by Zhang et al.127 and acetonitrile/water by Siparsky et al.110 The latter solvent had amuch higher dielectric constant (ε � 36) than acetone (ε � 21) or dioxane (ε � 2)so that the carboxylic acid groups were more ionized, as in water (ε � 79).Autocatalysis was observed in acetonitrile whereas it was not in the lowerdielectric solvents due to a lower extent of ionization of the carboxylic acid.
The solution hydrolysis rate constant (in dioxane solution) did not dependon the optical purity (OP) of the polymer whereas it does in solid-statedegradation (probably due to crystallinity differences).128 CaCO3 was
1741_C16.qxd 2/11/2005 9:57 AM Page 560

Polylactic Acid Technology 561
observed to slow the rate of hydrolysis due to its buffering capacity andwater insolubility.
16.5.4.4 Hydrolysis of Samples Exposed to HumiditySamples exposed to temperature and humidity best simulate the shelf-lifeenvironment for many PLA applications. The equilibrium moisture level asa function of humidity is known for PLA at room temperature. It has notbeen as thoroughly studied for elevated temperatures. Hydrophilic impuri-ties such as lactide and carboxylic acid end groups, whose concentrationincrease as degradation proceeds, increase the level of moisture.
Sharp et al.129 measured the water uptake of PDLLA using a quartz crys-tal microbalance in humid environments at various temperatures. Kinetics ofwater adsorption and equilibrium concentrations were measured, and theFlory Huggins interaction parameter X was found to be 3.5 at 20°C usingbinary mixing theory. The equilibrium swell level of water in PLA increaseswith decreasing molecular weight due to the strong affinity of acid andhydroxyl end groups for water. It was also shown that the Tg of high molec-ular weight PLA was depressed by 11°C at 7000 ppm water (the highest levelof swelling studied) compared to zero ppm water.
Witzke32 measured the swelling concentrations of water in PLA usingKarl-Fisher coulometric titration and arrived at an empirical model fit forestimating swell concentration:
Water (ppm) � 40.8RH(%)�6.6�10�7RH(%)5 (16.12)
where water is in parts per million by weight and RH is the relative humid-ity. A plot of this equation is shown in Figure 16.14.
0100020003000400050006000700080009000
10000110001200013000140001500016000
0 10 20 30 40 50 60 70 80 90 100Humidity (%RH)
Moi
stur
e co
nten
t (pp
m)
Low HighTypical
FIGURE 16.14Equilibrium swelling concentration of water in high molecular weight PLA in humidenvironments.
1741_C16.qxd 2/11/2005 9:57 AM Page 561
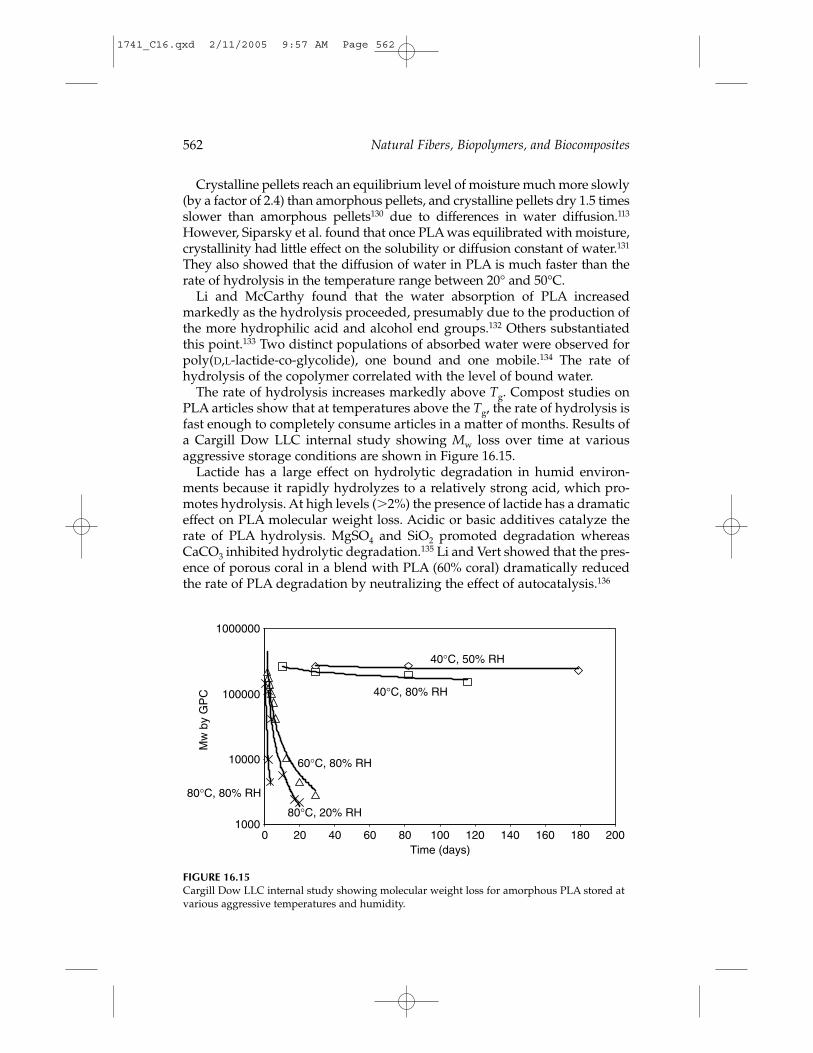
562 Natural Fibers, Biopolymers, and Biocomposites
Crystalline pellets reach an equilibrium level of moisture much more slowly(by a factor of 2.4) than amorphous pellets, and crystalline pellets dry 1.5 timesslower than amorphous pellets130 due to differences in water diffusion.113
However, Siparsky et al. found that once PLA was equilibrated with moisture,crystallinity had little effect on the solubility or diffusion constant of water.131
They also showed that the diffusion of water in PLA is much faster than therate of hydrolysis in the temperature range between 20° and 50°C.
Li and McCarthy found that the water absorption of PLA increasedmarkedly as the hydrolysis proceeded, presumably due to the production ofthe more hydrophilic acid and alcohol end groups.132 Others substantiatedthis point.133 Two distinct populations of absorbed water were observed forpoly(D,L-lactide-co-glycolide), one bound and one mobile.134 The rate ofhydrolysis of the copolymer correlated with the level of bound water.
The rate of hydrolysis increases markedly above Tg. Compost studies onPLA articles show that at temperatures above the Tg, the rate of hydrolysis isfast enough to completely consume articles in a matter of months. Results ofa Cargill Dow LLC internal study showing Mw loss over time at variousaggressive storage conditions are shown in Figure 16.15.
Lactide has a large effect on hydrolytic degradation in humid environ-ments because it rapidly hydrolyzes to a relatively strong acid, which pro-motes hydrolysis. At high levels (�2%) the presence of lactide has a dramaticeffect on PLA molecular weight loss. Acidic or basic additives catalyze therate of PLA hydrolysis. MgSO4 and SiO2 promoted degradation whereasCaCO3 inhibited hydrolytic degradation.135 Li and Vert showed that the pres-ence of porous coral in a blend with PLA (60% coral) dramatically reducedthe rate of PLA degradation by neutralizing the effect of autocatalysis.136
1000
10000
100000
1000000
0 20 40 60 80 100 120 140 160 180 200Time (days)
Mw
by
GP
C
40°C, 50% RH
40°C, 80% RH
60°C, 80% RH
80°C, 20% RH
80°C, 80% RH
FIGURE 16.15Cargill Dow LLC internal study showing molecular weight loss for amorphous PLA stored atvarious aggressive temperatures and humidity.
1741_C16.qxd 2/11/2005 9:57 AM Page 562

Polylactic Acid Technology 563
The crystalline regions of PLA were found to hydrolyze very slowly.112,125
The rate of molecular weight loss follows a two-step process, the first occur-ring very fast (interpreted as loss of the amorphous phase) and the secondoccurring much slowly (interpreted as loss of the crystalline phase). Theapparent percentage of crystallinity increases during the amorphous phase hydrolysis, consistent with the interpretation. The molecular weightof the remaining PLA after the amorphous phase was degraded was about5000 to 9000 (DP � 70 to 125), roughly that of the fold length of thelamella.74,137 Crystalline injection-molded samples lost physical propertiesfaster than amorphous samples, although the rate of Mw loss was similarbetween them.125 This was interpreted as the loss of amorphous tie chainsbetween the crystals causing catastrophic failure of the system. Also, chainfolds and tie chains connecting crystallites are thought to hydrolyze prefer-entially because they are stressed due to a “reeling in” force.138 These obser-vations may be the result of chain end groups and catalysts concentrating inthe amorphous regions.
Morphology also affects water permeation in another way. Microvoids orporosity allow water to migrate more freely into the material and therebyenhance the rate of hydrolysis. This is probably a small effect for samplesexposed to humidity because the rate of hydrolysis is slow compared to the water diffusion rate, as discussed above. Localized regions of high water absorption may cause mechanical stresses due to increased osmoticpressure.
16.5.5 Enzymatic Degradation
Several enzymes have been shown to catalyze PLA hydrolysis. Enzymatichydrolysis of PLLA with varied crystal morphologies was studied using pro-teinase K by Tsuji and Miyauchi.139,140 It was concluded that the enzymaticdegradation proceeded via mainly a surface erosion mechanism. Amorphousregions hydrolyzed more readily than crystalline regions. The enzymatichydrolysis rate (REH) was modeled using the following equation:
REH(µg/(mm2·h)�0.36(1�xc) (16.13)
where xc � the fraction of crystalline PLLA. Tie chains degraded more read-ily than folds or restricted amorphous chains.
16.6 Applications and Performance
Since PLA is an environmentally friendly polymer that can be designed tocontrollably biodegrade, it is ideally suited for many applications in the envi-ronment where recovery of the product is not practical, such as agricultural
1741_C16.qxd 2/11/2005 9:57 AM Page 563
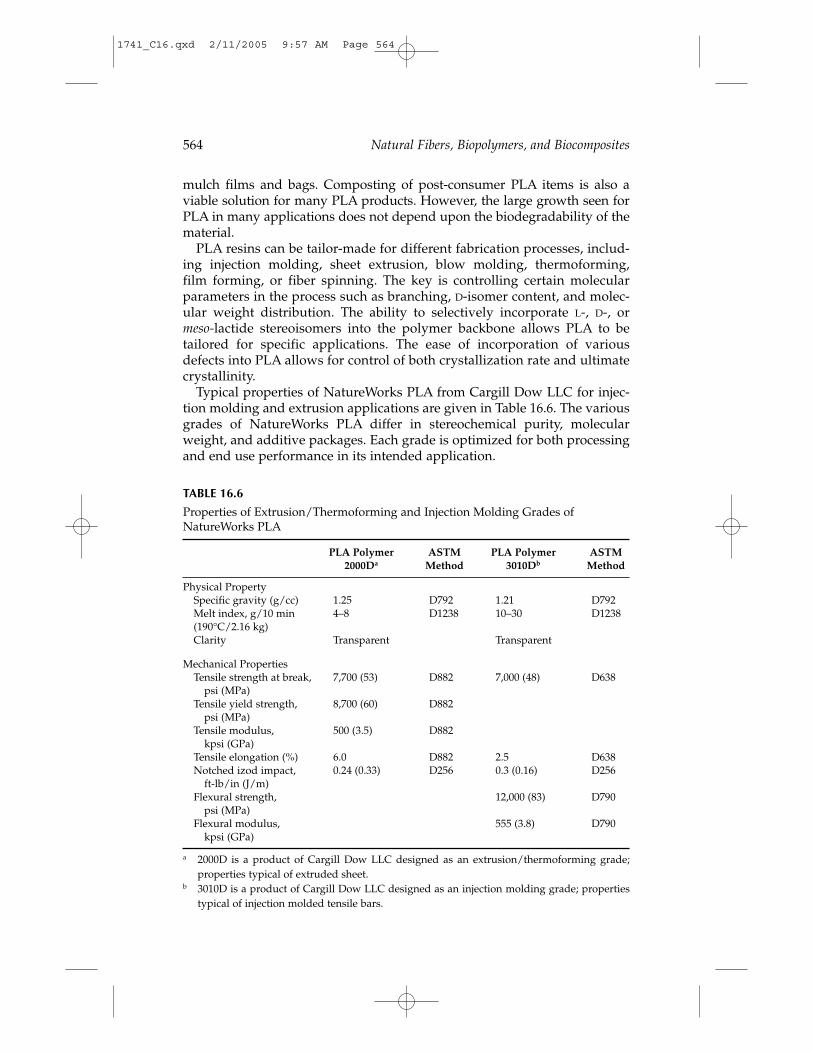
564 Natural Fibers, Biopolymers, and Biocomposites
mulch films and bags. Composting of post-consumer PLA items is also aviable solution for many PLA products. However, the large growth seen forPLA in many applications does not depend upon the biodegradability of thematerial.
PLA resins can be tailor-made for different fabrication processes, includ-ing injection molding, sheet extrusion, blow molding, thermoforming, film forming, or fiber spinning. The key is controlling certain molecularparameters in the process such as branching, D-isomer content, and molec-ular weight distribution. The ability to selectively incorporate L-, D-, ormeso-lactide stereoisomers into the polymer backbone allows PLA to betailored for specific applications. The ease of incorporation of variousdefects into PLA allows for control of both crystallization rate and ultimatecrystallinity.
Typical properties of NatureWorks PLA from Cargill Dow LLC for injec-tion molding and extrusion applications are given in Table 16.6. The variousgrades of NatureWorks PLA differ in stereochemical purity, molecularweight, and additive packages. Each grade is optimized for both processingand end use performance in its intended application.
TABLE 16.6
Properties of Extrusion/Thermoforming and Injection Molding Grades ofNatureWorks PLA
PLA Polymer ASTM PLA Polymer ASTM 2000Da Method 3010Db Method
Physical PropertySpecific gravity (g/cc) 1.25 D792 1.21 D792Melt index, g/10 min 4–8 D1238 10–30 D1238(190°C/2.16 kg)Clarity Transparent Transparent
Mechanical PropertiesTensile strength at break, 7,700 (53) D882 7,000 (48) D638
psi (MPa)Tensile yield strength, 8,700 (60) D882
psi (MPa)Tensile modulus, 500 (3.5) D882
kpsi (GPa)Tensile elongation (%) 6.0 D882 2.5 D638Notched izod impact, 0.24 (0.33) D256 0.3 (0.16) D256
ft-lb/in (J/m)Flexural strength, 12,000 (83) D790
psi (MPa)Flexural modulus, 555 (3.8) D790
kpsi (GPa)
a 2000D is a product of Cargill Dow LLC designed as an extrusion/thermoforming grade;properties typical of extruded sheet.
b 3010D is a product of Cargill Dow LLC designed as an injection molding grade; propertiestypical of injection molded tensile bars.
1741_C16.qxd 2/11/2005 9:57 AM Page 564

Polylactic Acid Technology 565
Injection molding of heat-resistant products requires rapid crystalli-zation rates that can be achieved by PLA, typically containing less than 1% D-isomer and often with the addition of nucleating agents.75 These composi-tions allow high levels of crystallinity to develop during the fast coolingcycle in the mold. Extrusion-thermoforming is optimized at a D-isomer con-tent that does not allow crystallization to occur during the melt processingsteps, with 4–8% D-content being the effective range. Branching can be intro-duced by a variety of methods,62–64, 66 thus enhancing melt strength duringfabrication and opening up new application opportunities in the areas offoams and extrusion coating. Examples of a number of articles fabricatedfrom PLA are shown in Figure 16.16. In short, the rheological characteristicsand physical properties of PLA can be tailored for use in a variety ofprocesses and applications.
Fiber is one of the largest potential application areas for PLA. PLA is read-ily melt spinnable, stress crystallizes upon drawing, and can be designed for
FIGURE 16.16Articles produced from PLA. (a) Injection stretch-blow molded bottles. (b) Films. (c)Extrusion-thermoformed containers. (d) Fiberfill products. (e) Carpet and coverings.
1741_C16.qxd 2/11/2005 9:57 AM Page 565
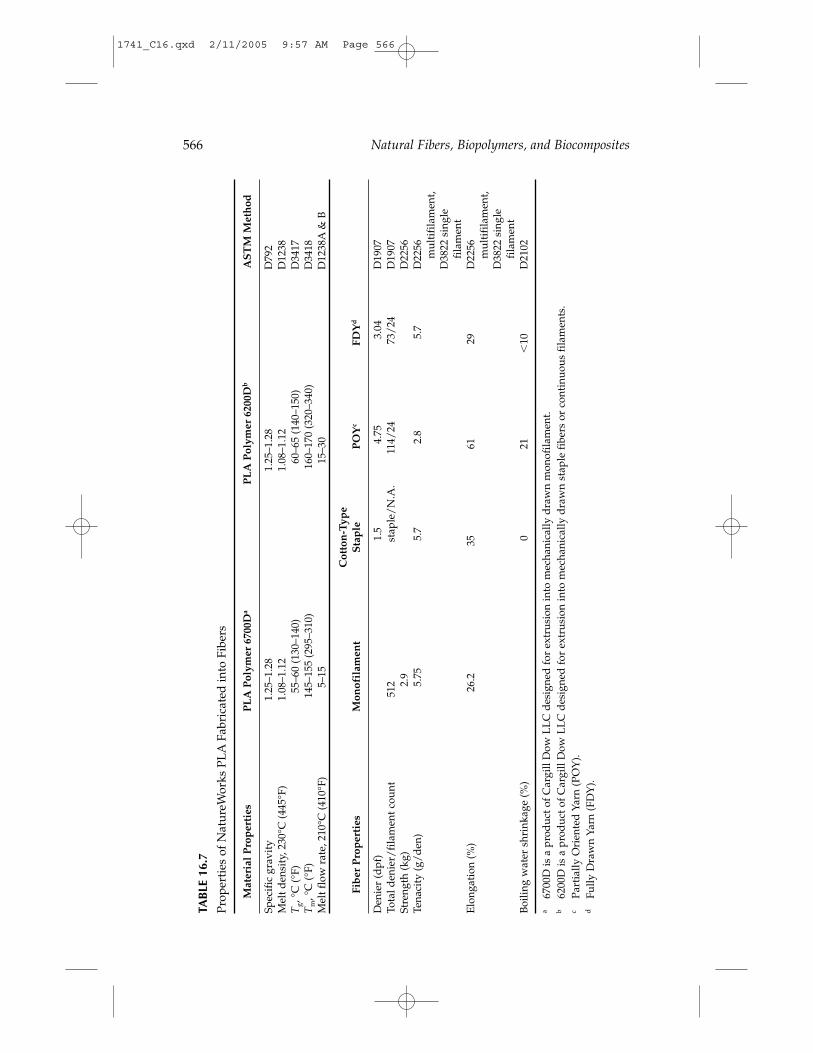
566 Natural Fibers, Biopolymers, and Biocomposites
TAB
LE 1
6.7
Prop
erti
es o
f N
atur
eWor
ks P
LA
Fabr
icat
ed in
to F
iber
s
Mat
eria
l P
rop
erti
es
PL
AP
olym
er 6
700D
aP
LA
Pol
ymer
620
0Db
AS
TM
Met
hod
Spec
ific
gra
vity
1.25
–1.2
81.
25–1
.28
D79
2M
elt d
ensi
ty, 2
30°C
(44
5°F)
1.08
–1.1
21.
08–1
.12
D12
38T
g, °
C (
°F)
55–6
0 (1
30–1
40)
60–6
5 (1
40–1
50)
D34
17T
m, °
C (
°F)
145–
155
(295
–310
)16
0–17
0 (3
20–3
40)
D34
18M
elt f
low
rat
e, 2
10°C
(41
0°F)
5–15
15–3
0D
1238
A&
B
Cot
ton
-Typ
e Fi
ber
Pro
per
ties
Mon
ofil
amen
tS
tap
leP
OY
cFD
Yd
Den
ier
(dpf
)1.
54.
753.
04D
1907
Tota
l den
ier/
fila
men
t cou
nt51
2st
aple
/N
.A.
114/
2473
/24
D19
07St
reng
th (
kg)
2.9
D22
56Te
naci
ty (
g/d
en)
5.75
5.7
2.8
5.7
D22
56
mul
tifi
lam
ent,
D38
22 s
ingl
e fi
lam
ent
Elo
ngat
ion
(%)
26.2
3561
29D
2256
m
ulti
fila
men
t, D
3822
sin
gle
fila
men
tB
oilin
g w
ater
shr
inka
ge (
%)
021
�10
D21
02
a67
00D
is a
pro
duc
t of
Car
gill
Dow
LL
C d
esig
ned
for
ext
rusi
on in
to m
echa
nica
lly d
raw
n m
onof
ilam
ent.
b62
00D
is a
pro
duc
t of
Car
gill
Dow
LL
C d
esig
ned
for
ext
rusi
on in
to m
echa
nica
lly d
raw
n st
aple
fib
ers
or c
onti
nuou
s fi
lam
ents
.c
Part
ially
Ori
ente
d Y
arn
(PO
Y).
dFu
lly D
raw
n Ya
rn (
FDY
).
1741_C16.qxd 2/11/2005 9:57 AM Page 566
many fiber applications. Some of the current fiber uses include hollow fiber-fill for pillows and comforters, bulk continuous filament (BCF) for carpet, fil-ament yarns, and spun yarns for apparel, spunbond, and other nonwovensand bicomponent fibers for binders and self-crimping fibers. Most fiberapplications require polymer with high optical purity (OP) to allow high lev-els of crystallinity to develop and to have adequate heat resistance in theapplication. Binder fibers are unique in that low crystallinity in the sheathlayer is desired to allow ease of melting and adhesion to other fibers; thus,high (8–20%) D- or meso-lactide content is incorporated. PLA can beprocessed on standard thermoplastic fiber spinning equipment with theappropriate temperature profiles relative to its crystal melting point. Melttemperatures of 200°–240°C are typically used. As with all melt processingof PLA, care must be taken to ensure that the material is dry and does notpick up moisture, otherwise unacceptable molecular weight loss will occur.Typical drying conditions are 2–4 h in a hopper dryer with – 40oC dew pointair with a resulting moisture content of less than 50 ppm. Some of the prop-erties of PLA fibers are shown in Table 16.7 and compared to other naturaland synthetic fibers in Table 16.8.
PLA fiber can be combined with natural or regenerated fibers includingcotton, wool, silk, viscose, lyocell, and others along with synthetic fibersmade from PET, nylon, and other petroleum-based synthetics. PLA can beincluded as a minor component (5–15%) or as the major fiber, depending onthe balance of properties and appearance desired. One of the fastest growingapplication areas is in nonwoven wipes containing 35% PLA and 65% viscose.PLA is replacing PET in these applications because of its superior perform-ance and the fact that the disposable products can be produced from fibersthat are from 100% renewable resources and are also 100% biodegradable.
Some of the beneficial characteristics of PLA fiber products include its nat-ural soft feel, ease of processing, and unique stain and soil resistance. PLAexcels at resistance to stain in standard tests with coffee, cola, tea, catsup, lip-stick, and mustard. PLA also burns with low smoke generation, has goodultraviolet resistance, is easily dyeable, and brings good wickability of mois-ture to applications.
Polylactic Acid Technology 567
TABLE 16.8
Properties of Synthetic and Natural Fibers
Fiber Property Nylon 6 PET Acrylics PLA Rayon Cotton Silk Wool
Specific gravity 1.14 1.39 1.18 1.25 1.52 1.52 1.34 1.31Tenacity (g/d) 5.5 6.0 4.0 6.0 2.5 4.0 4.0 1.6Moisture regain (%) 4.1 0.2–0.4 1.0–2.0 0.4–0.6 11 7.5 10 14–18Elastic recovery 89 65 50 93 32 52 52 69(5% strain)Flammability Medium High Medium Low Burns Burns Burns Burns
smoke smoke slowlyUV resistance Poor Fair Good Good Poor Fair Fair Fair
1741_C16.qxd 2/11/2005 9:58 AM Page 567

568 Natural Fibers, Biopolymers, and Biocomposites
Films are the second largest application area for PLA. Again, the ability tomodify the crystallization kinetics and physical properties for a broad range ofapplications by D- or meso-comonomer incorporation, branching, and molecu-lar weight change makes PLA extremely versatile. Films are transparent whenstress crystallized and have acceptance by customers for food contact. PLAfilms can be prepared by the blown double bubble technology or preferably,cast-tentering. Cast-tentered films have very low haze, excellent gloss, and gas(O2, CO2, and H2O) transmission rates desirable for consumer food packaging.PLA films also have superior dead fold or twist retention for twist wrap pack-aging. Some of the properties of PLA films are shown in Table 16.9.
16.7 Summary
Polymers of polylactic acid have finally become a commercial reality with theconstruction of a world-scale plant. The cost-performance balance of PLA has
TABLE 16.9
Properties of Biaxially Oriented NatureWorks PLA
Film Properties PLA Polymer 4040Da ASTM Method
Specific gravity (g/cc) 1.25 D1505Tensile strength (kpsi)
MDb 16 D882TD 21 D882
Tensile modulus (kpsi) MD 480 D882TD 560 D882
Elongation at break (%) MD 160 D882TD 100 D882
Elmendorf tear (g/mil)MD 15 D1922TD 13 D1922
Spencer impact (J) 2.5Transmission rates
O2, cc-mil/m2/24 h atm 550 D1434CO2, cc-mil/m2/24 h atm 3000 D1434H2O, g-mil/m2/24 h atm 325 E96
Optical characteristics Haze (%) 2.1 D1003Gloss, 20° 90 D1003
Thermal characteristics Tg, °C (°F) 52 (126) D3418 Tm, °C (°F) 135 (275) D1003
a 4040D is a product of Cargill Dow LLC suitable for preparation of biaxially oriented film.Properties on 1 mil film oriented 3.5� in MD and 5� in TD.
b MD refers to machine direction and TD refers to transverse direction.
1741_C16.qxd 2/11/2005 9:58 AM Page 568

Polylactic Acid Technology 569
resulted in its use in many applications, including packaging, paper coating,fibers, films, and a host of molded articles. PLA is not being used in theseapplications solely because of its degradability nor because it is made fromrenewable resources. PLA is in both durable and short-term applications. Theuse of PLA as a cost-effective alternative to commodity petrochemical-basedplastics will increase demand for agricultural products such as corn andsugar beets, and is an advanced example of sustainable technology.
One of the additional benefits of large commercial production of lacticacid for PLA is the increased availability and reduced cost of lactic acid orlactide for the production of lactic acid esters. These esters, especially ethyllactate, are finding increased use as environmentally friendly solvents,cleaning agents, and diluents. They are replacing more hazardous solventssuch as chlorinated organic and aromatic compounds.
As we move into the 21st century, increased utilization of renewableresources will be one of the strong drivers for sustainable products. Reducedenergy consumption, waste generation, and emission of greenhouse gaseswill take on greater emphasis. Polylactic acid is the first commodity plasticto incorporate these principles and Cargill Dow LLC was awarded the 2002Presidential Green Chemistry Award for their process to produceNatureWorks PLA.141 Future developments will include blends of PLA,copolymers, and impact-modified products, which will further expand theapplications where this unique polymer can be used.
Acknowledgments
The authors would like to thank Kevin McCarthy, Patrick Smith, and RayDrumright for their contributions to this manuscript, and Cargill Dow, LLCfor permission to publish it.
Reference
1. Lipinsky, E.S. and Sinclair, R.G., Chem. Eng. Prog., 82, 26–32, 1986.2. Carothers, H., Dorough, G. L., and Van Natta, F. J., J. Am. Chem. Soc., 54, 761, 1932.3. Nef, J. U., Liebigs. Ann. Chem., 403, 204, 1914; Pelouze, P. M. J., Ann. Chim. Phys.,
13, 257, 1845.4. Plastics Technology, January 1998, pp. 13–15.5. Enomoto, K., Ajioka, M., and Yamaguchi, A., U.S. Patent 5,310,865, 1995;
Kashima, T., Kameoka, T., Ajioka, M., and Yamaguchi, A., U.S. Patent 5,428,126,1995; Ichikawa, F., Kobayashi, M., Ohta, M., Yoshida, Y., Obuchi, S., and Itoh, H.,U.S. Patent 5,440,008, 1995; Ohta, M., Obuchi, S., and Yoshida, Y., U.S. Patent5,440,143, 1995.
1741_C16.qxd 2/11/2005 9:58 AM Page 569

570 Natural Fibers, Biopolymers, and Biocomposites
6. Gruber, P.R., Hall, E.S., Kolstad, J.J., Iwen, M.L., Benson, R.D., and Borchardt,R.L., U.S. Patent 5,142,023, 1992; Gruber, P.R., Hall, E.S., Kolstad, J.J., Iwen, M.L.,Benson, R.D., and Borchardt, R.L., U.S. Patent 5,247,058, 1992; Gruber, P.R., Hall,E.S., Kolstad, J.J., Iwen, M.L., Benson, R.D., and Borchardt, R.L., U.S. Patent5,247,059, 1993; Gruber, P.R., Hall, E.S., Kolstad, J.J., Iwen, M.L., Benson, R.D.,and Borchardt, R.L., U.S. Patent 5,258,488, 1993; Gruber, P.R., Hall, E.S., Kolstad,J.J., Iwen, M.L., Benson, R.D., and Borchardt, R.L., U.S. Patent 5,274,073, 1993;Gruber, P.R., Hall, E.S., Kolstad, J.J., Iwen, M.L., Benson, R.D., and Borchardt,R.L., U.S. Patent 5,357,035, 1994; Gruber, P.R., Hall, E.S., Kolstad, J.J., Iwen, M.L.,Benson, R.D., and Borchardt, R.L., U.S. Patent 5,484,881, 1996.
7. Chem. Week, May 2000, 162, pp. 24E.8. Hartmann, M.H., Biopolymers from Renewable Resources, Kaplan, D.L., Ed.,
Springer, Berlin, 1998, chap. 13; Dubois, P., Jerome, R., Lofgren, A., andAlbertsson, A.C., J.M.S. Rev. Macromol. Chem. Phys., 35, 379, 1995; Tsuji, H.,Polylactides in Biopolymers, Wiley-VCH, 2002, chap. 5; Drumright, R., Gruber, P.,and Henton, D. E., Adv. Mater., 12, 1841, 2000.
9. Holten, C.H., Lactic Acid, Properties and Chemistry of Lactic Acid and Derivatives,Verlag Chemie, GmbH, Weinheim/Bergstr., 1971.
10. Schopmeyer, H.H., Lactic Acid, in Industrial Fermentations, Underkofler, L. andHickley, R.J., Eds., Chemical Publishing Co. New York, NY, 1954, chap. 12.
11. Cox, G. and Macbean, R., Lactic Acid Recovery and Purification Systems, ResearchProject Series, No. 29, October, 1976.
12. Busche, R., Shimshick, E., and Yates, R., Biol. Bioeng. Symp., No. 12, 1982, pp.249–262.
13. Gruber, P., Keynote Address, Massachusetts Green Chemistry Symposium, Co-sponsored by National Environmental Technology Institute, UMASS and MAExecutive Office of Environmental Affairs Toxic Use Reduction Institute,Amherst, MA, Oct. 30, 2001.
14. Kricheldorf, H.R. and Krieser, I., Makromol. Chem., 187, 1861, 1986.15. Kurcok, P., Penczek, J., Franek, J., and Kedlinski, Z., Macromolecules, 25, 2285,
1992.16. Nijenhuis, A.J., Du, Y.J., Van Aert, H.A.M., and Bastiaansen, C., Macromolecules,
28, 2124, 1995; Kricheldorf, H.R., Kreiser-Saunders, I., and Boettcher, C.,Polymer, 36, 1253, 1995.
17. Sinclair, R.G., U.S. Patent 4,045,418, 1977; Sinclair, R.G., U.S. Patent 4,057,537,1977.
18. Feijen, J., J. Appl. Polym. Sci., 62, 1295, 1996.19. Ford, T.M., U.S. Patent 5,310,599, 1994.20. Jacobsen, S., Fritz, H.G., Degee, Ph., Dubois, Ph., and Jerome, R., Polymer, 41,
3395, 2000.21. Schmack, G., Jacobsen, S., Fritz, H.G., Vogel, R., Tandler, B., and Beyreuther, R.,
J. Biotechnol., 86, 15, 2001.22. Jacobsen, S., Fritz, H. G., Degee, Ph., Dubois, Ph., and Jerome, R., J. Polym. Sci.,
37, 2413, 1999.23. Jacobsen, S., Fritz, H.G., Degee, Ph., Dubois, Ph., and Jerome, R., Ind. Crops Prod.,
11, 265, 2000.
1741_C16.qxd 2/11/2005 9:58 AM Page 570

Polylactic Acid Technology 571
24. Jacobsen, S., Fritz, H.G., Degee, Ph., Dubois, Ph., and Jerome, R., Macromol.Symp., 144, 289, 1999.
25. Jacobsen, S., Fritz, H.G., Degee, Ph., Dubois, Ph., and Jerome, R., Macromol.Symp., 153, 261, 2000.
26. Fukushima, T., Intern. Polym. Process., XV, 4,2000.27. Miyoshi, R., Intern. Polym. Process., XI, 4, 1996.28. Miyoshi, R., et al., U.S. Patent 5,574,129, 1996.29. Narayan, R., et al., U.S. Patent 5,906,783, 1999.30. Reichert, D., et al., U.S. Patent 5,378,801, 1995.31. Jansson, K., Koskinen, J., and Selin, J.-F., U.S. Patent 6,214,967, 2001.32. Witzke, D.R., Introduction to Properties, Engineering, and Prospects of
Polylactide Polymer, Ph.D. thesis, Chemical Engineering, Michigan StateUniversity, 1997.
33. Garlotta, D., A literature review of poly(lactic acid), J. Polym. Environ., 9, 63,2002.
34. Tsuji, H. and Ikada, Y., Physical properties of polylactides, Curr. Trends Polym.Sci., 4, 27, 1999.
35. Tsuji, H., Polylactides, in Biopolymers 4 Polyesters III Applications and CommercialProducts, Wiley –VCH, Weinheim, Germany, 2002, chap. 5.
36. Hartmann, M.H., High molecular weight polylactic acid polymers, inBiopolymers from Renewable Resources, Springer, Berlin, pp. 367–411.
37. Thakur, K.A.M., Kean, R.T., Hall, E.S., Doscotch, M.A., and Munson, E.J., Aquantitative method for determination of lactide composition in poly(lactide)using 1H NMR, Anal. Chem., 69, 4303, 1997.
38. Thakur, K.M., Kean, R.T., Hall, E.S., Kolstad, J.J., and Munson, E.J.,Stereochemical aspects of lactide stereo-copolymerization investigated by 1HNMR: a case of changing stereospecificity, Macromolecules, 31, 1487, 1998.
39. Spassky, N., Wisniewski, M., Pluta, C., and Le Borgne, A., Highly stereoelectivepolymerization of rac-(D,L)-lactide with a chiral Schiff’s base/aluminum alkox-ide initiator, Macromol. Chem. Phys., 197, 2627, 1996.
40. Ovitt, T.M. and Coates, G.W., Stereochemistry of lactide polymerization withchiral catalysts: new opportunities for stereocontrol using polymer exchangemechanisms, J. Am. Chem. Soc., 124, 1316, 2002.
41. Ovitt, T.M. and Coates, G.W., Stereoselective ring-opening polymerization ofrac-lactide with a single-site, racemic aluminum alkoxide catalyst: synthesis ofstereoblock poly(lactic acid), J. Polym. Sci., Part A: Polym. Chem., 38, 4686, 2000.
42. Yui, N., Dijkstra, P., and Feijen, J., Stereo block copolymers of L- and D-lactides,Makromol. Chem., 191, 487–488, 1990.
43. Ikada, Y., Jamshidi, K., Tsuji, H., and Hyon, S.H., Stereocomplex formationbetween enantiomeric poly(lactides), Macromolecules 20, 904, 1987.
44. Fitz, B.D., Jamiolkowski, D.D., and Andjelic, S., Tg Depression in poly(L(-)-lac-tide) crystallized under partially constrained conditions, Macromolecules, 35,5869, 2002.
45. Tsuji, H. and Ikada, Y., Stereocomplex formation between enantiomericpoly(lactic acid)s. XI. Mechanical properties and morphology, Polymer, 40, 6699,1999.
1741_C16.qxd 2/11/2005 9:58 AM Page 571

46. Urayama, H., Kanamori, T., and Kimura, Y., Properties and biodegradability ofpolymer blends of poly(L-lactides) with different optical purity of the lactateunits, Macromole. Mater. Eng. 287, 116, 2002.
47. Janzen, J., Dorgan, J.R., Knauss, D.M., Hait, S.B., and Limoges, B.R.,Fundamental solution and single-chain properties of polylactides,Macromolecules, 2003.
48. Dorgan, J.R., Lehermeier, H., and Mang, M., Thermal and rheological propertiesof commercial-grade poly(lactic acid)s, J. Polym. Environ., 8, 1, 2000.
49. Janzen, J. and Dorgan, J.R., A novel approach to modeling viscoelastic proper-ties of thermoplastic polymer melts, with applications to polylactides, AnnualTechnical Conf.– Soc. Plastics Engineers, 60, Vol. 1, 2002, p. 929.
50. Palade, L., Lehermeier, H.J., and Dorgan, J.R., Melt rheology of high L- contentpoly(lactic acid), Macromolecules, 34, 1384, 2001.
51. Cooper-White, J.J. and MacKay, Michael, E., Rheological properties of poly(lac-tides). Effect of molecular weight and temperature on the viscoelasticity ofPoly(L-lactic acid), J. Polym. Sci., Part B: Polym. Phy., 37, 1803, 1999.
52. Ramkumar, D.H.S. and Bhattacharya, M., Steady shear and dynamic propertiesof biodegradable polyesters, Polym. Eng. Sci., 38, 1426, 1998.
53. Feng, Q. and Hanna, M.A., Rheological properties of amorphous and semicrys-talline polylactic acid polymers, Ind. Crops Prod., 10, 47, 1999.
54. Cicero, J.A., Dorgan, J.R., Dec, S.F., and Knauss, D.M., Phosphite stabilizationeffects on two-step melt-spun fibers of polylactide, Polym Degradation Stability,78, 95, 2002.
55. Gruber, P.R., Kolstad, J.J., Hall, E.S., Eichen-Conn, R.S., and Ryan, C.M., Melt-stable lactide polymer compositions and their manufacture, PCT Int. Appl., WO9407949, 1994.
56. Kang, S., Zhang, G., Aou, K., Hsu, S.L., Stidham, H.D., and Yang, X., An analysisof poly(lactic acid) with varying regio regularity, J. Chem. Phy., 118, 3430, 2003.
57. Schindler, A., and Harper, D., Polylactide. II., Viscosity-molecular weight rela-tionships and unperturbed chain dimensions., J. Polym. Sci., Polym. Chem. ed.,17, 2593, 1979.
58. Schindler, A., Hibionada, Y.M., and Pitt, C.G., Aliphatic polyesters. III.Molecular weight and molecular weight distribution in alcohol-initiated poly-merizations of ε-caprolactone, J. Polym. Sci., Polym. Chem., 20, 319, 1982.
59. Dorgan, J.R., Williams, J.S., and Lewis, D.N., Melt rheology of poly(lactic acid):Entanglement and chain architecture effects, J. Rheol., 43, 1141, 1999.
60. Zhao, Y., Cai, Q., Jiang, J., Shuai, X., Bei, J., Chen, C., and Xi, Fu., Synthesis andthermal properties of novel star-shaped poly(L-lactide)s with starburstPAMAM-OH dendrimer macroinitiator, Polymer, 43, 5819, 2002.
61. Drumright, R., Barker, H., Beaudoin, D., Berler, N., Broomall, C., Leibig, C.,Hartmann, M., Henton, D., McCarthy, K., Olson, M., Randall, J., and Syverud,K., Rheology modification of poly(lactic acid), ACS Regional Conference, GrandRapids, MI, 2001.
62. Tasaka, F., Ohya, Y., and Ouchi, T., One-pot synthesis of novel branched poly-lactide through the copolymerization of lactide with mevalonolactone,Macromol. Rapid Commun., 22, 820, 2002.
572 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:58 AM Page 572

63. Drumright, R.E., Hartmann, M., and Wolf, R., Copolymers of monocyclic estersand carbonates and methods for making same, Int. Appl., WO 02/100921A1,Dec. 19, 2002.
64. Gruber, P.R., Kolstad, J.J., Witzke, D.R., Hartmann, M.H., and Brosch, A.L.,Viscosity-Modified Lactide Polymer Composition and Process for TheirManufacture, U.S. Patent 5,594,095, 1997.
65. Gruber, P.R., Kolstad, J.J., Ryan, C.M., Hall, E.S., and Eichen-Conn, R.S., Paperhaving a melt-stable lactide polymer coating and process for manufacture, U.S.Patent 5,338,822, 1995.
66. Lehermeier, H.J. and Dorgan, J.R., Melt rheology of poly(lactic acid): conse-quences of blending chain architectures., Polym. Eng. Sci., 41, 2172, 2001.
67. Huda, M. S., Yasui, M., Mohri, N., Fujimura, T., and Kimura, Y., Dynamicmechanical properties of solution-cast poly(L-lactide) films, Mater. Sci. Eng., A:Structural Mater: Prop., Microstruct. Process., A333, 98, 2002.
68. Abe, H., Kikkawa, Y., Inoue, Y., and Doi, Y., Morphological and kinetic analysesof regime transition for poly[(S)-lactide] crystal growth, Biomacromolecules, 2,1007, 2001.
69. Baratian, S., Hall, E.S., Lin, J.S., Xu, R., and Runt, J., Crystallization and solid-state structure of random polylactide copolymers: poly(L-lactide-co-D-lac-tide)s, Macromolecules, 34, 4857, 2001.
70. Zell, M.T., Padden, B.E., Paterick, A.J., Hillmyer, M.A., Kean, R.T., Thakur,K.A.M., and Munson, E.J., Direct observation of stereodefect sites in semicrys-talline poly(lactide) using 13C solid-state NMR, J. Am. Chem. Soc., 120, 12672,1998.
71. Pyda, M., Buzin, A., Wunderlich, B., and Bopp, R.C., Morphology of poly(lacticacid) by AFM and calorimetry, Proceedings of the 30th NATAS Annual Conferenceon Thermal Analysis and Applications, 463, 2002.
72. Hoffman, D.J., Davis, G.T., and Lauritzen, J.I., Treatise on Solid State Chemistry,Vol. 3: Crystalline and Noncrystaline Solids, Plenum Press, New York, 1976, chap.7.
73. Tsuji, H. and Ikada, Y., Properties and morphologies of poly(L-lactide): 1.Annealing condition effects on properties and morphologies of poly(L-lactide),Polymer, 36, 2709, 1995.
74. Fischer, E.W., Sterzel, H.J., and Wegner, G., Investigation of the structure ofsolution grown crystals of lactide copolymers by means of chemicals reactions.,Kolloid Ze. Ze. fuer Polym., 251, 980, 1973.
75. Kolstad, J.J., Crystallization kinetics of poly(L-lactide-co-meso-lactide, J. Appl.Polym. Sci., 62, 1079, 1996.
76. Pluta, M. and Galeski, A., Crystalline and supermolecular structure of polylac-tide in relation to the crystallization method, J. Appl. Polym. Sci., 86, 1386, 2002.
77. Schmidt, S.C. and Hillmyer, M.A., Polylactide stereocomplex crystallites asnucleating agents for isotactic polylactide, J. Polym. Sci., Part B: Polym. Phy., 39,300, 2001.
78. Liao, K., Quan, D., Gao, J., Luo, B., and Lu, Z., Crystallization and Miscibility ofPoly(L-lactide)/Poly(DL-lactide) Blends, Institute of Polymer Science,Zhongshan University, Canton, People Republic of China.
Polylactic Acid Technology 573
1741_C16.qxd 2/11/2005 9:58 AM Page 573

79. Spinu, M., Jackson, C., Keating, M.Y., and Gardner, K.H., Material design inpoly(lactic acid) systems: block copolymers, star homo- and copolymers, andstereocomplexes, J. Macromol. Sci., Pure. Appl. Chem., A33, 1497, 1996.
80. Tsuji, H. and Ikada, Y., Crystallization from the melt of poly(lactide)s with dif-ferent optical purities and their blends, Macromol. Chem. Phy., 197, 3483, 1996.
81. Tsuji, H. and Ikada, Y., Stereocomplex formation between enantiomericpoly(lactic acid)s. XI. Mechanical properties and morphology of solution-castfilms, Polymer, 40, 6699, 1999.
82. Loomis, G.L. and Murdoch, J.R., Polylactide stereocomplexes, Polym. Prepr. 31,55, 1990.
83. Okihara, T., Kawaguchi, A., Tsuji, H., Hyon, S.H., Ikada, Y., and Katayama, K.,Lattice disorders in the stereocomplex of poly(L-lactide) and poly(D-lactide),Bull. Inst. Chem. Res., Kyoto Univ., 66, 271, 1988.
84. Tsuji, H., Horii, F., Hyon, S.H., and Ikada, Y., Stereocomplex formation betweenenantiomeric poly(lactic acid)s. 2. Stereocomplex formation in concentratedsolutions, Macromolecules, 24, 2719, 1991.
85. Okihara, T., Tsuji, M., Kawaguchi, A., Katayama, K., Tsuji, H., Hyon, S.H., andIkada, Y., Crystal structure of stereocomplex of poly(L-lactide) and poly(D-lac-tide), J. Macromol. Sci., Phy., 30, 119, 1991.
86. Tsuji, H., Hyon, S.H., and Ikada, Y., Stereocomplex formation between enan-tiomeric poly(lactic acid)s. 3. Calorimetric studies on blend films cast fromdilute solution, Macromolecules, 24, 5651, 1991.
87. Tsuji, H., Hyon, S.H., and Ikada, Y., Stereocomplex formation between enan-tiomeric poly(lactic acid)s. 4. Differential scanning calorimetric studies on pre-cipitates from mixed solutions of poly(D-lactic acid) and poly(L-lactic acid),Macromolecules, 24, 5657, 1991.
88. Tsuji, H., Hyon, S.H., and Ikada, Y., Stereocomplex formation between enan-tiomeric poly(lactic acids). 5. Calorimetric and morphological studies on thestereocomplex formed in acetonitrile solution, Macromolecules, 25, 2940, 1992.
89. Tsuji, H., Horii, F., Nakagawa, M., Ikada, Y., Odani, H., and Kitamaru, R.,Stereocomplex formation between enantiomeric poly(lactic acid)s. 7. Phasestructure of the stereocomplex crystallized from a dilute acetonitrile solution asstudied by high-resolution solid-state carbon-13 NMR spectroscopy,Macromolecules, 25, 4114, 1992.
90. Tsuji, H. and Ikada, Y., Stereocomplex formation between enantiomericpoly(lactic acid)s. 6. Binary blends from copolymers, Macromolecules, 25, 5719,1992.
91. Tsuji, H. and Ikada, Y., Stereocomplex formation between enantiomericpoly(lactic acids). 9. Stereocomplexation from the melt, Macromolecules, 26, 6918,1993.
92. Tsuji, H., Ikada, Y., Hyon, S.H., Kimura, Y., and Kitao, T., Stereocomplex forma-tion between enantiomeric poly(lactic acid). VIII. Complex fibers spun frommixed solution of poly (D-lactic acid) and poly (L-lactic acid), J. Appl. Polym.Sci., 51, 337, 1994.
93. Tsuji, H., In vitro hydrolysis of blends from enantiomeric poly(lactide)s part 1.Well-stereocomplexed blend and non-blended films, Polymer, 41, 3621, 2000.
574 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:58 AM Page 574

94. Tsuji, H. and Suzuki, M. In vitro hydrolysis of blends from enantiomericpoly(lactide)s. 2. Well-stereocomplexes fiber and film. Sen’i Gakkaishi, 57, 198,2001.
95. Kang, S., Aou, K., Pekrul, R., and Hsu, S.L., Crystallization of poly(lactic acid)stereocomplexes, 224th ACS National Meeting, Boston, MA, US, Abstr., Aug.18–22, 2002.
96. Kokturk, G., Serhatkulu, T.F., Cakmak, M., and Piskin, E., Evolution of phasebehavior and orientation in uniaxially deformed polylactic acid films, Polym.Eng. Sci., 42, 1619, 2002.
97. Puiggali, J., Ikada, Y., Tsuji, H., Cartier, L., Okihara, T., and Lotz, B., The frus-trated structure of poly(L-lactide), Polymer, 41, 8921, 2000.
98. Lee, J.K., Lee, K.H., and Jin, B.S., Structure development and biodegradabilityof uniaxially stretched poly(L-lactide), Eur. Polym. J., 37, 907, 2001.
99. Kister, G., Cassanas, G., Vert, M., Pauvert, B., and Terol, A., Vibrational analysisof poly(L-lactic acid), J. Raman Spectrosc., 26, 307, 1995.
100. Smith, P.B., Leugers, A., Kang, S., Yang, X., and Hsu, S.L., Raman characteriza-tion of orientation in poly(lactic acid) films, Macromol. Symp. PolymerizationProcesses Polym. Mater. II, 175, 81, 2001.
101. Smith, P.B., Leugers, A., Kang, S., Hsu, S.L., and Yang, X., An analysis of the cor-relation between structural anisotropy and dimensional stability for drawnpoly(lactic acid) films, J. Appl. Polym. Sci., 82, 2497, 2001.
102. Kang, S., Hsu, S.L., Stidham, H.D., Smith, P.B., Leugers, A.M., and Yang, X., ASpectroscopic analysis of poly(lactic acid) structure, Macromolecules, 34, 4542,2001.
103. Randall, J., McCarthy, K., Krueger, J., Smith, P., and Spruiell, J., Properties ofPolylactic Acid Optical Copolymers Achieved Through Stress InducedCrystallization, 58th Annual Technical Conference - Society of PlasticsEngineers, Vol. 3, 3728, 2000.
104. Cicero, J.A., Dorgan, J.R., Garrett, J., Runt, J., and Lin, J.S., Effects of moleculararchitecture on two-step, melt-spun poly(lactic acid) fibers, J. Appl. Polym. Sci.,86, 2839, 2002.
105. Cicero, J.A., Dorgan, J.R., Janzen, J., Garrett, J., Runt, J., and Lin, J.S.,Supramolecular morphology of two-step, melt-spun poly(lactic acid) fibers, J. Appl. Polym. Sci., 86, 2828, 2002.
106. Keller, A., Barham, P., and Wills, H., J. Polym. Sci., 13, 197, 1975.107. Lunt, J., Large-scale production, properties and commercial applications of
polylactic acid polymers, Polym. Degrad. Stability, 59, 145, 1998.108. Tsuji, H., Polylactides, in Biopolymers, Wiley-VCH, 2002, chap. 5.109. Pitt, C.G., Jeffcoat, A.R., and Zweidinger, R.A., Sustained drug delivery system.
I. The permeability of poly(caprolactone), poly (DL-lactic acid), and theircopolymers, J. Biomed. Mater. Res., 13, 497, 1979.
110. Siparsky, G., Voorhees, K., and Miao, F., J. Environ. Polym. Degrad., 6, 31, 1998.111. Lee, S., Kim, S.H., Han, Y., and Kim, Y., Synthesis and degradation of end-group-
functionalized polylactide, J. Polym. Sci., Part A: Polym. Chem., 39, 973, 2001.112. Joziasse, C.A.P., Grijpma, D.W., Bergsma, J.E., Cordewener, F.W., Bos, R.R.M.,
and Pennings, A.J., The influence of morphology on the hydrolytic degradation
Polylactic Acid Technology 575
1741_C16.qxd 2/11/2005 9:58 AM Page 575

of as-polymerized and hot-drawn poly(L-lactide), Colloid Polym. Sci., 276, 968,1998.
113. Grizzi, I., Garreau, H., Li, S., and Vert, M., Hydrolytic degradation of devicesbased on poly(DL-lactic acid) size-dependence, Biomaterials, 16, 305, 1995.
114. Nakamura, T., Hitomi, S., Watanabe, S., Shimizu, Y., Jamshidi, K., Hyon, S.H.,and Ikada, Y., Bioabsorption of polylactides with different molecular properties,J. Biomed. Mater. Res., 23, 1115, 1989.
115. Vert, M., Li, S., Garreau, H., Mauduit, J., Boustta, M., Schwach, G., Engel, R., andCoudane, J., Complexity of the hydrolytic degradation of aliphatic polyesters,Ange. Makromol. Chem., 247, 239, 1997.
116. Mauduit, J., Perouse, E., and Vert, M., Hydrolytic degradation of films preparedfrom blends of high and low molecular weight poly(DL-lactic acids), J. Biomed.Mater. Res., 30, 201, 1996.
117. Hyon, S., Jamshidi, K., and Ikada, Y., Effects of residual monomer on the degra-dation of DL-lactide polymer, Polym. Int., 46, 196, 1998.
118. Iwata, T. and Doi, Y., Alkaline hydrolysis of solution-grown poly(L-lactic acid)single crystals, Sen’i Gakkaishi, 57, 172, 2001.
119. Tsuji, H. and Nakahara, K., Poly(l-lactide). IX. Hydrolysis in acid media, J. Appl.Polym. Sci., 86, 186, 2002.
120. Makino, K. et al., Preparation and in vitro degradation properties of polylactidemicrocapsules, Chem. Pharm. Bull., 33, 1195, 1985.
121. Yuan, X., Mak, A., and Yao, K., In vitro degradation of poly(L-lactic acid) fibersin phosphate buffered saline, J. Appl. Polym. Sci., 85, 936, 2002.
122. Tsuji, H. and Suzuyoshi, K., Environmental degradation of biodegradable poly-esters 2. Poly(epsiln.-caprolactone), poly[(R)-3-hydroxybutyrate], and poly(L-lac-tide) films in natural dynamic seawater, Polym. Degrad. Stability, 75, 357, 2002.
123. Tsuji, H., Nakahara, K., and Ikarashi, K., Poly(L-lactide), 8a. High-temperaturehydrolysis of poly(L-lactide) films with different crystallinities and crystallinethicknesses in phosphate-buffered solution, Macromol. Mater. Eng., 286, 398,2001.
124. Iannace, S., Maffezzoli, A., Leo, G., and Nicolais, L., Influence of crystal andamorphous phase morphology on hydrolytic degradation of PLLA subjected todifferent processing conditions, Polymer, 42, 3799, 2001.
125. Amass, W., Amass, A., and Tighe, B., A review of biodegradable polymers: uses,current developments in the synthesis and characterization of biodegradablepolyesters, blends of biodegradable polymers and recent advances in biodegra-dation studies, Polym. Int., 47, 89, 1998.
126. Shih, C., A graphical method for the determination of the mode of hydrolysis ofbiodegradable polymers, Pharm. Res., 12, 2036, 1995.
127. Zhang, X., Wyss, U., Pichora, D., and Goosen, M., Investigation of poly(lacticacid) degradation, J. Bioact. Compat. Polym., 9, 80, 1994.
128. Li, S.M. and Vert, M., Degradable Polymers: Principles and Applications, Chapman& Hall, London, 1995.
129. Sharp, J. S., Forrest, J.A., and Jones, R.A.L., Swelling of poly(DL-lactide) andpolylactide-co-glycolide in humid environments. Macromolecules, 34, 8752,2001.
576 Natural Fibers, Biopolymers, and Biocomposites
1741_C16.qxd 2/11/2005 9:58 AM Page 576

130. McCarthy, K., Cargill Dow LLC product literature, Minnetonka, Minnesota,1998.
131. Siparsky, G.L., Voorhees, K.J., Dorgan, J.R., and Schilling, K., Water transport inpolylactic acid (PLA), PLA/polycaprolactone copolymers, and PLA/polyethyl-ene glycol blends, J. Environ. Polym. Degrad., 5, 125, 1997.
132. Li, S. and McCarthy, S., Further investigations on the hydrolytic degradation ofpoly (DL-lactide). Biomaterials, 20, 35, 1999.
133. Burg, K.J.L. and Shalaby, S.W., Water fugacity in absorbing polymers, J. Biomed.Mater. Res., 38, 337, 1997.
134. Schmitt, E.A., Flanagan, D.R., and Linhardt, R.J., Importance of distinct waterenvironments in the hydrolysis of poly(DL-lactide-co-glycolide),Macromolecules, 27, 743, 1994.
135. Renstad, R., Karlsson, S., Sandgren, A., and Albertsson, A., Influence of pro-cessing additives on the degradation of melt-pressed films of poly(ε-caprolac-tone) and poly(lactic acid), J. Environ. Polym. Degrad., 6, 209, 1998.
136. Li, S. and Vert, M., Hydrolytic degradation of coral/poly(DL-lactic acid) biore-sorbable material. J. Biomater. Sci., Polymer ed., 7, 817, 1996.
137. Fischer, E.W., Structure of partially crystalline polymer systems, Prog. ColloidPolym. Sci., 57, 149, 1975.
138. Nijenhuis, A.J., Grijpma, D.W., and Pennings, A.J., Highly crystalline as-poly-merized poly(L-lactide), Polym. Bull., 26, 71, 1991.
139. Tsuji, H. and Miyauchi, S., Poly(L-lactide): VI effects of crystallinity on enzy-matic hydrolysis of poly(L-lactide) without free amorphous region, Polym.Degrad. Stability, 71, 415, 2000.
140. Tsuji, H. and Miyauchi, S., Poly(L-lactide): 7. enzymatic hydrolysis of free andrestricted amorphous regions in poly(L-lactide) films with different crystallini-ties and a fixed crystalline thickness, Polymer, 42, 4463, 2001.
141. United States Environmental Protection Agency Presidential Green ChemistryChallenge, Alternate Solvents/Reaction Conditions Award, June 2002.
Polylactic Acid Technology 577
1741_C16.qxd 2/11/2005 9:58 AM Page 577

1741_C16.qxd 2/11/2005 9:58 AM Page 578


















