
Pneumonia and Gut Bacteria M2b Monocytes Provoke Bacterial
10
of April 9, 2018. This information is current as Sepsis in Alcoholics Associated - Pneumonia and Gut Bacteria M2b Monocytes Provoke Bacterial Suzuki Suzuki, Makiko Kobayashi, Kazuhide Higuchi and Fujio Ito, Tomoki Nishiguchi, Melanie C. Garcia, Sumihiro Yusuke Tsuchimoto, Akira Asai, Yasuhiro Tsuda, Ichiaki ol.1501369 http://www.jimmunol.org/content/early/2015/10/31/jimmun published online 2 November 2015 J Immunol average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2015 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on April 9, 2018 http://www.jimmunol.org/ Downloaded from by guest on April 9, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Pneumonia and Gut Bacteria M2b Monocytes Provoke Bacterial

of April 9, 2018.This information is current as
Sepsis in AlcoholicsAssociated−Pneumonia and Gut Bacteria
M2b Monocytes Provoke Bacterial
SuzukiSuzuki, Makiko Kobayashi, Kazuhide Higuchi and FujioIto, Tomoki Nishiguchi, Melanie C. Garcia, Sumihiro Yusuke Tsuchimoto, Akira Asai, Yasuhiro Tsuda, Ichiaki
ol.1501369http://www.jimmunol.org/content/early/2015/10/31/jimmun
published online 2 November 2015J Immunol
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2015 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
M2b Monocytes Provoke Bacterial Pneumonia and GutBacteria–Associated Sepsis in Alcoholics
Yusuke Tsuchimoto,* Akira Asai,* Yasuhiro Tsuda,* Ichiaki Ito,† Tomoki Nishiguchi,†
Melanie C. Garcia,† Sumihiro Suzuki,‡ Makiko Kobayashi,† Kazuhide Higuchi,* and
Fujio Suzuki†
Chronic alcohol consumption markedly impairs host antibacterial defense against opportunistic infections. g-irradiated NOD-SCID
IL-2Rgnull mice inoculated with nonalcoholic PBMCs (control PBMC chimeras) resisted Klebsiella pneumonia and gut bacteria-
associated sepsis, whereas the chimeras created with alcoholic PBMCs (alcoholic PBMC chimeras) were very susceptible to these
infections. M1 monocytes (IL-12+IL-102CD1632CD14+ cells), major effector cells in antibacterial innate immunity, were not induced
by a bacterial Ag in alcoholic PBMC cultures, and M2b monocytes (CCL1+CD163+CD14+ cells), which predominated in alcoholic
PBMCs, were shown to be inhibitor cells on the Ag-stimulated monocyte conversion from quiescent monocytes to M1 monocytes.
CCL1, which functions to maintain M2b macrophage properties, was produced by M2b monocytes isolated from alcoholic PBMCs.
These M2b monocytes reverted to quiescent monocytes (IL-122IL-102CCL12CD1632CD14+ cells) in cultures supplemented with
CCL1 antisense oligodeoxynucleotide, and the subsequent quiescent monocytes easily converted to M1 monocytes under bacterial Ag
stimulation. Alcoholic PBMC chimeras treated with CCL1 antisense oligodeoxynucleotide were resistant against pulmonary infec-
tion by K. pneumoniae and sepsis stemming from enterococcal translocation. These results indicate that a majority of monocytes
polarize to an M2b phenotype in association with alcohol abuse, and this polarization contributes to the increased susceptibility of
alcoholics to gut and lung infections. Bacterial pneumonia and gut bacteria-associated sepsis, frequently seen in alcoholics, can be
controlled through the polarization of macrophage phenotypes. The Journal of Immunology, 2015, 195: 000–000.
Alcohol abuse is associated with a variety of health prob-lems (e.g., liver diseases and cancer), and alcoholics havean increased risk for serious infectious complications,
such as pulmonary and gut bacteria-associated infections (1–5). Themajority of the causative pathogens in these infections in alcoholicsare microbes normally found in the upper respiratory tracts(staphylococci, Klebsiella) and the upper and lower intestine (en-terococci, Clostridium, Pseudomonas) (3–6). Because such infec-tions do not usually develop in healthy individuals, certain immunedysfunctions related to alcohol abuse are considered the underlyingcauses of the increased susceptibility of alcoholics to these infec-tions. In fact, various host antibacterial immune functions arestrongly influenced by chronic alcohol consumption (7–22). De-creased immune functions associated with chronic alcohol con-sumption include granulopoiesis (8), tissue recruitment of neutro-phils (9), TLR responsiveness of macrophages (10), and productionof IL-12 (11). In addition, decreased macrophage phagocytosis(12–14) and an increased Th2 response (11, 15, 16) were dem-
onstrated in alcoholics. A decrease in the Ag-presenting abilitiesof dendritic cells (17), perforin/granzyme expression, IFN-g pro-duction by conventional NK cells (18, 19), and Th1/Th17 responses(20–22) also were reported. All of these immune dysfunctions maybe involved in the increased susceptibility of alcoholics to theseinfections.Macrophages are highly plastic and heterogeneous (23). It has
been widely accepted that at least two types of macrophages withdistinct phenotypes can be found in the process of infection. M1macrophages (IL-102IL-12+CD1632CD14+ cells) are known tobe major antibacterial effector cells on the innate immunity.Therefore, in healthy subjects, gut bacteria–associated sepsis andbacterial pneumonia are commonly controlled by M1 macro-phages generated in association with invading pathogens (24–27).However, M1 macrophages are not easily generated in hostswhose M2 macrophages predominate (28). In our previous studies(29), M2b macrophages were predominantly isolated from thebacterial translocation sites (mesenteric lymph nodes [MLNs] andlamina propria [LP]) of chronic alcohol-consuming (CAC) mice,and M1 macrophages were not induced by bacterial Ags in cul-tures of F4/80+ cells derived from MLNs and LP of CAC mice.M2b macrophages were shown to inhibit the conversion of qui-escent macrophages to M1 macrophages. CCL1 was shown to beessential for the prolongation of M2b macrophages (30), and M2bmacrophages were reverted to quiescent macrophages posttreat-ment with CCL1 antisense oligodeoxynucleotide (ODN) (30, 31).The increased susceptibility of CAC mice to bacterial pneumoniaand sepsis stemming from enterococcal translocation was im-proved to a level shown in normal mice posttreatment with CCL1antisense ODN (29).These murine studies suggest the possibility that pulmonary and
gut bacteria–associated opportunistic infections are controllable inalcoholics through macrophage polarization from M2b macrophages
*Second Department of Internal Medicine, Osaka Medical College, Takatsuki 569-8686, Japan; †Department of Internal Medicine, The University of Texas MedicalBranch, Galveston, TX 77555; and ‡Department of Biostatistics, University of NorthTexas Health Science Center, Fort Worth, TX 76107
ORCID: 0000-0001-6028-9561 (A.A.).
Received for publication June 16, 2015. Accepted for publication September 10,2015.
Address correspondence and reprint requests to Dr. Fujio Suzuki, Department ofInternal Medicine, The University of Texas Medical Branch, 301 University Boule-vard, Galveston, TX 77555-0435. E-mail address: [email protected]
Abbreviations used in this article: CAC, chronic alcohol consuming; LP, laminapropria; MLN, mesenteric lymph node; NSG, NOD-SCID IL-2Rgnull; gNSGmice, NSG mice exposed to whole-body gamma radiation (4 Gy); ODN, oligo-deoxynucleotide.
Copyright� 2015 by The American Association of Immunologists, Inc. 0022-1767/15/$25.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1501369
Published November 2, 2015, doi:10.4049/jimmunol.1501369 by guest on A
pril 9, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

to M1 macrophages in local sites of bacterial infections. However,the functions and properties (phenotypes) of macrophages dis-tributed in alcoholics remain unclear. In this study, we investigatedthe cellular properties and antibacterial functions of macrophagesof alcoholics using peripheral blood monocytes and a chimeramodel of alcoholics against infectious complications caused byKlebsiella pneumonia and gut bacterial translocation. K. pneu-moniae is one of the causative pathogens of pneumonia, which isthe most common infection in alcoholics (32). Enterococcusfaecalis, a typical microbe in the gut of humans, translocates fromthe intestine and causes severe infections in immunocompromisedhosts (33, 34).
Materials and MethodsAlcoholics and nonalcoholics
The study was carried out in 25 individuals (23 male and 2 female, patients#1–25) who were diagnosed as alcoholics at one of five major hospitals inwestern Japan from June 2012 to March 2015. These patients, on average,consumed 257.4 6 34.3 g/d of ethanol for 12 to 56 y (35.3 6 3.0 y)(Table I). Alcoholics who had any malignancy, heart failure, chronickidney disease, liver cirrhosis, or infection were excluded from the study.Eligible patients gave written informed consent to participate in the study,and the protocol was approved by the ethics committees of the hospitals.Ten nonalcoholic individuals (7 male, 3 female; mean age: 36.1 y, healthydonors #1–10) were enrolled as controls (Table I). Nonalcoholics enrolledin the study did not consume $10 g/d ethanol for $20 y.
Bacteria, reagents, and media
K. pneumoniae (43816 strain) and E. faecalis (29212 strain) were pur-chased from the American Type Culture Collection (Manassas, VA). Inaccordance with the guidelines for assuring the quality of medical mi-crobiological culture media, bacteria were grown in Trypticase soy brothfor 18 h at 37˚C in aerobic conditions before being used in infectionexperiments. rIL-10, rIL-12, rCCL1, rCCL17, and rCXCL13 were ob-tained from PeproTech (San Diego, CA). mAbs for IL-10, IL-12, CCL1,CCL17, and CXCL13 were obtained from R&D Systems (San Diego, CA)and BD Biosciences (San Jose, CA). Biotin-conjugated anti-CD163,allophycocyanin-conjugated anti-CD163, FITC-conjugated anti-CD14,anti-CCL1, anti-CCL17, anti-CXCL13, and anti–IL-10 mAbs; AlexaFluor 647 anti-human CD163 Ab; Alexa Fluor 488 anti-human CD14 Ab;and DAPI Fluoromount-G were purchased from eBioscience (San Diego,CA). Streptavidin Particles Plus - DM, Cytofix/Cytoperm solution, isotype-control mAbs, and IMag buffer were purchased from BD Biosciences.Heat-killed K. pneumoniae was prepared by heating bacteria at 65˚C for30 min. Their inactivated cell properties were confirmed by culturingthe pathogen on agar plates. This Ag was stored at 280˚C until needed.Phosphorothioated single-stranded nucleic acid that inhibits the synthesis ofhuman CCL1 (59-CAGCTAGCAGCAAGCACA-39) and phosphorothioatedscrambled ODN (59-ACCACTTGGTGTATTTGC-39) were purchased fromSigma-Proligo (The Woodlands, TX). For cell cultivation, RPMI 1640
medium supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine,and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin) (com-plete medium) was used.
Humanized murine chimeras
Nine- to twelve-week-old pathogen-free, male NOD-SCID IL-2Rgnull
(NSG) mice, purchased from The Jackson Laboratory (Bar Harbor, ME),were used. NSG mice are described as mice lacking functional T cells,B cells, and NK cells (35–39). Also, NSG mice are carriers of macro-phages with defective functions for phagocytosis, digestion, and Ag pre-sentation (35–39). All NSG mice were exposed to whole-body gammaradiation (4 Gy) to deplete neutrophils (gNSG mice). Neutrophils did notrecover in these mice for 2 wk after the irradiation. These mice were usedin all experiments throughout the study. PBMCs were isolated from theheparinized blood of alcoholic patients and healthy donors by Ficoll-Hypaque sedimentation. These PBMC preparations (1 3 106 cells) wereinoculated i.v. into gNSG mice; mice are designated as alcoholic PBMCchimeras (gNSG mice inoculated with alcoholic PBMCs) and controlPBMC chimeras (gNSG mice inoculated with nonalcoholic PBMCs).Commonly, 4.1–13.4 3 106 PBMCs were isolated from 8 ml blood drawnfrom alcoholics and nonalcoholics; there was not a significant difference inthe numbers of PBMCs in blood between the two groups. In chimeras, theinoculated cells spread throughout the body within 2 d of inoculation andfunctioned for $2 wk. In some experiments, humanized murine chimeraswere created with PBMCs from nonalcoholics #8, #9, and #10 and weredepleted previously of CD3+, CD19+, CD56+, or CD14+ cells, respectively.For the depletion of specific cell populations, 5 3 106 cells/ml PBMCssuspended in MagCellect buffer were incubated with magnetic beadscoated with anti-human CD3, anti-human CD19, anti-human CD56, oranti-human CD14 mAb for 30 min at 4˚C and then certain cell populationswere magnetically removed. After washing, the above procedure (mAbtreatment and magnetic removal) was repeated one more time. By thisprocedure, $92% of the cell populations were depleted from PBMCs.Also, humanized murine chimeras were created with CD14+ cells isolatedfrom PBMCs of nonalcoholics #1–#7. These chimeras were used in theappropriate experiments.
Preparation of human monocytes
In all experiments, peripheral blood monocytes were used as effector cellsin host antibacterial innate immunity. Monocytes (CD14+ cells) wereisolated from PBMCs as follows: 5 3 106 PBMCs/ml suspended inMagCellect buffer were incubated with magnetic beads coated with anti-human CD14 mAb for 30 min at 4˚C and then CD14+ cells were mag-netically harvested. The number of monocytes isolated from 8 ml bloodwas 1–23 106 cells in nonalcoholics and 0.5–1.83 106 cells in alcoholics.The purity of CD14+ cells isolated by this procedure was routinely .97%,and the viability of these cells was .99%.
Characterization of CD14+ cells isolated from alcoholicPBMCs
To determine the cellular properties of peripheral blood CD14+ cells fromalcoholics, 1 3 106 CD14+ cells/ml isolated from alcoholic #1–22 PBMCsand nonalcoholic #1–10 PBMCs were cultured for 48 h with (for the assayof IL-12) or without (for the assay of IL-10, CCL17, CCL1, and CXCL13)105 heat-killed K. pneumoniae. Culture fluids were harvested and assayedfor IL-12, IL-10, CCL17, CCL1, and CXCL13 using ELISA. IL-12 isa biomarker of M1 macrophages; IL-10 is a biomarker of M2a, M2b, andM2c macrophages; CCL17 is a biomarker of M2a macrophages; CCL1 isa biomarker of M2b macrophages; and CXCL13 is a biomarker of M2cmacrophages (40, 41). The minimum detection limits for the above cyto-kines were 4–13 pg/ml. A portion of CD14+ cell preparations derived frompatient #1–10 PBMCs and healthy donor #1–10 PBMCs was stained withPE-conjugated anti-human CD163 mAb and fixed in 2% paraformaldehydeto determine the percentage of CD163+ cells (M2 monocytes). For intra-cellular staining, CD14+ cells were permeabilized with Cytofix/Cytopermsolution at 4˚C for 20 min, washed with a Perm/Wash solution, and stainedwith FITC-conjugated anti-human CCL17 mAb, anti-human CCL1 mAb,or anti-human CXCL13 mAb. CCL17+CD163+ cells, CCL1+CD163+ cells,and CXCL13+CD163+ cells in the CD14+ cell preparation were analyzedusing FACSCanto (Becton-Dickinson) and FlowJo software. Also, a por-tion of CD14+ cells isolated from patient #22 PBMCs and healthy donor#10 PBMCs was stained with Alexa Fluor 647 anti-human CD163 Ab,Alexa Fluor 488 anti-human CD14 Ab, or DAPI Fluoromount-G, andCD14+CD163+ cells were analyzed by fluorescence microscopy. M2bmonocytes were identified as IL-10–producing IL-12–nonproducingCCL1+CD163+CD14+ cells.
Table I. Alcoholics and nonalcoholics enrolled in this study
AlcoholicsNonalcoholics
(Healthy Donors)
Total no. of subjects 25 10Male/female (n) 23/2 7/3Age (y)a 56.8 6 14.7 36.1 6 4.2Consumption of
ethanol (g/d)a257.4 6 34.3 6.6 6 1.2
Duration of alcoholconsumption (y)a
35.3 6 3.0 NA
Albumin (g/dl)a 3.9 6 0.2 4.4 6 0.1Total bilirubin (mg/dl)a 1.1 6 0.2 0.7 6 0.2AST (IU/l)a 101.6 6 23.7 18.3 6 5.0ALT (IU/l)a 62.2 6 10.7 15.9 6 5.7g-GTP (IU/l)a 223.7 6 38.7 21.6 6 5.6Platelet count (3 104 ml)a 20.2 6 1.8 27.3 6 5.3
aData are mean 6 SD.ALT, alanine aminotransferase; AST, aspartate aminotransferase; GTP, glutamyl
transpeptidase; NA, not applicable.
2 M2b MONOCYTES PROVOKE INFECTIONS IN ALCOHOLICS
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
In some experiments, 5 3 105 CD14+ cells/ml isolated from alcoholic#23, #24, and #25 PBMCs or nonalcoholic #8, #9, and #10 PBMCs werecultured with 5 mg/ml CCL1 antisense ODN or scrambled ODN for 48 h.Harvested cells were washed and stimulated with heat-killed K. pneumo-niae (105 cells/ml) for 24 h. Culture fluids were harvested and assayed forIL-10 and IL-12 by ELISA, and the remaining cells were tested for theirbactericidal activities, as follows: cells were suspended in antibiotic-freeRPMI 1640 medium supplemented with 10% FBS and exposed to 5 3 104
CFU/well K. pneumoniae. Three hours postincubation, cells were lysedwith 0.1% Triton X-100 (Sigma-Aldrich). Serial 10-fold dilutions of thesehomogenates were plated on tryptic soy agar. As a control, pathogen wasincubated alone. The number of colonies was counted postincubation for24 h at 37˚C. The following formula was applied to determine the cytocidalpercentages of cells: (1 2 test group CFU)/(control group CFU) 3 100.M1 macrophages were identified as IL-12–producing IL-10–nonproducingCD14+ cells.
Infection experiments in chimeras
K. pneumoniae pulmonary infection. In the first series of experiments, threehumanized murine chimeras were created with 104, 105, or 106 cells/mouseof nonalcoholic #1 PBMCs. Two days after the cell inoculation, thesechimeras were anesthetized with ketamine and xylazine. Their tracheaswere exposed, and 30 ml bacterial suspension (102 CFU) was injected viaa 26-gauge needle. To determine the percentage of survival, chimeras weremonitored twice a day for 7 d postinfection. This set of experiments wasrepeated six more times using PBMCs from nonalcoholics #2–7. Theresults obtained from seven experiments were combined, and survival rateswere calculated. In the next series of experiments, three control PBMCchimeras were created with 106 cells/mouse of nonalcoholic #1 PBMCsand infected with 102, 103, or 104 CFU K. pneumoniae, as described above.This set of experiments was repeated six more times using nonalcoholic#2–7 PBMCs, and the results obtained from seven experiments werecombined. To test the susceptibility of alcoholics to Klebsiella pneumonia,PBMCs were isolated from the blood of alcoholic #1 when first arriving atthe hospital and inoculated into a gNSG mouse (106 cells/mouse, an al-coholic PBMC chimera). At same time, the same number of PBMCsisolated from nonalcoholic #1 was inoculated into a gNSG mouse (13 106
cells/mouse, control PBMC chimera). Then, these chimeras were infectedwith 102 CFU K. pneumoniae, as shown above. This pathogenic dosecorresponds to ,0.01 LD50 in control PBMC chimeras and 10 LD50 inalcoholic PBMC chimeras. This set of experiments was repeated two moretimes using alcoholic #2 and #3 PBMCs or nonalcoholic #2 and #3PBMCs. All chimeras in each group were sacrificed 1 d postinfection, andthe numbers of pathogens in the lung, spleen, and kidneys of the chimeraswere determined as described below. Also, experiments were repeatedusing alcoholic #4–9 PBMCs and nonalcoholic #4–9 PBMCs to determinethe bacterial growth in the chimeras 2 d (patients #4, #5, and #6) and 3 d(patients #7, #8, and #9) postinfection. The results obtained from three
chimeras each for 1, 2, and 3 d postinfection were combined. To determinethe effect of CCL1 antisense ODN treatment on the antibacterial resistanceof alcoholic PBMC chimeras against K. pneumoniae infection, two chi-meras were created using alcoholic #20 PBMCs. One chimera was treatedwith CCL1 antisense ODN (10 mg, s.c.) twice a day for 3 d, and anotherchimera was treated with scrambled ODN in the same fashion. For datainterpretation (additional controls), two control PBMC chimeras (createdwith nonalcoholic #8 PBMCs) were treated with CCL1 antisense ODN(10 mg, s.c.) and scrambled ODN (10 mg, s.c.), respectively, twice a dayfor 3 d. Then, these four chimeras were infected with 102 CFU K. pneu-moniae and continuously treated with CCL1 antisense ODN or scrambledODN on the same day and 1 d postinfection. This set of experiments wasrepeated two more times using alcoholic #21 and #22 PBMCs and non-alcoholic #9 and #10 PBMCs. The results obtained from three chimeraswere combined. The efficacy of the treatment was evaluated by bacterialgrowth in organs (lung and kidneys) of alcoholic PBMC chimeras treatedwith CCL1 antisense ODN compared with those of alcoholic PBMCchimeras treated with scrambled ODN. To measure the quantity of bac-teria, organ specimens were weighed and disrupted in 2 ml PBS usinga Bruikman homogenizer. A serial 10-fold dilution of the organ homo-genates was plated onto blood agar plates and incubated for 24 h at 37˚C.The colonies were counted, and the number of bacteria/g of organ wasdetermined.
Enterococcal translocation. Both groups of chimeras were infected orallywith E. faecalis after decontamination with a mixture of three antibioticsand oral treatment with a proton-pump inhibitor (lansoprazole), as pre-viously described (29, 31). In decontaminated mice, the numbers ofmicrobiota in the gut were negligibly small. A proton-pump inhibitorwas used for the stabilization of oral enterococcal infection. AlcoholicPBMC chimeras were created with alcoholic #10, #11, and #12 PBMCs,and control PBMC chimeras were created with nonalcoholic #4, #5, and#6 PBMCs, as described for K. pneumoniae infection. These chimeraswere orally exposed to E. faecalis at a dose of 106 CFU. Two dayspostinfection, all chimeras were sacrificed, MLNs and liver were re-moved, and the numbers of pathogens in these organs were determinedby the colony-counting method. For experiments with CCL1 antisenseODN treatment, chimeras created with alcoholic #17, #18, and #19PBMCs or nonalcoholic #8, #9, and #10 PBMCs were used, under thesame protocol as the experiments of K. pneumoniae infection. The se-verity of infectious complications caused by E. faecalis translocation wasevaluated by growth of bacteria in the primary infection site organ(MLNs) and liver of alcoholic PBMC chimeras treated with CCL1 an-tisense ODN versus scrambled ODN.
Statistical analyses
The mean 6 SE was computed, and data were analyzed using a Studentt test or Kaplan–Meier curve with a log-rank test. The results were con-sidered significant at p , 0.05.
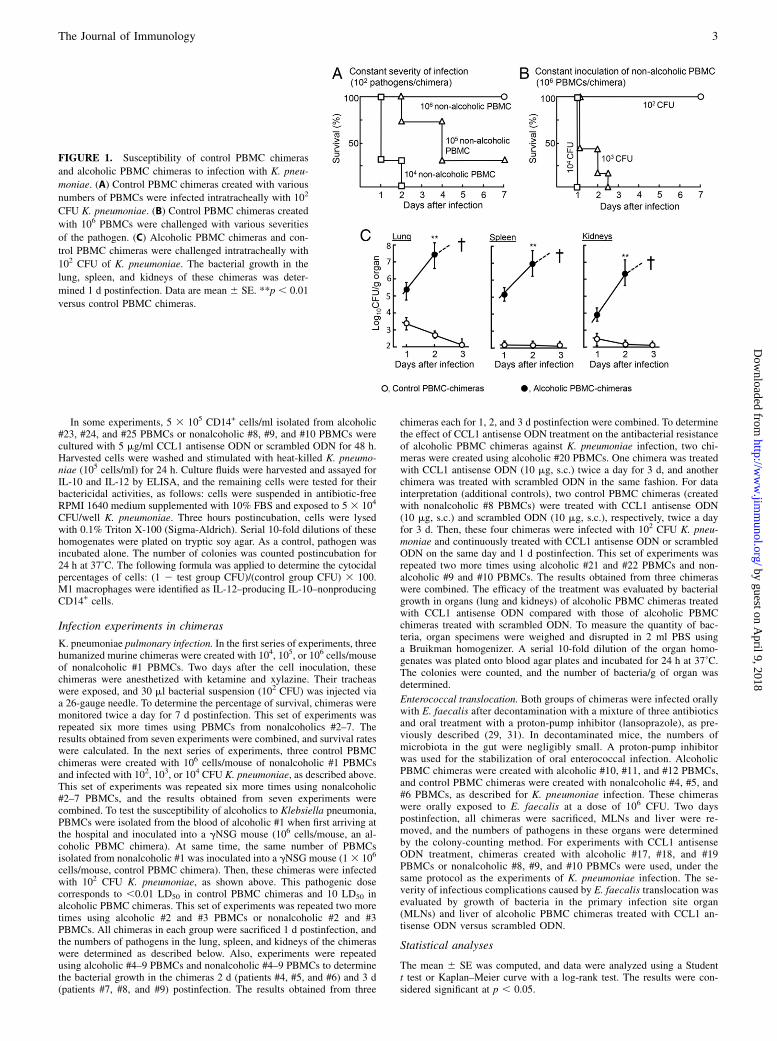
FIGURE 1. Susceptibility of control PBMC chimeras
and alcoholic PBMC chimeras to infection with K. pneu-
moniae. (A) Control PBMC chimeras created with various
numbers of PBMCs were infected intratracheally with 102
CFU K. pneumoniae. (B) Control PBMC chimeras created
with 106 PBMCs were challenged with various severities
of the pathogen. (C) Alcoholic PBMC chimeras and con-
trol PBMC chimeras were challenged intratracheally with
102 CFU of K. pneumoniae. The bacterial growth in the
lung, spleen, and kidneys of these chimeras was deter-
mined 1 d postinfection. Data are mean 6 SE. **p , 0.01
versus control PBMC chimeras.
The Journal of Immunology 3
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
ResultsSusceptibility of alcoholic PBMC chimeras to bacterialpneumonia and gut bacteria–associated sepsis
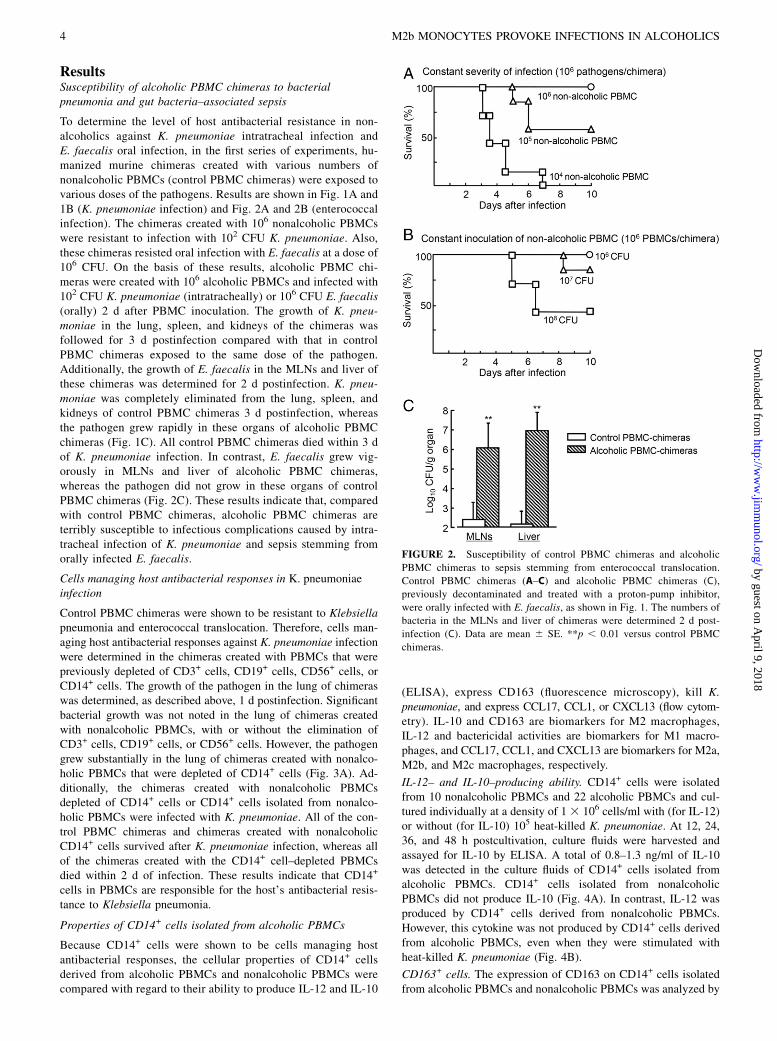
To determine the level of host antibacterial resistance in non-alcoholics against K. pneumoniae intratracheal infection andE. faecalis oral infection, in the first series of experiments, hu-manized murine chimeras created with various numbers ofnonalcoholic PBMCs (control PBMC chimeras) were exposed tovarious doses of the pathogens. Results are shown in Fig. 1A and1B (K. pneumoniae infection) and Fig. 2A and 2B (enterococcalinfection). The chimeras created with 106 nonalcoholic PBMCswere resistant to infection with 102 CFU K. pneumoniae. Also,these chimeras resisted oral infection with E. faecalis at a dose of106 CFU. On the basis of these results, alcoholic PBMC chi-meras were created with 106 alcoholic PBMCs and infected with102 CFU K. pneumoniae (intratracheally) or 106 CFU E. faecalis(orally) 2 d after PBMC inoculation. The growth of K. pneu-moniae in the lung, spleen, and kidneys of the chimeras wasfollowed for 3 d postinfection compared with that in controlPBMC chimeras exposed to the same dose of the pathogen.Additionally, the growth of E. faecalis in the MLNs and liver ofthese chimeras was determined for 2 d postinfection. K. pneu-moniae was completely eliminated from the lung, spleen, andkidneys of control PBMC chimeras 3 d postinfection, whereasthe pathogen grew rapidly in these organs of alcoholic PBMCchimeras (Fig. 1C). All control PBMC chimeras died within 3 dof K. pneumoniae infection. In contrast, E. faecalis grew vig-orously in MLNs and liver of alcoholic PBMC chimeras,whereas the pathogen did not grow in these organs of controlPBMC chimeras (Fig. 2C). These results indicate that, comparedwith control PBMC chimeras, alcoholic PBMC chimeras areterribly susceptible to infectious complications caused by intra-tracheal infection of K. pneumoniae and sepsis stemming fromorally infected E. faecalis.
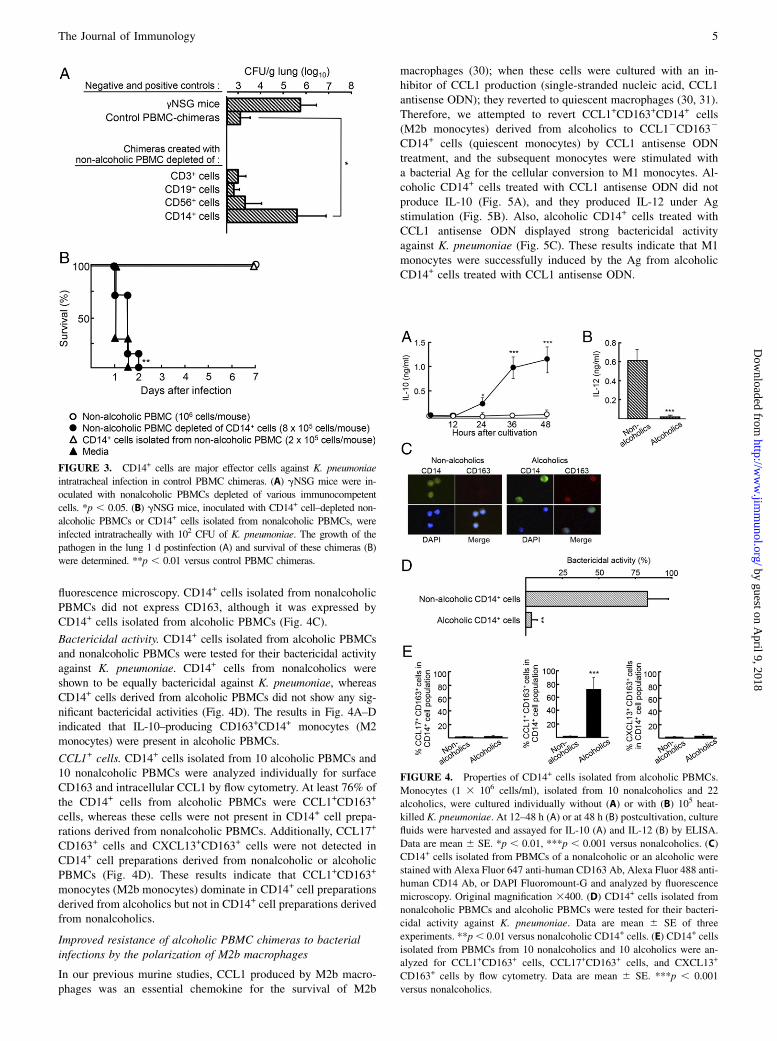
Cells managing host antibacterial responses in K. pneumoniaeinfection
Control PBMC chimeras were shown to be resistant to Klebsiellapneumonia and enterococcal translocation. Therefore, cells man-aging host antibacterial responses against K. pneumoniae infectionwere determined in the chimeras created with PBMCs that werepreviously depleted of CD3+ cells, CD19+ cells, CD56+ cells, orCD14+ cells. The growth of the pathogen in the lung of chimeraswas determined, as described above, 1 d postinfection. Significantbacterial growth was not noted in the lung of chimeras createdwith nonalcoholic PBMCs, with or without the elimination ofCD3+ cells, CD19+ cells, or CD56+ cells. However, the pathogengrew substantially in the lung of chimeras created with nonalco-holic PBMCs that were depleted of CD14+ cells (Fig. 3A). Ad-ditionally, the chimeras created with nonalcoholic PBMCsdepleted of CD14+ cells or CD14+ cells isolated from nonalco-holic PBMCs were infected with K. pneumoniae. All of the con-trol PBMC chimeras and chimeras created with nonalcoholicCD14+ cells survived after K. pneumoniae infection, whereas allof the chimeras created with the CD14+ cell–depleted PBMCsdied within 2 d of infection. These results indicate that CD14+
cells in PBMCs are responsible for the host’s antibacterial resis-tance to Klebsiella pneumonia.
Properties of CD14+ cells isolated from alcoholic PBMCs
Because CD14+ cells were shown to be cells managing hostantibacterial responses, the cellular properties of CD14+ cellsderived from alcoholic PBMCs and nonalcoholic PBMCs werecompared with regard to their ability to produce IL-12 and IL-10
(ELISA), express CD163 (fluorescence microscopy), kill K.pneumoniae, and express CCL17, CCL1, or CXCL13 (flow cytom-etry). IL-10 and CD163 are biomarkers for M2 macrophages,IL-12 and bactericidal activities are biomarkers for M1 macro-phages, and CCL17, CCL1, and CXCL13 are biomarkers for M2a,M2b, and M2c macrophages, respectively.
IL-12– and IL-10–producing ability. CD14+ cells were isolatedfrom 10 nonalcoholic PBMCs and 22 alcoholic PBMCs and cul-tured individually at a density of 1 3 106 cells/ml with (for IL-12)or without (for IL-10) 105 heat-killed K. pneumoniae. At 12, 24,36, and 48 h postcultivation, culture fluids were harvested andassayed for IL-10 by ELISA. A total of 0.8–1.3 ng/ml of IL-10was detected in the culture fluids of CD14+ cells isolated fromalcoholic PBMCs. CD14+ cells isolated from nonalcoholicPBMCs did not produce IL-10 (Fig. 4A). In contrast, IL-12 wasproduced by CD14+ cells derived from nonalcoholic PBMCs.However, this cytokine was not produced by CD14+ cells derivedfrom alcoholic PBMCs, even when they were stimulated withheat-killed K. pneumoniae (Fig. 4B).
CD163+ cells. The expression of CD163 on CD14+ cells isolatedfrom alcoholic PBMCs and nonalcoholic PBMCs was analyzed by
FIGURE 2. Susceptibility of control PBMC chimeras and alcoholic
PBMC chimeras to sepsis stemming from enterococcal translocation.
Control PBMC chimeras (A–C) and alcoholic PBMC chimeras (C),
previously decontaminated and treated with a proton-pump inhibitor,
were orally infected with E. faecalis, as shown in Fig. 1. The numbers of
bacteria in the MLNs and liver of chimeras were determined 2 d post-
infection (C). Data are mean 6 SE. **p , 0.01 versus control PBMC
chimeras.
4 M2b MONOCYTES PROVOKE INFECTIONS IN ALCOHOLICS
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
fluorescence microscopy. CD14+ cells isolated from nonalcoholicPBMCs did not express CD163, although it was expressed byCD14+ cells isolated from alcoholic PBMCs (Fig. 4C).
Bactericidal activity. CD14+ cells isolated from alcoholic PBMCsand nonalcoholic PBMCs were tested for their bactericidal activityagainst K. pneumoniae. CD14+ cells from nonalcoholics wereshown to be equally bactericidal against K. pneumoniae, whereasCD14+ cells derived from alcoholic PBMCs did not show any sig-nificant bactericidal activities (Fig. 4D). The results in Fig. 4A–Dindicated that IL-10–producing CD163+CD14+ monocytes (M2monocytes) were present in alcoholic PBMCs.
CCL1+ cells. CD14+ cells isolated from 10 alcoholic PBMCs and10 nonalcoholic PBMCs were analyzed individually for surfaceCD163 and intracellular CCL1 by flow cytometry. At least 76% ofthe CD14+ cells from alcoholic PBMCs were CCL1+CD163+
cells, whereas these cells were not present in CD14+ cell prepa-rations derived from nonalcoholic PBMCs. Additionally, CCL17+
CD163+ cells and CXCL13+CD163+ cells were not detected inCD14+ cell preparations derived from nonalcoholic or alcoholicPBMCs (Fig. 4D). These results indicate that CCL1+CD163+
monocytes (M2b monocytes) dominate in CD14+ cell preparationsderived from alcoholics but not in CD14+ cell preparations derivedfrom nonalcoholics.
Improved resistance of alcoholic PBMC chimeras to bacterialinfections by the polarization of M2b macrophages
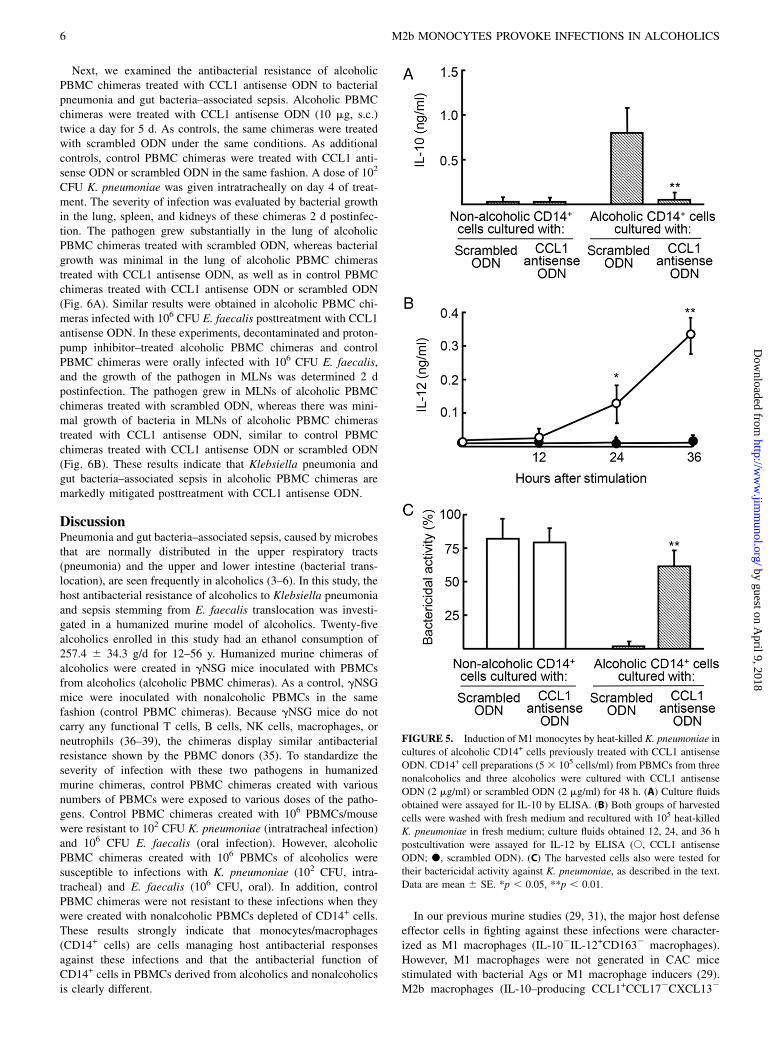
In our previous murine studies, CCL1 produced by M2b macro-phages was an essential chemokine for the survival of M2b
macrophages (30); when these cells were cultured with an in-hibitor of CCL1 production (single-stranded nucleic acid, CCL1antisense ODN); they reverted to quiescent macrophages (30, 31).Therefore, we attempted to revert CCL1+CD163+CD14+ cells(M2b monocytes) derived from alcoholics to CCL12CD1632
CD14+ cells (quiescent monocytes) by CCL1 antisense ODNtreatment, and the subsequent monocytes were stimulated witha bacterial Ag for the cellular conversion to M1 monocytes. Al-coholic CD14+ cells treated with CCL1 antisense ODN did notproduce IL-10 (Fig. 5A), and they produced IL-12 under Agstimulation (Fig. 5B). Also, alcoholic CD14+ cells treated withCCL1 antisense ODN displayed strong bactericidal activityagainst K. pneumoniae (Fig. 5C). These results indicate that M1monocytes were successfully induced by the Ag from alcoholicCD14+ cells treated with CCL1 antisense ODN.
FIGURE 3. CD14+ cells are major effector cells against K. pneumoniae
intratracheal infection in control PBMC chimeras. (A) gNSG mice were in-
oculated with nonalcoholic PBMCs depleted of various immunocompetent
cells. *p , 0.05. (B) gNSG mice, inoculated with CD14+ cell–depleted non-
alcoholic PBMCs or CD14+ cells isolated from nonalcoholic PBMCs, were
infected intratracheally with 102 CFU of K. pneumoniae. The growth of the
pathogen in the lung 1 d postinfection (A) and survival of these chimeras (B)
were determined. **p , 0.01 versus control PBMC chimeras.
FIGURE 4. Properties of CD14+ cells isolated from alcoholic PBMCs.
Monocytes (1 3 106 cells/ml), isolated from 10 nonalcoholics and 22
alcoholics, were cultured individually without (A) or with (B) 105 heat-
killed K. pneumoniae. At 12–48 h (A) or at 48 h (B) postcultivation, culture
fluids were harvested and assayed for IL-10 (A) and IL-12 (B) by ELISA.
Data are mean 6 SE. *p , 0.01, ***p , 0.001 versus nonalcoholics. (C)
CD14+ cells isolated from PBMCs of a nonalcoholic or an alcoholic were
stained with Alexa Fluor 647 anti-human CD163 Ab, Alexa Fluor 488 anti-
human CD14 Ab, or DAPI Fluoromount-G and analyzed by fluorescence
microscopy. Original magnification 3400. (D) CD14+ cells isolated from
nonalcoholic PBMCs and alcoholic PBMCs were tested for their bacteri-
cidal activity against K. pneumoniae. Data are mean 6 SE of three
experiments. **p, 0.01 versus nonalcoholic CD14+ cells. (E) CD14+ cells
isolated from PBMCs from 10 nonalcoholics and 10 alcoholics were an-
alyzed for CCL1+CD163+ cells, CCL17+CD163+ cells, and CXCL13+
CD163+ cells by flow cytometry. Data are mean 6 SE. ***p , 0.001
versus nonalcoholics.
The Journal of Immunology 5
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Next, we examined the antibacterial resistance of alcoholicPBMC chimeras treated with CCL1 antisense ODN to bacterialpneumonia and gut bacteria–associated sepsis. Alcoholic PBMCchimeras were treated with CCL1 antisense ODN (10 mg, s.c.)twice a day for 5 d. As controls, the same chimeras were treatedwith scrambled ODN under the same conditions. As additionalcontrols, control PBMC chimeras were treated with CCL1 anti-sense ODN or scrambled ODN in the same fashion. A dose of 102
CFU K. pneumoniae was given intratracheally on day 4 of treat-ment. The severity of infection was evaluated by bacterial growthin the lung, spleen, and kidneys of these chimeras 2 d postinfec-tion. The pathogen grew substantially in the lung of alcoholicPBMC chimeras treated with scrambled ODN, whereas bacterialgrowth was minimal in the lung of alcoholic PBMC chimerastreated with CCL1 antisense ODN, as well as in control PBMCchimeras treated with CCL1 antisense ODN or scrambled ODN(Fig. 6A). Similar results were obtained in alcoholic PBMC chi-meras infected with 106 CFU E. faecalis posttreatment with CCL1antisense ODN. In these experiments, decontaminated and proton-pump inhibitor–treated alcoholic PBMC chimeras and controlPBMC chimeras were orally infected with 106 CFU E. faecalis,and the growth of the pathogen in MLNs was determined 2 dpostinfection. The pathogen grew in MLNs of alcoholic PBMCchimeras treated with scrambled ODN, whereas there was mini-mal growth of bacteria in MLNs of alcoholic PBMC chimerastreated with CCL1 antisense ODN, similar to control PBMCchimeras treated with CCL1 antisense ODN or scrambled ODN(Fig. 6B). These results indicate that Klebsiella pneumonia andgut bacteria–associated sepsis in alcoholic PBMC chimeras aremarkedly mitigated posttreatment with CCL1 antisense ODN.
DiscussionPneumonia and gut bacteria–associated sepsis, caused by microbesthat are normally distributed in the upper respiratory tracts(pneumonia) and the upper and lower intestine (bacterial trans-location), are seen frequently in alcoholics (3–6). In this study, thehost antibacterial resistance of alcoholics to Klebsiella pneumoniaand sepsis stemming from E. faecalis translocation was investi-gated in a humanized murine model of alcoholics. Twenty-fivealcoholics enrolled in this study had an ethanol consumption of257.4 6 34.3 g/d for 12–56 y. Humanized murine chimeras ofalcoholics were created in gNSG mice inoculated with PBMCsfrom alcoholics (alcoholic PBMC chimeras). As a control, gNSGmice were inoculated with nonalcoholic PBMCs in the samefashion (control PBMC chimeras). Because gNSG mice do notcarry any functional T cells, B cells, NK cells, macrophages, orneutrophils (36–39), the chimeras display similar antibacterialresistance shown by the PBMC donors (35). To standardize theseverity of infection with these two pathogens in humanizedmurine chimeras, control PBMC chimeras created with variousnumbers of PBMCs were exposed to various doses of the patho-gens. Control PBMC chimeras created with 106 PBMCs/mousewere resistant to 102 CFU K. pneumoniae (intratracheal infection)and 106 CFU E. faecalis (oral infection). However, alcoholicPBMC chimeras created with 106 PBMCs of alcoholics weresusceptible to infections with K. pneumoniae (102 CFU, intra-tracheal) and E. faecalis (106 CFU, oral). In addition, controlPBMC chimeras were not resistant to these infections when theywere created with nonalcoholic PBMCs depleted of CD14+ cells.These results strongly indicate that monocytes/macrophages(CD14+ cells) are cells managing host antibacterial responsesagainst these infections and that the antibacterial function ofCD14+ cells in PBMCs derived from alcoholics and nonalcoholicsis clearly different.
In our previous murine studies (29, 31), the major host defenseeffector cells in fighting against these infections were character-ized as M1 macrophages (IL-102IL-12+CD1632 macrophages).However, M1 macrophages were not generated in CAC micestimulated with bacterial Ags or M1 macrophage inducers (29).M2b macrophages (IL-10–producing CCL1+CCL172CXCL132
FIGURE 5. Induction of M1 monocytes by heat-killed K. pneumoniae in
cultures of alcoholic CD14+ cells previously treated with CCL1 antisense
ODN. CD14+ cell preparations (5 3 105 cells/ml) from PBMCs from three
nonalcoholics and three alcoholics were cultured with CCL1 antisense
ODN (2 mg/ml) or scrambled ODN (2 mg/ml) for 48 h. (A) Culture fluids
obtained were assayed for IL-10 by ELISA. (B) Both groups of harvested
cells were washed with fresh medium and recultured with 105 heat-killed
K. pneumoniae in fresh medium; culture fluids obtained 12, 24, and 36 h
postcultivation were assayed for IL-12 by ELISA (s, CCL1 antisense
ODN; d, scrambled ODN). (C) The harvested cells also were tested for
their bactericidal activity against K. pneumoniae, as described in the text.
Data are mean 6 SE. *p , 0.05, **p , 0.01.
6 M2b MONOCYTES PROVOKE INFECTIONS IN ALCOHOLICS
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

CD163+CD14+ monocytes), which inhibit conversion from qui-escent macrophages to M1 macrophages, were predominant in thebronchoalveolar lavage fluid, MLNs, and LP of CAC mice (29).Therefore, CAC mice became highly susceptible to pulmonaryinfections and gut bacteria–associated infections.In this study, IL-10–producing, IL-12–nonproducing CCL1+
CCL172CXCL132CD163+CD14+ monocytes (M2b monocytes)were predominantly isolated from the peripheral blood of alco-holics. IL-12+CD1632CD14+ cells (M1 macrophages) were notgenerated in alcoholic PBMC cultures supplemented with a bac-terial Ag. CCL1+CD163+CD14+ cells (M2b monocytes) pre-dominated in alcoholic PBMCs that reverted to quiescentmonocytes when they were cultured with CCL1 antisense ODN,an inhibitor of CCL1 production. CCL1 produced by M2b mac-rophages was characterized as an essential chemokine for themaintenance of cellular properties of M2b macrophages (30). M1macrophages were induced by a bacterial Ag in cultures of al-coholic CCL1+CD163+CD14+ cells previously treated with CCL1antisense ODN. The resistance of alcoholic PBMC chimeras toKlebsiella pneumonia and sepsis stemming from enterococcaltranslocation was improved to the levels shown in control PBMCchimeras posttreatment with CCL1 antisense ODN. These results
indicate that Klebsiella pneumonia and sepsis stemming fromenterococcal translocation in alcoholic PBMC chimeras can becontrolled by the polarization of macrophages using a M2bmacrophage reverter (CCL1 antisense ODN) and a M1 macro-phage inducer (a bacterial Ag).Recently, many articles reported that macrophages exist along
a spectrum in a given context or anatomical location (42–44), andthey readily switch from one functional phenotype to another inresponse to new microenvironmental signals (45, 46). Althoughdistinct subpopulations of macrophages have been described (47,48), it is understood that macrophages represent a spectrum ofactivated phenotypes rather than discrete stable subpopulations.Many studies on macrophage plasticity were performed on mac-rophages under stimulation with LPS and IFN-g (to cause polar-ization to M1 macrophages) or IL-4 and IL-13 (to causepolarization to M2a macrophages). Plasticity of M2b macro-phages was not discussed in these articles. M2b macrophagespersist longer than M2a macrophages (3 w–1 mo versus a fewdays) in hosts exposed to burn injuries, gamma radiation, andchronic alcohol consumption. Plastic properties of M2a macro-phages and M2b macrophages may be different. More informationis needed on M2b macrophage plasticity. It was reported that M2bmacrophages are generated from quiescent macrophages aftercombined stimulation with immune complexes (FcRs) and IL-1b(IL-1R) or LPS (TLR) (40, 49, 50). A high level of immunecomplexes was detected in the circulation of alcoholics (51, 52).Alcoholic hyaline, which results from the destruction of tissues, isresponsible for the formation of immune complexes (52). How-ever, the sources of IL-1b and LPS required for M2b macrophage/monocyte generation remain unknown. In recent studies, NLRP3inflammasome activation was observed in ethanol-fed mice (53).Pyroptosis is explained by NLRP3 inflammasome activation, andcaspase-1 in the activated NLRP3 inflammasome cleaves pro–IL-1b into mature IL-1b (54–56). Additionally, endotoxemia wasdemonstrated in hosts with chronic alcohol consumption (53, 57,58). With a high level of serum immune complex and IL-1b (and/or LPS) produced by these cellular reactions, M2b macrophages(or M2b monocytes) are predominant in alcoholics. Detailedstudies are needed to assess the distribution, stability, and re-versibility of M2b macrophages, as well as the mechanism of theirgeneration in alcoholics.In contrast, alcoholic liver diseases (59), acute respiratory dis-
order syndrome (60), and inflammatory bowel disease (61) areoften seen in alcoholics. M2a macrophages that appear in the liverof ethanol-fed mice produce arginase (56), extracellular matrixcomponents (e.g., collagen, fibronectin, and bIG-H3) (57, 58), andTGF-b (62), which play a role in the healing/repair responsesagainst inflamed tissues. As anti-inflammatory cells, M2 macro-phages play an important role in controlling excessive or pro-longed inflammation in organs (63–66). M1 macrophages weredetected in injured liver tissues (inflammation sites) of ethanol-fedmice (63–65). M1 macrophages are eliminated from the injuredarea through the induction of M2 macrophages, and hepatocellularinjury in ethanol-fed mice improved (66). These facts suggest thatthe elimination of M2b macrophages may play a role in the ac-celeration of hepatocellular injuries that develop in alcoholics.Further studies are required to determine the pathogenic role forM1 and M2 macrophages in alcoholics.The majority of human peripheral monocytes are strongly
positive for CD14 and negative for FcgRIII (CD16). Human mono-cytes were recently divided into three subsets based on surfaceCD14 and CD16 expression: CD14++CD162 monocytes (classicalmonocytes), CD14+CD16++ monocytes (nonclassical monocytes),and CD14++CD16+ monocytes (intermediate monocytes) (67). In
FIGURE 6. Antibacterial resistance of alcoholic PBMC chimeras
treated with CCL1 antisense ODN. (A) Two groups of three alcoholic
PBMC chimeras and two groups of three control PBMC chimeras were
treated with CCL1 antisense ODN or scrambled ODN, respectively, and
infected with K. pneumoniae as in Fig. 1. (B) Two other groups of alco-
holic PBMC chimeras (n = 3) and control PBMC chimeras (n = 3) were
treated with CCL1 antisense ODN or scrambled ODN and infected with
E. faecalis, as in Fig. 2. Two days postinfection, bacterial growth in the
lung, spleen, and kidneys of the chimeras infected with K. pneumoniae or
in the MLNs and liver of the chimeras infected with E. faecalis was de-
termined individually by a standard colony-counting method. Data are
mean 6 SE. **p , 0.01.
The Journal of Immunology 7
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

healthy subjects, CD14+CD16+ monocytes compose #10% of themononuclear cell population; however, they increase during in-fection (67). Although M2 monocytes may refer to CD14+CD16++
monocytes, this remains unclear. Future studies are required forthe detailed classification of M2a, M2b, and M2c monocytes basedon surface CD16 expression.In conclusion, the majority of monocytes in alcoholics exhibit an
M2b phenotype, which plays a role in their increased susceptibilityto Klebsiella pneumonia and sepsis stemming from enterococcaltranslocation, which is frequently seen in alcoholics. The impairedantibacterial resistance of humanized murine chimeras createdwith alcoholic PBMCs is markedly improved through macrophagepolarization using an M2b macrophage reverter, CCL1 antisenseODN. Macrophage polarization from the M2b to the M1 pheno-type may improve the resistance of alcoholics to these infections.
AcknowledgmentsWe thank Shin-Abuyama Hospital, Shinai Hospital, Sousei Hospital, and
Tesseikai Neurosurgical Hospital, as well as Drs. Hideko Ohama and Shinya
Fukunishi for help in collecting alcoholic patient samples.
DisclosuresThe authors have no financial conflicts of interest.
References1. Shigemitsu, H., and K. Afshar. 2007. Aspiration pneumonias: under-diagnosed
and under-treated. Curr. Opin. Pulm. Med. 13: 192–198.2. Szabo, G., and P. Mandrekar. 2009. A recent perspective on alcohol, immunity,
and host defense. Alcohol. Clin. Exp. Res. 33: 220–232.3. Boe, D. M., R. W. Vandivier, E. L. Burnham, and M. Moss. 2009. Alcohol abuse
and pulmonary disease. J. Leukoc. Biol. 86: 1097–1104.4. Bhatty, M., S. B. Pruett, E. Swiatlo, and B. Nanduri. 2011. Alcohol abuse and
Streptococcus pneumoniae infections: consideration of virulence factors andimpaired immune responses. Alcohol 45: 523–539.
5. Tuomisto, S., T. Pessi, P. Collin, R. Vuento, J. Aittoniemi, and P. J. Karhunen.2014. Changes in gut bacterial populations and their translocation into liver andascites in alcoholic liver cirrhotics. BMC Gastroenterol. 14: 40.
6. Tabata, T., T. Tani, Y. Endo, and K. Hanasawa. 2002. Bacterial translocation andpeptidoglycan translocation by acute ethanol administration. J. Gastroenterol.37: 726–731.
7. Szabo, G. 1999. Consequences of alcohol consumption on host defence. AlcoholAlcohol. 34: 830–841.
8. Siggins, R. W., J. N. Melvan, D. A. Welsh, G. J. Bagby, S. Nelson, and P. Zhang.2011. Alcohol suppresses the granulopoietic response to pulmonary Strepto-coccus pneumoniae infection with enhancement of STAT3 signaling. J. Immunol.186: 4306–4313.
9. Bagby, G. J., P. Zhang, D. A. Stoltz, and S. Nelson. 1998. Suppression of thegranulocyte colony-stimulating factor response to Escherichia coli challenge byalcohol intoxication. Alcohol. Clin. Exp. Res. 22: 1740–1745.
10. Pascual, M., S. Fernandez-Lizarbe, and C. Guerri. 2011. Role of TLR4 in eth-anol effects on innate and adaptive immune responses in peritoneal macro-phages. Immunol. Cell Biol. 89: 716–727.
11. Zisman, D. A., R. M. Strieter, S. L. Kunkel, W. C. Tsai, J. M. Wilkowski,K. A. Bucknell, and T. J. Standiford. 1998. Ethanol feeding impairs innate im-munity and alters the expression of Th1- and Th2-phenotype cytokines in murineKlebsiella pneumonia. Alcohol. Clin. Exp. Res. 22: 621–627.
12. Mørland, H., J. Johnsen, A. Bjørneboe, G. E. Bjørneboe, C. A. Drevon,J. Mørland, and B. Mørland. 1988. Reduced IgG Fc-receptor-mediated phago-cytosis in human monocytes isolated from alcoholics. Alcohol. Clin. Exp. Res.12: 755–759.
13. Bautista, A. P. 2002. Chronic alcohol intoxication primes Kupffer cells andendothelial cells for enhanced CC-chemokine production and concomitantlysuppresses phagocytosis and chemotaxis. Front. Biosci. 7: a117–a125.
14. Boe, D. M., T. R. Richens, S. A. Horstmann, E. L. Burnham, W. J. Janssen,P. M. Henson, M. Moss, and R. W. Vandivier. 2010. Acute and chronic alcoholexposure impair the phagocytosis of apoptotic cells and enhance the pulmonaryinflammatory response. Alcohol. Clin. Exp. Res. 34: 1723–1732.
15. Domınguez-Santalla, M. J., C. Vidal, J. Vinuela, L. F. Perez, and A. Gonzalez-Quintela. 2001. Increased serum IgE in alcoholics: relationship with Th1/Th2cytokine production by stimulated blood mononuclear cells. Alcohol. Clin. Exp.Res. 25: 1198–1205.
16. Liu, W., J. Li, W. Tian, T. Xu, and Z. Zhang. 2011. Chronic alcohol consumptioninduces cardiac remodeling in mice from Th1 or Th2 background. Exp. Mol.Pathol. 91: 761–767.
17. Laso, F. J., J. M. Vaquero, J. Almeida, M. Marcos, and A. Orfao. 2007. Chronicalcohol consumption is associated with changes in the distribution, immuno-
phenotype, and the inflammatory cytokine secretion profile of circulating den-dritic cells. Alcohol. Clin. Exp. Res. 31: 846–854.
18. Pan, H. N., R. Sun, B. Jaruga, F. Hong, W. H. Kim, and B. Gao. 2006. Chronicethanol consumption inhibits hepatic natural killer cell activity and acceleratesmurine cytomegalovirus-induced hepatitis. Alcohol. Clin. Exp. Res. 30: 1615–1623.
19. Laso, F. J., P. Lapena, J. I. Madruga, J. F. San Miguel, A. Orfao, M. C. Iglesias,and M. Alvarez-Mon. 1997. Alterations in tumor necrosis factor-alpha,interferon-gamma, and interleukin-6 production by natural killer cell-enrichedperipheral blood mononuclear cells in chronic alcoholism: relationship with liverdisease and ethanol intake. Alcohol. Clin. Exp. Res. 21: 1226–1231.
20. Starkenburg, S., M. E. Munroe, and C. Waltenbaugh. 2001. Early alteration inleukocyte populations and Th1/Th2 function in ethanol-consuming mice. Alco-hol. Clin. Exp. Res. 25: 1221–1230.
21. Shellito, J. E., M. quan Zheng, P. Ye, S. Ruan, M. K. Shean, and J. Kolls. 2001.Effect of alcohol consumption on host release of interleukin-17 during pulmo-nary infection with Klebsiella pneumoniae. Alcohol. Clin. Exp. Res. 25: 872–881.
22. Latif, O., J. D. Peterson, and C. Waltenbaugh. 2002. Alcohol-mediated polari-zation of type 1 and type 2 immune responses. Front. Biosci. 7: a135–a147.
23. Sica, A., and A. Mantovani. 2012. Macrophage plasticity and polarization:in vivo veritas. J. Clin. Invest. 122: 787–795.
24. Sester, D. P., K. J. Stacey, M. J. Sweet, S. J. Beasley, S. L. Cronau, andD. A. Hume. 1999. The actions of bacterial DNA on murine macrophages. J.Leukoc. Biol. 66: 542–548.
25. Rothfuchs, A. G., D. Gigliotti, K. Palmblad, U. Andersson, H. Wigzell, andM. E. Rottenberg. 2001. IFN-a b-dependent, IFN-g secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J. Immunol.167: 6453–6461.
26. Rosenberger, C. M., and B. B. Finlay. 2003. Phagocyte sabotage: disruption ofmacrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 4: 385–396.
27. Tsuda, Y., K. Shigematsu, M. Kobayashi, D. N. Herndon, and F. Suzuki. 2008.Role of polymorphonuclear neutrophils on infectious complications stemmingfrom Enterococcus faecalis oral infection in thermally injured mice. J. Immunol.180: 4133–4138.
28. Katakura, T., T. Yoshida, M. Kobayashi, D. N. Herndon, and F. Suzuki. 2005.Immunological control of methicillin-resistant Staphylococcus aureus (MRSA)infection in an immunodeficient murine model of thermal injuries. Clin. Exp.Immunol. 142: 419–425.
29. Ohama, H., A. Asai, I. Ito, S. Suzuki, M. Kobayashi, K. Higuchi, and F. Suzuki.2015. M2b macrophage elimination and improved resistance of mice withchronic alcohol consumption to opportunistic infections. Am. J. Pathol. 185:420–431.
30. Asai, A., K. Nakamura, M. Kobayashi, D. N. Herndon, and F. Suzuki. 2012.CCL1 released from M2b macrophages is essentially required for the mainte-nance of their properties. J. Leukoc. Biol. 92: 859–867.
31. Kobayashi, M., K. Nakamura, M. Cornforth, and F. Suzuki. 2012. Role of M2bmacrophages in the acceleration of bacterial translocation and subsequent sepsisin mice exposed to whole body [137Cs] g-irradiation. J. Immunol. 189: 296–303.
32. Ko, W. C., D. L. Paterson, A. J. Sagnimeni, D. S. Hansen, A. Von Gottberg,S. Mohapatra, J. M. Casellas, H. Goossens, L. Mulazimoglu, G. Trenholme, et al.2002. Community-acquired Klebsiella pneumoniae bacteremia: global differ-ences in clinical patterns. Emerg. Infect. Dis. 8: 160–166.
33. Sekirov, I., S. L. Russell, L. C. Antunes, and B. B. Finlay. 2010. Gut microbiotain health and disease. Physiol. Rev. 90: 859–904.
34. Kamada, N., S. U. Seo, G. Y. Chen, and G. Nunez. 2013. Role of the gut microbiotain immunity and inflammatory disease. Nat. Rev. Immunol. 13: 321–335.
35. Shultz, L. D., F. Ishikawa, and D. L. Greiner. 2007. Humanized mice in trans-lational biomedical research. Nat. Rev. Immunol. 7: 118–130.
36. Piganelli, J. D., T. Martin, and K. Haskins. 1998. Splenic macrophages from theNODmouse are defective in the ability to present antigen.Diabetes 47: 1212–1218.
37. O’Brien, B. A., Y. Huang, X. Geng, J. P. Dutz, and D. T. Finegood. 2002.Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced.Diabetes 51: 2481–2488.
38. Maree, A. F., M. Komba, D. T. Finegood, and L. Edelstein-Keshet. 2008. Aquantitative comparison of rates of phagocytosis and digestion of apoptotic cellsby macrophages from normal (BALB/c) and diabetes-prone (NOD) mice. J.Appl. Physiol. 104: 157–169.
39. Takenaka, K., T. K. Prasolava, J. C. Wang, S. M. Mortin-Toth, S. Khalouei,O. I. Gan, J. E. Dick, and J. S. Danska. 2007. Polymorphism in Sirpa modulatesengraftment of human hematopoietic stem cells. Nat. Immunol. 8: 1313–1323.
40. Sironi, M., F. O. Martinez, D. D’Ambrosio, M. Gattorno, N. Polentarutti,M. Locati, A. Gregorio, A. Iellem, M. A. Cassatella, J. Van Damme, et al. 2006.Differential regulation of chemokine production by Fcgamma receptor engage-ment in human monocytes: association of CCL1 with a distinct form of M2monocyte activation (M2b, Type 2). J. Leukoc. Biol. 80: 342–349.
41. Edwards, J. P., X. Zhang, K. A. Frauwirth, and D. M. Mosser. 2006. Biochemicaland functional characterization of three activated macrophage populations. J.Leukoc. Biol. 80: 1298–1307.
42. Murray, P. J., and T. A. Wynn. 2011. Protective and pathogenic functions ofmacrophage subsets. Nat. Rev. Immunol. 11: 723–737.
43. Wynn, T. A., A. Chawla, and J. W. Pollard. 2013. Macrophage biology in de-velopment, homeostasis and disease. Nature 496: 445–455.
44. Hussell, T., and T. J. Bell. 2014. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14: 81–93.
45. Vergadi, E., K. Vaporidi, E. E. Theodorakis, C. Doxaki, E. Lagoudaki,E. Ieronymaki, V. I. Alexaki, M. Helms, E. Kondili, B. Soennichsen, et al. 2014.Akt2 deficiency protects from acute lung injury via alternative macrophage ac-tivation and miR-146a induction in mice. J. Immunol. 192: 394–406.
8 M2b MONOCYTES PROVOKE INFECTIONS IN ALCOHOLICS
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

46. Das, A., K. Ganesh, S. Khanna, C. K. Sen, and S. Roy. 2014. Engulfment ofapoptotic cells by macrophages: a role of microRNA-21 in the resolution ofwound inflammation. J. Immunol. 192: 1120–1129.
47. Gordon, S., and F. O. Martinez. 2010. Alternative activation of macrophages:mechanism and functions. Immunity 32: 593–604.
48. Biswas, S. K., and A. Mantovani. 2010. Macrophage plasticity and interactionwith lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11: 889–896.
49. Martinez, F. O., A. Sica, A. Mantovani, and M. Locati. 2008. Macrophage ac-tivation and polarization. Front. Biosci. 13: 453–461.
50. Nimmerjahn, F., and J. V. Ravetch. 2008. Fcgamma receptors as regulators ofimmune responses. Nat. Rev. Immunol. 8: 34–47.
51. Thomas, H. C., D. De Villiers, B. Potter, H. Hodgson, S. Jain, D. P. Jewell, andS. Sherlock. 1978. Immune complexes in acute and chronic liver disease. Clin.Exp. Immunol. 31: 150–157.
52. Penner, E., B. Albini, and F. Milgrom. 1978. Detection of circulating immunecomplexes in alcoholic liver disease. Clin. Exp. Immunol. 34: 28–31.
53. Lippai, D., S. Bala, J. Petrasek, T. Csak, I. Levin, E. A. Kurt-Jones, and G. Szabo.2013. Alcohol-induced IL-1b in the brain is mediated by NLRP3/ASC inflam-masome activation that amplifies neuroinflammation. J. Leukoc. Biol. 94: 171–182.
54. Bergsbaken, T., S. L. Fink, and B. T. Cookson. 2009. Pyroptosis: host cell deathand inflammation. Nat. Rev. Microbiol. 7: 99–109.
55. von Moltke, J., J. S. Ayres, E. M. Kofoed, J. Chavarrıa-Smith, and R. E. Vance.2013. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 31: 73–106.
56. Latz, E., T. S. Xiao, and A. Stutz. 2013. Activation and regulation of theinflammasomes. Nat. Rev. Immunol. 13: 397–411.
57. Hritz, I., P. Mandrekar, A. Velayudham, D. Catalano, A. Dolganiuc, K. Kodys,E. Kurt-Jones, and G. Szabo. 2008. The critical role of toll-like receptor (TLR) 4in alcoholic liver disease is independent of the common TLR adapter MyD88.Hepatology 48: 1224–1231.
58. Keshavarzian, A., A. Farhadi, C. B. Forsyth, J. Rangan, S. Jakate, M. Shaikh,A. Banan, and J. Z. Fields. 2009. Evidence that chronic alcohol exposure pro-
motes intestinal oxidative stress, intestinal hyperpermeability and endotoxemiaprior to development of alcoholic steatohepatitis in rats. J. Hepatol. 50: 538–547.
59. Jaurigue, M. M., and M. S. Cappell. 2014. Therapy for alcoholic liver disease.World J. Gastroenterol. 20: 2143–2158.
60. Moss, M., B. Bucher, F. A. Moore, E. E. Moore, and P. E. Parsons. 1996. Therole of chronic alcohol abuse in the development of acute respiratory distresssyndrome in adults. JAMA 275: 50–54.
61. Swanson, G. R., S. Sedghi, A. Farhadi, and A. Keshavarzian. 2010. Pattern ofalcohol consumption and its effect on gastrointestinal symptoms in inflammatorybowel disease. Alcohol 44: 223–228.
62. Lu, J., Q. Cao, D. Zheng, Y. Sun, C. Wang, X. Yu, Y. Wang, V. W. Lee, G. Zheng,T. K. Tan, et al. 2013. Discrete functions of M2a and M2c macrophage subsetsdetermine their relative efficacy in treating chronic kidney disease. Kidney Int.84: 745–755.
63. Wan, J., M. Benkdane, F. Teixeira-Clerc, S. Bonnafous, A. Louvet, F. Lafdil,F. Pecker, A. Tran, P. Gual, A. Mallat, et al. 2014. M2 Kupffer cells promote M1Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalco-holic fatty liver disease. Hepatology 59: 130–142.
64. Louvet, A., F. Teixeira-Clerc, M. N. Chobert, V. Deveaux, C. Pavoine,A. Zimmer, F. Pecker, A. Mallat, and S. Lotersztajn. 2011. Cannabinoid CB2receptors protect against alcoholic liver disease by regulating Kupffer cell po-larization in mice. Hepatology 54: 1217–1226.
65. Mandal, P., B. T. Pratt, M. Barnes, M. R. McMullen, and L. E. Nagy. 2011.Molecular mechanism for adiponectin-dependent M2 macrophage polarization:link between the metabolic and innate immune activity of full-length adipo-nectin. J. Biol. Chem. 286: 13460–13469.
66. Curtis, B. J., A. Zahs, and E. J. Kovacs. 2013. Epigenetic targets for reversingimmune defects caused by alcohol exposure. Alcohol Res. 35: 97–113.
67. Shi, C., and E. G. Pamer. 2011. Monocyte recruitment during infection andinflammation. Nat. Rev. Immunol. 11: 762–774.
The Journal of Immunology 9
by guest on April 9, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from




![alarme-maison-m2b[1] Copy](https://static.fdocuments.net/doc/165x107/5571fe9649795991699bb5c8/alarme-maison-m2b1-copy.jpg)













