
Plasma Polymerization Mechanism - Universität...
45
Polymer Technique / Polymer Physics Polymer Surfaces Plasma Polymerization Mechanism J. Friedrich J. Friedrich Bundesanstalt für Materialforschung und –prüfung 12200 Berlin J. Friedrich, 03-2011, Klingenthal-Mühlleithen, Germany
Transcript of Plasma Polymerization Mechanism - Universität...

Polymer Technique / Polymer PhysicsPolymer Surfaces
Plasma Polymerization Mechanismy
J. FriedrichJ. Friedrich
Bundesanstalt für Materialforschung und –prüfung12200 Berlin
J. Friedrich, 03-2011, Klingenthal-Mühlleithen, Germany
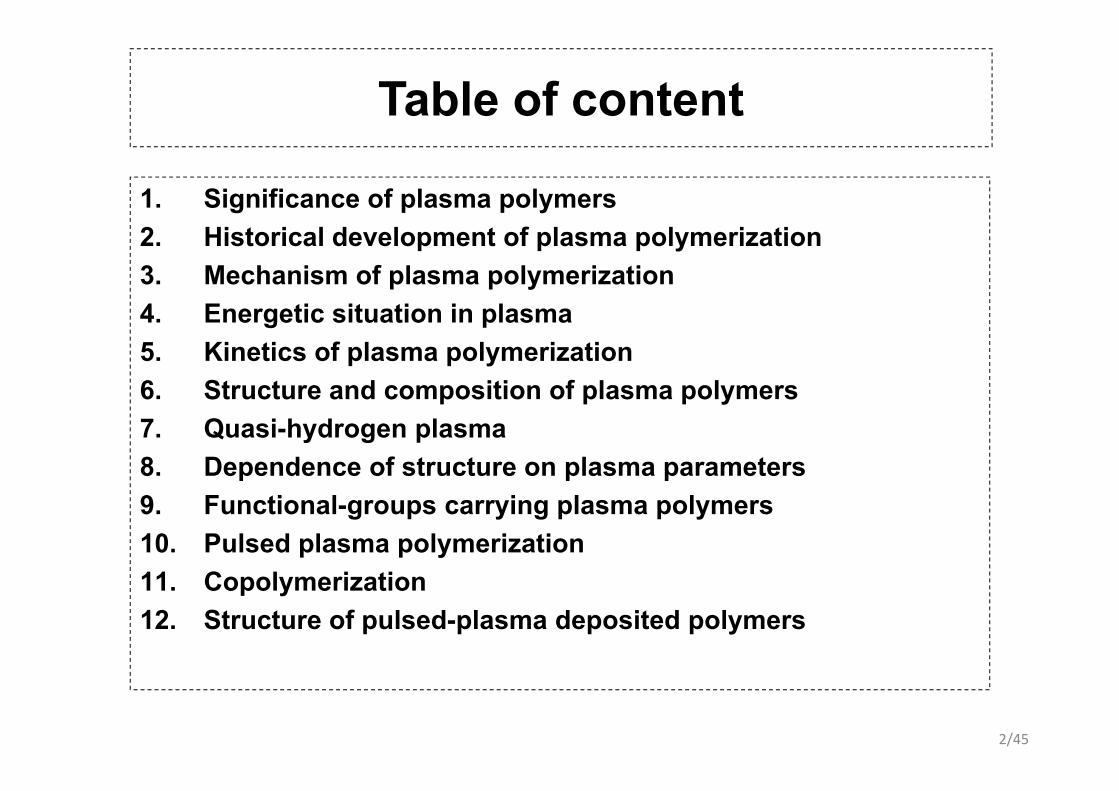
Table of content
1. Significance of plasma polymers2. Historical development of plasma polymerization3. Mechanism of plasma polymerization4 E ti it ti i l4. Energetic situation in plasma5. Kinetics of plasma polymerization6. Structure and composition of plasma polymers6. Structure and composition of plasma polymers7. Quasi-hydrogen plasma8. Dependence of structure on plasma parameters9. Functional-groups carrying plasma polymers10. Pulsed plasma polymerization11 C l i ti11. Copolymerization12. Structure of pulsed-plasma deposited polymers
2/45

1 Significance of plasma polymersGeneral intention is to produce ultra-thin and pinhole-free polymer layers of variable composition but with defined and regular structure
bl t th f l i l d th f d bl dcomparable to those of classic polymers and therefore durable and resistant towards ageing, oxidation, shrinking etc.
monomer molecules
plasmapolymer
plasma polymer layer
real
Using monomers with functional groups polymer films with monosort
polymer polymerideal
Using monomers with functional groups polymer films with monosort functional groups can be deposited.
XX
monomer molecules
X X OX O X
lpolymer layer with monosort functional groups
OH
real
XX
X
X X X X Xplasma
polymer layer with monosort functional groups
polymer
ideal
3/45
degree of retention f of functional groups : f= [cfgM]n / n cfgM, where M=monomer, n=number of monomers, cfg=concentrationof functional groups per monomer or per monomer unit, where the resulting concentration of X is most often <1 (<100%).
polymer polymerpolymer layer with monosort functional groups

1 Significance of plasma polymersApplications
• Plasma polymers are well-suited for coating of solids such as membranes, semiconductors metals textiles or polymers with minimal thickness of 10 nmsemiconductors, metals, textiles or polymers, with minimal thickness of 10 nm and maximal thickness of a few 100 nm. Clever choice of “monomer” allows to produce corrosion-inhibition, chemical, scratch, abrasion resistance, lubrification permeability biocomp antistatic optical properties It must belubrification, permeability, biocomp., antistatic, optical properties. It must be noted that the substrate properties are not influenced strongly by this deposition process.
• First application for polymer pinhole-free thin layers as separator membranes in nuclear battery devices by Goodman in 1960 [J. Goodman, J. Polym. Sci. 44 (1960) 551], f th t til [ ] i f i l t ifurther textiles [A. Bradley, J. D. Fales, Chem. Technol. 4 (1971) 232], capping of microelectronicdevices [J. J. Licari, Plastic Coatings for Electronics, McGraw-Hill Book Co., New York, 1970], PET tire cord [E. L. Lawton, J. Appl. Polym. Sci. 18 (1974) 1557], barrier layers-membranes [H. K. Yasuda, J. Membr. Sci. 18 (1984) 273-284; N. Inagaki, N. Kobayashi, M. Matsushima, J. Membr. Sci. 38 (1988) 85-95; L. Agres, Y. Segui, R. del Sol, P. Raynaud, J. Appl. Polym. Sci. 61 (1996) 2015-2022; F. Fracassi, R. d'Agostino, P. Favia, M. van Sambeck,
Plasma Sources Sci. Tech. 2 (1993) 106], corrosion-inhibiting coatings on aluminium components in car industry [H Stein E Zehender B Blaich DE pat 2 625 448 (1976); G Dittmer Ind
4/45
components in car industry [H. Stein, E. Zehender, B. Blaich, DE pat. 2 625 448 (1976); G. Dittmer, Ind. Res. Devel. Sept. 1978, 169-183; H. Grünwald, M. Jung, R. Kukla; R. Adam, J. Krempel-Hesse, in, Metallized Plastics 5-
6: Fundamental and Applied Aspects (Ed. K. L. Mittal), VSP, Utrecht, 1998].

2 Historical on plasma polymers
1796 - 1950
• First reference 1796Bondt, Deimann, Paets van Trostwijk, Lauwerenburg, cited in J. Fourcroy, Ann. Chem. 21 (1796) 58
• More detailed research was performed by well-known chemists in theMore detailed research was performed by well known chemists in the19th century such as M. Berthelot, who has developed arc synthesis of acetylene M. Berthelot, Ann. Chim. Phys. 6/7 (1863) 53; 9 (1866) 413; 12 (1867) 5; Compt. Red., 67 (1869) 1141, 62 (1876)1283
• P. de Wilde P. de Wilde, Ber. 7 (1874) 352
• P. and A. Thenard P. and A. Thenard, Compt. Rend. 78 (1874) 219.
• More extensive research on plasma technique and polymerization wereMore extensive research on plasma technique and polymerization were published by German scientists in 1930ies and 1950iesE. Linder, A. Davis, J. Phys. Chem. 35 (1931) 3649, H. Schüler, L. Reinebeck, Z. Naturforsch. 6a (1951) 271, 7a (1952) 285, 9a (1954) 350, H. Schüler, M. Stockburger 14a (1959) 981,
S f ( )H. Schüler, K. Prchal, E. Kloppenburg, Z. Naturforsch. 15a (1960) 308,H. König, G. Hellwig, Z. Physik 129 (1951) 491
5/45

2 Historical on plasma polymers2 Historical on plasma polymers
1960 - 19701960 1970
• In 1960-1970ies some extensive and also controversial discussion on mechanism of polymerization were heldmechanism of polymerization were held - radical mechanism- ionic (ion-molecule reaction)
fragmentation and dissociation ( atomic polymerization“ Yasuda)- fragmentation and dissociation („atomic polymerization - Yasuda). G. Möllenstedt, R. Speidel, Z. Angew. Phys., 13 (1961) 231 A. Bradley, J. P. Hammes, J. Electrochem. Soc., 110 (1963) 15 D. T. Williams, M. W. Hayes, Nature, 209 (1966) 769 V. M. Kolotyrkin, A. B. Gilman, A. K. Tsapuk, Uspechii chimii, 8 (1967) 1380; A. R. Denaro, P. A. Owens, A. Crawshaw, Europ. Polym. J., 4 (1968) 93 und 5 (1969) 471A. R. Westwood, Europ. Polym. J., 7 (1971) 363 L. F. Thompson, K. G. Mayhan, J. Appl. Polym. Sci., 16 (1972) 2291H. Yasuda, Plasma Polymerization, Orlando, Florida, Academic Press, 1986
• Jesch reported on the structure of plasma polymers. He describesformation of unsaturations (C=C, C≡C), occurrence of insolubility ( li ki ) tt h t t di l t t i d th(crosslinking), oxygen attachment to radicals at exposure to air, and the very important fact, the absence of IR absorptions in the 720-750 cm-1
range for each plasma polymer (indicator for the existence of [CH2-CH2]>2 sequences as minimal retention of monomer structure in the
6/45
[CH2 CH2]>2 sequences as minimal retention of monomer structure in the polymer molecule). K. Jesch, J. E. Bloor, P. L. Kronick, J. Polym. Sci. A1 (1966) 1487P. L. Kronick, K. Jesch, J. E. Bloor, J. Polym. Sci. A1/7 (1969) 767

2 Historical on plasma polymers2 Historical on plasma polymers1970 – 2011• In 1971, A. R. Westwood found that the structure of plasma polymers
is more regularly and defined in the sense of classic polymers the l th i t t th llower the power input to the plasmaA. R. Westwood, Eur. Polym. J. 7 (1971) 363-375
• On the other hand the low plasma power induced the inclusion of monomer and oligomer molecules into the polymer structure Thusmonomer and oligomer molecules into the polymer structure. Thus, the thermal stability of polymer layers becomes low because of the evaporation of these components at temperatures lower than 100°CL. F. Thompson, K. G. Mayhan, J. Appl. Polym. Sci. 16 (1972) 2291L. F. Thompson, K. G. Mayhan, J. Appl. Polym. Sci. 16 (1972) 2291
• Tibbitt summarizes all knowledge on structure of plasma polymers and designed models of soluble and crosslinked ethylene plasma polymers another for plasma polymerized toluenepolymers another for plasma polymerized tolueneJ. M. Tibbitt, A. T. Bell, M. Shen, J. Macromol. Sci., Chem. A10, (1976) 513;J. Friedrich, H. Wittrich, J. Gähde, Faserforsch. Textiltechn. / Z. Polymerenforsch. 29 (1978) 481
• Recently, this debate on mechanism was revitalized by comments of
7
Short, Hegemann and d´ AgostinoR. d´Agostino, P. Favia, R. Förch, C. Oehr, M. R. Wertheimer, Plasma Proc. Polym. 7 (2010) 363; D. Hegemann, D. A. Steele, R. D. Short, Plasma Proc. Polym. 7 (2010) 365]
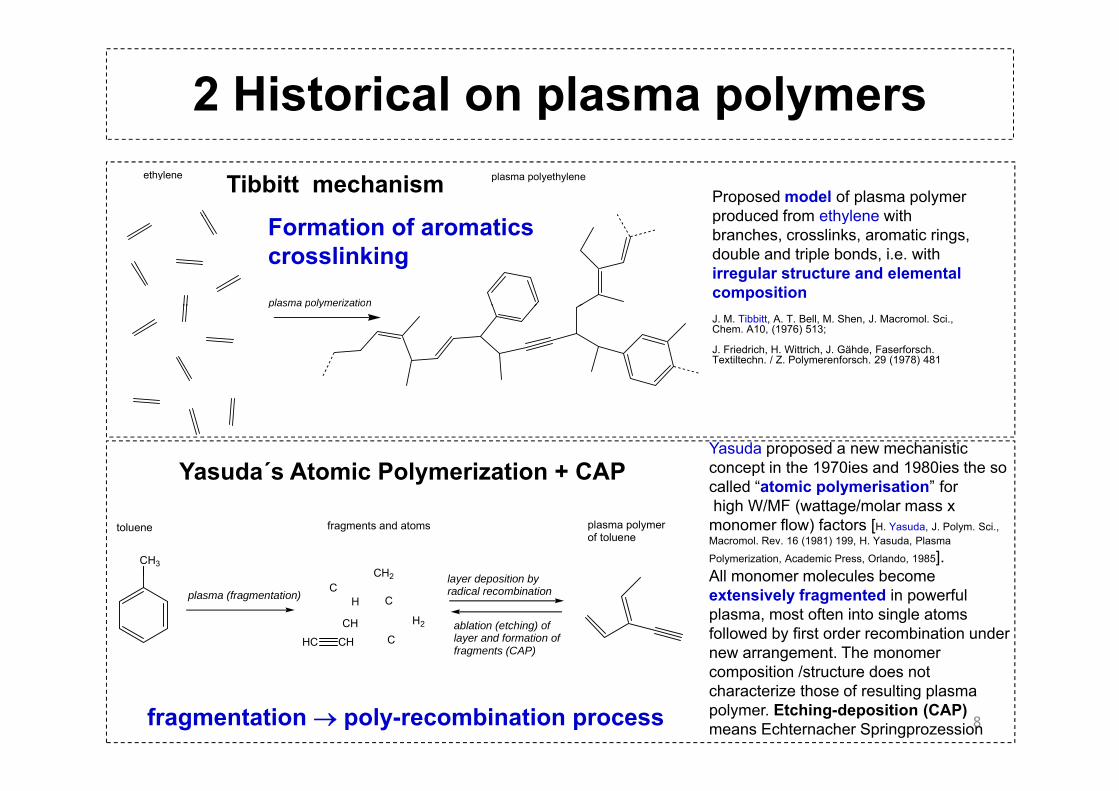
2 Historical on plasma polymersethylene plasma polyethylene
Proposed model of plasma polymer produced from ethylene with
Tibbitt mechanism
plasma polymerization
produced from ethylene with branches, crosslinks, aromatic rings, double and triple bonds, i.e. with irregular structure and elemental composition
Formation of aromaticscrosslinking
plasma polymerizationJ. M. Tibbitt, A. T. Bell, M. Shen, J. Macromol. Sci., Chem. A10, (1976) 513;
J. Friedrich, H. Wittrich, J. Gähde, Faserforsch. Textiltechn. / Z. Polymerenforsch. 29 (1978) 481
Yasuda proposed a new mechanistic concept in the 1970ies and 1980ies the so called “atomic polymerisation” for
Yasuda´s Atomic Polymerization + CAP
CH3CH2 l d iti b
toluene fragments and atoms plasma polymerof toluene
called atomic polymerisation forhigh W/MF (wattage/molar mass x monomer flow) factors [H. Yasuda, J. Polym. Sci., Macromol. Rev. 16 (1981) 199, H. Yasuda, Plasma
Polymerization, Academic Press, Orlando, 1985]. All monomer molecules become
plasma (fragmentation)C
C
CHC
CH2
HC CH
H
H2
layer deposition byradical recombination
ablation (etching) of layer and formation offragments (CAP)
All monomer molecules become extensively fragmented in powerful plasma, most often into single atoms followed by first order recombination under new arrangement. The monomer
8
composition /structure does not characterize those of resulting plasma polymer. Etching-deposition (CAP) means Echternacher Springprozessionfragmentation → poly-recombination process
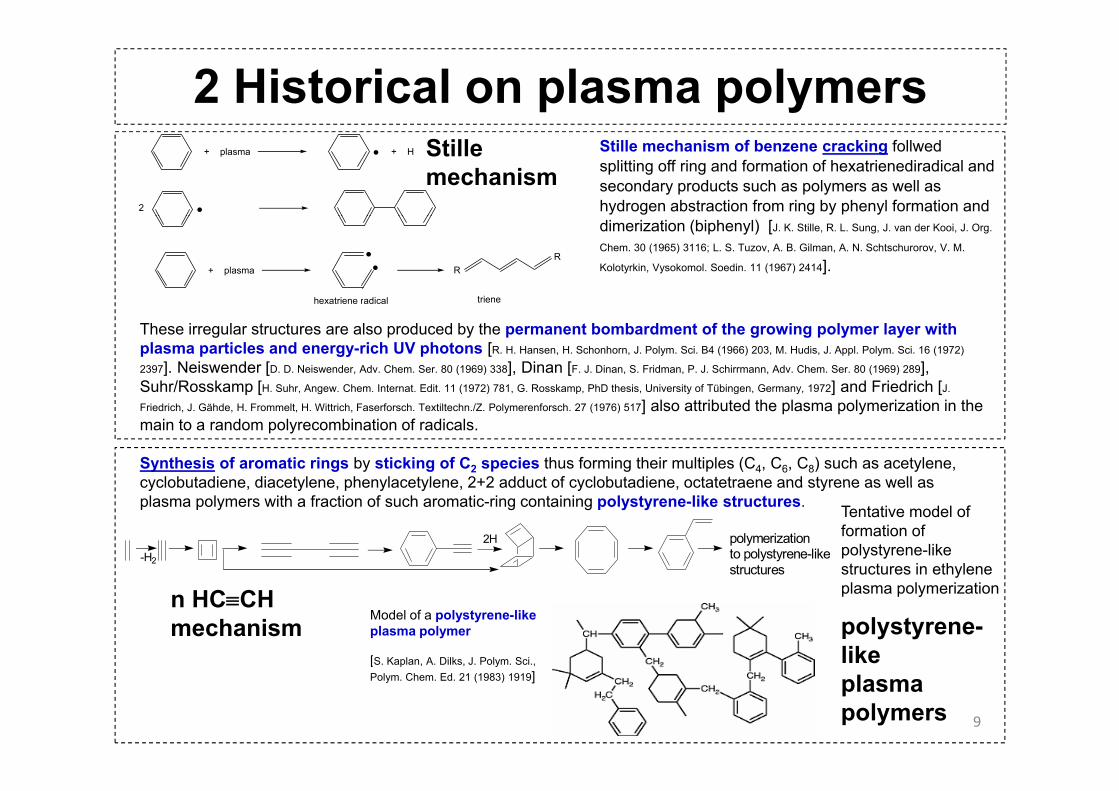
2 Historical on plasma polymers+ plasma + H
2
Stille mechanism of benzene cracking follwed splitting off ring and formation of hexatrienediradical and secondary products such as polymers as well as hydrogen abstraction from ring by phenyl formation and
Stillemechanism
+ plasma RR
hexatriene radical triene
dimerization (biphenyl) [J. K. Stille, R. L. Sung, J. van der Kooi, J. Org.
Chem. 30 (1965) 3116; L. S. Tuzov, A. B. Gilman, A. N. Schtschurorov, V. M.
Kolotyrkin, Vysokomol. Soedin. 11 (1967) 2414].
These irregular structures are also produced by the permanent bombardment of the growing polymer layer with plasma particles and energy-rich UV photons [R. H. Hansen, H. Schonhorn, J. Polym. Sci. B4 (1966) 203, M. Hudis, J. Appl. Polym. Sci. 16 (1972)
2397]. Neiswender [D. D. Neiswender, Adv. Chem. Ser. 80 (1969) 338], Dinan [F. J. Dinan, S. Fridman, P. J. Schirrmann, Adv. Chem. Ser. 80 (1969) 289], Suhr/Rosskamp [H. Suhr, Angew. Chem. Internat. Edit. 11 (1972) 781, G. Rosskamp, PhD thesis, University of Tübingen, Germany, 1972] and Friedrich [J.
Friedrich, J. Gähde, H. Frommelt, H. Wittrich, Faserforsch. Textiltechn./Z. Polymerenforsch. 27 (1976) 517] also attributed the plasma polymerization in the main to a random polyrecombination of radicals.
Synthesis of aromatic rings by sticking of C2 species thus forming their multiples (C4, C6, C8) such as acetylene, cyclobutadiene diacetylene phenylacetylene 2+2 adduct of cyclobutadiene octatetraene and styrene as well as
polymerizationto polystyrene-likestructures
2H-H2
Tentative model of formation of polystyrene-like structures in ethylene
cyclobutadiene, diacetylene, phenylacetylene, 2+2 adduct of cyclobutadiene, octatetraene and styrene as well as plasma polymers with a fraction of such aromatic-ring containing polystyrene-like structures.
yplasma polymerization
Model of a polystyrene-like plasma polymer
[S. Kaplan, A. Dilks, J. Polym. Sci.,
polystyrene-like
n HC≡CHmechanism
9
[ p , , y ,Polym. Chem. Ed. 21 (1983) 1919] plasma
polymers

2 Historical on plasma polymersV M Kolot rkin A B Gilman A K Tsap k Uspechii Chimii 8 (1967) 1380B V Tkach k V V B shin V M Kolot rkin N P Smetankina V sokomol Soedin V. M. Kolotyrkin, A. B. Gilman, A. K. Tsapuk, Uspechii Chimii 8 (1967) 1380;A. Bradley, J. Hammes, J. Electrochem. Soc. 110 (1963) 59; T. Williams, M. W. Hayes, Nature 209 (1966) 769; I. Haller, P. White, J. Phys. Chem. 9 (1967) 1784; A. M. Mearns, Thin Solid Films 3 (1969) 201; A. B. Gilman, V. M. Kolotyrkin, V. N. Tunizki, Kinetika i Katalis 10 (1970) 1267; J. R. Hollahan, R. P. McKeever, Adv. Chem. Ser. 80 (1969) 272; J. R. Hollahan, Makromol. Chem. 154 (1972) 303
B. V. Tkachuk, V. V. Bushin, V. M. Kolotyrkin, N. P. Smetankina, Vysokomol. Soedin. A9 (1967) 2018; P. M. Hay, Adv. Chem. Ser. 80 (1969) 350; M. J. Vasile, G. Smolinsky, J. Electrochem. Soc., 119 (1972) 451; H. Yasuda, C. E. Lamaze, J. Appl. Polym. Sci., 17 (1973) 1519; H. Yasuda, C. E. Lamaze, J. Appl. Polym. Sci., 17 (1973) 1533;Cr. Simionescu, N. Asandei, F. Dénes, M. Sandulovici, Gh. Popa, Europ. Polym. J., 5 (1969) 4275 (1969) 427
gas phase
"condensed"plasma polymers("dust")
deposition
gas phase d
gas phase model
substrate
g p
monomer molecules
adsorption
substrate substrate and adsorption layer model
adsorption layer model
substratepolymerization
substratefor location of plasma polymerization
hνhν
hν
hνhν
hν
hνhν
plasma
Effect of plasma-UV irradiation and monomer
substrate substrate substrate substrate substrate
untreated ideal layer layer deposited plasma-deposited plasma polymer layer
grafting after switching-off the discharge on plasma polymer structure shown schematically for radiation
10/45
untreated ideal layer layer depositedfrom plasma-disso-ciated monomer frag-ments or atoms
plasma depositedlayer additionallycrosslinked anddegraded byUV-irradiation
plasma polymer layerreact with monomermolecules after swit-ching off the plasma("skin")
schematically for radiation induced crosslinking and “skin” formation

3 Mechanism Ion-Molecule Reactions
I l l ti l i th l Th l i iti t• Ion-molecule reactions may occur also in the plasma. The plasma can initiate an ionic polymerization similar to that of radicals:Fragment+ + M → Pi
+
P + + M → P+
+ + M +
Pi+ + M → P+
i+1
• Ordinary ion-molecule reactions are not simple monomer additions as with radical or ionic polymerization. Simplest reaction is charge transfer without change of molecular weight from A to A [H Drost Plasmachemistry Akademie Verlag Berlin 1978]:
+ + M + etc.
weight from A to A [H. Drost, Plasmachemistry, Akademie-Verlag, Berlin, 1978]:A+ + A → A + A+
or for different atoms A and B:A+ + B → A + B+ + ΔE (energy surplus)A + B → A + B + ΔE (energy surplus)
• More probable is the dissociative charge transfer, for example:He+ + CH4 → C(H4-x)+ + 4-x H + He0
• Ion molecule reaction can also produce an increasing in molecular weight of the• Ion-molecule reaction can also produce an increasing in molecular weight of the main product. The reaction enthalpy is contained in the side-product such as methane ion-molecule reactions [H. Drost, Plasmachemistry, Akademie-Verlag, Berlin, 1978]:CH3
+ + CH4 → C2H5+ + H2
11/45
CH3 CH4 → C2H5 H2
C2H5+ + CH4 → C3H7
+ + H2 etc.

3 Mechanism Radical Chain-Growth Polymerization
• Plasma-produced radicals, radical fragments, radical-sites at solid surfaces etc. initiate a classic chain- growth polymerization to polymer molecules P by continuousa classic chain- growth polymerization to polymer molecules P by continuous addition of monomer molecules M, which can be undergo a classic radical polymerization such as acrylic or vinyl monomers:fragment• + M → Pi• + Mg i
Pi• + M → •Pi+1
Free radical terminations (recombination) dominate the ending of chain growth:+ M etc.
( ) g g2 Pi• → Pi-Pi
Disproportionation and radical transfer are further termination mechanism [H.-G. Elias,
+
Disproportionation and radical transfer are further termination mechanism [H. G. Elias, An Introduction to Polymer Science, VCH, Weinheim, Germany, 1997]. All processes need collisions→pressure dependent!
P l i ti ti ilib i ti hi h t ll d b
+ + + +
• Polymerization reactions are equilibrium reactions, which are controlled by thermodynamics. They occur when the standard Gibbs energy is negative:ΔG0
p=ΔH0p-TΔS0
p=-RT logeKn,h ΔH0 i th t d d l i ti th l ΔS0 th t d d l i ti
12/45
where ΔH0p is the standard polymerization enthalpy, ΔS0
p the standard polymerization entropy, T temperature and Kn the equilibrium constant. Therefore, all introduced plasma energy is NOT needed, only for initiation minimal energy.
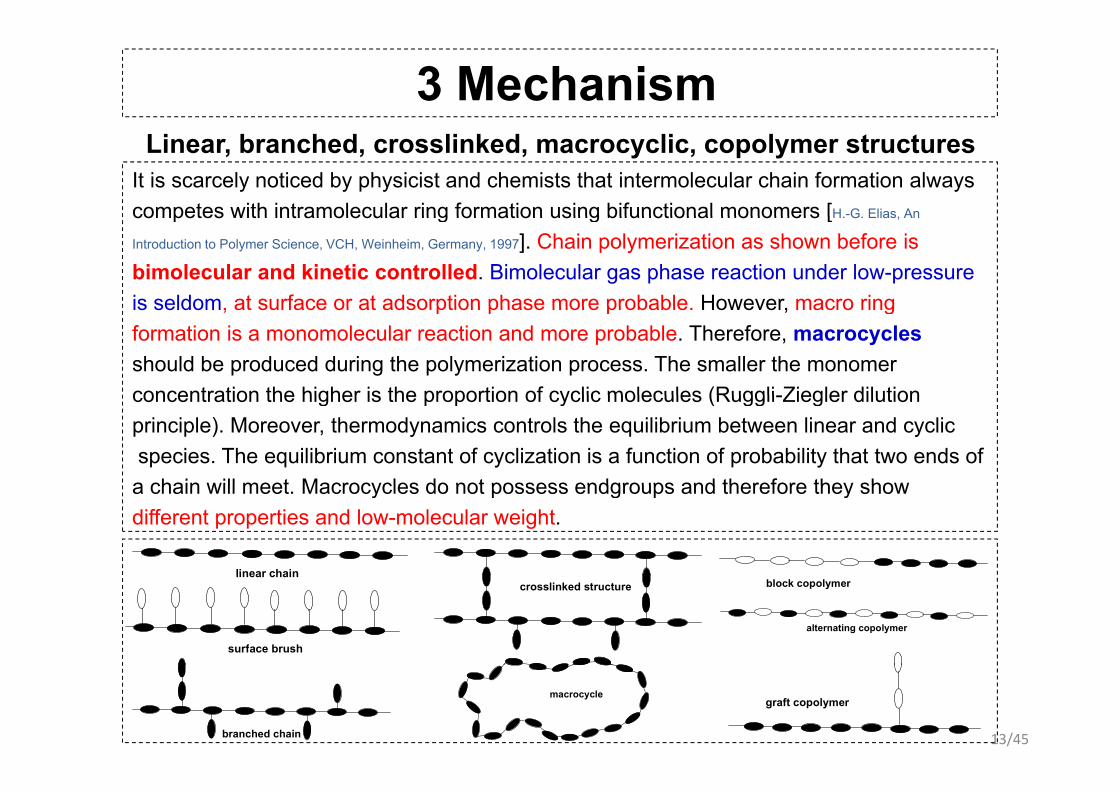
3 Mechanism Linear, branched, crosslinked, macrocyclic, copolymer structures
It is scarcely noticed by physicist and chemists that intermolecular chain formation always competes with intramolecular ring formation using bifunctional monomers [H -G Elias Ancompetes with intramolecular ring formation using bifunctional monomers [H. G. Elias, An
Introduction to Polymer Science, VCH, Weinheim, Germany, 1997]. Chain polymerization as shown before is bimolecular and kinetic controlled. Bimolecular gas phase reaction under low-pressureis seldom at surface or at adsorption phase more probable However macro ringis seldom, at surface or at adsorption phase more probable. However, macro ring formation is a monomolecular reaction and more probable. Therefore, macrocycles should be produced during the polymerization process. The smaller the monomer concentration the higher is the proportion of cyclic molecules (Ruggli-Ziegler dilutionconcentration the higher is the proportion of cyclic molecules (Ruggli-Ziegler dilution principle). Moreover, thermodynamics controls the equilibrium between linear and cyclicspecies. The equilibrium constant of cyclization is a function of probability that two ends of a chain will meet Macrocycles do not possess endgroups and therefore they showa chain will meet. Macrocycles do not possess endgroups and therefore they show different properties and low-molecular weight.
linear chainlinear chain
surface brush
block copolymercrosslinked structure
alternating copolymer
13/45branched chain
macrocyclegraft copolymer
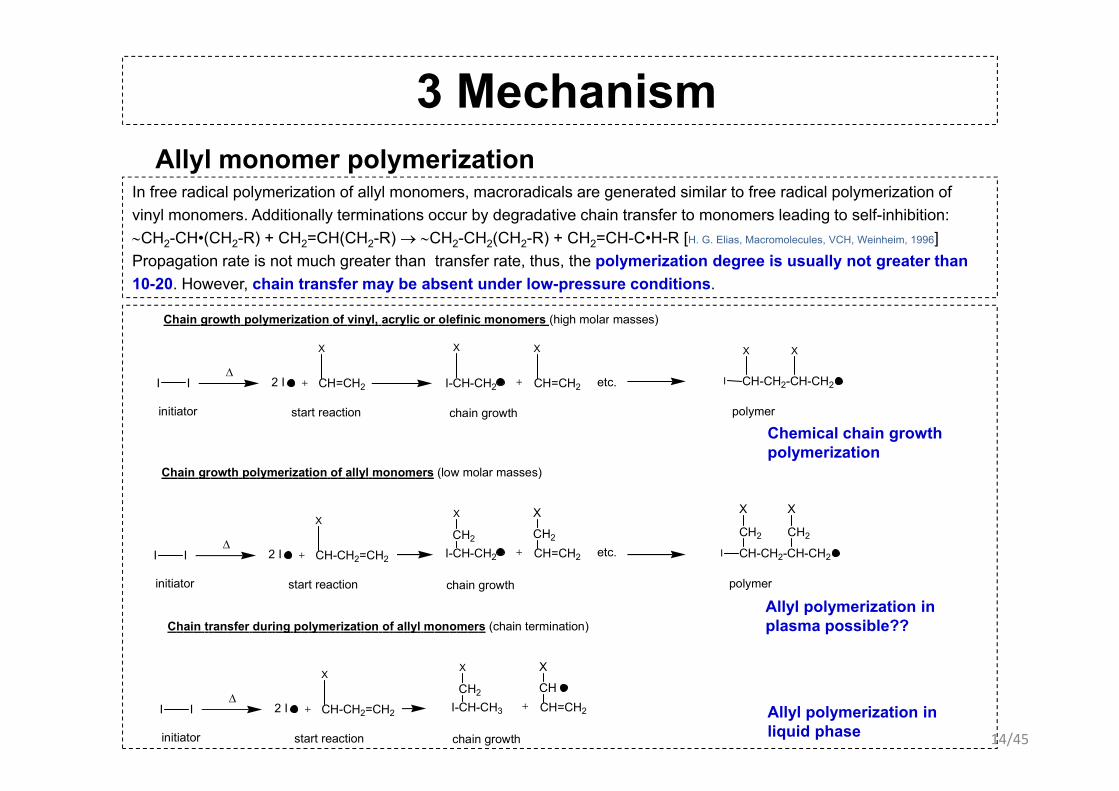
3 Mechanism Allyl monomer polymerization
In free radical polymerization of allyl monomers, macroradicals are generated similar to free radical polymerization of vinyl monomers Additionally terminations occur by degradative chain transfer to monomers leading to self-inhibition:vinyl monomers. Additionally terminations occur by degradative chain transfer to monomers leading to self-inhibition:∼CH2-CH•(CH2-R) + CH2=CH(CH2-R) → ∼CH2-CH2(CH2-R) + CH2=CH-C•H-R [H. G. Elias, Macromolecules, VCH, Weinheim, 1996]Propagation rate is not much greater than transfer rate, thus, the polymerization degree is usually not greater than 10-20. However, chain transfer may be absent under low-pressure conditions.
CH=CH2
X
CH=CH2
X
+ CH-CH2-CH-CH2
X X
etc.I IΔ
2 I +
X
I
Chain growth polymerization of vinyl, acrylic or olefinic monomers (high molar masses)
I-CH-CH2
initiator start reaction chain growth polymer
Chain growth polymerization of allyl monomers (low molar masses)
Chemical chain growth polymerization
CH-CH2=CH2
X
CH-CH2-CH-CH2
CH2 CH2
I IΔ
2 I + ICH=CH2
CH2
+ etc.CH2
I-CH-CH2
X X X X
initiator start reaction chain growth polymer
Chain transfer during polymerization of allyl monomers (chain termination)
X X
Allyl polymerization in plasma possible??
14/45
CH-CH2=CH2
X
I IΔ
2 I +
initiator start reaction chain growth
CH=CH2
CH+
CH2
I-CH-CH3
X X
Allyl polymerization in liquid phase

3 Mechanism Fragmentation-recombination and energy dose per monomer• Furthermost to the chemical chain-growth polymerization the continuous-wave (cw) plasma polymerization is far
from chemistry. The basic process is the fragmentation of monomer molecules to atoms or fragments:from chemistry. The basic process is the fragmentation of monomer molecules to atoms or fragments:n (ABCDEF) + plasma → n (A + B + CD + E + F)
• The plasma presents a source of “unlimited” energy/enthalpy and it produces also particle and photon energies much higher then chemical bonds in polymers (ca. 3.7 eV). Therefore, the exposure of monomer and polymer can be characterized as a permanent shower high-energy particles and radiation onto the molecules and the polymer surface. Therefore, it is negligible if the “monomer” can easily undergo a classic polymerization or not. Dienes, vinyl or acrylic monomers are not necessary for such plasma polymerization. Moreover, saturated monomers, monomers without reactive groups or inorganic gases and vapours form (easily) plasma polymers.
• The fragmentation is enforced for all types of chemical substances. The fragments and atoms form a statistically randomly structured plasma polymer of highly irregular structure by polyrecombination:randomly structured plasma polymer of highly irregular structure by polyrecombination: n (A + B + CD + E + F) → [FCABDE]n
• Hydrogen abstraction, CO-, CO2- and H2O scission, cracking of aromatic rings produce additionally unsaturated film-forming intermediates, which also contribute to the plasma polymer formation (acetylene etc.). These intermediates produce a few defined structure elements within the polymer structure. However, the influence of this p p y ,slightly more defined pathway on intermediates is dependent on the energy absorption of monomer molecules during their residence in the plasma zone. The higher the energy dose per monomer molecule the more dissociation occurs, the lower the energy dose the more units with monomer structure survive the plasma polymerization process. The energy dose per monomer can be expressed by the Yasuda-factor (YF): YF W (W tt ) / M (M l M ) F (M Fl )YF = W (Wattage) / M (Molar Mass) • F (Monomer Flow) [H. K. Yasuda, T. Hirotsu, J. Polym. Sci.: Polym. Chem. Ed. 16 (1973) 743; H. K. Yasuda, J. Polym. Sci., Macromol. Rev. 16 (1981) 119].
• Similar calculations were performed by Friedrich who referenced the consumed energy in Ws/mol or eV/mol to one monomer molecule during its residence time in the plasma [J. Friedrich, J. Gähde, H. Frommelt, H. Wittrich, Faserforsch. Textiltechn. / Z.
Polymerenforsch 27 (1976) 517] Easily polymerizable (classic) monomers needs about 10 to 100 eV per molecule to convert
15/45
Polymerenforsch. 27 (1976) 517]. Easily polymerizable (classic) monomers needs about 10 to 100 eV per molecule to convert the monomer to nearly 100% to polymer layer. Low-molecular-weight alkanes (aliphates) such as n-hexane need about 100-1000 eV for a 60% conversion to plasma polymer [J. Friedrich, H. Wittrich, J. Gähde, Faserforsch. Textiltechn. / Z. Polymerenforsch.
29 (1978) 481].
electrical field
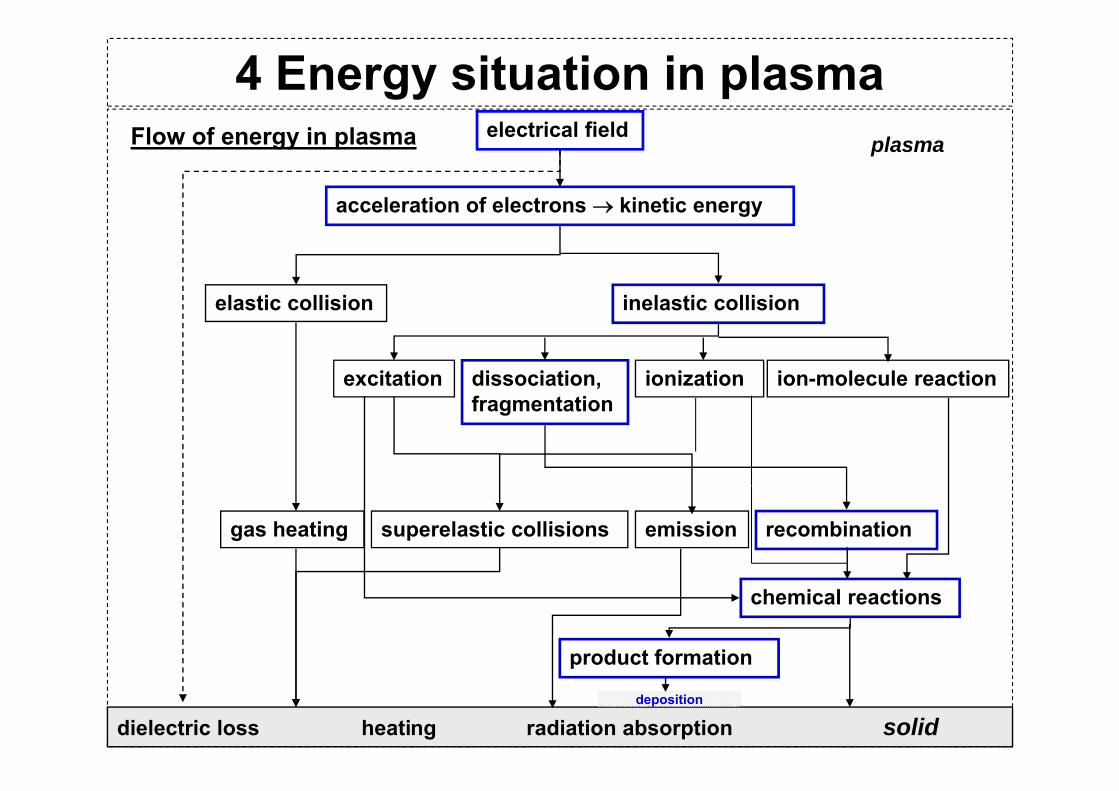
4 Energy situation in plasma electrical field
acceleration of electrons → kinetic energy
plasmaFlow of energy in plasma
gy
elastic collision inelastic collisionelastic collision inelastic collision
dissociation, ionization ion-molecule reactionexcitationfragmentation
gas heating superelastic collisions emission recombination
chemical reactions
product formation
16
p
dielectric loss heating radiation absorption soliddeposition

4 Energy situation in plasma
0,7
0,8 ectron on f(ε)
7
8Electron energy distribution
0,5
0,6
capture crosibutio
n fun
ctio
high ergetic "tail"of electron energy distribution
5
6ε = 1.0 eV too high energy →
no chemical selectivity
0 2
0,3
0,4
ss-section σ(nene
rgy d
istr
2
3
4Electron energy distribution function (EEDF) of low-pressure glow discharge plasmas plotted for average
0 1 2 3 4 5 6 7 8 9 10 11 120,0
0,1
0,2 (ε) [10-18cm
2]
electr
on e
0
1
2ε = 3.0 eV
g p p gelectron energies of 1.0 eV and 3.0 eV as well as showing electron capturing function of benzene. Gray= high energy tail of EEDF and
activation energy for initiation of chain growth polymerization ≈ 1 eV
0 1 2 3 4 5 6 7 8 9 10 11 12electron energy ε [eV] Gray= high-energy tail of EEDF and
dissociation energies of C-C and C-H bonds in polymers
C-H- and C-C-dissociation ≈ 3.7 eV (→ radicals→ oxidation/crosslinking
activation energy for initiation of chain growth polymerization ≈ 1 eV
ring scission ≈ 8 eV
ionisation 5-10 eV
17/45
ring scission ≈ 8 eV
CH3‐CH3 (CH3)3C‐H (CH3)2CH‐H CH3‐CH2‐H CH3‐H C6H5‐H bond
370 385 396 411 435 458 SDE [kJ/mol]

5 Kinetics of plasma polymerizationProposed „pseudo“ kinetics based on radical mechanism
Kassel interpreted the plasma-electrical conversion pf methane to acetylene [L. S. Kassel, J. Amer. Chem. Soc., 54 (1932) 3949] asf C C C C C C C C C C C Cfollows: CH4 k1→C2H6, C2H6 k2→C2H4, C2H4 k3→C2H2, C2H4 k4→Endprodukte, CH4 k5→C2H2, C2H6 k6→C2H2, CH4k7→C2H4, C2H4 k8→end products. Methane cannot directly form a polymer layer, therefore, it must be converted tointermediates (acytylene and ethylene), which are able to form plasma polymer layers
Neiswender-mechanism: monomer fragmentation H →C H →C H →C H →C Hmonomer fragmentation – H2 →C2H2→C4H4→C6H6→C8H8.
Denaro proposed 3 steps in a radical mechanism (1969): a) chain growing Rn• + M → Rn+1• b) radical recombination Rn• + Rm• → Pn+m
c) trapping of radicals (loss of radicals) R R (trapped)c) trapping of radicals (loss of radicals) Rn• → Rn• (trapped). Using the BET-equation for multilayer adsorption and other assumptions following rate equation was derived: R= r p (p+A+[A2+Br]-1/2)-1, with r-radical formation rate, p-monomer pressure, A and B constants [A. R. Denaro, P. A. Owens, A. Crawshaw, Europ. Polym. J. 4 (1968) 93; A. R. Denaro, P. A. Owens, A. Crawshaw, Europ. Polym. J. 5 (1969) 471].
In 1971 H K Yasuda creates an improved and widely accepted kinetic model considering the role of inert gas (X) additionsIn 1971, H. K. Yasuda creates an improved and widely accepted kinetic model considering the role of inert gas (X) additions as energy transmitter to organic monomers (M) [H. Yasuda, Plasma Polymerization, Orlando, Florida, Academic Press, 1986].M → M* (k1) andX → X*(k2), energy transferX* + M →X + M* (k ) known as Penning ionization followed by the initiation step M*→ M• (k ) and chain growthX + M →X + M (k3), known as Penning ionization, followed by the initiation step M → M• (ki) and chain growth M• + M → MM• (kp) or Mn• + M →Mn+1•. Following equation was derived fort the deposition rate: R=kikpk1[M]2(1+k2k3/k1[X]).This equation agrees with an empirical created function
18/45
This equation agrees with an empirical created function R= a[pM]2[1+b(pgas)], where a and b are constants.
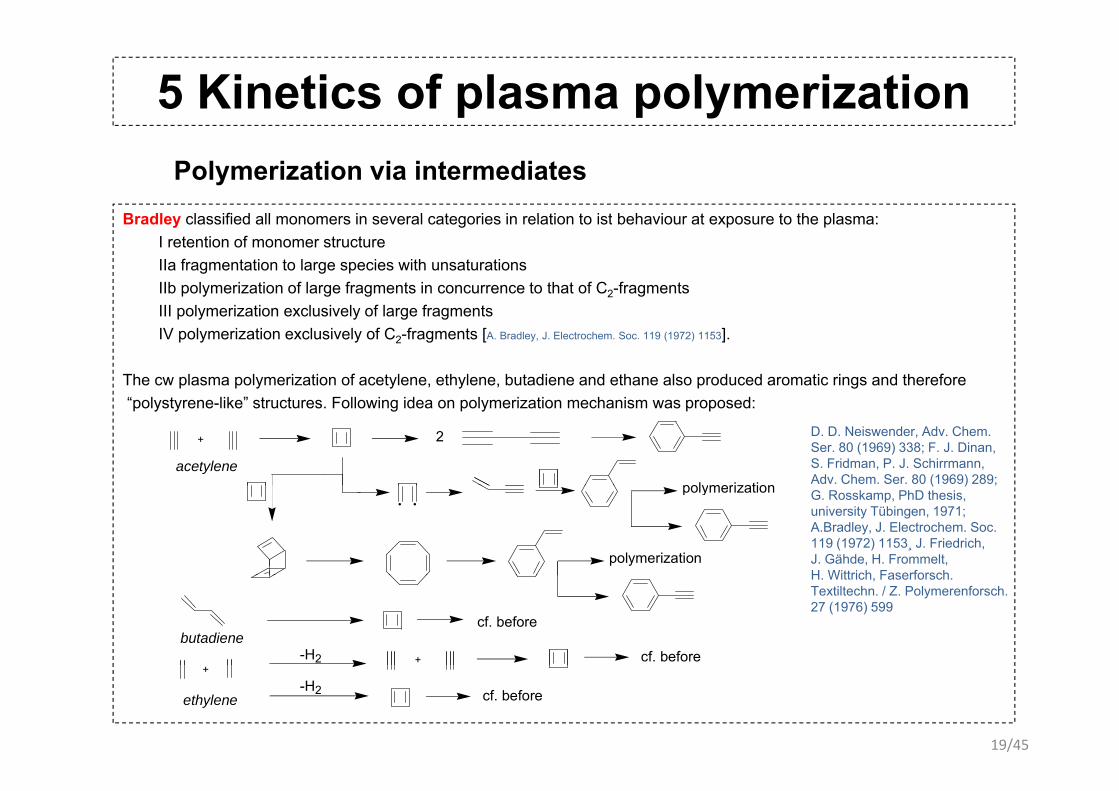
5 Kinetics of plasma polymerizationPolymerization via intermediates
Bradle classified all monomers in se eral categories in relation to ist beha io r at e pos re to the plasmaBradley classified all monomers in several categories in relation to ist behaviour at exposure to the plasma:I retention of monomer structureIIa fragmentation to large species with unsaturationsIIb polymerization of large fragments in concurrence to that of C2-fragments III l i ti l i l f l f tIII polymerization exclusively of large fragmentsIV polymerization exclusively of C2-fragments [A. Bradley, J. Electrochem. Soc. 119 (1972) 1153].
The cw plasma polymerization of acetylene, ethylene, butadiene and ethane also produced aromatic rings and therefore“polystyrene-like” structures. Following idea on polymerization mechanism was proposed:
+
polymerizationacetylene
2 D. D. Neiswender, Adv. Chem.Ser. 80 (1969) 338; F. J. Dinan, S. Fridman, P. J. Schirrmann, Adv. Chem. Ser. 80 (1969) 289; polymerization
polymerization
( ) ;G. Rosskamp, PhD thesis, university Tübingen, 1971; A.Bradley, J. Electrochem. Soc. 119 (1972) 1153¸ J. Friedrich, J. Gähde, H. Frommelt, H Wittrich Faserforsch
+
cf. before
cf. beforebutadiene
-H2
H. Wittrich, Faserforsch. Textiltechn. / Z. Polymerenforsch. 27 (1976) 599
19/45
+-H2 cf. beforeethylene

5 Kinetics of plasma polymerizationProposed „Pseudo“ Kinetics (radical mechanism)
Tibbitt has also created a kinetic model for a mechnism that explains the structure of two different ethylene plasma polymerTibbitt has also created a kinetic model for a mechnism that explains the structure of two different ethylene plasma polymerspecies, a low-molecular weight oligomer and a crosslinked polymer in gas phase as well as in adsorption layer [J. M. Tibbitt,
A. T. Bell, M. Shen, J. Macromol. Sci. Chem. A10 (1976) 513]: e + Mg + ki→ 2 Rg• + e´ initiationS + R • + k → R • radical-adsorptionS + Rg + kpg→ Rs radical adsorptionRg• + Mg + kpg→ 2 Rg+1• homogeneous chain growth (chain propagation)Rs• + Mg + kps→ 2 Rs+1• heterogeneous chain growth
e-electron, M-monomer, R-radical, S-substrate, G-gas, k-rate constant. The polymerization rate was then: r =(0 5dk +K k ) [M ][R ]rp=(0,5dkpg+KRkps) [Mg][Rg],KR-adsorption coefficient of radicals at surface of electrodes [Rs]=KR[Rg], d-distance between electrodes. The equation can be solved if monomer and radical concentrations are known: rp=[Mg]0
2 2ac[1-exp-(a-b)τ]/[(a-b)exp(a+b)τ], where is a=k [e] b=(2/d)k [S] c=(0 5d)k +kp τ=V/Q and Q= monomer flow and V=volume of plasma zonewhere is a=ki[e], b=(2/d)ka[S], c=(0,5d)ki+kps, τ=V/Q and Q= monomer flow and V=volume of plasma zone.
Lam and Baddour assumed that activation of molecules take place in the gas phase and chain growing in the adsorptionlayer [D. K. Lam, R. F. Baddour, A. F. Stancell, J. Macromol. Sci.-Chem. 10 (1976) 421].
Morita postulated also a radical mechanism in radio-frequency (rf) plasmas. Under the influence of the rf field the charge carriers oscillate. Under low-frequency conditions charge carriers are oscillating but in low-frequency powered plasmaionic mechanism dominates [S. Morita, G. Sawa, M. Ieda, J. Macromol. Sci.-Chem. 10 (1976) 501].
20/45
Shen and Bell considered bond dissociation energies and a radical mechanism because of the 104 higher concentrationof radicals than that of ions in the plasma [M. Shen, A. T. Bell, Plasma Polymerization, ACS Symp. Ser. 108 (1979) 1].

5 Kinetics of plasma polymerizationProposed „pseudo“ kinetics (radical mechanism)
Tiller and Friedrich have distinguished between chemical chain growth in analogy to the classic radical chain growth mechanism (A) and the polyrecombination of plasma-produced radical fragments (B) [J. Friedrich, J. Gähde, H. Frommelt, H. Wittrich, Faserforsch. Textiltechn. / Z. Polymerenforsch. 27 (1976) 599-603; H.-J. Tiller, U. Wagner, P. Fink, K. Meyer, Plaste u. Kautschuk 24 (1977)
619]. Moreover, formation of plasma polymers in the gas phase was also considered (M0-monomer concentration and B2
deposition of polymer layer via gas phase) or in the adsorption layer (Mads-monomer concentration and B1-formation of plasma polymer in the adsorption layer). Tiller expressed the layer deposition rate Pr as follows, considering sub-reaction pathways: Pr= PrA+ PrB1+ PrB2.
Tiller proposed following elementary processes:g yA) Radiation and plasma particle activation of monomer molecules, chemical chain growth processes and recombination of
radicals within the adsorption layer: Mads→R• (k1); R•+Mads→R2• (k2); Rn• + Rm•→Pn+m (k3) B1) Irradiative and plasma particle activation, fragmentation and recombination in the adsorption layer:
Mads→R• (k4); R•+R´•→P (k5); B2) Irradiative and plasma particle activation, fragmentation and recombination in the gas phase layer:
Aads→R• (k4); M0→R0•; R•+R0•→P For PrA and chemical chain growth: PrA=k2[R•][Mads]=k2(k1/k3 [hν])-1/2[M]3/2
and for PrB1 and layer growing by recombination in the adsorption layer is: PrB1=k5[R•]2=k4k5[Ihν][Mads] and in gas phase: rB1 y g g y y rB1 5 4 5 hν ads gPrB2=k7[I0hν][M0]. The total polymerization rate is: Pr=k2(k1/k3)1/2[Mads]3/2[Ihν]1/2+ k4k5[Ihν][Mads]+ k7[I0hν][M0].
21/45

Kinetics based on ionic mechanism5 Kinetics of plasma polymerization
Kinetics based on ionic mechanismFirst model for plasma polymerization basing on an ionic mechanism of polymerization within the adsorption layer was created by Williams and Hayes, in 1966 [D. T. Williams, M. W. Hayes, Nature 209 (1966) 769]. The polymerizsation rate was written as m=α • I • t, where m the deposited mass of polymer per area is, α a conctant depending on adsorption, I current density at, p p y p , p g p , yelectrodes (ac, kHz) and t deposition time. 1970 Poll qualified this model of ionic mechanism by consideration of transport rates of ions to surface by ambipolar diffusionand control of the plasma boundary layer [H.-U. Poll, Z. Angew. Phys. 4 (1970) 260], also supplemented by Carchano [H. Carchano, J.
Chem. Phys. 61 (1974) 3634]. The model proposed an ionic initiation step: P + e- → P+ + e-´, followed also by radical steps. Next,Chem. Phys. 61 (1974) 3634]. The model proposed an ionic initiation step: P e P e , followed also by radical steps. Next,the positively charged ions should be rapidly [H. Drost, Plasmachemie, Akademie-Verlag, Berlin, 1978]: P+ + e- → P* → 2 P•.It follows: dP•/dP=k1j2 (j-current density). The polymer growing is product of radical and monomer concentration. Adapting the Langmuir-adsorption model isotherm it is: dn/dt=γΦ(1-n/n0)-n/τ, where: γ-sticking coefficient, Φ-average collision number of thermal fluctuation n0-coverage at saturation τ-average residence time (in plasma) τ0-10-13 s p-gas pressure Consideringthermal fluctuation, n0 coverage at saturation, τ average residence time (in plasma), τ0 10 s, p gas pressure. Considering the cross-section of monomer particles for polymerization the following polymer growing rule for P can be derived:P=γΦ(1+Ze γΦ /J0 2qn∞)-1 mit 1/n∞=1/n0+1/γΦZ. Westwood considered exclusively an ionic (kationic) chain propagation mechanism for the plasma polymerization of vinyl chloride as published in 1971 [A R Westwood Europ Polym J 7 (1971) 363] He interpret radical reactions observed by Denaro [Achloride as published in 1971 [A. R. Westwood, Europ. Polym. J. 7 (1971) 363]. He interpret radical reactions observed by Denaro [A.
R. Denaro, P. A. Owens, A. Crawshaw, Europ. Polym. J. 4 (1968) 93] as concurrent reaction. Because of preferred polymer deposition onto the cathode in direct current glow discharges he concluded also on cation mechanism.Thompson and Mayhan presented 1972 also a kationic mechanism as most probable mechanism [L. F. Thompson, K. G.
Ma han J Appl Pol m Sci 16 (1972) 2317] They admixed radical scavenger to the plasma and found no change in polymerMayhan, J. Appl. Polym. Sci. 16 (1972) 2317]. They admixed radical scavenger to the plasma and found no change in polymer deposition rate. They proposed an excitation step at first: M + e- → M** + e-´ followed by formation of M+, and also M-, M•+, M•-
or ion pair formation P+ and Q- (which need much energy, > 10 eV). These species were also formed directly by inelastic collisions with electrons, such as: M + e- → M+ + e-´ + e-´´. Chain propagation occurs following a kationic ion-molecule
h i M+ + M MM+ d MM+ + M M[M ]M+ ll b hi MMM[M ]M+ + ∆E MM+M[M ]M+ d id
22/45
mechanism: M+ + M → MM+ and MM+ + n M → M[Mn]M+ as well as branching MMM[Mn]M+ + ∆E → MM+M[Mn]M+ and side-chain formation:MM+M[Mn]M+ + M → MM(M+)M[Mn]M+. Vasile and Smolinski investigated the plasma phase for charged and neutral reactive intermediates [M. J. Vasile, G. Smolinsky J.
Appl. Polym. Sci., 16 (1972) 2317; G. Smolinsky, M. J. Vasile, J. Electrochem. Soc. 119 (1972) 451].
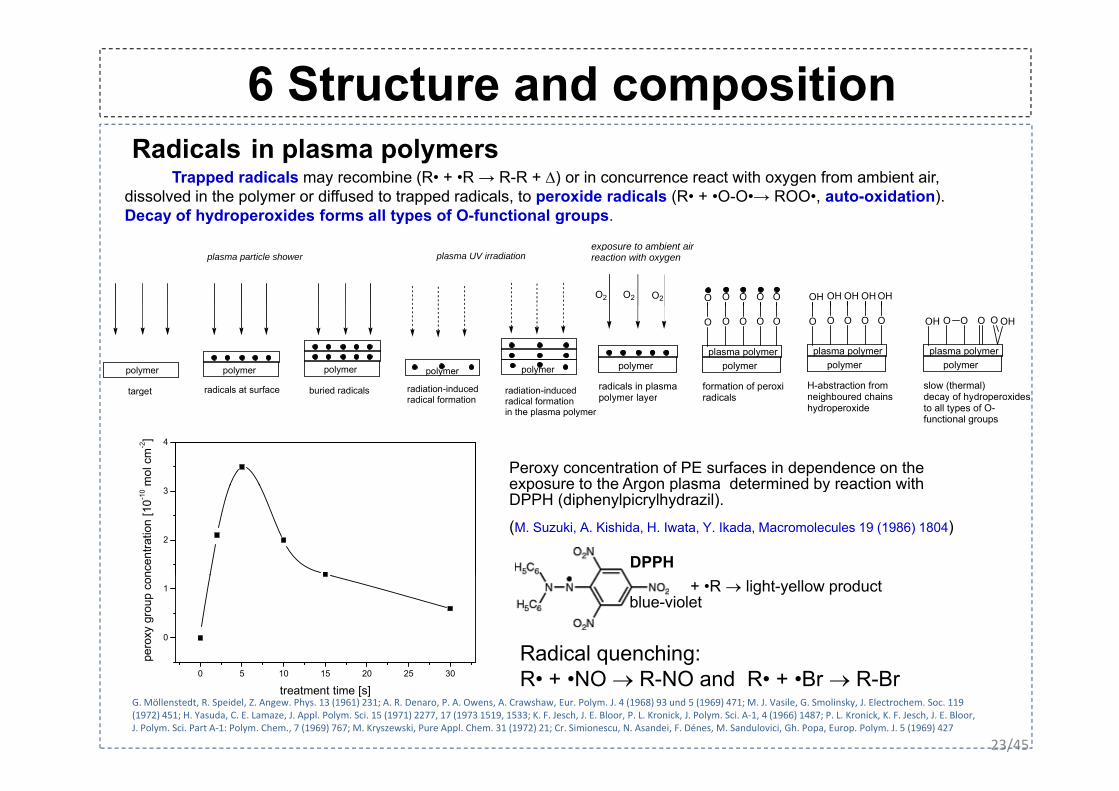
6 Structure and composition Radicals in plasma polymers
Trapped radicals may recombine (R• + •R → R-R + Δ) or in concurrence react with oxygen from ambient air, dissolved in the polymer or diffused to trapped radicals, to peroxide radicals (R• + •O-O•→ ROO•, auto-oxidation). Decay of hydroperoxides forms all types of O-functional groupsDecay of hydroperoxides forms all types of O-functional groups.
plasma particle shower plasma UV irradiationexposure to ambient airreaction with oxygen
O2 O2 O2 O O O O O OH OH OH OH OH
polymer
radicals at surfacetarget
polymer polymer
buried radicals
polymer
radiation-induced
polymer
radiation-induced radicals in plasmapol mer la er
polymer
formation of peroxiradicals
polymer
O O O O O
plasma polymer
H-abstraction from neighboured chains
polymer
O O O O O
plasma polymer
slow (thermal)decay of hydroperoxides
polymer
OH O O O O
plasma polymer
OH
4
mol
cm
-2]
Peroxy concentration of PE surfaces in dependence on the exposure to the Argon plasma determined by reaction with
gradical formation radical formation
in the plasma polymerpolymer layer radicals neighboured chains
hydroperoxidedecay of hydroperoxidesto all types of O-functional groups
2
3
ncen
tratio
n [1
0-10 m exposure to the Argon plasma determined by reaction with
DPPH (diphenylpicrylhydrazil).
(M. Suzuki, A. Kishida, H. Iwata, Y. Ikada, Macromolecules 19 (1986) 1804)
DPPH
0
1
pero
xy g
roup
con + •R → light-yellow product
blue-violet
Radical quenching:
23/45
G. Möllenstedt, R. Speidel, Z. Angew. Phys. 13 (1961) 231; A. R. Denaro, P. A. Owens, A. Crawshaw, Eur. Polym. J. 4 (1968) 93 und 5 (1969) 471; M. J. Vasile, G. Smolinsky, J. Electrochem. Soc. 119 (1972) 451; H. Yasuda, C. E. Lamaze, J. Appl. Polym. Sci. 15 (1971) 2277, 17 (1973 1519, 1533; K. F. Jesch, J. E. Bloor, P. L. Kronick, J. Polym. Sci. A‐1, 4 (1966) 1487; P. L. Kronick, K. F. Jesch, J. E. Bloor, J. Polym. Sci. Part A‐1: Polym. Chem., 7 (1969) 767; M. Kryszewski, Pure Appl. Chem. 31 (1972) 21; Cr. Simionescu, N. Asandei, F. Dénes, M. Sandulovici, Gh. Popa, Europ. Polym. J. 5 (1969) 427
0 5 10 15 20 25 30
treatment time [s]
gR• + •NO → R-NO and R• + •Br → R-Br

6 Structure and composition Plasma polymers and classic polymers show strong differences in bulk properties and structure. A very high polydispersity (broadness of molar mass di t ib ti ) i h t i ti C li k d f ti li t i di d
distribution) is characteristic. Crosslinked fractions, oligomers, tri-, di- and monomers, cyclic structures as well macrocycles produce the complex structure of plasma polymers, which is not easy to separate by chromatographic methods [M. V. Hudis, J. Appl. Polym. Sci., 16 (1972) 2397; R. Mix,
V. Gerstung, J. Falkenhagen, J. Friedrich, J. Adhes. Sci. Technol., 21 (2007), S. 487-507].
-1
pulsed plasma polymerized poly(1,2-butadiene)
poly(1,2-butadiene)
104
106
108
[%]
3001ν(CH)ar
3026
ν(CH)ar
3080ν(CH)ar
3059
poly(styrene)
-2
log ε''
p p p y p y( , )
98
100
102
trans
mitt
ance
[
νs(CH2)2850
νs(CH3)2873
νas(CH3)2962
dielectric relaxation spectroscopy
IR
-100 -50 0 50 100 150 200
-3
temperature [°C]
reference-poly(1,2-butadiene)
3200 3100 3000 2900 2800
94
96 PS standard pp-PS (100 W)
wavenumber [cm-1]
νas(CH2)2923 pp-PS (50 W)
p [ ]
Dielectric loss ε´´ vs. temperature at frequency of 1000 Hz for pp-PB ( )and anionic polymerized commercial PB poly(1,2-butadiene) ( ), arrows mark peaksThe concentration of dipoles is increased by
[ ]
Comparison of IR-spectra of pulsed plasma PS (cw-rf plasma, 50 W; 0.015 ms plasma on / 0.085 ms plasma off; 4.5 Pa; 80 sccm) and commercial PS standard (150 000 g/mol) using the Grazing –
24/45
The concentration of dipoles is increased by 2 orders of magnitude (defects)Heating induces radical recombination & polar gr.oups
PS standard (150,000 g/mol) using the Grazing Incidence-Reflectance-FTIR (α= 70°) normalized to γCH=701 cm-1. Retention of aromatic νCH but loss in νas
CH2 and new νasCH3.

6 Structure and composition 6
NEXAFS (Near Edge X-ray Absorption Fine Structure) CK edge spectra of pulsed plas
4n
Yie
ld [a
.u.]
continuous-wave PS pulsed plasma PS reference PS
Surface compositionmay roughly correspond to reference polymers as shown for polystyreneNEXAFS spectra of pulsed plas-
ma polymerized poly-styrene and reference polystyrene0
2
Par
tial E
lect
ron shown for polystyrene
„skin-effect“NEXAFS
All spectra show polystyrene( i l )
Comparison of C1s-, h k t llitu.
]
VBBA
C 1sx10
u.]
p y y280 290 300 310 320 330
photon energy [eV]
Valence
(vinyl monomer)
shake-up-satellites-and XPS (X-ray Photoelectron Spectroscopy) valence
inte
nsity
[a.u
π
C
pulsed plasma PS
Shake-up
inte
nsity
[a.u
Core-level XPS
bandXPS
Spectroscopy) valence band spectra (VB) of pulsed plasma PS and commercial reference PS (A B C t
30 25 20 15 10 5 0
pulsed plasma PS
binding energy [eV]295 290 285 280
binding energy [eV]
B VB
PS (A, B=C- atom orbitals, C=C-, molecular orbitals from σ- and π-bonds) with
Shake-up
C 1sx10
inte
nsity
[a.u
.]
inte
nsiy
[a.u
.]
π
C
BA VB
25/45
)rough conformance
295 290 285 280
PSreference
binding energy [eV]30 25 20 15 10 5 0
PSreference
binding energy [eV]
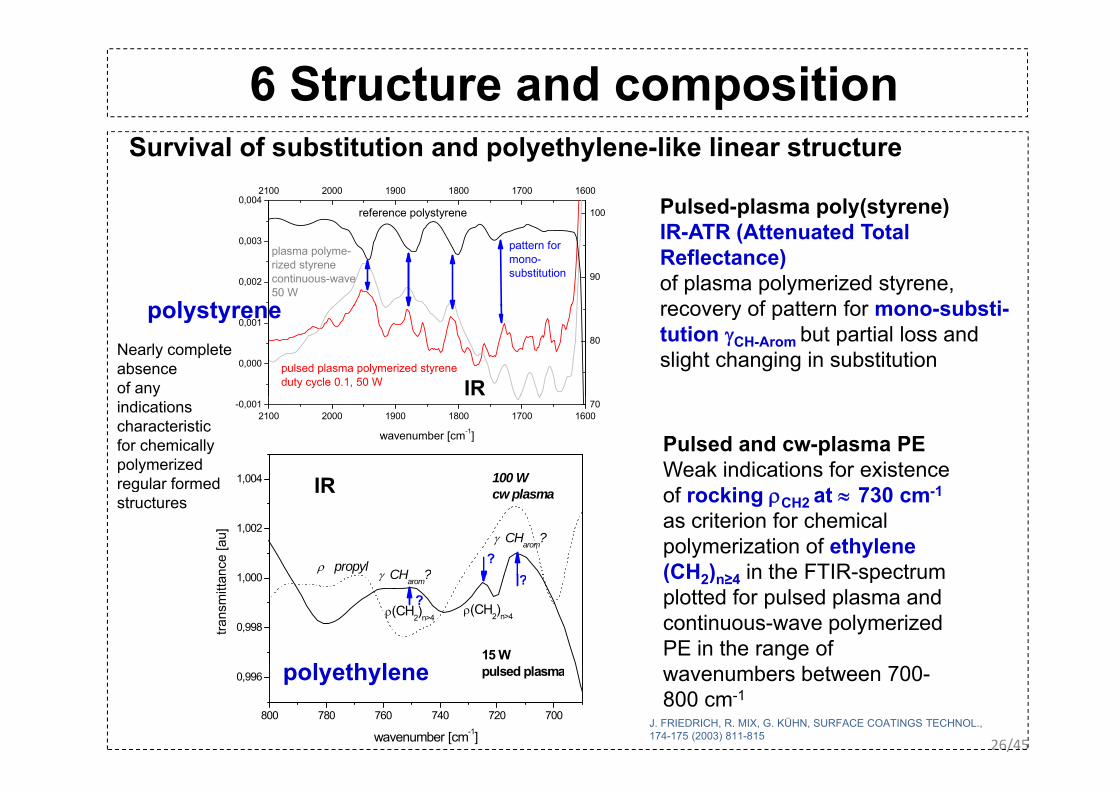
6 Structure and composition
0,0042100 2000 1900 1800 1700 1600
100reference polystyrene Pulsed-plasma poly(styrene)
Survival of substitution and polyethylene-like linear structure
0,002
0,003
90
reference polystyrene
plasma polyme-rized styrenecontinuous-wave 50 W
pattern formono-substitution
p p y( y )IR-ATR (Attenuated Total Reflectance)of plasma polymerized styrene, recovery of pattern for mono substipolystyrene
Nearly complete absence of any
0,000
0,001
80
pulsed plasma polymerized styreneduty cycle 0.1, 50 W
recovery of pattern for mono-substi-tution γCH-Arom but partial loss and slight changing in substitution
IR
polystyrene
yindications characteristicfor chemically polymerizedregular formed 1,004
100 W
Pulsed and cw-plasma PEWeak indications for existence
2100 2000 1900 1800 1700 1600-0,001 70
wavenumber [cm-1]
IRregular formed structures
1 000
1,002
??γ CHarom?
γ CH ?ρ propyl
cw plasma
nce
[au]
of rocking ρCH2 at ≈ 730 cm-1
as criterion for chemical polymerization of ethylene (CH2) ≥4 in the FTIR-spectrum
IR
0,998
1,000 ? γ CHarom?
ρ(CH2)n>4ρ(CH2)n>4
15 W
trans
mitt
a
?
(CH2)n≥4 in the FTIR spectrum plotted for pulsed plasma and continuous-wave polymerized PE in the range of
l th l
26/45
800 780 760 740 720 700
0,996 pulsed plasma
wavenumber [cm-1]
wavenumbers between 700-800 cm-1
J. FRIEDRICH, R. MIX, G. KÜHN, SURFACE COATINGS TECHNOL.,174-175 (2003) 811-815
polyethylene

7 Quasi-hydrogen plasmaDuring plasma polymerization of non-polymerizable and also polymerizable monomers considerable percentage of hydrogen is not deposited in the plasma polymer but H is transferred to gas phase There the hydrogen surplus increases significantly the (inner)transferred to gas phase. There, the hydrogen surplus increases significantly the (inner) plasma parameters such as electron temperature
Changing of plasma parameters during polymerization
120
140
er 1
00 C
theoretical value for the 1:1 styrene - allyl alcohol copolymer
continuous wave24%15
20
Ar+toluene plasma (Ar + H2)
e [a
u]
decrease in Te because of enrichment of Ar
100
conc
entra
tion
pe
hydrogen
continuous-waveplasmapulsed plasma
-24%
10
ctro
n te
mpe
ratu
re
pure Ar plasma
great electron capture cross-section of toluene molecules
enrichment of Arwith H and increasingTe
C
0 0 0 1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 1 00,0
0,4
0,880
nitrogen
H a
nd N
c
0 20 40 60 80 1000
5
pumptoluene injectionrela
tive
elecCH analysis electron temperature
(Langmuir probe)
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
duty cycle
Theoretical and CHN analysis-measured hydrogen (nitrogen)
0 20 40 60 80 100position in tube-type reactor [cm]
Differences in electron temperature as measured by Langmuir probe between
27/45
ratio in a plasma-deposited styrene-allyl alcohol (1:1) copolymer
pure Ar- and Ar + toluene plasmaCooperation: L. Weixelbaum, H. Wittrich

7 Quasi-hydrogen plasma
1 0
1,2
1,4
1,6
1,8
of p
lasm
a po
lym
er
D+H
Htheoretical H/C ratio of toluene
Dependence of D↔H exchange in the plasma polymer deposited in d8toluene + 3 H2 dc low-
Hydrogen loss in plasma polymers layers of toluene in dependence on
1,0
1,2
1,4
1,6
ted
plas
ma
poly
mer
toluene + 3 H2
theoretical H:C composition
0 0
0,2
0,4
0,6
0,8
1,0
C-H
/D c
ompo
sitio
n o
D
d8 toluene introduction pump
pressure plasma atmosphere on tube length
dependence on locus of deposition
CH analysisCH analysis
0,0
0,2
0,4
0,6
0,8
C-H
ratio
of d
epos
i t
toluene
monomer introduction pump
0 20 40 60 80 100 120
0,0
length of discharge tube [cm]
layer target gas probing for GC Tube-type reactor for plasma polymerization under low-pressure
0 20 40 60 80 100 120
,
length of discharge tube [cm]
positive column
dc-discharge
reactor length
polymerization under low pressure conditions (10 Pa) in a direct-current glow discharge equipped with several ports for Langmuir probe, gas chromatography (GC), layer deposition for H/C analysis and measuring the deposition rate
toluene pump
80
100
CH3
toluene
referenced to 100% Loss in aromatic rings in h8-toluene
d d t l80
100CH3
toluene
mono substitutionToluene plasma polymerization in low pressure dc
0 10 20 30 40 50 60 70 80 90 cmand measuring the deposition rate
20
40
60
80 toluene
1
545 cm-1
trans
mitt
ance
[%]
700 cm-1
and d8-toluene plasma polymer layers in dependence on the locus of deposition
20
40
60
perc
enta
ge [%
]
low-pressure dc discharge and changing of substitution at aromatic ring
IR IR
28/45
0 20 40 60 80 100 120
0
20
1580 cm-1
1600 cm-1
length of discharge tube [cm]
toluene introduction pump
p
0 20 40 60 80 100 120
0
20 para substitution
length of discharge tube [cm]
toluene injection pump
structures
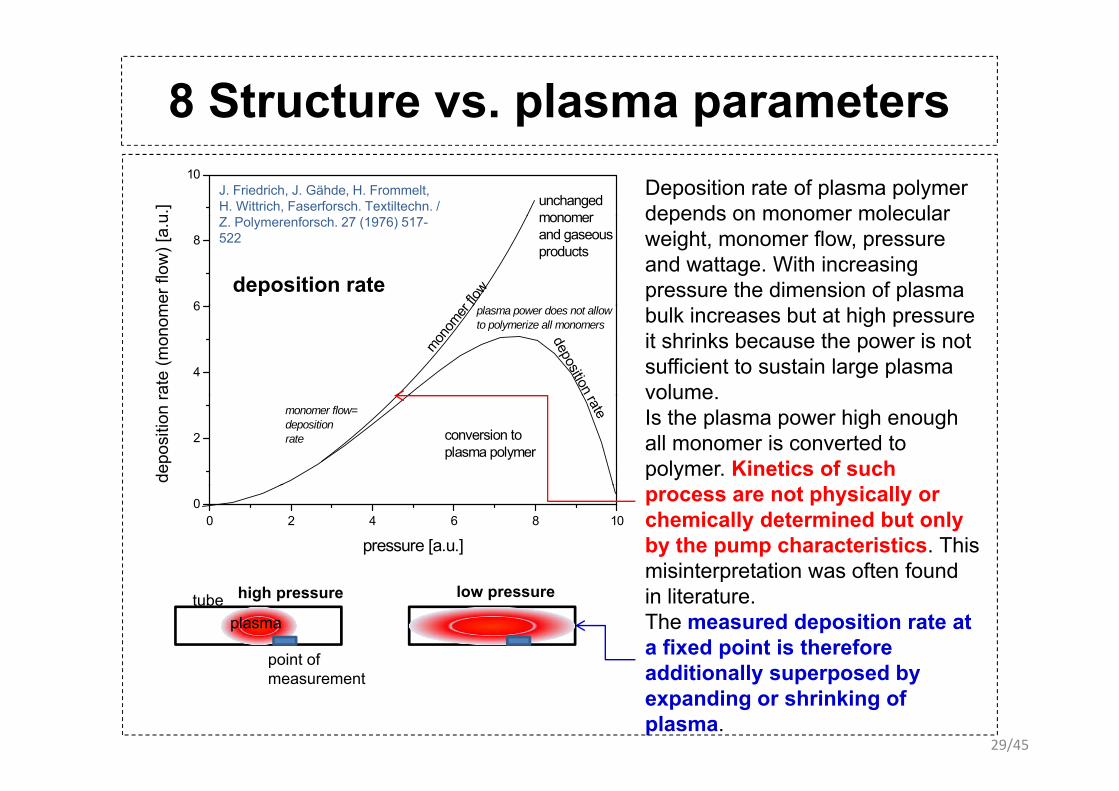
8 Structure vs. plasma parametersp p10
u.
] unchanged monomer
Deposition rate of plasma polymer depends on monomer molecular
J. Friedrich, J. Gähde, H. Frommelt, H. Wittrich, Faserforsch. Textiltechn. /
6
8
mer
flow
) [a.
u monomerand gaseousproducts
r flow
depends on monomer molecular weight, monomer flow, pressure and wattage. With increasing pressure the dimension of plasma
Z. Polymerenforsch. 27 (1976) 517-522
deposition rate
4
6
rate
(mon
om
plasma power does not allowto polymerize all monomers
monom
er
deposition
bulk increases but at high pressure it shrinks because the power is not sufficient to sustain large plasma volume
2
depo
sitio
n r
conversion toplasma polymer
monomer flow=depositionrate
rate
volume. Is the plasma power high enough all monomer is converted to polymer. Kinetics of such
0 2 4 6 8 100
pressure [a.u.]
process are not physically or chemically determined but only by the pump characteristics. This misinterpretation was often foundmisinterpretation was often found in literature.The measured deposition rate at a fixed point is therefore ddi i ll d b
tubeplasma
point of
high pressure low pressure
29/45
additionally superposed by expanding or shrinking of plasma.
po omeasurement
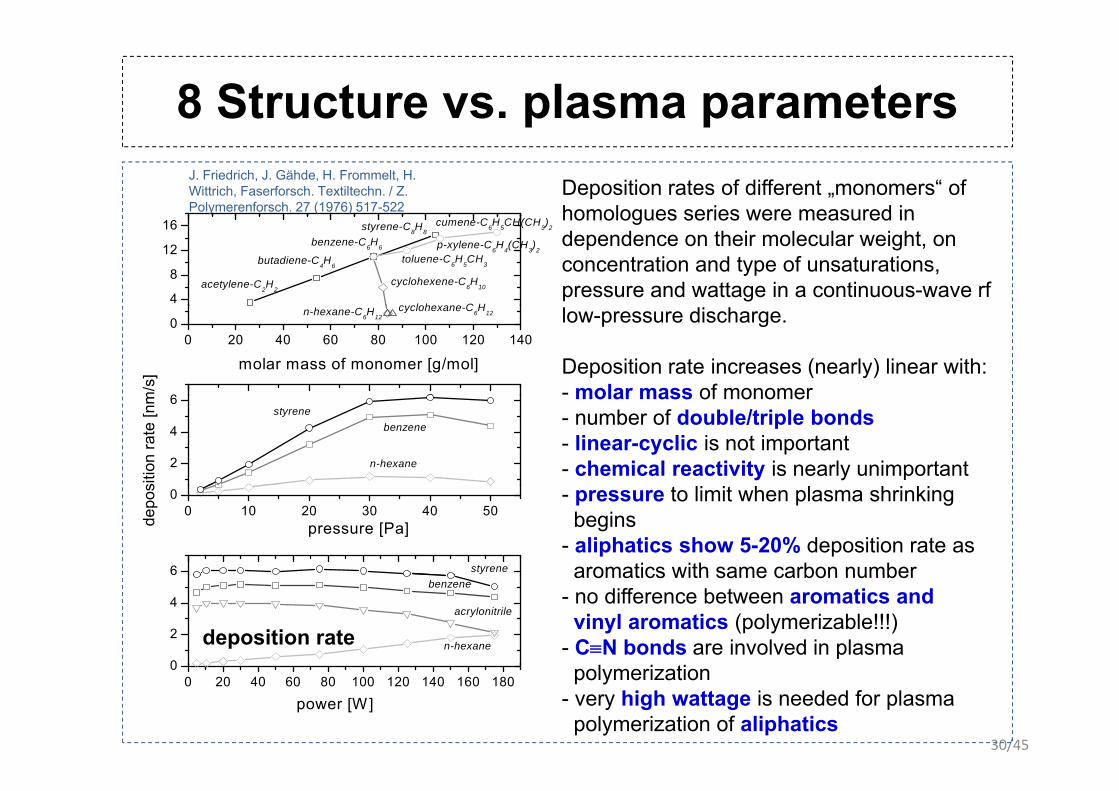
8 Structure vs. plasma parametersp pDeposition rates of different „monomers“ of homologues series were measured in
J. Friedrich, J. Gähde, H. Frommelt, H. Wittrich, Faserforsch. Textiltechn. / Z. Polymerenforsch. 27 (1976) 517-522
p-xylene-C6H4(CH3)2toluene-C6H5CH3
styrene-C8H8
benzene-C6H6
4
8
12
16 cumene-C6H5CH(CH3)2
cyclohexane C H
cyclohexene-C6H10
butadiene-C4H6
acetylene-C2H2
homologues series were measured in dependence on their molecular weight, on concentration and type of unsaturations, pressure and wattage in a continuous-wave rf
n-hexane-C6H12
molar mass of monomer [g/mol]
m/s
]
6
0 20 40 60 80 100 120 1400
cyclohexane-C6H12 low-pressure discharge.
Deposition rate increases (nearly) linear with: - molar mass of monomer
n-hexane
benzenestyrene
sitio
n ra
te [n
m
2
4
6 molar mass of monomer- number of double/triple bonds - linear-cyclic is not important- chemical reactivity is nearly unimportant
6 styrene
depo
s
0 10 20 30 40 500
pressure [Pa]
- pressure to limit when plasma shrinking begins
- aliphatics show 5-20% deposition rate as aromatics with same carbon number
0
2
4acrylonitrile
n-hexane
benzenearomatics with same carbon number
- no difference between aromatics and vinyl aromatics (polymerizable!!!)
- C≡N bonds are involved in plasma l i i
deposition rate
30/45
0 20 40 60 80 100 120 140 160 1800
power [W]
polymerization- very high wattage is needed for plasma
polymerization of aliphatics

9 Plasma polymers + functional groups
CH2 CHtheor. stoichiometric ratio 75 C and 25 Omeasured stoich. ratio 74 C and 24 O
poly(allyl alcohol) = OH
IR
OH and COOH functionalization
2000
3000
OH OH
OH OH
CH2
OHn
ty [c
ts.]
CH, CH299
100
C-H2C=Oance
[%]
IR
0
1000
l ( ll l l h l) OH
OH OH
inte
nsit
O-C-O/>C=O (28%)
OH (72%)
98
C-OH
C-H2C-H2, C-H
O Hpulsed plasma poly(allyl alcohol)
100 W, duty cycle 0.1, 10 Hz
trans
mitt
a
XPS-C1s
290 288 286 284
0 poly(allyl alcohol) = OH
binding energy [eV]
3500 3000 2500 2000 1500 1000 500
O-H 100 W, duty cycle 0.1, 10 Hz
wavenumber [cm-1]
0,550 W, cw plasmapoly(acrylic acid) at PE
poly(acrylic acid) = COOH16000 O1sC1s
0,3
0,4
0,5
50 W, 1000 Hz, dc 0.1poly(acrylic acid) at PE
poly(acrylic acid) at PE
ce [a
u]
50 W, 1000 Hz, 0.5 dcpoly(acrylic acid) at PE
10000
12000
14000
160005 40 53 8 536 5 34 53 2 530
inte
nsity
[cp
s.]
b in ding e ne rg y [e V]29 6 292 2 88 28 4 280
inte
nsit
y [c
ps.]
bi ndi ng e ne rgy [ eV]
O1sC1s
cps.
]
IR
0,0
0,1
0,2
abso
rban
2000
4000
6000
8000
inte
nsity
[c
XPS
31/45
4000 3500 3000 2500 2000 1500 1000-0,1
wavenumber [cm-1]800 700 600 500 400 300 200
0poly(acylic acid) = COOH
binding energy [eV]

9 Plasma polymers + functional groups
FTIR-ATR spectra of plasma polymerized allylamine with
Side- and post-plasma reactions using allylamine
νCC
100
νCN
IR
NH-/ NH2-specific bands (in grey) undesired C≡N/C≡C bands
0,8
1,0
[au]
Plasma polymerized allylamine30W , 5Pa, 0.5 duty cycle undesired
oxygenintroduction
νCC-H
δN-Hallylamine
100 W duty cycle 0 1; 10 Hz
98
99
trans
mitt
ance
[%]
νN-H
νC=O
0 2
0,4
0,6
"in situ" without contact to air
3 months aged
orm
aliz
ed in
tens
ity
C1s
introduction
XPS
3500 3000 2500 2000 1500 1000
poly(allylamine) = NH2
100 W, duty cycle 0,1; 10 Hz
97
wavenumber [cm-1] 20
292 290 288 286 284 282 280
0,0
0,2
poly(allylamine) = NH2
n
binding energy [eV]
IR
0,10
0,12
0,14
0,16 exposure of pulsed plasma polymerized allylamineto oxygen from ambient air:
0.5 h air 2.0 h air 16.0 h air
e [a
u]
>C=O
IR
12
14
16
18
20
cw-plasma polyallyamine pulsed plasma polyallylamineO
/100
C]
poly(allylamine) = NH2XPSundesiredoxygenintroduction
undesiredoxygenintroductionfound forcw plasma
0,02
0,04
0,06
0,08
abso
rban
ce
NH2
-OH
2
4
6
8
10 cw-plasma polystyrene pulsed plasma polystyrene
O c
once
ntra
tion
[O
l l th l
cw plasmamode
XPS
32/45
4000 3500 3000 2500 2000 1500 1000 500-0,02
0,00
wavenumber [cm-1]
poly(allylamine) = NH2
commercial poly(allylamine)
0 10 20 30 40
0
2
time of storage [d]
cw-plasma polyethylene pulsed plasma polyethyleneIR

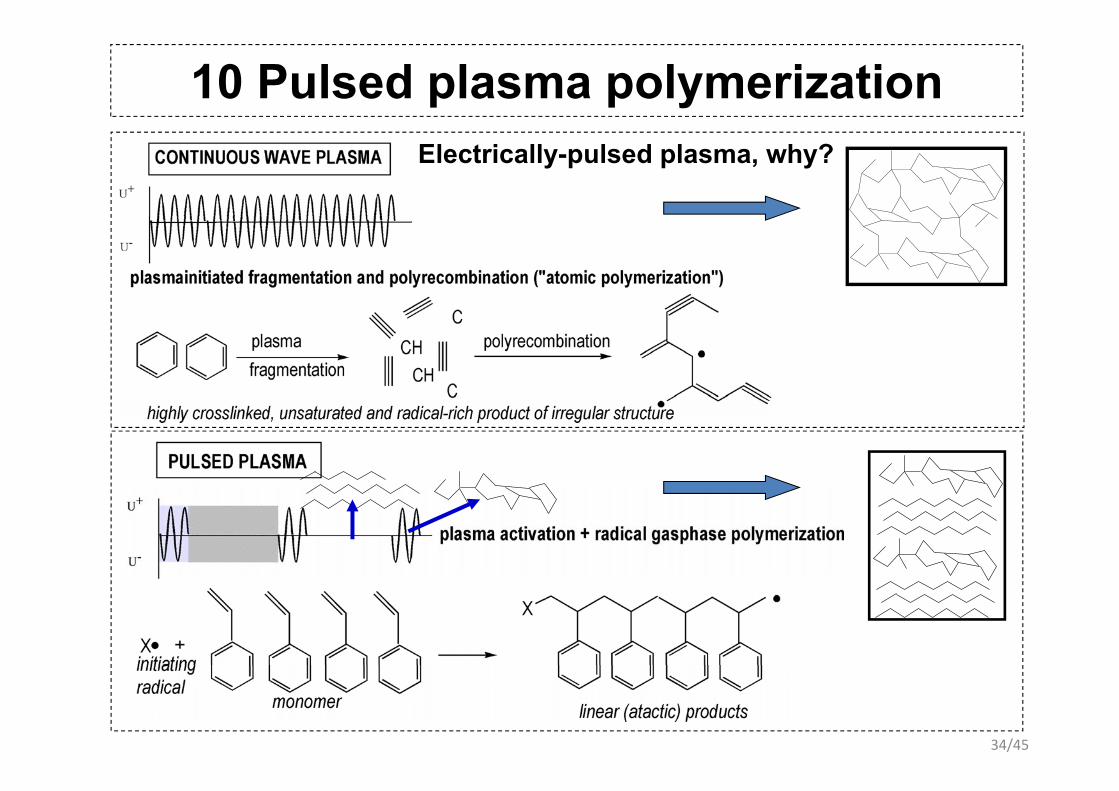
10 Pulsed plasma polymerizationPlasma polymerization modes1. plasma-induced (chemical or radical) polymerization to linear or branched products:
A•+nA→ An+1•
2. polymerization in the continuous-wave plasma by monomer fragmentation and their (radical) polyrecombination to strongly irregularly formed polymers : ABCDE→A+B+C+D+E→DFACBABCDE→A B C D E→DFACB
3. co-polymerization in the continuous-wave plasma by comonomer fragmentation and their (radical) polyrecombination to randomly formed “copolymers”: AB + CD → DACB
4. pulsed-plasma polymerisation and pressure-pulsed plasma polymerization to p p p y p p p p yproducts with higher contents of linear products: A•+nA→ An+1•
5. pulsed-plasma co-polymerisation and pressure-pulsed plasma co-polymerization to copolymers with higher contents of linear sequences: AB + CD → ABCD
6. plasma polymerization in underwater plasma and glow-discharge electrolysis:OH•+nA→ HO-An•
7. polymerization in adsorption layers initiated by electrospray: [AB]n → [AB]n
8. chemical grafting onto radicals or functional groups of plasma polymers or plasma-exposed polymer surfaces [V. V. Korschak, Dokl. Akad. Nauk 151 (1963) 1332]: [A(NH2)]n + nHOC(CH2)3-CHO → [A(N=CH(CH2)3-CHO)]n
33/45
9. polymerization in the after glow by neutral excited species produced in the plasma [L. S. Tuzov, A. B. Gilman, A. N. Schtschurorov, V. M. Kolotyrkin, Vysokomol. Soedin. 11 (1967) 2414; S. E. Kuprianov, J. eksp. i teor. Fisikii 48
(1965) 468] : Ar* + A → Ar + A• → A•+nA→ An+1•
10 Pulsed plasma polymerizationElectrically-pulsed plasma, why?
34/45

10 Pulsed plasma polymerizationpressure pulse (300 ms)
after plasma pulse300ms
Pressure- and electrically pulsed plasma, why?
isolated chain propagationcentre (radical)
low pressure
desactivated chain propagationcentre (disproportionation, re-combination, chain transfer) 1000 ms plasma pulse
ton =100ms
high pressure
ton =100 ms, toff =900 ms
.]
chain propagation centre (radical) in continuouscontact with monomer molecules
growing chain
real plasma pulse
inte
nsity
[a.u ton =100ms
di b d h i i i h
high pressure100 ms opening of
the vacuum valve
after the pressure pulse
100 msplasma ignition at low pressureformation of new start radicals
undisturbed chain propagation in theplasma-off period at high pressure
Schematic view on the correlation betweenpurpose of high pressure, and high sticking rate to and the chain propagation centre forduring the chemical gas phase polymerization. In Principle of plasma and pressure pulse
h i ti d d f th
time [ms]
100 ms
0
35/45
g g ythe last line the principal particle densities are depicted for the low-pressure plasma ignition and the high-pressure chemical chain propagation
synchronization and measured response of the plasma systemMeasured by A. Meyer-Plath
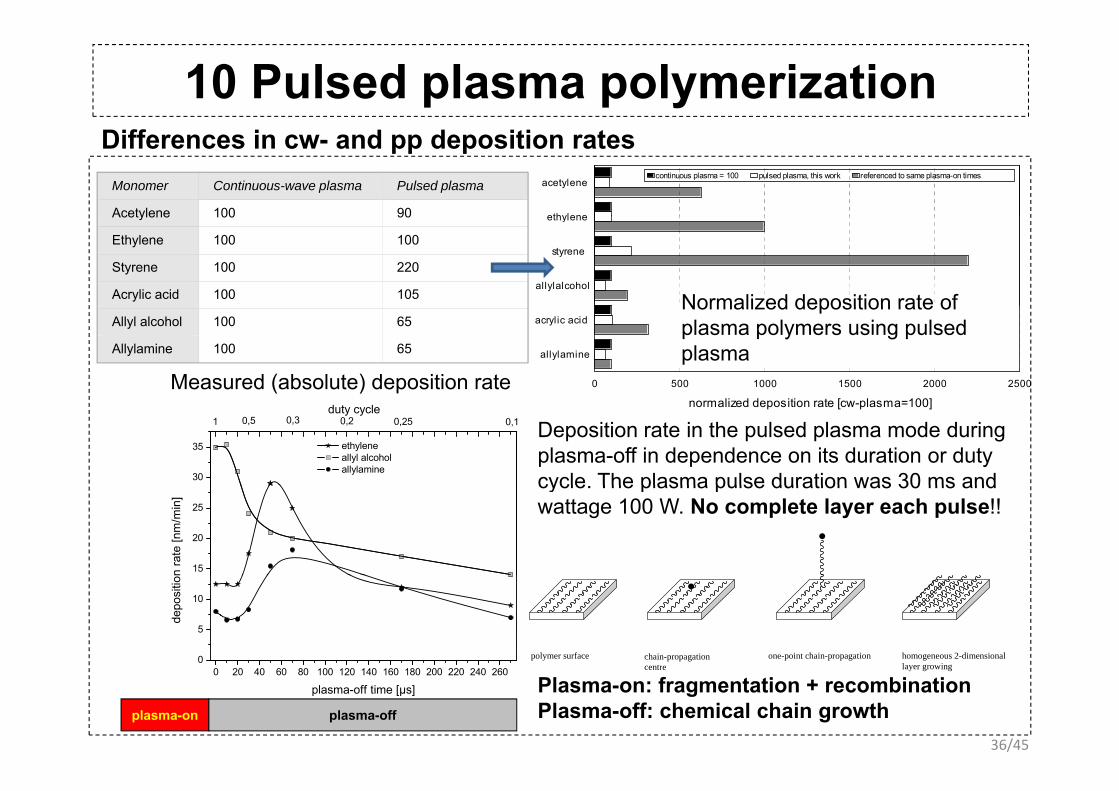
10 Pulsed plasma polymerizationDifferences in cw- and pp deposition rates
Monomer Continuous-wave plasma Pulsed plasma
Acetylene 100 90
acetylene
ethylene
continuous plasma = 100 pulsed plasma, this work referenced to same plasma-on times
Acetylene 100 90
Ethylene 100 100
Styrene 100 220
Acrylic acid 100 105
ethylene
styrene
allylalcohol
Normalized deposition rate ofAllyl alcohol 100 65
Allylamine 100 65
acrylic acid
allylamine
0 500 1000 1500 2000 2500
Normalized deposition rate of plasma polymers using pulsed plasma
Measured (absolute) deposition rate
A duty cycle of 0.1 was applied, e.g. the plasma was burning only during 10% of the total treatment time
normalized deposition rate [cw-plasma=100]
30
35 ethylene allyl alcohol allylamine
1 0,5 0,3 0,10,2duty cycle
0,25 Deposition rate in the pulsed plasma mode during plasma-off in dependence on its duration or duty cycle The plasma pulse duration was 30 ms and
15
20
25
n ra
te [n
m/m
in]
cycle. The plasma pulse duration was 30 ms and wattage 100 W. No complete layer each pulse!!
0
5
10
depo
sitio
n
polymer surface chain-propagationt
one-point chain-propagation homogeneous 2-dimensionall i
36/45
0 20 40 60 80 100 120 140 160 180 200 220 240 260
plasma-off time [µs] Plasma-on: fragmentation + recombinationPlasma-off: chemical chain growthplasma-on plasma-off
centre layer growing

11 Pulsed plasma copolymerizationPrinciple of copolymerization
plasma polymerizationmonomer A
homopolymer AXXXX Theor. homopolymer compos.
All l l h l 33 OH/100 C
plasma polymerizationmonomer B
homopolymer B
Allyl alcohol 33 OH/100 CAllybromide 33 Br/100 CVinylbromide 50 Br/100 CAllylamine 33 NH /100 C
+plasma co-polymerization
alt-copolymer A-BX X
Allylamine 33 NH2/100 CAcrylic acid 33 COOH/100 CStyrene 70 aryl/100 C
Measured plasma polymersAllyl alcohol 30 OH/100 CAllybromide 26 Br/100 C
alternating (alt) copolymer A-B
X
B B
B
A
X
A
h l B Allybromide 26 Br/100 CVinylbromide 24 Br/100 CAllylamine 15 NH2/100 CAcrylic acid 27 COOH/100 C
X
B B B B
A
X
A
X
A
X
A
X X
homopolymer B
homopolymer A
yStyrene 50 aryl/100 C
Copolymers
X
A
X
A B B
X
block (b) copolymer A-B
37/45
yBetween 0 and theor. value
B B B B
XX
graft(g) copolymer A-B

Aus Datenschutzgründen wurde das automatische Herunterladen dieser externen Grafik von PowerPoint verhindert. Klicken Sie auf der Statusleiste auf 'Optionen', und klicken Sie dann auf 'Externe Inhalte aktiv ieren', um diese Grafik herunterzuladen und anzuzeigen.
11 Pulsed plasma copolymerizationDeposition rate of copolymer in dependence on comonomer ratio
p p y
Representative diagram (Wikipedia) for copolymerization. Dependence of copolymer composition on comonomer
10
12
/min
]
5
6
allylalcohol-ethylene10
allyl alcohol-butadiene
14
16
p y pratio
6
8
10
posi
tion
rate
[nm
/
2
3
4
2
4
6
8
4
6
8
10
12
allyl alcohol-acetylene
0 20 40 60 80 100
4dep
allyl alcohol [mole%]
allyl alcohol-styrene0 20 40 60 80 100
0
1
allyl alcohol [mole%]0 20 40 60 80 100
0
2
allyl alcohol [mole%]0 20 40 60 80 100
0
2allyl alcohol-acetylene
allyl alcohol [mole%]
Dependence of copolymer deposition rates on types of comonomers and their ratio in the comonomer mixture for the example of allyl alcohol and and olefins and acetylene. Most often deviation of linearity. In chemistry of copolymers such observations were also made. For description of copolymerization kinetics the Fineman Ross approch is often used Here this theory was also
38/45
copolymerization kinetics the Fineman-Ross approch is often used. Here, this theory was also applied successfully to plasma copolymerization. The same is true for the Alfred-Price Q,e scheme.

11 Pulsed plasma copolymerization
100 80 60 40 20 0ethylene or butadiene [mol%]
OH- or COOH yield vs. comonomer ratio
28
32
36100 80 60 40 20 0
CO
OH
/100
C]
maximal stoichiometric conc.: 33 COOH / 100 C
copolymerization homopolymerization28
32
36 maximal stoichiometric conc. 33 OH/100 C
[OH
/100
C]
100 W
300 Wethylene
16
20
24
conc
entra
tion
[C
ethylene
12
16
20
24
conc
entra
tion
[
300 W
butadiene
0
4
8
12
CO
OH
-gro
up c
butadiene
0
4
8
12
OH
-gro
up
styrene
allyl alcoholOH
acrylic acidCOOH
0 20 40 60 80 1000
acrylic acid in comonomer mixture [mol%]
0 20 40 60 80 1000
allyl alcohol in comonomer mixture [mol%]
Yield in OH groups for plasma-initiated copolymerization of allyl alcohol and butadiene ethylene and styrene
Yield in COOH groups for plasma-initiated copolymerization of acrylic acid and butadiene or ethylenebutadiene, ethylene and styrene butadiene or ethylene
Pulsed plasma copolymerisation follows shows non-linearity, similar to chemical copolymerization Selected comonomer type comonomer ratio consumption etc
39/45
copolymerization. Selected comonomer type, comonomer ratio, consumption etc.determine composition and structure of copolymer.

12 Structure of pp-homo-/copolymersComparison of pulsed plasma polymerized polystyrene with reference polystyrene (standard, MW=226,000 g/mol)W
581
376263
DTGTG
16,5 %2,1 %
3,5 100 W, cw-plasma 3 W, cw-plasma3 W (30 W pulsed duty cycle=0 1
100 W, cw-mode
350 670
TG
GD
TG
16,4 %
67,4 %
2,0
2,5
3,03 Weffective (30 W pulsed, duty cycle=0.1,
pulse frequency=1000 Hz) PS (standard, Mn= 226 kg/mol) PS (standard, Mn= 226 kg/mol) x 5
n [a
u]
7,2 %92,8 %
3 W; cw-mode
86 1 %
670350
G
TG
DTG
0,5
1,0
1,5
extin
ctio
n
415
30 W; DuCy 0.1; 1000 Hz
residue 5.4 %
100 %
8,5 %
86,1 %
G
TG
DTG
200 250 300 350 400
0,0 referenced to c = 40 µg/ml THF
wavelength [nm]
100 200 300 400 500 600 700 800
PS-standard
temperature [°C]airnitrogen
G
UV spectra plasma-produced PS
Strong structural deviations from PS
40/45
TG-and DTG-plots of cw-, pp-and reference PS
Strong structural deviations from PSstandard are evident if comparingthermal degradation and UV spectroscopy
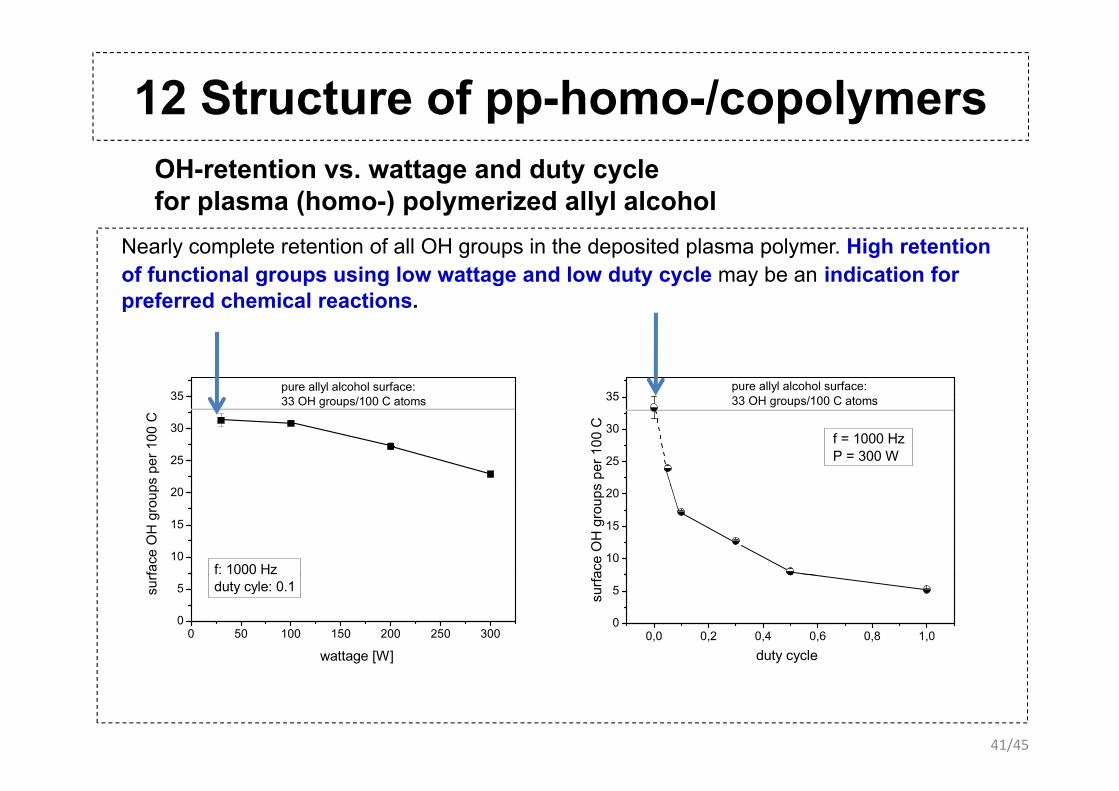
12 Structure of pp-homo-/copolymersOH-retention vs. wattage and duty cyclefor plasma (homo-) polymerized allyl alcohol
Nearly complete retention of all OH groups in the deposited plasma polymer. High retention of functional groups using low wattage and low duty cycle may be an indication for preferred chemical reactions.
35
pure allyl alcohol surface:35
pure allyl alcohol surface:
preferred chemical reactions.
25
30
35 33 OH groups/100 C atoms
ps p
er 1
00 C
25
30
35 33 OH groups/100 C atoms
f = 1000 HzP = 300 W
s pe
r 100
C
10
15
20
f: 1000 Hz
fa
ce O
H g
roup
10
15
20
ace
OH
gro
ups
0 50 100 150 200 250 3000
5 duty cyle: 0.1surf
wattage [W]0,0 0,2 0,4 0,6 0,8 1,0
0
5surfa
duty cycle
41/45
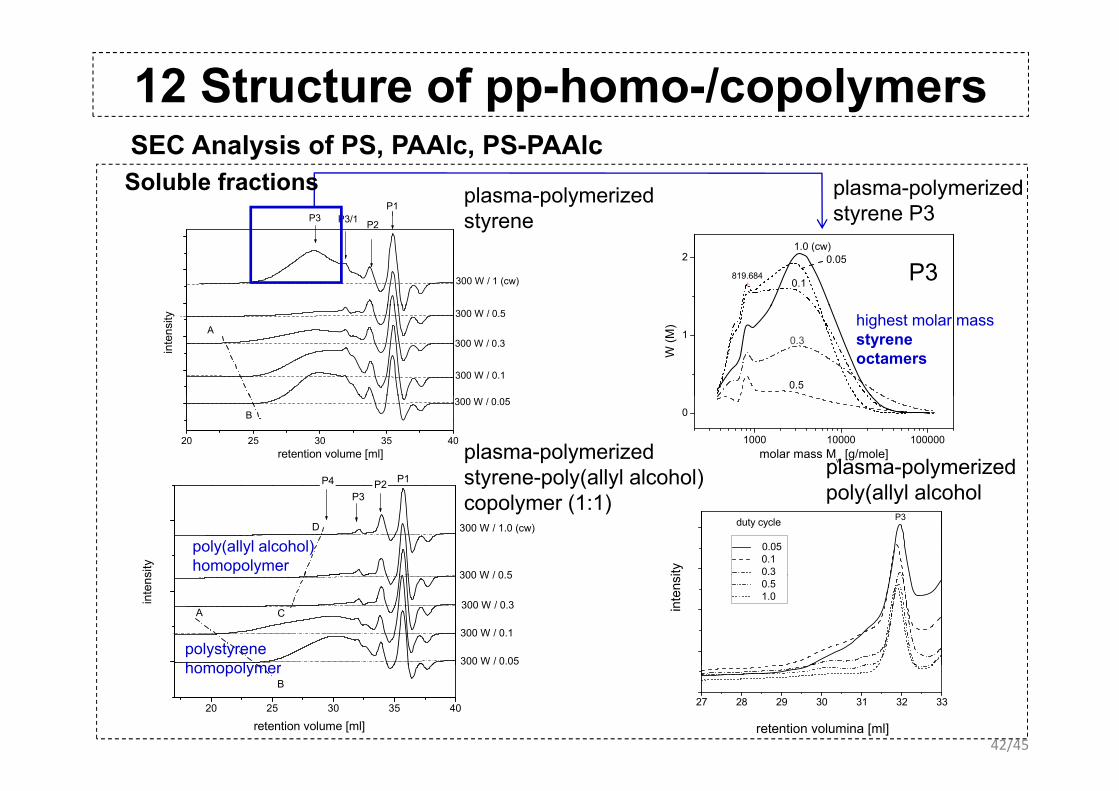
12 Structure of pp-homo-/copolymersSEC Analysis of PS, PAAlc, PS-PAAlc
P3 P3/1P1
plasma-polymerizedt
plasma-polymerizedstyrene P3
Soluble fractionsP3 P3/1 P2
300 W / 1 (cw) 819.684
21.0 (cw)
0.05
0.1 P3
styrene styrene P3
A300 W / 0.3
300 W / 0.5
300 W / 0.1
inte
nsity
1
W (M
)
0.3
0.5
highest molar massstyrene octamers
20 25 30 35 40
B300 W / 0.05
retention volume [ml]
P4 P1P2
1000 10000 100000
0
molar mass Mw [g/mole]plasma-polymerizedstyrene-poly(allyl alcohol) plasma-polymerized
l ( ll l l h lD
P3P2
300 W / 0 5
300 W / 1.0 (cw)
sity
y p y( y )copolymer (1:1)
P3
ity
0.05 0.1 0.3
duty cycle
poly(allyl alcohol
poly(allyl alcohol)homopolymer
CA300 W / 0.3
300 W / 0.5
300 W / 0.05
300 W / 0.1
inte
ns
inte
nsi
0.5 1.0
polystyrenehomopol mer
42/45
20 25 30 35 40
B
retention volume [ml]
27 28 29 30 31 32 33
retention volumina [ml]
homopolymer

Perspectives• Plasma polymerization is a fragmentation – polyrecombination process• This process involves the fragmentation of „monomers“ to atoms and small fragmentssmall fragments
• These fragments recombine randomly to complete irregular structuresfar from known chemical structures of classic polymers
• The deposition rate of such plasma polymerization is derived from p p p yfragmentation, intermediate formation and layer formation and was brought into a few pseudo kinetics theories
• Nevertheless, chemical chain growth polymerization occurs to low extentusing classic monomers, such as vinyl or acrylic precursors
• Surprisingly, allyl monomers were also plasma polymerized with asignificant retention of functional groups
• It can be summarized that the retention of functional groups is much more successful than the retention of macromolecular structure
• Pulsed and pressure-pulsed plasma improve the chance for more regulart t if i l i ith t t d f h i l lstructures if using classic monomers with great tendency of chemical poly-
merization• Suppressing of undesired plasma fragmentation by new developments are not to e pect beca se plasma prod ces too high enrgies
43/45
are not to expect because plasma produces too high enrgies• The only way out is to use prefabricated polymers for deposition withoutplasma (electro spray - ESI) or with soft plasma (APCI)

AcknowledgementsAuthor´s investigations on plasma polymerization processes started in 1972.After exmatriculation from Humboldt university and finishing his PhD work
ft 1 b f liti l th th t t PhDafter 1 year because of political reasons the author starts a new PhD studentship German Academy of Sciences from 1972-1991…1994. There (Berlin-Adlershof, Central Institute for Organic Chemistry, Institute for Macro-Molec lar Compo nds) the research began on plasma processes applied toMolecular Compounds), the research began on plasma processes applied to polymers.A large number of coworker had coworked with him in this time such as Dr. Gähde Mrs Dr Loeschcke Mrs Dr Pohl Mrs Dr Müller Dr Throl Mrs MGähde, Mrs. Dr. Loeschcke, Mrs. Dr. Pohl, Mrs. Dr. Müller, Dr. Throl, Mrs. M. Hannemann, Prof. Dr. Frommelt, Dr. Wittrich, Dr. Weixelbaum, Dr. John……
Beginning from 1995 the author started a new career at Federal Institute forBeginning from 1995 the author started a new career at Federal Institute forMaterials Research and Testing in Berlin as „director and professor“ and as professor at Technical university Berlin (Institute of Material Sciences). Many coworker are to mention such as Mrs. G. Hidde, Mrs. Dr. J. Falkenhagen, Mrs.coworker are to mention such as Mrs. G. Hidde, Mrs. Dr. J. Falkenhagen, Mrs. Dr. Mix, Mrs. Dr. Vogel, Mrs. Altmann, Mrs. Dr. Retzko, Mrs. Dr. Gerstung, Dr. Kühn, Dr. Weidner, Dr. Wettmarshausen, Dr. Koprinarov, Dr. Geng, Dr. Unger,Dr. Meyer-Plath, Dr. Mach, Dr. Klotz, A. Mohamed, Prof. Dr. Schönhals, Prof.
44/45
y , , , , ,Hennecke…….

AcknowledgementsDr. R. Mix
G. Hidde
Dr. A. Meyer-Plath Prof. Dr. A. Schönhals(deputy chief)
Dr. S. Wettmarshausen
Dr. R. D. Schulze
Prof. Dr. J. Friedrich(head)Thanks to:
Y. Huajie
45/45


















