
PHSS Welsh QP Forum Conference 2015 - c.ymcdn.com · o Traditional process validation o Continuous...
109
-
Upload
vuongduong -
Category
Documents
-
view
220 -
download
0
Transcript of PHSS Welsh QP Forum Conference 2015 - c.ymcdn.com · o Traditional process validation o Continuous...


PHSS Welsh QP Forum Conference 2015
Di Morris
Changes in the EU Guidance – Impact
of Changes

Changes already into force
2
Chapter 3:
Premises and Equipment
1st March 2015
Chapter 5: Production
1st March 2015
Chapter 8: Complaints,
Quality Defects and Product
Recalls 1st March 2015
Annex 15: Qualification and
Validation 1st October 2015

Regulations to prepare for
3
Annex 16:
Certification by a Qualified Person and Batch Release
15 April 2016
Annex 1: Manufacture of Sterile Products
Still under review
Annex 17: Parametric
Release Still under review
Annex 21: Importers of
Medicinal Products
1st Draft January 2016

Why so much Change?
• Changes happen as a consequence of:
– Poor GMP practices found on inspection
– Catastrophic events – Heparin
– Changes in technology - sterile manufacture; AMTPs; gene therapy
– Combination of all
– What is the driver of the biggest change
Falsified Medicines Directive
4

Chapter 3 and Chapter 5 – Key Changes
5
• Chapter 3
Point
3. 6
• part of improved guidance on prevention of cross-contamination
• Chapter 5
Points 3.17-21
• improved guidance on prevention of cross-contamination & to refer to toxicological assessment.
With transitional arrangement for toxicological evaluation

Chapter 3 and Chapter 5 – Key Changes cont.
6
Points
3.27-30
• includes new section, on the qualification of suppliers to reflect the legal obligation of Manufacturing Authorisation holders to ensure active substances are produced to GMP.
• Formalised Risk Assessment for the GMP of excipients
Points
3.35 & 3.36
• inserted to clarify / harmonise expectations of manufacturers regarding the testing of starting materials
• Rationale for outsourcing testing – justified and documented
Point
3.71
• introduces the requirement of notification of restrictions in supply to the competent authority.

Impact on the QP?
• Have you changed your audit frequency of own and overseas manufacturers?
• Have you increased the time on site performing audits?
• Have you changed what you are looking at?
• Have you checked whether the cleaning validation calculations have been amended?
• Have toxicological studies been performed / planned?
• Have you assessed both technical and operational measures?
• Has your batch disposition procedure changed to include a review of the sites:
– Supply chain – do you comply with Chapter 1 requirements also
– Agreements in place
– Excipient GMPs Risk Assessment
7

Dedicated Facilities Debate
8
Regulator’s View
Its a highly potent product of a type that would previously have been manufactured in dedicated
facilities.
Therefore that would appear to be a good starting point
Manufacturer’s View
Its a highly potent product of a type that would previously have been manufactured in dedicated
facilities.
I want you “techies” to find me a way to safely manufacture it in
shared facilities

Impact on the QP?
• Have you had training on performing Risk Assessments and evaluating them?
– Remember you cannot Risk Assess bad practices
– There are some things that are just bad GMP
9

Chapter 8 - Complaints, Quality Defects and Product Recalls – Key Additions
31 points now – previous version had only 16 • Principle
o Quality Risk Management principles should be applied to the investigation and assessment of quality defects and to the decision-making.
o competent authorities should be informed in a timely manner in case of a confirmed quality defect
• Personnel and Organisation
o Should be independent of Sales and Marketing
o Include the QP
• Procedure for handling and investigating complaints including possible quality defects
o Lists all aspects to be considered
10

Chapter 8 - Complaints, Quality Defects and Product Recalls – Key Additions
• Investigation and decision making
o Ensure that appropriate risk-reducing actions are taken
o Timely notification to competent authority if a quality defect may result in the recall of the product or in an abnormal restriction in the supply of the product
• Root cause analysis and Corrective and Preventive Actions
o Links directly to the wording in Chapter 1
• Product recalls and other potential risk-reducing agents
o All concerned Competent Authorities should be informed in advance in cases where products are intended to be recalled
o The rationale for any decision to rework recalled products should be documented and discussed with the relevant competent authority
11

Impact on the QP?
• Have to be part of or fully informed of any complaints and recalls
• Have to be aware of complaint trends
• More detailed reports for complaints and recalls
• Falsification now used rather than counterfeiting
12

Annex 15: Qualification and Validation
• Additional sections covering: o Traditional process validation
o Continuous process verification
o Hybrid approach
o Ongoing process verification during Lifecycle
o Verification of transportation
o Validation of packaging
o Qualification of utilities
o Validation of test methods
12

Annex 15: Qualification and Validation – Key aspects
• Risk assessment throughout
• Includes appropriate checks for data integrity
• Requalification is performed at an appropriate frequency.... Which is justified.....using defined criteria
• If validation batches are to be released this must be defined upfront so that it is a planned event
• Introduction of control strategy – quality attributes of incoming materials and components
• Ongoing process verification – the extent and frequency should be reviewed
• Links to Chapter 3 and 5 cleaning validation and toxicological data
13

Impact on QPs
• Remember
o 5.26 Processes and procedures should undergo periodic critical re-validation to ensure that they remain capable of achieving the intended results.
• Have you had training on performing Risk Assessments and evaluating them?
– Remember you cannot Risk Assess bad practices
– There are some things that are just bad GMP
– Are you confident enough to challenge decisions in risk assessments
• Quality oversight is required; ensure you have confidence in who is looking at the data for you
14

Changes in the EU Guidance – Impact of Changes - what else?
Data Integrity
15

Data Integrity
• Are concerns over data integrity something new?
– No
• Is it just an issue for Quality Control
– No its all departments in a company
• Is it happening more
– No, we are more aware
• Is it always intentional acts
– Not always; a lot are due bad GMP practices
2

Definitions – Data Integrity
• The extent to which all data are complete, consistent and accurate throughout the data lifecycle. This means: – Data integrity arrangements must ensure that the accuracy,
completeness, content and meaning of data is retained throughout the data lifecycle.
• Ref MHRA Data Integrity Definitions and Guidance March 2015
• Data integrity refers to maintaining and assuring the accuracy and consistency of data over its entire life-cycle, and is a critical aspect to the design, implementation and usage of any system which stores, processes or retrieves data.
• (From Wikipedia and GAMP Data Integrity SIG preferred definition)
3

Definitions – Data Integrity
The Quality, Safety and Efficacy of the medicines we make is assured by applying the regulations and best practice
If we don’t......
Would you be happy taking those medicines
5

Definitions – Data Integrity
• ALCOA
– Attributable
– Legible/Permanent
– Contemporaneous
– Original
– Accurate
• ALCOA+
– Complete
– Consistent
– Enduring
– Available Ref MHRA Symposium Dec 2014 and has been used by FDA for many years
This applies to both Paper and Electronic Systems
The following are examples 6

Data Integrity - Attributable
Paper Record
• Initials
• Hand written signature
They have to be equivalent.
Electronic Record
• Log-on User identification
• Electronic signature
7
Traceable to a UNIQUE person
You should be able to tell who created, modified, or
deleted a record. AND, you can judge if that person was
appropriately authorized to do it!

Data Integrity - Legible
Paper Record
• Legible
• No Pencil
• No Correction Fluid
• Permanent Ink
• Correct Amendment of an Error
• Appropriate Archival
Electronic Record
• No data annotation tools enabled
• No Over Writing
• No Deletions
• No Hidden Fields
• Changes captured in Audit Trail
• Back-up and Archive
• Enforced Saving
8
Readable, Traceable Changes, Permanent
You should be able to read all the entries on the paper record. If a change
was made, the original value was crossed out with a single line, and the
change was dated and initialled.
You should be able to see in an electronic files data changes and deletions.
Is it clear what the original value was? When it was created and who created
it?

Data Integrity - Contemporaneous
Paper Record
• No back- dating
• No pre-completion of a record
• Record the date (and time) of the activity
Electronic Record
• Records saved immediately the data is entered
• Synchronised clocks
• Locked date and time stamps
9
For a paper record the creation, modification, and deletion of data
happens at the right time in the process.
For an electronic record when it was created in a process, the audit
trail should confirm that.
Record the activity at the time it occurred

Data Integrity - Original
Paper Record
• Original Document – Manufacturing record; laboratory record;
Electronic Record
• Original electronic file including metadata – a UV spectra, FTIR spectra
10
The first record made by the appropriate person
if not original should be exact copy - Original records and true copies must
preserve the integrity (accuracy, completeness, content and meaning) of
the record. Exact (true) copies of original records may be retained in place
of the original record (e.g. scan of a paper record), provided that a
documented system is in place to verify and record the integrity of the
copy.
You must be able to reconstruct the activity from the data

Data Integrity – Certified Copy
• A certified copy is: – Verified by a second person
– Compares the copy to the original
– Confirms the copy is accurate and complete
– Documents the verification
– Stamped a ‘true copy’
• What is not a certified copy – A pdf or printout – these are flat files of for example
• A FTIR spectra
• A HPLC chromatogram
– They do not preserve the content and meaning
11

Data Integrity - Accurate
• Accurate, consistent and real representation of facts.
• No editing without documented amendments /audit trail entries by authorised personnel
– Make sure of the information that you are recording is correct, honest and transparent
– Record the data directly into the controlled unique blank record/bound book/electronic programme
– Where possible use automatic data capture
– Are the electronic record the same as the paper record
12

Data Integrity – ALCOA+
• Complete – All data from an analysis, from the start of analysis to the end and any
repeated or reanalysis performed on the sample.
– For electronic systems, the paper output must be linked to the underlying electronic records used to produce it.
• Consistent – All elements of the analysis, such as the sequence of events, follow on
and data files are date (all processes) and time (when using electronic systems) stamped in the expected order
• Enduring – Recorded on authorised media e.g. laboratory notebooks, numbered
worksheets, for which there is accountability or electronic media
• Available – The complete collection of records can be accessed or retrieved for
review over the lifetime of the record. 13

Data Integrity – Data Governance
14
• MHRA Definition:
• The sum total of arrangements to ensure that data, irrespective of the format in which it is generated, is recorded, processed, retained and used to ensure a complete, consistent and accurate record throughout the data lifecycle.
– It should be integral part of the Pharmaceutical Quality System
– There should be clear Management Responsibilities
IT should be part of Management Reviews/Quality Councils
Everyone should be trained to understand the terms and implications of IT systems
— There should be data owners such as process owners in computer systems
— There has to be data integrity training
— There has to be procedures and processes in place
— Create a blame free, open and transparent culture

Data Integrity - Consequences
• So what does this mean:
– What happens when data integrity is breached? The worst case scenario is impact on patient safety and the loss of lives.
– the recent New England Compounding Pharmacy incident in the United States can be used as an example of the consequences of fraudulent activity. Here, 64 patients died and over 750 were sickened from fungal meningitis as a result of sterility negligence and data integrity issues. In this case, a FDA official said pharmacy technicians were instructed to lie on cleaning logs, showing rooms as being properly cleaned when they had not been
18

Data Integrity - Conclusion
• The message for data integrity is clear.
• It’s not a new concept; it’s about getting back to the roots of training all staff on the importance of data integrity in cGMP documentation and honesty.
• It is critical to ensure employees understand the accountability and traceability requirements for retention of raw data and the consequences of data manipulation.
• Training operators and analysts to document the performance of a task by recording what happened at the time it occurs, including information about the person who performed it along with clearly documenting and investigating deviations, is vital for patient safety and product efficacy.
• This is achievable by providing the training and creating a company culture that promotes and rewards ethical behaviour as a core value from top to
bottom of an organization.
26

Impact on the QPs?
• All decisions are now based on Quality Risk Management Principles
• Have you had training on performing Risk Assessments and evaluating them?
– Remember you cannot Risk Assess bad practices
– There are some things that are just bad GMP
– Are you confident enough to challenge decisions in risk assessments
• Data Integrity is important throughout the manufacture and testing of medicinal products
– Have you changed the way you audit
– Are you confident in the batch files scanned from an overseas facility
• Are your companies up to date on incorporating the changes – if not have you documented your Risk Assessment when you made your certification decision
17

27
Thank you

1
PHSS Welsh QP Forum Conference 2015
Tim Holliday
NDC QA Manager
Norgine Ltd.
Experiences in Implementing the
Principles of Quality by Design

Contents
• Overview of Quality by Design
• Strategy for Implementation
• Practical Examples
2

OVERVIEW OF QUALITY BY DESIGN
3

ICH Q8 ‘Pharmaceutical Development’
• First published November 2005 as guidance to content of CTD module 3.2.P.2 (Pharmaceutical Development report)
• Introduced concept of ‘Design Space’, but little guidance on how to implement
• Annex published in November 2008 detailing principles of ‘Quality by Design’ as ‘enhanced’ (systemic) approach to Pharmaceutical Development
• Updated document ICH(R2) incorporating Annex published in August 2009
• FDA Manual ‘Applying ICH Q8(R2), Q9, and Q10 Principles to CMC Review’ published August 2011 here
4

ICH Q8 ‘Pharmaceutical Development’
Definition of Quality by Design:
‘Quality by Design is a systemic approach to product
development that begins with predefined objectives and
emphasises product and process understanding and process
control, based on sound science and quality risk management’
5

Benefits of Quality by Design
• Structured, lifecycle-based approach
• Focus on risk management allows efficient use of available resources
• Focus on product and process knowledge allows clear understanding of impact of process changes on Critical Quality Attributes (‘knowns’ and ‘unknowns’)
• Potential to deliver robust, reproducible, capable processes
• Potential for registration of ‘Design Space’
6

Key Stages
• Establish Quality Target Product Profile
• Determine Critical Quality Attributes (CQAs) for product
• Link Material Attributes and Process Parameters to CQAs
• Optimisation of manufacturing process (e.g. ‘Design of Experiments’)
• Establish Control Strategy
• Lifecycle Management
7

Key Stages
8
PLAN
DO

Establish Quality Target Product Profile
• Derived from Target Product Profile – ref. FDA draft guidance here
• Takes into account:– Dose form/ strength
– Route of administration
– Container closure system
– Required pharmacokinetic characteristics (e.g. controlled release)
– Quality criteria appropriate for marketed product (e.g. Ph. Eur. General Monographs, stability)
• Dependant on Target Product Profile capturing all critical product characteristics!
9

Determine Critical Quality Attributes for Product
• Attributes identified on Quality Target Product Profile are assessed to determine impact on product quality.
• Determination of whether an individual attribute is critical or not should be based on consideration of the following criteria:– Would failure to meet the target value for the attribute compromise
the safety or efficacy of the product, or cause the product not to function correctly?
– Does the failure mode directly impact the attribute measured, or is the failure detected through failure of another attribute?
• Critical Quality Attributes should be re-evaluated as product/ process knowledge increases
10

Example Target Quality Product Profile
11
Quality Attribute Target CQA
Dosage Form Powder for Oral Solution No
Packaging
ALU/PE laminate sachet
Meets the requirements of
16 CFR 1700
Yes
Shelf life 3 years at 25oC No
AppearanceWhite to yellow free flowing
powderYes
ID and Assay95-105 % release
90-110 % stabilityYes
ImpuritiesLimits to be determined in
accordance with ICH Q3BYes
Uniformity
Complies with Ph. Eur.
requirements for uniformity
of dosage units
Yes
Moisture Limit to be determined Yes
Reconstitution ≤5 minutes Yes
Microbiological quality
TAMC ≤103 cfu/ml
TYMC ≤102 cfu/ml
Absence of E. coli
Yes
Organoleptic Acceptable odour/taste Yes

Link Material Attributes and Process Parameters to Critical Quality Attributes
• Map process, identifying material attributes and process parameters that may impact on Critical Quality Attributes
• As complex as your process!
• Use of Ishikawa (fishbone) diagram recommended
• Perform risk assessment on mapped process (e.g. FMEA) to determine impact of identified material attributes and process parameters:– High risk: further investigation required to identify appropriate control
measures
– Low risk: further investigation may not be required (with justification!)
– Unknown/ insufficient information: further investigation required
12

Examples
• Raw materials– Particle size Uniformity
– Moisture content Appearance, Moisture, Reconstitution
– Morphology Uniformity
• Blending– Time Uniformity
– Speed Uniformity
– Environment Appearance, Assay, Uniformity, Moisture, Microbiological quality
• Filling– Line speed Assay, Uniformity
– Seal settings Appearance, Moisture, Reconstitution
13

Optimisation of Manufacturing Process
• Investigate effects of variation of factors identified in previous stage
• May include Design of Experiments, ‘a statistical technique for the exploration of multiple factors (including interactions between factors) that may influence product quality using multivariate analysis’
• Other approaches may be used
• ‘Design Space’ may be established
• ‘Fixed factors’ may not require investigation (but may rule out possibility of establishing design space!)
• Update risk assessment based on outcome of investigations
14

Establish Control Strategy
• Ensure that product of required quality will be produced based on confirmed CQAs and knowledge gathered during optimisation of manufacturing process
• Examples:– Control of input material attributes
– Product specifications
– Controls for unit operations that have an impact on downstream processing or product quality
– In-process testing (‘At line’/ Process Analytical Testing)
– Real-Time Release Testing (ref EU GMP annex 17 draft update here)
– Statistical models (e.g. Statistical Process Control/ process capability)
15

Lifecycle Management
• Update Risk Assessment and confirm that all identified risks have been managed appropriately
• Ensure that development data can be accessed by those responsible for lifecycle management – ’knowns’ and ‘unknowns’ (Module 3.2.P.2 and supporting data)
• Verify effectiveness of control strategy
• Use data to assess post-marketing changes
• Where development data is insufficient, use Quality by Design methodology to manage change
16

STRATEGY FOR IMPLEMENTATION
17

Strategy for Implementation
Key Considerations
• Senior Management engagement
• Stakeholder management
• Resource concerns
• Timing
• Process definition
18

Strategy for Implementation
• Worked with stakeholders to identify resource requirements to implement principles of Quality by Design– Stakeholders identified by RACI
– No significant additional resource required!
– ‘Pareto effect’
• Workshop held with key stakeholders – Establish common understanding of principles of Quality By Design
– Develop policy defining high-level process to be followed within Norgine
– Confirmed timing for formal Quality by Design activities (prior to manufacture of Phase III/ Registration batches)
– Buy-in to agreed process by key stakeholders (Pharmaceutical Development, M&S Technical/ Quality, Regulatory Affairs)
19

Strategy for Implementation
• Policy presented to Senior Management for approval– Senior management commitment to principles of Quality by Design
confirmed
– Pilot project proposed and approved
• Online toolkit developed to ensure consistent application of principles of Quality by Design across organisation– Hosted on company intranet
– Incorporates ‘lessons learnt’ from pilot project
– Includes stage outlines, templates, worked examples and reference documents
– Flexible approach to allow incorporation of additional material as experience with Quality by Design increases
20

Strategy for Implementation
21

PRACTICAL EXAMPLES
22

Example 1 – Adequacy of TPP
• Single dose product in laminated sachet
• TPP stated ’25g single dose sachet (or similar)’
• Quality by Design process identified seal settings as critical parameter, with vacuum leak test as additional control measure
• Process developed using existing filling machine. Pack dimensions selected to optimise filling process (e.g. line speed, seal quality)
However…
23

Example 1 – Adequacy of TPP
• During preparations for launch, more detailed requirements for pack size were identified
• TPP should have reflected this, e.g. ’25g single dose sachet with dimensions 30-40mm x 130-150mm’
• May have had impact on selection of filling machine/ process
24

Example 2 – Constraints to Process Optimisation
• New powder product developed using established API and excipients
• Potential content uniformity issue identified during Phase II development
• Full risk assessment performed on proposed manufacturing process prior to Phase III/ Registration batches
• Constraints to process optimisation:– Use of existing manufacturing and filling equipment
– Existing material grades to be used
– Batch size limited by commercial constraints
– Limited time to complete Quality by Design activities!
25

Example 2 – Constraints to Process Optimisation
• Uniformity issue linked to particle size of API; screening of material prior to manufacture proposed
• ‘Design of Experiments’ study performed to investigate effects of variation of process parameters (blend time and speed) and use of screened/ unscreened API
• Results confirmed optimum blend parameters and use of screened API to obtain uniform blend
• Risk assessment updated to document that effects of variation of other material attributes and process parameters was not investigated due to constraints of the selected materials and process
26

Example 3 – Linking Material Attributes to Pharmacokinetic Properties
• New semi-solid product (suspension) developed with novel API
• Only 2 batches of API supplied prior to Phase III (used in Phase II clinical trial supplies), with limited particle size distribution information (non-uniform distribution):
• Risk assessment identified particle size as potential critical API material attribute, with impact on product pharmacokinetic properties
27

Example 3 – Linking Material Attributes to Pharmacokinetic Properties
• Phase II Pharmacokinetic data shows satisfactory absorption profile:
28

Example 3 – Linking Material Attributes to Pharmacokinetic Properties
• Microscopy performed on Phase II clinical trial supplies to investigate particle size distribution in formulated product
• Even particle size and distribution observed, considered to result from mixing during product manufacture
29

Example 3 – Linking Material Attributes to Pharmacokinetic Properties
• Process optimisation strategy to duplicate particle size distribution of Phase II clinical trial supplies
• Particle distribution of API to be fully characterised during process validation (pre-Phase III)
• Design of Experiments study to be performed to investigate impact of variation of manufacturing parameters (mix time/ speed, temperature) and API particle size (screened/ unscreened)
30

Summary
• Quality by Design provides a formal structure for planning and executing Pharmaceutical Development activities
• Focus on risk management allows resources to be targeted towards areas of greatest benefit
• Constraints of selected process should be recognised
• Knowledge is power!
31

32

1
PHSS Welsh QP Forum Conference 2015
Sue Batten, Spheriqual Ltd
The impact of the
revised Annex 16
on the QP

Introduction
Sue Batten
Registered Pharmacist in 1984
Previously worked in NHS at North West Thames Regional QC, as lab manager, then Martindale Pharmaceuticals, as QP and Specials QA manager.
Eligible for QP status 1991 under transitional arrangements
Started with my own company Spheriqual Ltd in 2006 as contract QP, auditor and consultant
www.spheriqual.co.uk
2

Introduction
• What is “Annex 16” ?
• Why did it come into being originally?
• Comparison of the two:
what’s new
what’s not
• What does this mean to me as a contract QP?
3

What is “Annex 16” ? (1)
Directive 2001/83 Article 51:
The QP must:
• Ensure each batch made within a Member State has been manufactured and checked in accordance with the laws of that Member State and in accordance with the MA.
• Ensure each production batch coming from third countries has undergone a full qualitative analysis, quantitative analysis of at least the APIs and all other tests and checks necessary to ensure quality and compliance with the MA.
• Certify in a register that each released batch complies with this Article.
4

5

What is “Annex 16” ? (2)
Directive 2001/83 Article 51 item 2:
IF…..
• The 3rd country applies GMP standards at least as good as EU
• The Community makes appropriate arrangements
• The range of tests have been carried out in the 3rd country…
The QP may be “relieved of responsibility”
for carrying out those controls.
6

What is “Annex 16” ? (3)
The Annexes:
- more detail
- more easily updated(!)
- 19 in total but only 9 main Chapters
- numbered simply in order of initial publication.
7

A simple supply chain…..
8

…. became complex
9

The old Annex 16
A series of Scenarios:
- Manufacture in Europe x 7
- Manufacture in 3rd country x 2
… Sampling
… MRAs.
…and a reminder of the Routine Duties of a QP.
10

But what happens when….
11

12

13

14

The New Annex 16 can be found here:
http://ec.europa.eu/health/files/eudralex/vol-4/v4_an16_201510_en.pdf
15

The New Annex 16
What has changed?
- Reflects globalisation of the Supply Chain
- New QC strategies
- Falsified Medicines Directive now included
- Implements ICH Q8, Q9 and Q10
- Clarifies inconsistencies of interpretation
16

The New Annex 16
DOES apply to:
- All licensed products
- Products made for export
- IMPs for human use
- QP certification of blood / immunological products.
17

The New Annex 16
The Process of Certification: not new
The Batch Release process:
- Checking manufacture and testing
- QP Certification – batch is in compliance with GMP and the MA
- Recording certification in a register
- Transfer to saleable stock / export
BUT there is now a Batch Certificate Template in Appendix II.
18

What QPs need to do now: (1)
- PROVE continuous training on product type, processes, technical advances and GMP changes. (1.2)
- We can share defined responsibilities with other QPs in the EU supply chain
- CHECK storage AND transport conditions of sample AND batch
- Make sure any requirements of national legislation are complied with
- KNOW the supply chain in its entirety AND have a diagram of it, from API onwards
19

What QPs need to do now: (2)
Section 1.7 has 21 steps:
The QP has responsibility for “ensuring they are secured”
BUT CAN DELEGATE….
20

The 21 Steps…
- In accordance with GMP
- Documented supply chain - as a diagram
- Audits reports to be available
- Sites of manufacture, testing and certification in MA
- All manufacture and testing activities as in MA and validated
- Source and specifications of starting materials as in MA
- APIs / excipients manufactured and imported according to GMP
- TSE status as in MA
- Personnel trained
21

More 21 Steps….
- Finished Product testing complies with Specification in MA
- Post-marketing commitments
- Stability data must support certification
- Changes have been evaluated
- OOS and OOT have been evaluated
- Complaints, recall investigations completed
- Technical agreements are in place
- Self-inspection programme is active and current
- Shipment and distribution arrangements
- Safety features
22

Summary of 21 Steps:
The QP needs to know that the Quality System is not only in place….
but is working
23

Other points (1)
Sampling
- VERY detailed: items 1.5.5 and 1.5.6, plus several sub-points
- Samples MUST be fully representative of the batch
- Now allows sampling at either end BUT if not in EU:
> Technically justified, documented approach
> Audit
> Written responsibilities
> Proof of equivalent shipping conditions
> Random periodic analysis
24

Other points (2)
QP Certification
- The Register must be kept for at least 5 years
- The release certificate must be available to other Member States
25

Other points (3)
- Parallel Imports (1.9)
- Audits:
Written assessment of third party audits
Risk assess the need for repeat audits
26

Deviations
CAN certify a batch with a manufacturing deviation
CAN certify a batch with a QC method deviation
MUST be risk assessed
MUST be investigated and root cause corrected.
MUST meet registered specifications in MA!
27

Summary
What’s new?
- Lots more detail
- Clarification: no scenarios
- Quality Risk Management principles
- Expectation regarding audits
What’s not?
- The role of the QP
- The batch release process
- No QP discretion. A batch still MUST meet the MA specification
28

29

Where do we go from here?
Make a detailed release checklist from the “21 Steps”
Risk assess everything untoward
Good luck!
Any questions?
30

PHSS talk
29 October 2015

Definition of an ATMP - Regulation 1394/2007
Somatic Cell Therapy Medicinal Product
• substantially manipulation of cells and tissues or not intended to be used for the same essential function(s)
• administered to humans with a view to treating, preventing or diagnosing a disease through the pharmacological, immunological or metabolic action
Tissue Engineered Product
• engineered cells or tissues and administered to human beings with a view to regenerating, repairing or replacing a human tissue
Gene Therapy Medicinal Product
• contains an active substance which contains or consists of a recombinant nucleic acid used or administered to human beings with a view to regulating, repairing, replacing, adding or deleting a genetic sequence
• the therapeutic, prophylactic or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains or to the product of genetic expression of this sequence
2

An emerging area for expertise and regulation
3
Medicaldevices
Tissueengineering
Celltherapy
Genetherapy
Biologicals Chemicals

ReNeuron’s ATMPs
4
CTX cell line
One singlestem cell Potential to broaden therapeutic pipeline
beyond cell-based programs
CTX platform
Exosome platform
Clinical and pre-clinical pipeline in vascular and neurological indications
hRPCs Targeting retinal regenerative diseases
In-licensed technology (Harvard, Boston)
Retinal platform

Example ATMPS to Google…………..
5
Somatic Cell Therapy Medicinal Products
• Provenge sipuleucel-T (Dendreon) - autologous call based oncology immunotherapy
• Autologous CAR-T (Novartis, Juno, etc ) - oncology, in development
Tissue Engineered Products
• ChondroCelect(TiGenix) and Carticell ( Genzyme)– autologous cultured chondrocytes
• HoloClar (Chiesi) autologous limbal epithelial stem cells
• Apligraf, Dermagraft (Organogenesis) – allogeneic wound healing
Gene Therapy Medicinal Products
• Glybera (UniQure) – AAV based product for familial hyperchylomicronemia
• Talimogene laherparepvec (BioVex/Amgen) - oncolytic immunotherapy
• Allogeneic (Cellectis) CAR-T vaccine

A dynamic and evolving environment
• Need to constantly apply principles of quality assurance across whole development process from research to clinic .
• GMP-GCP integration is key. Guidance is Guidance.
• Cell and tissue products rely on aseptic handling, no opportunity to sterilise
• Autologous ( and small scale allogeneic) – limited product for testing
• Multiple modes of action given challenges for development of suitable potency assays which correlate to clinical outcome
• Frequent interfaces with regulators
• Opportunities for fast track and accelerated approvals move CMC development and scale up to critical path.
• Often short shelf life, so need to design appropriate batch release strategies
6

A dynamic and evolving environment
• More diversity of platform approaches than for other biologics
• Limited pool of experience to recruit ( Great opportunities for QPs in this area!)
• Can have significant manipulations at clinical sites, and there is limited experience of handling cell products at clinical sites, need to build it in.
• Complex logistics supply chains and need for full traceability. Often using LN2.
• Build in process controls and checks and balances as much as possible upstream to minimise activities downstream.
7

Example life of an ATMP, expansion to treatment
88
Harvest and formulate cells and fill DP. Fill diluent if needed
IPCs – Rapid mycoplasma, viability & cell counts
TIME OF MANUFACTURE SET (start of fill)
Source and expand cell IPCs - % viability and cell counts
Pack diluent and DP – start Temp logger.
Transfer to Warehouse – pack into shipper - Quarantine release
QC testing and CofA (viable cell count, mycoalert, endotoxin,
gram stain, appearance, non-viable EM data)
Interim QA release for clinic
Day X-1 :23:00 to Day
X 07:00Shipment collection 23:00
and transfer to site at 2-8C
Day x : 07:00 to 07:30 Delivery receipt at Clinic Pharmacy 07:00. Send Temp / tipNtell data to CMO
Dilution and Surgery within 60 minutes of dilution
Surgery to take place within fixed time window before shelf life expiry.
Day x : 07:30 to 08:00
Week 3 :
18 to 21 days later
QC testing and CofA (sterility, mycoplasma, Viable EM data)
Final Release
Escalation, if appropriate according to clinical protocol.
Day 0 to Day X - 1
Day X - 1 : 17:00 to
22:00
(t=0 at 21:00)
Day X-1 : 22:00 to
23:00
Day x : 08:00 to 13:00

Arthurian
9
• Arthurian Life Sciences Ltd was established by Europe’s leading biotech entrepreneur Professor Sir Chris Evans OBE to act as General Partner of the Wales Life Sciences Investment Fund, a £100 million fund and key part of the Welsh Government’s Life Sciences initiative
• The Fund had its first closing with £50 million on 28th February 2013.
• Arthurian Life Sciences Ltd will deliver the expectations of the Welsh Government in further developing a successful, vibrant, growing, profitable and socially meaningful Life Sciences sector in Wales. The Arthurian team has been specially assembled to make a major difference and generate a real excitement within Welsh Life Sciences both domestically and internationally.

ReNeuron Commitment to Wales
10
Press release July 2013: £7.8m to be provided through a grant package from the Welsh government to establish a cell manufacturing and development facility in South Wales for late stage clinical and commercial product requirements. The company will move it’s principal operations to this facility as it is phased in over the next two years.
Press release July 2015: ReNeuron raised £68.4M raised through new and existing shareholders
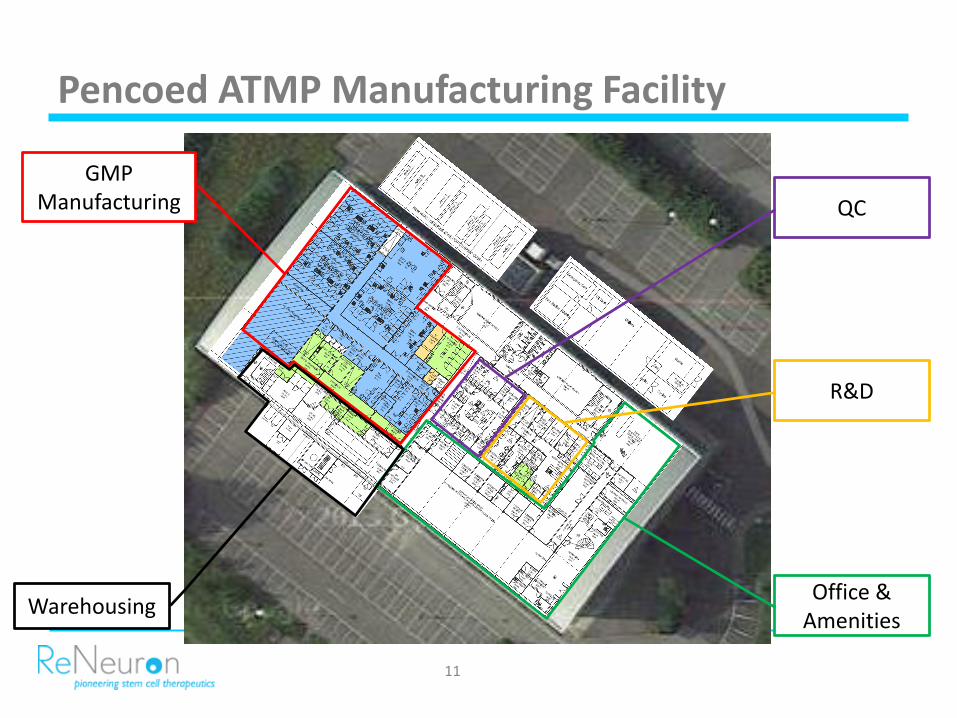
Pencoed ATMP Manufacturing Facility
11
GMP Manufacturing
WarehousingOffice &
Amenities
R&D
QC

Life Sciences in Wales…..recent news
• Life Science sector in Wales employs around 11,000 people at more than 350 companies turning over £2bn/yr
• Life Sciences hub opened in Cardiff Bay
• CeQur – established subsidiary in Wales. Series C financing raised £100m in September 2015 with investment co-led by Arthurian
• Simbec – Contract Research Organisation. Management buy out in 2013 as maiden investment of WLSIF. Subsequently acquired Orion Clinical Services in 2015
• Proton Partners – July 2015 secures new site in Newport for first proton beam therapy centre.
12

Useful resources
EMA website www.emea.europa.euAdvanced Therapies
www.emea.europa.eu/htms/human/advancedthereapies/intro.htm
Cell Therapy Catapultwww.ct.catapult.org.uk
London Regenerative Medicine Networkwww.lrmn.com
Wales Life Sciences Hubwww.lifescienceshubwales.com
13

9.00am - 10.00amRegistration and coffee
10.00am - 10.15pmWelcome and program for the dayKay O’HaganSenior QP, Hospira UK Ltd
10.15am - 11.15amSteam Sterilization – are you really in controlAlan HeaveyConsultant & Managing Director, Sterilization Solutions Gail HeaveyDirector of Time Design51 Ltd
11.15am - 11.30am Coffee
11.30am - 12.15pmChanges in the EU guidance - impact of changesDi MorrisGSK Global auditor, Ex-MHRA senior inspector
12:15pm - 13.00pmExperiences in implementing the principles of QbDTim HollidayNDC QA Manager, Norgine B.V.13.00pm - 14.00pm Lunch
14.00pm - 14.45pmA day in the life of Advanced Therapeutic Medicinal productsSharon GrimsterGeneral Manager, Wales,Reneuron Group PLC
14.45pm - 15.00pm Coffee
15.00pm - 15.45pmAnnex 16 changes and the impact on the Qualified PersonSue BattenContract Qualified Person and Director of Spherical Ltd
15.45am - 16.00pmConferenece closureKay O’HaganSenior QP, Hospira UK Ltd
Schedule


















