
Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against...
11
www.elsevier.com/locate/brainres Available online at www.sciencedirect.com Research Report Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice Li Zhao a , Xu Liu a , Jing Liang a , Song Han a , Yue Wang b , Yanling Yin a , Yanlin Luo a , Junfa Li a,n a Department of Neurobiology and Beijing Institute for Brain Disorders, Capital Medical University, Beijing 100069, China b Department of Neurology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing 100069, China article info Article history: Accepted 28 January 2013 Available online 8 February 2013 Keywords: Hypoxic preconditioning Middle cerebral artery occlusion Phosphorylation of p38 mitogen- activated protein kinase Bcl-xL Apoptosis Neuroprotection abstract Hypoxic preconditioning (HPC) initiates intracellular signaling pathway to provide protec- tion, but the role of p38 mitogen-activated protein kinase (p38 MAPK) in HPC-induced neuroprotection against cerebral ischemic injuries is a matter of debate. In this study, we found that HPC could reduce 6 h middle cerebral artery occlusion (MCAO)-induced infarct volume, edema ratio and cell apoptosis, as well as enhancing the up-regulated p38 MAPK phosphorylation (P-p38 MAPK) levels in the peri-infarct region of mice after 6 h MCAO. However, intracerebroventricular injection of p38 MAPK inhibitor SB203580 abolished this HPC-induced neuroprotection. HPC significantly increased the translocation of anti- apoptotic Bcl-2-related protein Bcl-xL from the cytosol to the mitochondria in the peri- infarct region of MCAO mice. Interestingly, the results of reciprocal immunoprecipitation showed that Bcl-xL and P-p38 MAPK were coimmunoprecipitated reciprocally only in the peri-infarct region of HPC and MCAO treated mice, while Bcl-xL and total p38 (T-p38 MAPK), not P-p38 MAPK, could be coimmunoprecipited by each other in the brain of normal control mice. In addition, we found SB203580 significantly decreased P-p38 MAPK levels, and inhibited HPC-induced mitochondria translocation of Bcl-xL in the brain of HPC and MCAO treated mice. Taken together, our findings suggested that P-p38 MAPK mediates HPC-induced neuroprotection against cerebral ischemic injury via mitochondria transloca- tion of Bcl-xL, which might be a key anti-cell apoptotic mechanism of HPC. & 2013 Elsevier B.V. All rights reserved. 0006-8993/$ - see front matter & 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.brainres.2013.01.051 n Correspondence to: Department of Neurobiology and Beijing Institute for Brain Disorders Capital Medical University #10 You An Men Wai Xi Tou Tiao, Beijing 100069, China. Fax: þ8610 8395 0060. E-mail address: [email protected] (J. Li). brain research 1503 (2013) 78–88
Transcript of Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against...

Available online at www.sciencedirect.com
www.elsevier.com/locate/brainres
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8
0006-8993/$ - see frohttp://dx.doi.org/10
nCorrespondenceWai Xi Tou Tiao, Be
E-mail address:
Research Report
Phosphorylation of p38 MAPK mediates hypoxicpreconditioning-induced neuroprotection againstcerebral ischemic injury via mitochondria translocationof Bcl-xL in mice
Li Zhaoa, Xu Liua, Jing Lianga, Song Hana, Yue Wangb, Yanling Yina,Yanlin Luoa, Junfa Lia,n
aDepartment of Neurobiology and Beijing Institute for Brain Disorders, Capital Medical University, Beijing 100069, ChinabDepartment of Neurology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing 100069, China
a r t i c l e i n f o
Article history:
Accepted 28 January 2013
Hypoxic preconditioning (HPC) initiates intracellular signaling pathway to provide protec-
tion, but the role of p38 mitogen-activated protein kinase (p38 MAPK) in HPC-induced
Available online 8 February 2013
Keywords:
Hypoxic preconditioning
Middle cerebral artery occlusion
Phosphorylation of p38 mitogen-
activated protein kinase
Bcl-xL
Apoptosis
Neuroprotection
nt matter & 2013 Elsevie.1016/j.brainres.2013.01.0
to: Department of Neuroijing 100069, China. Fax:[email protected] (J. L
a b s t r a c t
neuroprotection against cerebral ischemic injuries is a matter of debate. In this study, we
found that HPC could reduce 6 h middle cerebral artery occlusion (MCAO)-induced infarct
volume, edema ratio and cell apoptosis, as well as enhancing the up-regulated p38 MAPK
phosphorylation (P-p38 MAPK) levels in the peri-infarct region of mice after 6 h MCAO.
However, intracerebroventricular injection of p38 MAPK inhibitor SB203580 abolished this
HPC-induced neuroprotection. HPC significantly increased the translocation of anti-
apoptotic Bcl-2-related protein Bcl-xL from the cytosol to the mitochondria in the peri-
infarct region of MCAO mice. Interestingly, the results of reciprocal immunoprecipitation
showed that Bcl-xL and P-p38 MAPK were coimmunoprecipitated reciprocally only in the
peri-infarct region of HPC and MCAO treated mice, while Bcl-xL and total p38 (T-p38
MAPK), not P-p38 MAPK, could be coimmunoprecipited by each other in the brain of
normal control mice. In addition, we found SB203580 significantly decreased P-p38 MAPK
levels, and inhibited HPC-induced mitochondria translocation of Bcl-xL in the brain of HPC
and MCAO treated mice. Taken together, our findings suggested that P-p38 MAPK mediates
HPC-induced neuroprotection against cerebral ischemic injury via mitochondria transloca-
tion of Bcl-xL, which might be a key anti-cell apoptotic mechanism of HPC.
& 2013 Elsevier B.V. All rights reserved.
r B.V. All rights reserved.51
biology and Beijing Institute for Brain Disorders Capital Medical University #10 You An Menþ8610 8395 0060.i).

b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8 79
1. Introduction
Stroke is one of the leading causes of morbidity and mortality in
the world. Among several protection strategies directed against
ischemic injury, hypoxic preconditioning (HPC) demonstrated a
powerful protective potential against ischemic cell death
(Ostrowski et al., 2008; Tsai et al., 2004, 2008). Studies suggested
that mitogen-activated protein kinases (MAPK) had been
involved in this biological process (Heidbreder et al., 2008).
MAPKs are serine–threonine kinases that mediate intracellular
signaling associated with a variety of cellular activities including
cell proliferation, differentiation, survival, death, and transfor-
mation (Dhillon et al., 2007; McCubrey et al., 2006; Torii et al.,
2006). p38 MAPK, a member of MAPK super-family, was activated
by the MAPK kinases 3 and 6 via dual phosphorylation of the
Thr-Gly-Tyr motif in the activation loop (Cohen, 1997; Cuenda
and Rousseau, 2007). Several studies in animal models had
shown that the activation of p38 MAPK increased in ischemic
brain tissue (Irving et al., 2000; Irving and Bamford, 2002; Sugino
et al., 2000). Pretreatment with SB203580, a p38 MAPK inhibitor
may aggravate ischemic brain injury and cerebral vascular
leakage in the model of transient ischemia (Lennmyr et al.,
2003). In this study, we used mouse HPC and middle cerebral
artery occlusion (MCAO)-induced focal cerebral ischemia
models to explore the effect of p38 MAPK on HPC-induced
neuroprotection.
Cell apoptosis has been considered to be one of future
targets for neuroprotective strategies in recent years
(Bielewicz et al., 2010; Ferrer, 2006). Compelling evidences
indicate that apoptosis may occur after transient cerebral
ischemia (MacManus and Buchan, 2000; Islam et al., 1995).
Moreover, recent cerebral ischemia studies in vivo have
revealed that dysregulation of Bcl-2 family proteins can exacer-
bate ischemic neuronal injury and that the interaction between
Bcl-2 family members that suppress (such as Bcl-2 and Bcl-xL)
and those that promote (such as Bax) apoptosis determined
whether cells underwent survival or apoptosis (Billen et al.,
2008; Doeppner et al., 2009; Youle and Strasser, 2008). Bcl-xL was
abundantly expressed in adult neurons and predominantly
localized to the outer mitochondrial membrane and cytosol
(Edlich et al., 2011; Huang and Strasser, 2000; Kaufmann et al.,
2003). Bcl-xL preserved mitochondrial integrity and prevented
the subsequent release of apoptogenic molecules such as
cytochrome c (Gollapudi et al., 2003). In a mouse model,
transcription and expression of Bcl-2 and Bcl-xL were increased
after sublethal forebrain ischemia, which attenuated the DNA
fragmentation induced by lethal ischemia (Wu et al., 2003).
Inhibition of p38 MAPK protected cardiomyocytes from
apoptosis during simulated ischemia in vitro (Cheng, 2006).
Activation of p38 MAPK could protect brain endothelial cells
from apoptosis in stroke (Pfeilschifter et al., 2010). Immuno-
precipitation experiments revealed p38 MAPK-dependent
serine–threonine phosphorylation of Bcl-xL in tumour necrosis
factor (TNF)-treated cells (Sugawara et al., 2004). However,
whether p38 MAPK participate in the HPC-induced neuropro-
tection by reduced neural apoptosis through Bcl-xL remains
unclear. In this study, we explored the role of p38 MAPK-Bcl-xL
antiapoptotic pathway in HPC-induced neuroprotection in the
peri-infarct region of ischemic cortex.
2. Results
2.1. HPC attenuated ischemia-induced injuries in mice
With the increasing times of hypoxia, the tolerance time of
mice increased significantly (n¼30, Fig. 1A). The tolerance
time of hypoxia four times was almost double
(34.0070.80 min) that of hypoxia once (16.9071.20 min). So
mice were exposed to auto-hypoxia for four times in the
following study.
MCAO-induced cerebral ischemic injuries were evaluated
by edema, 2, 3, 5-triphenyltetrazolium chloride (TTC) and
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP
nick end labeling (TUNEL) staining. Fig. 1B showed represen-
tative TTC-stained brain sections from each group. We found
that there was no infarction in the Sham group. A large area
of infarction was seen in the cerebral cortex and striatum in
the Ischemia group (Fig. 1B and C). Similarly, the edema ratio
remarkably increased at 6 h MCAO (Fig. 1D). However, HPC
could significantly decrease the infarct volume and edema
ratio compared with those of the Ischemia group (Fig. 1B–D),
respectively. In concert with these results, the percentage of
apoptotic cells significantly increased in the peri-infarct
region of ischemic cortex but was remarkably reduced by
HPC (Fig. 2).
2.2. Effect of p38 MAPK phosphorylation on HPC-inducedneuroprotection in the ischemic cortex of mice
To reveal the involvement of p38 MAPK in HPC-induced
neuroprotection, we detected the levels of p38 MAPK phos-
phorylation (P-p38 MAPK) and protein expression (T-p38
MAPK) both in the ischemic core and peri-infarct region. As
shown in Fig. 3, we found that the level of P-p38 MAPK in the
peri-infarct region was enhanced in the Ischemia group
compared to that of the Sham group. HPC could further
increase the P-p38 MAPK level in the ischemic peri-infarct
region (Fig. 3). However, there were no significant differences
in the level of P-p38 MAPK between Ischemia and HPCþI
groups whether in the ischemic core or contralateral cortex.
We also found that the level of T-p38 MAPK remarkably
decreased in the ischemic core of Ischemia group. But
combined HPC treatment did not further change T-p38 MAPK
level in the ischemic core as well as peri-infarct region. These
results suggested that p38 MAPK activation by phosphoryla-
tion might be involved in the neuroprotective effect of HPC.
To explore whether p38 MAPK pathway was necessary for
the neuroprotective effect of HPC, we used SB203580, a p38
MAPK inhibitor, before ischemia in this study. We found that
SB203580 treatment in the HPCþI group significantly
increased infarct volume, edema ratio and apoptotic cells at
6 h MCAO compared to DMSO treated controls (Fig. 4A–E).
These results indicated that p38 MAPK inhibitor could abolish
the HPC-induced neuroprotective effects. At the same time,
we found that intracerebroventricular injection of 1 mM
SB203580 significantly inhibited the p38 MAPK phosphoryla-
tion level (Fig. 4F and G).

Fig. 1 – Effects of HPC on edema and infarct volume of mice after MCAO. (A) Statistical results of tolerance time of mice
subjected to auto-hypoxia from 1 to 4 times which were named H1, H2, H3 and H4, respectively (n¼30). �po0.05 compared
with H1. (B) Representative photographs of TTC-stained coronal brain sections of mice from Sham, Ischemia and HPCþI
groups. The white areas were cerebral infarcts. (C) Statistical results of infarct volume from Sham, Ischemia and HPCþI
groups (n¼6 per group). �po0.05 compared with Sham group, #po0.05 compared with Ischemia group, respectively.
(D) Statistical results of edema ratio from Sham, Ischemia and HPCþI groups (n¼6 per group). �po0.05 compared with Sham
group, #po0.05 compared with Ischemia group, respectively.
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 880
2.3. Effect of HPC on Bcl-xL distribution in cytosol andmitochondria in the ischemic cortex of mice
To reveal the involvement of Bcl-xL in HPC-induced neuro-
protection, we observed the level of Bcl-xL in cytosol and
mitochondria. As shown in Fig. 5, we found that Bcl-xL was
localized to both cytosol and mitochondria in the Sham
group. In the ischemic core of cortex, the level of Bcl-xL
decreased in both cytosol and mitochondria significantly at
6 h MCAO and was not affected by HPC. In the peri-infarct
region of cortex, Bcl-xL protein level only decreased in
mitochondrial fraction but did not change in cytosolic frac-
tion at 6 h MCAO. The Bcl-xL level increased remarkably
in mitochondrial fraction, meanwhile decreased significantly
in cytosolic fraction after HPC pretreatment in the ischemic
peri-infarct region (Fig. 5A and B). The ratio of cytosolic
Bcl-xL to mitochondrial Bcl-xL decreased significantly
in the HPCþI group, suggesting that HPC induced the
translocation of Bcl-xL from the cytosol to the mitochondria
(Fig. 5C).
2.4. Co-immunoprecipitation of the T-p38 MAPK or P-p38MAPK with Bcl-xL in the cortex of mice
In order to test whether T-p38 MAPK or P-p38 MAPK inter-
acted with Bcl-xL, we performed the immunoprecipitations
by using the antibodies of anti- Bcl-xL, anti-p38 MAPK and
anti-P-p38 MAPK in the mouse cortex. The results of recipro-
cal immunoprecipitation showed that Bcl-xL could bind T-p38
MAPK in the brain of normal control mice (Fig. 6A). We did
not observe any co-immunoprecipitation of Bcl-xL and P-p38
MAPK in the normal mice (Fig. 6B). But after HPC and MCAO
treatments, the co-immunoprecipitation results actually
showed Bcl-xL could interact with P-p38 MAPK in peri-
infarct region (Fig. 6C). Whole cell lysate and IgG were used
as positive and negative control, respectively (Fig. 6A–C).
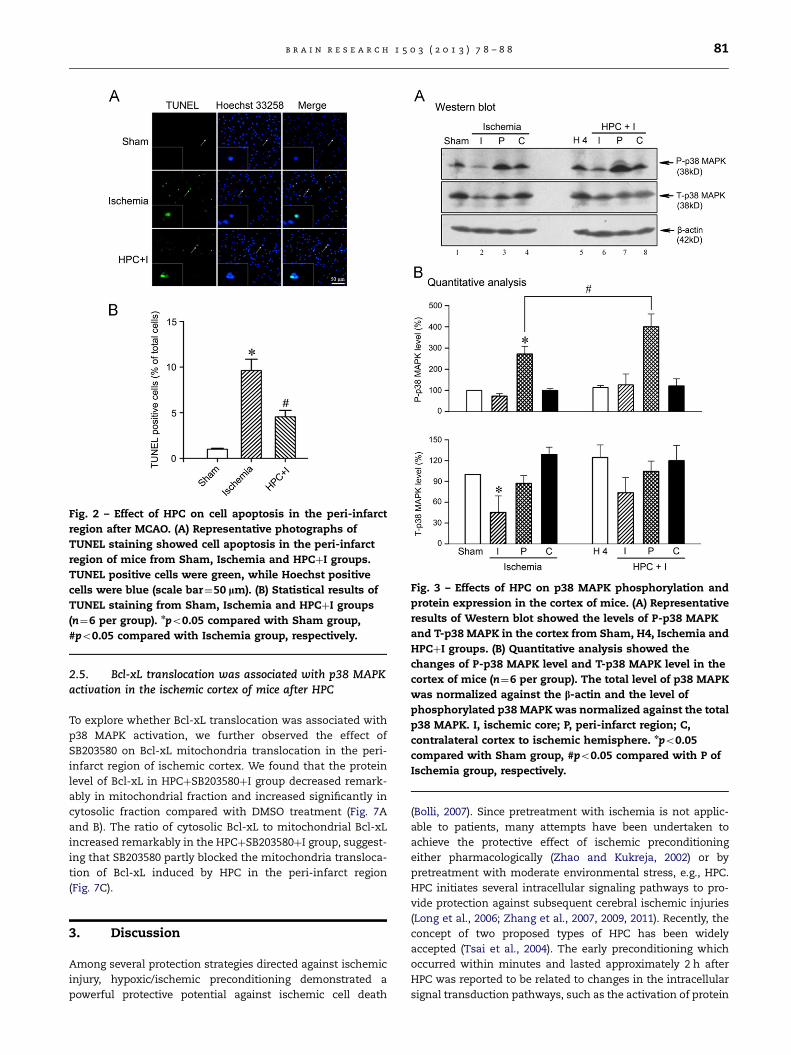
Fig. 2 – Effect of HPC on cell apoptosis in the peri-infarct
region after MCAO. (A) Representative photographs of
TUNEL staining showed cell apoptosis in the peri-infarct
region of mice from Sham, Ischemia and HPCþI groups.
TUNEL positive cells were green, while Hoechst positive
cells were blue (scale bar¼50 lm). (B) Statistical results of
TUNEL staining from Sham, Ischemia and HPCþI groups
(n¼6 per group). npo0.05 compared with Sham group,
#po0.05 compared with Ischemia group, respectively.
Fig. 3 – Effects of HPC on p38 MAPK phosphorylation and
protein expression in the cortex of mice. (A) Representative
results of Western blot showed the levels of P-p38 MAPK
and T-p38 MAPK in the cortex from Sham, H4, Ischemia and
HPCþI groups. (B) Quantitative analysis showed the
changes of P-p38 MAPK level and T-p38 MAPK level in the
cortex of mice (n¼6 per group). The total level of p38 MAPK
was normalized against the b-actin and the level of
phosphorylated p38 MAPK was normalized against the total
p38 MAPK. I, ischemic core; P, peri-infarct region; C,
contralateral cortex to ischemic hemisphere. npo0.05
compared with Sham group, #po0.05 compared with P of
Ischemia group, respectively.
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8 81
2.5. Bcl-xL translocation was associated with p38 MAPKactivation in the ischemic cortex of mice after HPC
To explore whether Bcl-xL translocation was associated with
p38 MAPK activation, we further observed the effect of
SB203580 on Bcl-xL mitochondria translocation in the peri-
infarct region of ischemic cortex. We found that the protein
level of Bcl-xL in HPCþSB203580þI group decreased remark-
ably in mitochondrial fraction and increased significantly in
cytosolic fraction compared with DMSO treatment (Fig. 7A
and B). The ratio of cytosolic Bcl-xL to mitochondrial Bcl-xL
increased remarkably in the HPCþSB203580þI group, suggest-
ing that SB203580 partly blocked the mitochondria transloca-
tion of Bcl-xL induced by HPC in the peri-infarct region
(Fig. 7C).
3. Discussion
Among several protection strategies directed against ischemic
injury, hypoxic/ischemic preconditioning demonstrated a
powerful protective potential against ischemic cell death
(Bolli, 2007). Since pretreatment with ischemia is not applic-
able to patients, many attempts have been undertaken to
achieve the protective effect of ischemic preconditioning
either pharmacologically (Zhao and Kukreja, 2002) or by
pretreatment with moderate environmental stress, e.g., HPC.
HPC initiates several intracellular signaling pathways to pro-
vide protection against subsequent cerebral ischemic injuries
(Long et al., 2006; Zhang et al., 2007, 2009, 2011). Recently, the
concept of two proposed types of HPC has been widely
accepted (Tsai et al., 2004). The early preconditioning which
occurred within minutes and lasted approximately 2 h after
HPC was reported to be related to changes in the intracellular
signal transduction pathways, such as the activation of protein
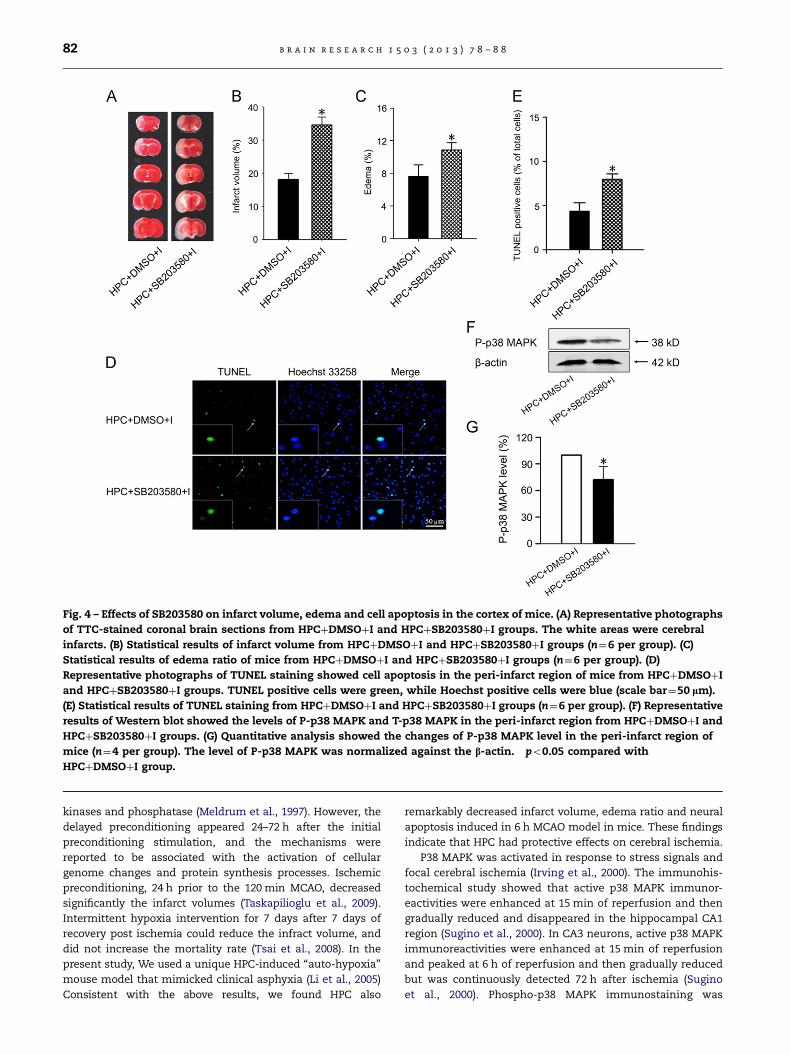
Fig. 4 – Effects of SB203580 on infarct volume, edema and cell apoptosis in the cortex of mice. (A) Representative photographs
of TTC-stained coronal brain sections from HPCþDMSOþI and HPCþSB203580þI groups. The white areas were cerebral
infarcts. (B) Statistical results of infarct volume from HPCþDMSOþI and HPCþSB203580þI groups (n¼6 per group). (C)
Statistical results of edema ratio of mice from HPCþDMSOþI and HPCþSB203580þI groups (n¼6 per group). (D)
Representative photographs of TUNEL staining showed cell apoptosis in the peri-infarct region of mice from HPCþDMSOþI
and HPCþSB203580þI groups. TUNEL positive cells were green, while Hoechst positive cells were blue (scale bar¼50 lm).
(E) Statistical results of TUNEL staining from HPCþDMSOþI and HPCþSB203580þI groups (n¼6 per group). (F) Representative
results of Western blot showed the levels of P-p38 MAPK and T-p38 MAPK in the peri-infarct region from HPCþDMSOþI and
HPCþSB203580þI groups. (G) Quantitative analysis showed the changes of P-p38 MAPK level in the peri-infarct region of
mice (n¼4 per group). The level of P-p38 MAPK was normalized against the b-actin. �po0.05 compared with
HPCþDMSOþI group.
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 882
kinases and phosphatase (Meldrum et al., 1997). However, the
delayed preconditioning appeared 24–72 h after the initial
preconditioning stimulation, and the mechanisms were
reported to be associated with the activation of cellular
genome changes and protein synthesis processes. Ischemic
preconditioning, 24 h prior to the 120 min MCAO, decreased
significantly the infarct volumes (Taskapilioglu et al., 2009).
Intermittent hypoxia intervention for 7 days after 7 days of
recovery post ischemia could reduce the infract volume, and
did not increase the mortality rate (Tsai et al., 2008). In the
present study, We used a unique HPC-induced ‘‘auto-hypoxia’’
mouse model that mimicked clinical asphyxia (Li et al., 2005)
Consistent with the above results, we found HPC also
remarkably decreased infarct volume, edema ratio and neural
apoptosis induced in 6 h MCAO model in mice. These findings
indicate that HPC had protective effects on cerebral ischemia.
P38 MAPK was activated in response to stress signals and
focal cerebral ischemia (Irving et al., 2000). The immunohis-
tochemical study showed that active p38 MAPK immunor-
eactivities were enhanced at 15 min of reperfusion and then
gradually reduced and disappeared in the hippocampal CA1
region (Sugino et al., 2000). In CA3 neurons, active p38 MAPK
immunoreactivities were enhanced at 15 min of reperfusion
and peaked at 6 h of reperfusion and then gradually reduced
but was continuously detected 72 h after ischemia (Sugino
et al., 2000). Phospho-p38 MAPK immunostaining was

Fig. 5 – Effects of HPC on Bcl-xL protein expression in
cytosol and mitochondria of cortex. (A) Representative
results of Western blot showed Bcl-xL protein expression in
cytosol and mitochondria in the cortex from Sham, H4,
Ischemia and HPCþI groups. (B) Quantitative analysis
showed the changes of Bcl-xL distribution in cytosol and
mitochondria in the cortex of mice (n¼6 per group). The Bcl-
xL level in cytosol was normalized against the b-actin level.
The Bcl-xL level in mitochondria was normalized against
the COX-IV level. (C) Quantitative analysis showed the ratio
of cytosolic Bcl-xL to mitochondrial Bcl-xL. �po0.05
compared with Sham group, #po0.05 compared with P of
Ischemia group, respectively.
Fig. 6 – Co-immunoprecipitation of the T-p38 MAPK or P-p38
MAPK with Bcl-xL in the cortex of mice.
(A) Immunoprecipitation assay showed that the association
between Bcl-xL and T-p38 MAPK existed in the cortex of
normal mice. (B) Immunoprecipitation assay showed no co-
immunoprecipitation of the P-p38 MAPK with Bcl-xL in the
cortex of normal mice. (C) Immunoprecipitation assay
showed that the association between Bcl-xL and P-p38
MAPK existed in the peri-infarct region from HPCþI group.
WCL and IgG were used as positive and negative control,
respectively. WCL, whole cell lysate.
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8 83
markedly increased in cells with astrocyte-like morphology in
both ischemic core and penumbra-like regions (Irving et al.,
2000). In this study, immunoblot analysis demonstrated that T-
p38 MAPK level decreased significantly at 6 h MCAO in the
ischemic core of cortex. More neural cell necrosis in the ischemic
core might contribute to these results. On the other hand, in the
ischemic peri-infarct region, the level of P-p38 MAPK increased
significantly but the T-p38 MAPK level did not change.
Moreover, several reports found that p38 MAPK could be
implicated in the preconditioning effects (Moolman et al.,
2006; Sun et al., 2006, 2010; Yamashita et al., 2009). In the
apoptosis model using global ischemia, ischemic precondi-
tioning resulted in a significant decrease in p38 MAPK
activation at the end of reperfusion when compared to non-
preconditioned isolated rat hearts (Moolman et al., 2006).
Expression of P-p38 MAPK was increased after limb ischemic
preconditioning in the CA1, CA3 and dentate gyrus regions of
hippocampus (Sun et al., 2006). The level of P-p38 MAPK was
up-regulated at 6 h and reached its peak 12 h in the CA1
region of the hippocampus after limb ischemic precondition-
ing (Sun et al., 2010). However, the pretreatment of
3.5-atomsphere absolute (3.5 ATA) and hyperbaric oxygen
significantly decreased the level of P-p38 MAPK at 10-min
reperfusion (Yamashita et al., 2009). In the present study, we
found that HPC could further increase the level of P-p38
MAPK in the ischemic peri-infarct region, but not affect the
T-p38 MAPK level both in the ischemic core and peri-infarct

Fig. 7 – Effects of SB203580 on Bcl-xL protein expression in
cytosol and mitochondria of cortex. (A) Representative results
of Western blot showed Bcl-xL protein expression in cytosol
and mitochondria in the cortex from HPCþDMSOþI and
HPCþSB203580þI groups. (B) Quantitative analysis showed the
changes of Bcl-xL protein level in cytosol and mitochondria
after SB203580 or DMSO administration (n¼6 per group).
The Bcl-xL protein level in cytosol was normalized against the
b-actin. The Bcl-xL protein level in mitochondria was
normalized against the COX-IV. (C) Quantitative analysis
showed the ratio of cytosolic Bcl-xL to mitochondrial Bcl-xL
after SB203580 or DMSO administration. �po0.05 compared
with P of HPCþDMSOþI group.
b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 884
region. These results suggested that p38 MAPK phosphoryla-
tion, not expression level, involved in the HPC-induced
neuroprotection.
However, the role of p38 MAPK in preconditioning effect
has not been defined unequivocally. SB203580 was first
described as an inhibitor of p38MAPK activity by competing
with ATP for binding (Young et al., 1997). However, it was later
demonstrated that SB203580 also prevented p38MAPK
phosphorylation (Galan et al., 2000). In this study we really
observed that intracerebroventricular injection of 1 mM
SB203580 significantly inhibited p38MAPK phosphorylation.
SB203580 could reduce preconditioning effects of sublethal-
ischemia, isoflurane-exposure, or limb-ischemia (Sun et al.,
2006; Zheng and Zuo, 2004). Several reports suggested that
the cardioprotective effects of preconditioning could be
abolished by SB203580 (Fryer et al., 2001; Nakano et al.,
2000; Sato et al., 2000). SB203580 blocked the protective effect
of limb ischemic preconditioning against delayed neuronal
death that was normally induced by lethal brain ischemic
insult (Sun et al., 2010). But Lochner et al. (2003) found that in
the isolated perfused rat heart improved functional recovery
and reduction in infarct size were associated with attenua-
tion of p38 MAPK activation by SB203580 during sustained
ischemia-reperfusion. SB203580, when given between admin-
istration of 3.5 ATA and hyperbaric oxygen treatment,
remarkably increased the number of survived neurons
(Yamashita et al., 2009). Bell et al. (2008) reported that
SB203580 blocked p38 MAPK activity, but did not block
ischemic preconditioning and was not protective when given
during ischemia in rat heart . The different effects of
SB203580 may be attributable to the use of different species,
models, protocols and measured end-points. Using our estab-
lished mouse HPC and MCAO models of mice, we found that
SB203580 could abolish the neuroprotective effects induced
by HPC. Taken together, our results suggested that HPC might
attenuate MCAO-induced cerebral ischemic injuries of mice
through the phosphorylation of p38 MAPK.
The ischemic penumbra or peri-infarct region is poten-
tially salvageable and thus represents an opportunity for
therapeutic intervention although it is under threat of infarc-
tion. Apoptosis is believed to contribute to loss of neurons in
the ischemic penumbra. Bcl-xL is a potent inhibitor of
programmed cell death and acts by heterodimerization with
other Bcl-2 family members to inhibit caspase activation,
presumably by blocking release of cytochrome c (Antignani
and Youle, 2006). Bcl-xL promoted cell survival by inhibiting
the function of the proapoptotic Bcl-2 proteins (Chipuk and
Green, 2008; Youle and Strasser, 2008). Bcl-xL protected lung
endothelial cells against hypoxia/reoxygenation-induced cell
death by blocking Bax and Bid mitochondrial translocation
and inhibiting mitochondrial cytochrome c release. A large
number of studies have shown that HPC attenuated apoptosis
through changing the expression of Bcl-xL (Rybnikova et al.,
2006; Wang et al., 2010). HPC protected neurons in rat
hippocampus and neocortex from the post-hypoxic apoptotic
transformations via persistent overexpression of Bcl-xL
(Rybnikova et al., 2006). HPC suppressed group III secreted
phospholipase A2-induced apoptosis via Janus-activated
kinase 2/signal transducers and activators of transcription 3
signal pathways that regulated the expression of Bcl-xL in
cultured cortical neurons (Wang et al., 2010). In the present
study, we found that HPC triggered Bcl-xL translocation to
mitochondria from cytosol in the peri-infarct region of

b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8 85
ischemic cortex. Our results suggested that Bcl-xL in mito-
chondria might participate in antiapoptosis effect induced
by HPC.
Various upstream signaling mechanisms can stimulate
transcription of Bcl-xL or suppress the inhibitory effect of
Bad on Bcl-xL, such as Janus-activated kinase 2, extracellular
signal-regulated kinase 1/2, Akt, and protein kinase A path-
ways (Basu and Haldar, 2003; Kharbanda et al., 2000; Saito
et al., 2003; Sugawara et al., 2004; Upreti et al., 2008). P38
MAPK was found to play a crucial role in the ischemic
preconditioning-induced cardioprotection through activation
of Bcl-2 (Kilic et al., 2005). P38 MAPK could mediate
TNF-induced apoptosis in endothelial cells via phosphoryla-
tion and down-regulation of Bcl-xL (Saito et al., 2003). In
this study, we found that Bcl-xL and T-p38 MAPK could
be coimmunoprecipited by each other in the cortex of
normal control mice. We did not observe any co-
immunoprecipitation of Bcl-xL and P-p38 MAPK in the nor-
mal group. But Bcl-xL and P-p38 MAPK were coimmunopre-
cipitated reciprocally in the peri-infarct region of HPC and
MCAO treated mice possibly because of the quadrupled P-p38
MAPK level. Using SB203580, the effect of HPC on mitochon-
dria translocation of Bcl-xL was inhibited: the protein level of
Bcl-xL decreased remarkably in mitochondrial fraction but
increased significantly in cytosolic fraction.
In summary, our findings suggested that P-p38 MAPK
mediates HPC-induced neuroprotection against cerebral
ischemic injury via mitochondria translocation of Bcl-xL,
which might be a key anti-apoptotic mechanism of HPC.
Further studies about the Bcl-xL phosphorylation sites by p38
MAPK during cerebral ischemia and HPC and the relations
between Bcl-xL phosphorylation and translocation will be
required.
4. Experimental procedures
4.1. Animals
The experimental procedures were carried out in accordance
with the Chinese regulations involving animal protection and
approved by the animal ethics committee of the China
Capital Medical University (Permit Number: 2011-X-026).
Experiments were performed at room temperature
(20–22 1C) on male BALB/c mice (12–14 W, 18–22 g). All surgery
was performed under sodium pentobarbital anesthesia, and
all efforts were made to minimize suffering. Animals were
randomly divided into six groups, namely Sham group (no
MCAO and HPC treatment), H4 group (auto-hypoxia four
times), Ischemia group (MCAO-induced cerebral ischemia),
HPCþI group (ischemia with H4 treatment), HPCþDMSOþI
group (ischemia with HPC and DMSO treatments) and
HPCþSB203580þI group (ischemia with HPC and SB203580
treatments).
4.2. Animal models
The auto-hypoxia-induced HPC mouse model was prepared
as our reported (Li et al., 2005; Niu et al., 2005). In brief, mice
were placed individually in a 125 ml airtight jar, which was
full of fresh air and sealed with a rubber plug to duplicate a
progressive auto-hypoxia environment. Mice were removed
as soon as the first signs of gasping appeared. A minimum of
30 min was allowed for recovery in normoxic condition, and
then the mice were switched to another hermetically sealed
jar of the same volume. All of the HPC mice underwent 4
times repetitive hypoxic exposures. It was designated as
H1–H4 when the mice were exposed to hypoxia once, twice,
three time and four times, respectively.
MCAO was performed 1 h after pretreatment of HPC. The
MCAO-induced cerebral focal ischemia mouse model was
conducted as described previously with minor modification
(Chen et al., 2007). An arteriotomy was made in common
carotid artery allowing the introduction of a 5-0 surgical
filament with its tip (0.23 mm in diameter) rounded by heat
(#1622-A, Shadong Biotechnology Company, Beijing, China).
The filament was gently advanced into the internal carotid
artery to occlude the middle cerebral artery. In the Sham
group, the mice received the same surgical exposure of
carotid artery without occlusion. During the surgery, body
temperature was maintained with the use of a heating lamp
and thermal blanket.
4.3. Drug treatment
Just before MCAO, the mice received an intracerebroventri-
cular injection of 5 ml SB203580 (1 mM, #S8307-1MG, Sigma-
Aldrich, St. Louis, MO, USA) or DMSO (0.1%) in the ischemic
side. Drug administration procedures followed the method
described by Munoz et al. (2003). The location of each
injection was 0.5 mm posterior to the bregma, 1.0 mm lateral
to the midline, and 3.5 mm below the skull surface. The mice
underwent MCAO as described above.
4.4. Measurement of infarct size, edema ratio and TUNELstaining
Infarct volume, edema ratio and cell apoptosis were assessed
at 6 h after MCAO, although at this time point cerebral
ischemic injuries were still under development, ischemic
penumbra or peri-infarct region exited more apparently and
the p38 MAPK mediated molecular changes in this region at
early stage could be studied.
Mice were sacrificed at 6 h after MCAO, and then the brain
was quickly removed and cut into 1.5 mm-thickness coronal
sections. Brain sections were incubated for 20 min in a
solution of 0.5% TTC in 10 mM phosphate buffered saline at
37 1C, and then the slices were photographed by using
scanner. According to ischemic volume evaluation procedure
reported by Wexler et al. (2002), the infarct size was analyzed.
Edema ratio (E) was calculated using the following equation:
E¼ (SVL�SVR)/(SVLþSVR)�100%. SVL and SVR are the
volume of left (ischemic) and right (non-ischemic) hemi-
sphere volume, respectively.
Mouse brains were cut into 10 mm-thickness sections for
TUNEL staining. The TUNEL staining kit (#G3250, DeadEndTM-
Fluorometric TUNEL system, Promega, Madison, WI, USA)
was used to assess cell apoptosis. In brief, the brain sections
were placed in equilibration buffer and incubated with
nucleotide mix and rTdT enzyme at 37 1C for 1 h. Using 2X

b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 886
saline–sodium citrate buffer stopped the reaction. Hoechst
33258 was used to stain neural nuclei. The images were
visualized by fluorescence microscope (Leica TCS SP5, Wet-
zlar, Germany). The TUNEL positive cells in peri-infarct region
of cortex were counted.
4.5. Sample preparation and Western blot analysis
As previously reported (Jiang et al., 2009; Li et al., 2005; Zhang
et al., 2011), the tissues from ischemic core (I), peri-infarct
region (P) and contralateral cortex (C) were collected. The
frozen samples were rapidly thawed and homogenized at 4 1C
in buffer A (5 mM Tris–Cl, pH 7.5, containing 2 mM DTT, 2 mM
EDTA, 1 mM EGTA, 5 mg/ml each of leupeptin, aprotinin,
pepstatin A and chymostatin, 50 mM potassium fluoride,
50 mM okadaic acid, 5 mM sodium pyrophosphate). Homoge-
nates were centrifuged at 30,000g for 30 min at 4 1C.
The supernatants were collected as cytosolic fraction. The
mitochondrial fraction was obtained from the pellets accord-
ing to manufacture instruction of mitochondria isolation kit
(#GMS10006.2, GENMED SCIENTIFICS INC, Boston, MA, USA).
In order to get whole tissue homogenate, frozen samples
were homogenized at 4 1C in buffer C [(Buffer A mixed with
2% sodium dodecyl sulfate (SDS)] and sonicated to dissolve
the tissue completely. The protein concentration was deter-
mined by BCA kit (Pierce Company, Rockford, IL, USA) with
bovine serum albumin diluted in lysis buffer as standard.
The tissue extracts with equal amount of total protein
were separated by 10% SDS polyacrylamide gel electrophor-
esis (SDS-PAGE) and the proteins were transferred to poly-
vinylidene difluoride membranes, and probed with anti-
phospho-p38 MAPK (#P1491-1VL, Sigma-Aldrich, St. Louis,
MO, USA), anti-p38 MAPK (#9212, Cell Signalling Technology,
Danvers, MA, USA) and anti-Bcl-xL (#2764, Cell Signaling
Technology, Danvers, MA, USA) antibodies, respectively.
The horseradish peroxidase-conjugated goat anti-rabbit or
anti-mouse IgG (Stressgen Biotechnologies Corporation, Vic-
toria, BC, Canada) was used as second antibodies. Following
incubation with the primary and secondary antibodies, the
Enhanced Chemiluminescence kit (Pierce Company, Rock-
ford, IL, USA) was employed to detect the signals. To verify
equal loading of protein, the blots were reprobed with
primary monoclonal antibodies against b-actin (#60008-1-Ig,
Proteintech Company, Chicago, IL, USA) or COX-IV (#11242-1-
AP, Proteintech Company, Chicago, IL, USA).
4.6. Coimmunoprecipitation
Nondenaturing lysis buffer extracted 500 mg protein of mouse
cortex was immunoprecipitated with 2 mg of Bcl-xL antibody,
p38 MAPK antibody, P-p38 MAPK antibody and IgG, respec-
tively (Bu et al., 2011). After 3 h incubation, protein
G sepharose was added for 12 h at 4 1C and centrifuged for
1 min at 12,000g. The precipitates were rinsed with IP buffer
(0.5% NP-40, Tris–Cl pH 8.0, 0.15 M NaCl) four times to remove
nonspecific interaction products. The immunoprecipitates
were resolved in loading buffer for SDS-PAGE.
4.7. Statistics
Quantitative analysis for immunoblotting was done by
Quantitative-One software (Gel Doc 2000 imaging system,
Bio-Rad Company, CA, USA). For protein level, the ratio of p38
MAPK or Bcl-xL (band density of protein/band density of
b-actin or COX-IV) was expressed as 100% in the control
group. Both bands of Bcl-xL were quantitative analyzed. The
data from other groups were expressed as percentage of that
from control group. For p38 MAPK phosphorylation, the ratio
of P-p38 MAPK to T-p38 MAPK in sham group was recognized
as 100%, and the values of ischemic groups were expressed as
a percentage of sham group. The presented values were
expressed as mean7SE. Statistical analysis was conducted
by one-way analysis of variance (ANOVA) followed by
Student–Newma–Keuls post hoc analysis. In all cases,
po0.05 was considered statistically significant.
Acknowledgments
This work was supported by the following grants: National
Natural Science Foundation of China (31071048, 31171147 and
31140005), China 973 Pre-program (2011CB512109), Ph.D.
Programs Foundation of Ministry of Education of China
(20091107110001), Basic and clinical research joint program
of Capital Medical University (11JL34 and 11JL44), Research
fund of Capital Medical University (2011ZR02) and the Scien-
tific Research Foundation for the Returned Overseas Chinese
Scholars, State Education Ministry. The funders had no role in
study design, data collection and analysis, decision to pub-
lish, or preparation of the manuscript.
r e f e r e n c e s
Antignani, A., Youle, R.J., 2006. How do Bax and Bak lead topermeabilization of the outer mitochondrial membrane?.Curr. Opin. Cell Biol. 18, 685–689.
Basu, A., Haldar, S., 2003. Identification of a novel Bcl-xLphosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 538, 41–47.
Bell, J.R., Eaton, P., Shattock, M.J., 2008. Role of p38-mitogen-activated protein kinase in ischaemic preconditioning in ratheart. Clin. Exp. Pharmacol. Physiol. 35, 126–134.
Bielewicz, J., Kurzepa, J., Lagowska-Lenard, M., Bartosik-Psujek, H.,2010. The novel views on the patomechanism of ischemicstroke. Wiad. Lek. 63, 213–220.
Billen, L.P., Kokoski, C.L., Lovell, J.F., Leber, B., Andrews, D.W.,2008. Bcl-XL inhibits membrane permeabilization bycompeting with Bax. PLoS Biol. 6, e147.
Bolli, R., 2007. Preconditioning: a paradigm shift in the biology ofmyocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 292,H19–H27.
Bu, X., Zhang, N., Yang, X., Liu, Y., Du, J., Liang, J., Xu, Q., Li, J.,2011. Proteomic analysis of cPKCbetaII-interacting proteinsinvolved in HPC-induced neuroprotection against cerebralischemia of mice. J. Neurochem. 117, 346–356.
Chen, A., Liao, W.P., Lu, Q., Wong, W.S., Wong, P.T., 2007.Upregulation of dihydropyrimidinase-related protein 2,spectrin alpha II chain, heat shock cognate protein 70pseudogene 1 and tropomodulin 2 after focal cerebral

b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 8 87
ischemia in rats–a proteomics approach. Neurochem. Int. 50,1078–1086.
Cheng, T.O., 2006. Danshen: a versatile Chinese herbal drug forthe treatment of coronary heart disease. Int. J. Cardiol. 113,437–438.
Chipuk, J.E., Green, D.R., 2008. How do BCL-2 proteins inducemitochondrial outer membrane permeabilization?. TrendsCell Biol. 18, 157–164.
Cohen, P., 1997. The search for physiological substrates of MAPand SAP kinases in mammalian cells. Trends Cell Biol. 7,353–361.
Cuenda, A., Rousseau, S., 2007. p38 MAP-kinases pathwayregulation, function and role in human diseases. Biochim.Biophys. Acta 1773, 1358–1375.
Dhillon, A.S., Hagan, S., Rath, O., Kolch, W., 2007. MAP kinasesignalling pathways in cancer. Oncogene 26, 3279–3290.
Doeppner, T.R., Dietz, G.P., El, A.M., Gerber, J., Witte, O.W., Bahr, M.,Weise, J., 2009. TAT-Bcl-x(L) improves survival of neuronalprecursor cells in the lesioned striatum after focal cerebralischemia. Neurobiol. Dis. 34, 87–94.
Edlich, F., Banerjee, S., Suzuki, M., Cleland, M.M., Arnoult, D.,Wang, C., Neutzner, A., Tjandra, N., Youle, R.J., 2011. Bcl-x(L)retrotranslocates Bax from the mitochondria into the cytosol.Cell 145, 104–116.
Ferrer, I., 2006. Apoptosis: future targets for neuroprotectivestrategies. Cerebrovasc. Dis. 21 (Suppl. 2), 9–20.
Fryer, R.M., Patel, H.H., Hsu, A.K., Gross, G.J., 2001. Stress-activated protein kinase phosphorylation duringcardioprotection in the ischemic myocardium. Am. J. Physiol.Heart Circ. Physiol. 281, H1184–H1192.
Galan, A., Garcia-Bermejo, M.L., Troyano, A., Vilaboa, N.E., de, B.E.,Kazanietz, M.G., Aller, P., 2000. Stimulation of p38 mitogen-activated protein kinase is an early regulatory event for thecadmium-induced apoptosis in human promonocytic cells. J.Biol. Chem. 275, 11418–11424.
Gollapudi, S., McCormick, M.J., Gupta, S., 2003. Changes inmitochondrial membrane potential and mitochondrial massoccur independent of the activation of caspase-8 and caspase-3 during CD95-mediated apoptosis in peripheral blood T cells.Int. J. Oncol. 22, 597–600.
Heidbreder, M., Naumann, A., Tempel, K., Dominiak, P.,Dendorfer, A., 2008. Remote vs. ischaemic preconditioning:the differential role of mitogen-activated protein kinasepathways. Cardiovasc. Res. 78, 108–115.
Huang, D.C., Strasser, A., 2000. BH3-Only proteins-essentialinitiators of apoptotic cell death. Cell 103, 839–842.
Irving, E.A., Bamford, M., 2002. Role of mitogen- and stress-activated kinases in ischemic injury. J. Cereb. Blood FlowMetab. 22, 631–647.
Irving, E.A., Barone, F.C., Reith, A.D., Hadingham, S.J., Parsons, A.A.,2000. Differential activation of MAPK/ERK and p38/SAPK inneurones and glia following focal cerebral ischaemia in the rat.Brain Res. Mol. Brain Res. 77, 65–75.
Islam, N., Aftabuddin, M., Moriwaki, A., Hori, Y., 1995. Detectionof DNA damage induced by apoptosis in the rat brainfollowing incomplete ischemia. Neurosci. Lett. 188, 159–162.
Jiang, J., Yang, W., Huang, P., Bu, X., Zhang, N., Li, J., 2009.Increased phosphorylation of Ets-like transcription factor-1in neurons of hypoxic preconditioned mice. Neurochem. Res.34, 1443–1450.
Kaufmann, T., Schlipf, S., Sanz, J., Neubert, K., Stein, R., Borner, C.,2003. Characterization of the signal that directs Bcl-x(L), butnot Bcl-2, to the mitochondrial outer membrane. J. Cell Biol.160, 53–64.
Kharbanda, S., Saxena, S., Yoshida, K., Pandey, P., Kaneki, M.,Wang, Q., Cheng, K., Chen, Y.N., Campbell, A., Sudha, T., Yuan,Z.M., Narula, J., Weichselbaum, R., Nalin, C., Kufe, D., 2000.Translocation of SAPK/JNK to mitochondria and interaction
with Bcl-x(L) in response to DNA damage. J. Biol. Chem. 275,322–327.
Kilic, E., Kilic, U., Soliz, J., Bassetti, C.L., Gassmann, M., Hermann,D.M., 2005. Brain-derived erythropoietin protects from focalcerebral ischemia by dual activation of ERK-1/-2 and Aktpathways. FASEB J. 19, 2026–2028.
Lennmyr, F., Ericsson, A., Gerwins, P., Ahlstrom, H., Terent, A.,2003. Increased brain injury and vascular leakage afterpretreatment with p38-inhibitor SB203580 in transientischemia. Acta Neurol. Scand. 108, 339–345.
Li, J., Niu, C., Han, S., Zu, P., Li, H., Xu, Q., Fang, L., 2005. Identificationof protein kinase C isoforms involved in cerebral hypoxicpreconditioning of mice. Brain Res. 1060, 62–72.
Lochner, A., Genade, S., Hattingh, S., Marais, E., Huisamen, B.,Moolman, J.A., 2003. Comparison between ischaemic andanisomycin-induced preconditioning: role of p38 MAPK.Cardiovasc. Drugs Ther. 17, 217–230.
Long, C., Gao, Y., Gao, G., Han, S., Zu, P., Fang, L., Li, J., 2006.Decreased phosphorylation and protein expression of ERK1/2in the brain of hypoxic preconditioned mice. Neurosci. Lett.397, 307–312.
MacManus, J.P., Buchan, A.M., 2000. Apoptosis after experimentalstroke: fact or fashion?. J. Neurotrauma 17, 899–914.
McCubrey, J.A., Lahair, M.M., Franklin, R.A., 2006. Reactive oxygenspecies-induced activation of the MAP kinase signalingpathways. Antioxid. Redox. Signal. 8, 1775–1789.
Meldrum, D.R., Cleveland Jr., J.C., Rowland, R.T., Banerjee, A.,Harken, A.H., Meng, X., 1997. Early and delayedpreconditioning: differential mechanisms and additiveprotection. Am. J. Physiol. 273, H725–H733.
Moolman, J.A., Hartley, S., Van, W.J., Marais, E., Lochner, A., 2006.Inhibition of myocardial apoptosis by ischaemic and beta-adrenergic preconditioning is dependent on p38 MAPK.Cardiovasc. Drugs Ther. 20, 13–25.
Munoz, A., Nakazaki, M., Goodman, J.C., Barrios, R., Onetti, C.G.,Bryan, J., guilar-Bryan, L., 2003. Ischemic preconditioning inthe hippocampus of a knockout mouse lacking SUR1-basedK(ATP) channels. Stroke 34, 164–170.
Nakano, A., Cohen, M.V., Critz, S., Downey, J.M., 2000. SB 203580,an inhibitor of p38 MAPK, abolishes infarct-limiting effect ofischemic preconditioning in isolated rabbit hearts. Basic Res.Cardiol. 95, 466–471.
Niu, C., Li, J., Cui, X., Han, S., Zu, P., Li, H., Xu, Q., 2005. Changes incPKC isoform-specific membrane translocation and proteinexpression in the brain of hypoxic preconditioned mice.Neurosci. Lett. 384, 1–6.
Ostrowski, R.P., Graupner, G., Titova, E., Zhang, J., Chiu, J., Dach, N.,Corleone, D., Tang, J., Zhang, J.H., 2008. The hyperbaric oxygenpreconditioning-induced brain protection is mediated by areduction of early apoptosis after transient global cerebralischemia. Neurobiol. Dis. 29, 1–13.
Pfeilschifter, W., Czech, B., Hoffmann, B.P., Sujak, M., Kahles, T.,Steinmetz, H., Neumann-Haefelin, T., Pfeilschifter, J., 2010.Pyrrolidine dithiocarbamate activates p38 MAPK and protectsbrain endothelial cells from apoptosis: a mechanism for theprotective effect in stroke?. Neurochem. Res. 35, 1391–1401.
Rybnikova, E., Sitnik, N., Gluschenko, T., Tjulkova, E., Samoilov, M.O.,2006. The preconditioning modified neuronal expression ofapoptosis-related proteins of Bcl-2 superfamily following severehypobaric hypoxia in rats. Brain Res. 1089, 195–202.
Saito, A., Hayashi, T., Okuno, S., Ferrand-Drake, M., Chan, P.H.,2003. Overexpression of copper/zinc superoxide dismutase intransgenic mice protects against neuronal cell death aftertransient focal ischemia by blocking activation of the Bad celldeath signaling pathway. J. Neurosci. 23, 1710–1718.
Sato, M., Cordis, G.A., Maulik, N., Das, D.K., 2000. SAPKsregulation of ischemic preconditioning. Am. J. Physiol. HeartCirc. Physiol. 279, H901–H907.

b r a i n r e s e a r c h 1 5 0 3 ( 2 0 1 3 ) 7 8 – 8 888
Sugawara, T., Fujimura, M., Noshita, N., Kim, G.W., Saito, A.,Hayashi, T., Narasimhan, P., Maier, C.M., Chan, P.H., 2004.Neuronal death/survival signaling pathways in cerebralischemia. NeuroRx 1, 17–25.
Sugino, T., Nozaki, K., Takagi, Y., Hattori, I., Hashimoto, N.,Moriguchi, T., Nishida, E., 2000. Activation of mitogen-activated protein kinases after transient forebrain ischemia ingerbil hippocampus. J. Neurosci. 20, 4506–4514.
Sun, X.C., Li, W.B., Li, Q.J., Zhang, M., Xian, X.H., Qi, J., Jin, R.L., Li, S.Q.,2006. Limb ischemic preconditioning induces brain ischemictolerance via p38 MAPK. Brain Res. 1084, 165–174.
Sun, X.C., Xian, X.H., Li, W.B., Li, L., Yan, C.Z., Li, Q.J., Zhang, M.,2010. Activation of p38 MAPK participates in brain ischemictolerance induced by limb ischemic preconditioning by up-regulating HSP 70. Exp. Neurol. 224, 347–355.
Taskapilioglu, M.O., Alkan, T., Goren, B., Tureyen, K., Sahin, S.,Taskapilioglu, O., Korfali, E., 2009. Neuronal protective effectsof focal ischemic pre- and/or postconditioning on the modelof transient focal cerebral ischemia in rats. J. Clin. Neurosci.16, 693–697.
Torii, S., Yamamoto, T., Tsuchiya, Y., Nishida, E., 2006. ERK MAP kinasein G cell cycle progression and cancer. Cancer Sci. 97, 697–702.
Tsai, B.M., Wang, M., March, K.L., Turrentine, M.W., Brown, J.W.,Meldrum, D.R., 2004. Preconditioning: evolution of basicmechanisms to potential therapeutic strategies. Shock 21,195–209.
Tsai, Y.W., Yang, Y.R., Chen, G.H., Chang, H.C., Wang, R.Y., 2008.The time window of intermittent hypoxia intervention aftermiddle cerebral artery occlusion. Chin. J. Physiol. 51, 324–328.
Upreti, M., Galitovskaya, E.N., Chu, R., Tackett, A.J., Terrano, D.T.,Granell, S., Chambers, T.C., 2008. Identification of the majorphosphorylation site in Bcl-xL induced by microtubuleinhibitors and analysis of its functional significance. J. Biol.Chem. 283, 35517–35525.
Wang, G., Zhou, D., Wang, C., Gao, Y., Zhou, Q., Qian, G., DeCoster,M.A., 2010. Hypoxic preconditioning suppresses group IIIsecreted phospholipase A2-induced apoptosis via JAK2-STAT3activation in cortical neurons. J. Neurochem. 114, 1039–1048.
Wexler, E.J., Peters, E.E., Gonzales, A., Gonzales, M.L., Slee, A.M.,Kerr, J.S., 2002. An objective procedure for ischemic area
evaluation of the stroke intraluminal thread model in themouse and rat. J. Neurosci. Methods 113, 51–58.
Wu, C., Fujihara, H., Yao, J., Qi, S., Li, H., Shimoji, K., Baba, H.,2003. Different expression patterns of Bcl-2, Bcl-xl, and Baxproteins after sublethal forebrain ischemia in C57Black/Crj6mouse striatum. Stroke 34, 1803–1808.
Yamashita, S., Hirata, T., Mizukami, Y., Cui, Y.J., Fukuda, S., Ishida, K.,Matsumoto, M., Sakabe, T., 2009. Repeated preconditioning withhyperbaric oxygen induces neuroprotection against forebrainischemia via suppression of p38 mitogen activated proteinkinase. Brain Res. 1301, 171–179.
Youle, R.J., Strasser, A., 2008. The BCL-2 protein family: opposingactivities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9,47–59.
Young, P.R., McLaughlin, M.M., Kumar, S., Kassis, S., Doyle, M.L.,McNulty, D., Gallagher, T.F., Fisher, S., McDonnell, P.C., Carr, S.A.,Huddleston, M.J., Seibel, G., Porter, T.G., Livi, G.P., Adams, J.L., Lee,J.C., 1997. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J. Biol. Chem. 272,12116–12121.
Zhang, N., Gao, G., Bu, X., Han, S., Fang, L., Li, J., 2007. Neuron-specific phosphorylation of c-Jun N-terminal kinase increasedin the brain of hypoxic preconditioned mice. Neurosci. Lett.423, 219–224.
Zhang, N., Yin, Y., Han, S., Jiang, J., Yang, W., Bu, X., Li, J., 2011.Hypoxic preconditioning induced neuroprotection againstcerebral ischemic injuries and its cPKCgamma-mediatedmolecular mechanism. Neurochem. Int. 58, 684–692.
Zhang, Q.G., Wang, R.M., Han, D., Yang, L.C., Li, J., Brann, D.W.,2009. Preconditioning neuroprotection in global cerebralischemia involves NMDA receptor-mediated ERK-JNK3crosstalk. Neurosci. Res. 63, 205–212.
Zhao, T.C., Kukreja, R.C., 2002. Late preconditioning elicited byactivation of adenosine A(3) receptor in heart: role of NF-kappa B, iNOS and mitochondrial K(ATP) channel. J. Mol. Cell.Cardiol. 34, 263–277.
Zheng, S., Zuo, Z., 2004. Isoflurane preconditioning inducesneuroprotection against ischemia via activation of P38mitogen-activated protein kinases. Mol. Pharmacol. 65,1172–1180.


















