
PHCH 402: Analytical Quality Control - WordPress.com · PHCH 402: Analytical Quality Control ......
43
PHCH 402: Analytical Quality Control Different methods of analysis of some groups of drugs in common use in Nigeria (8 hrs) Course Content: 01 Introduction to the problems of drug quality control (2hrs) 2 Official methods of drug analysis with examples (5 hrs) 3 Analytical methods used by the manufacturer with examples (6 hrs) 4 Importance of standards in drug quality control, their use and storage (4 hrs) 5 Limit tests (12 hrs): Presence of impurities in pharmaceuticals and their sources, limit tests and factors considered in their design, negative and comparison tests. Classification of limit tests: limits of soluble and insoluble matter; moisture; volatile matter; residual solvents; nonvolatile matter. Residue on ignition, loss on ignition, ash values, limit tests for metallic impurities, other limit tests
Transcript of PHCH 402: Analytical Quality Control - WordPress.com · PHCH 402: Analytical Quality Control ......

PHCH 402: Analytical Quality Control Different methods of analysis of some groups of drugs in
common use in Nigeria (8 hrs)
Course Content:
01 Introduction to the problems of drug quality control (2hrs)
2 Official methods of drug analysis with examples (5 hrs)
3 Analytical methods used by the manufacturer with examples (6 hrs)
4 Importance of standards in drug quality control, their use and storage (4 hrs)
5 Limit tests (12 hrs):
Presence of impurities in pharmaceuticals and their sources, limit tests and factors considered in their design, negative and comparison tests.
Classification of limit tests: limits of soluble and insoluble matter; moisture; volatile matter; residual solvents; nonvolatile matter. Residue on ignition, loss on ignition, ash values, limit tests for metallic impurities, other limit tests

Course Content:
6 Different methods of analysis of some groups of drugs in common use in
Nigeria (8 hrs)
i. Antimalarials
ii. Non-narcotic analgesics
iii. Antibiotics
iv. Antihelminthics and amoebecides

Official Books (Pharmacopoeias):
• These are a collection of specifications for the determination of the quality
of a drug, meant for the use of manufacturers, pharmacists and
controlling bodies.
• They provide specifications on purity and potency (the legal basis for the
control of pharmaceutical products), in addition to standardised
methodology of testing and evaluation that are publicly available.
• They allow independent evaluation of drug quality to be made at any
stage after the drug has left the manufacturer.
• They are used for the establishment of identity, purity and ultimately
composition of the substances, and confirm the absence of traces of
foreign substance beyond a specified limit
• Examples include the BP, USP, EuP, JP, IP, AfrP, WHO-P, ChP etc
• They are usually reviewed every five years

ASSAYS:
• Assays in the monographs consist of tests for identity and purity
• Tests are determined by many factors including dosage form, method of
manufacture, types and sources of raw materials, chemical nature of the
drug, etc
• Broadly divided into instrumental and non-instrumental methods
• The following drugs will be discussed:
i. Chloroquine – anti malarial
ii. Aspirin – non narcotic analgesic
iii. Chloramphenicol – antibiotic
iv. Metronidazole – amoebicide
All assays are from the British Pharmacopoeia 2002

CHLOROQUINE: exists as phosphate (formulated as tablets) and sulphate (formulated
as tablets, syrup and injections)
Mpt 208oC Exists in two forms with mpts
195oC and 218oC
N4-(7-chloroquinolin-4-yl)-N1,N1-diethylpentane-1,4-diamine -

CHLOROQUINE SULPHATE
DEFINITION: Chloroquine sulphate contains not less than 98.5 per cent and not more than the equivalent of 101.0 per cent of N4-(7-chloroquinolin-4-yl)-N1,N1-diethylpentane-1,4-diamine sulphate, calculated with reference to the anhydrous substance.
IDENTIFICATION:
• First identification: B, D.
• Second identification: A, C, D.
A. Dissolve 0.100 g in water R and dilute to 100.0 ml with the same solvent. Dilute 1.0 ml of this solution to 100.0 ml with water R. Examined between 210 nm and 370 nm (2.2.25), the solution shows absorption maxima at 220 nm, 235 nm, 256 nm, 329 nm and 342 nm. The specific absorbances at the maxima are respectively 730 to 810, 430 to 470, 370 to 410, 400 to 440 and 430 to 470.

CHLOROQUINE SULPHATE
B. Examine by infrared absorption spectrophotometry (2.2.24), comparing
with the spectrum obtained with the base isolated from chloroquine
sulphate CRS. Record the spectra using solutions prepared as follows:
dissolve separately 0.1 g of the substance to be examined and of the
reference substance in 10 ml of water R, add 2 ml of dilute sodium
hydroxide solution R and shake with two quantities, each of 20 ml, of
chloroform R; combine the chloroform layers, wash with water R, dry
over anhydrous sodium sulphate R, evaporate to dryness and dissolve the
residues separately each in 2 ml of chloroform R.
C. Dissolve 25 mg in 20 ml of water R and add 8 ml of picric acid solution R1.
The precipitate, washed with water R, with alcohol R and finally with ether
R, melts (2.2.14) at 206°C to 209°C.
D. It gives reaction (a) of sulphates (2.3.1)

CHLOROQUINE SULPHATE
CHARACTERS
• A white or almost white, crystalline powder, freely soluble in water and in methanol, very slightly soluble in alcohol, practically insoluble in ether.
• It melts at about 208°C (instantaneous method).
TESTS:
• Heavy metals (2.4.8). Dissolve 2.0 g in 10 ml of water R. Add 5 ml of concentrated ammonia R and shake with 40 ml of ether R. Filter the aqueous layer and neutralise the filtrate with glacial acetic acid R. Heat on a water-bath to eliminate ether, allow to cool and dilute to 20.0 ml with water R. 12 ml of this solution complies with limit test A for heavy metals (20 ppm). Prepare the standard using lead standard solution (2 ppm Pb) R.
• Water (2.5.12): 3.0 per cent to 5.0 per cent, determined on 0.500 g by the semi-micro determination of water.
• Sulphated ash (2.4.14). Not more than 0.1 per cent, determined on 1.0 g.
ASSAY
• Dissolve 0.400 g in 50 ml of anhydrous acetic acid R. Titrate with 0.1M perchloric acid determining the end-point potentiometrically (2.2.20).
• 1 ml of 0.1M perchloric acid is equivalent to 41.8 mg of C18H28ClN3O4S.

Chloroquine Sulphate Injection
Definition: Chloroquine Sulphate Injection is a sterile solution of Chloroquine Sulphate in Water for Injections
Identification
• A. To a volume containing the equivalent of 60 mg of chloroquine add 2 ml of 2M sodium hydroxide and extract with two 20-ml quantities of chloroform. Wash the chloroform extracts with water, dry with anhydrous sodium sulphate, evaporate to dryness and dissolve the residue in 2 ml of chloroform. The infrared absorption spectrum of the resulting solution, Appendix II A, is concordant with the reference spectrum of chloroquine (RS 054).
• B. Dilute a volume containing the equivalent of 15 mg of chloroquine to 20 ml with water and add 8 ml of picric acid solution R1. The melting point of the precipitate, after washing successively with water, ethanol (96%) and ether, is about 207°, Appendix V A.
• C. Yields the reactions characteristic of sulphates, Appendix VI.
• Acidity pH, 4.0 to 5.5, Appendix V L.

Chloroquine Sulphate Injection
Assay To a volume containing the equivalent of 0.4 g of chloroquine add 20 ml
of 1M sodium hydroxide and extract with four 25 ml quantities of
chloroform. Combine the chloroform extracts and evaporate to a volume
of about 10 ml. Add 40 ml of anhydrous acetic acid and carry out Method I
for non-aqueous titration, Appendix VIII A, determining the end point
potentiometrically. Each ml of 0.1M perchloric acid VS is equivalent to
15.99 mg of C18H26ClN3.

Chloroquine Sulphate Tablets
Definition: Chloroquine Sulphate Tablets contain Chloroquine Sulphate. They are coated.
Content of chloroquine sulphate, C18H26ClN3,H2SO4,H2O 92.5 to 107.5% of the stated amount.
Identification
• A. Dissolve a quantity of the powdered tablets containing 0.1 g of Chloroquine Sulphate in a mixture of 10 ml of water and 2 ml of 2M sodium hydroxide and extract with two 20-ml quantities of chloroform. Wash the chloroform extracts with water, dry with anhydrous sodium sulphate, evaporate to dryness and dissolve the residue in 2 ml of chloroform IR. The infrared absorption spectrum of the resulting solution, Appendix II A, is concordant with the reference spectrum of chloroquine (RS 054).
• B. Shake a quantity of the powdered tablets containing 0.1 g of Chloroquine Sulphate with 10 ml of water and 1 ml of 2M hydrochloric acid and filter. To the filtrate add 1 ml of barium chloride solution. A white precipitate is produced.

Chloroquine Sulphate Tablets
Dissolution Comply with the dissolution test for tablets and capsules,
Appendix XII D, using as the medium 900 ml of 0.1M hydrochloric acid and
rotating the basket at 100 revolutions per minute. Withdraw a sample of
10 ml of the medium. Measure the absorbance of a layer of suitable
thickness of the filtered sample, suitably diluted if necessary, at the
maximum at 344 nm, Appendix II B. Calculate the total content of
chloroquine sulphate, C18H26ClN3,H2SO4,H2O, in the medium taking 450 as
the value of A(1%, 1 cm) at the maximum at 344 nm.

Chloroquine Sulphate Tablets
• Assay Weigh and powder 20 tablets. Dissolve a quantity of the powder
containing 0.5 g of Chloroquine Sulphate in 20 ml of 1M sodium hydroxide
and extract with four 25 ml quantities of chloroform. Combine the
chloroform extracts and evaporate to a volume of about 10 ml. Add 40 ml
of anhydrous acetic acid and carry out Method I for non-aqueous titration,
Appendix VIII A, determining the end point potentiometrically. Each ml of
0.1M perchloric acid VS is equivalent to 20.90 mg of C18H26ClN3,H2SO4.
• 200 mg of Chloroquine Sulphate is approximately equivalent to 146 mg of
chloroquine.
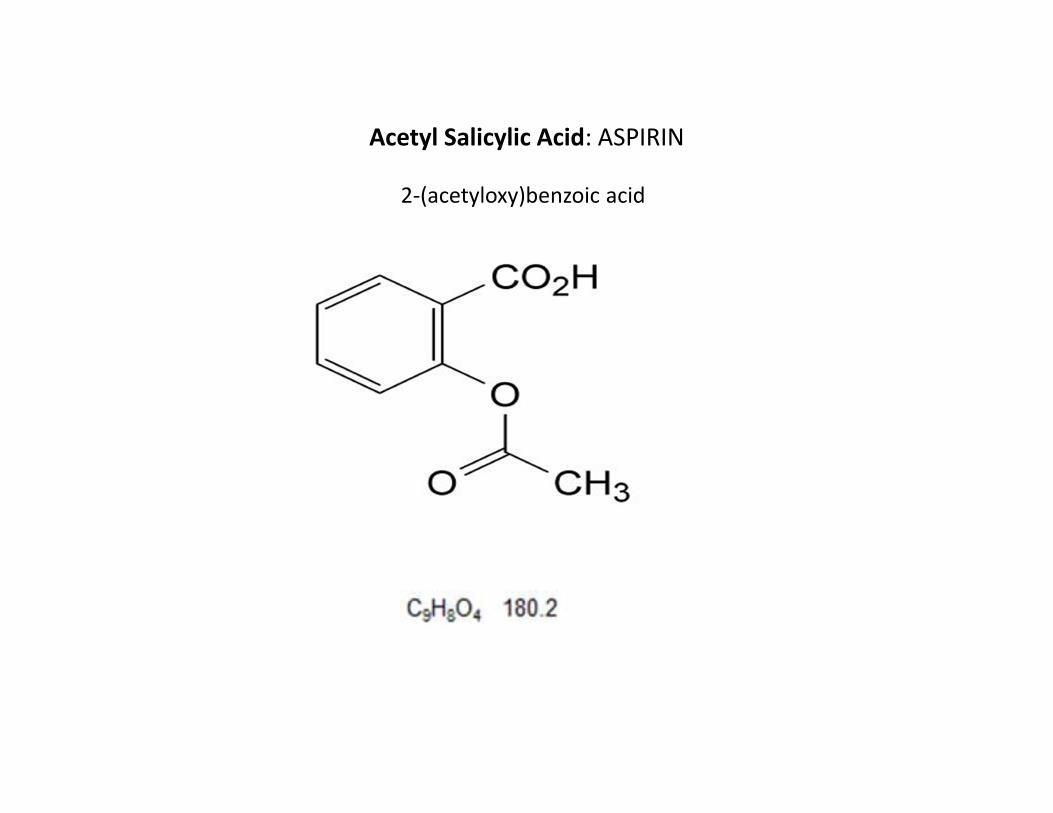
Acetyl Salicylic Acid: ASPIRIN
2-(acetyloxy)benzoic acid

Acetyl Salicylic Acid
Preparations
• Aspirin Tablets
• Dispersible Aspirin Tablets
• Effervescent Soluble Aspirin Tablets
• Enteric-coated Aspirin Tablets
• Aspirin and Caffeine Tablets
• Co-codaprin Tablets
• Dispersible Co-codaprin Tablets

Acetyl Salicylic Acid
DEFINITION
• Acetylsalicylic acid contains not less than 99.5 per cent and not more than
the equivalent of 101.0 per cent of 2-(acetyloxy)benzoic acid, calculated
with reference to the dried substance.
CHARACTERS
• A white, crystalline powder or colourless crystals, slightly soluble in water,
freely soluble in alcohol, soluble in ether.
• It melts at about 143°C (instantaneous method).

Acetyl Salicylic Acid
IDENTIFICATION
• First identification: A, B.
• Second identification: B, C, D.
• A. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with acetylsalicylic acid CRS.
• B. To 0.2 g add 4 ml of dilute sodium hydroxide solution R and boil for 3 min. Cool and add 5 ml of dilute sulphuric acid R. A crystalline precipitate is formed. Filter, wash the precipitate and dry at 100°C to 105°C. The melting point (2.2.14) is 156°C to 161°C.
• C. In a test tube mix 0.1 g with 0.5 g of calcium hydroxide R. Heat the mixture and expose, to the fumes produced, a piece of filter paper impregnated with 0.05 ml of nitrobenzaldehyde solution R. A greenish-blue or greenish-yellow colour develops on the paper. Moisten the paper with dilute hydrochloric acid R. The colour becomes blue.
• D. Dissolve with heating about 20 mg of the precipitate obtained in identification test B in 10 ml of water R and cool. The solution gives reaction (a) of salicylates (2.3.1).

Acetyl Salicylic Acid
Related substances: Salicylic acid as a product of degradation of aspirin. NMT 0.1%, but carried out using HPLC
Heavy metals (2.4.8). Dissolve 1.0 g in 12 ml of acetone R and dilute to 20 ml with water R. 12 ml of this solution complies with limit test B for heavy metals (20 ppm). Prepare the standard using lead standard solution (1 ppm Pb) obtained by diluting lead standard solution (100 ppm Pb) R with a mixture of 6 volumes of water R and 9 volumes of acetone R.
Loss on drying (2.2.32). Not more than 0.5 per cent, determined on 1.000 g by drying in vacuo.
Sulphated ash (2.4.14). Not more than 0.1 per cent, determined on 1.0 g.
ASSAY
• In a flask with a ground-glass stopper, dissolve 1.000 g in 10 ml of alcohol R. Add 50.0 ml of 0.5M sodium hydroxide. Close the flask and allow to stand for 1 h. Using 0.2 ml of phenolphthalein solution R as indicator, titrate with 0.5M hydrochloric acid. Carry out a blank titration.
• 1 ml of 0.5M sodium hydroxide is equivalent to 45.04 mg of C9H8O4.

Acetyl Salicylic Acid
IMPURITIES
• A. R = H: 4-hydroxybenzoic acid,
• B. R = CO2H: 4-hydroxybenzene-1,3-dicarboxylic acid (4-
hydroxyisophthalic acid),

Acetyl Salicylic Acid
IMPURITIES
C. salicylic acid,

Acetyl Salicylic Acid
IMPURITIES
D. R = O-CO-CH3: 2-[[2-(acetyloxy) benzoyl]oxy]benzoic acid
(acetylsalicylsalicylic acid),
E. R = OH: 2-[(2-hydroxybenzoyl)oxy]benzoic acid (salicylsalicylic acid),

Acetyl Salicylic Acid
IMPURITIES
F. 2-(acetyloxy)benzoic anhydride (acetylsalicylic anhydride).

Acetyl Salicylic Acid tabs
Content of aspirin, C9H8O4 95.0 to 105.0% of the stated amount.
Identification Boil 0.5 g of the powdered tablets for 2 to 3 minutes with 10 ml
of 5M sodium hydroxide, cool and add an excess of 1M sulphuric acid; a
crystalline precipitate is produced. To a solution of the precipitate in water
add iron(III) chloride solution R1; a deep violet colour is produced.
Salicylic acid Shake a quantity of the powdered tablets containing 0.20 g of
Aspirin with 4 ml of ethanol (96%) and dilute to 100 ml with water at a
temperature not exceeding 10°. Filter immediately, transfer 50 ml of the
filtrate to a Nessler cylinder, add 1 ml of freshly prepared ammonium
iron(III) sulphate solution R1, mix and allow to stand for 1 minute. Any
violet colour produced is not more intense than that obtained by adding 1
ml of freshly prepared ammonium iron(III) sulphate solution R1 to a
mixture of 3 ml of a freshly prepared 0.010% w/v solution of salicylic acid,
2 ml of ethanol (96%) and sufficient water to produce 50 ml contained in a
second Nessler cylinder (0.3%).

Acetyl Salicylic Acid tabs
• Dissolution Comply with the dissolution test for tablets and capsules, Appendix XII D, using as the medium 500 ml of a pH 4.5 buffer prepared by mixing 29.9 g of sodium acetate and 16.6 ml of glacial acetic acid with sufficient water to produce 10 litres and rotating the basket at 50 revolutions per minute. Withdraw a sample of 20 ml of the medium and filter. Immediately measure the absorbance of the filtrate, Appendix II B, diluted with the dissolution medium if necessary, at 265 nm using dissolution medium in the reference cell. Measure the absorbance of a suitable solution of aspirin BPCRS in the dissolution medium and calculate the total content of aspirin, C9H8O4, in the medium using the declared content of C9H8O4 in aspirin BPCRS.
• Assay Weigh and powder 20 tablets. To a quantity of the powder containing 0.5 g of Aspirin add 30 ml of 0.5M sodium hydroxide VS, boil gently for 10 minutes and titrate the excess of alkali with 0.5M hydrochloric acid VS using phenol red solution as indicator. Repeat the operation without the substance being examined. The difference between the titrations represents the amount of sodium hydroxide required. Each ml of 0.5M sodium hydroxide VS is equivalent to 45.04 mg of C9H8O4.

Chloramphenicol, Chlorampheniclol Palmitate and Chloramphenicol
Succinate
2,2-dichloro-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)-2-(4-nitrophenyl)ethyl]acetamide

Chloramphenicol
Preparations
• Chloramphenicol Capsules
• Chloramphenicol Ear Drops
• Chloramphenicol Eye Drops
• Chloramphenicol Eye Ointment
DEFINITION
• Chloramphenicol is 2,2-dichloro-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)-
2-(4-nitrophenyl)ethyl]acetamide, produced by the growth of certain
strains of Streptomyces venezuelae in a suitable medium. It is normally
prepared by synthesis. It contains not less than 98.0 per cent and not more
than the equivalent of 102.0 per cent of C11H12Cl2N2O5, calculated with
reference to the dried substance.

Chloramphenicol
IDENTIFICATION
• First identification: A, B.
• Second identification: A, C, D, E.
• A. Melting point (2.2.14): 149°C to 153°C.
• B. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with chloramphenicol CRS.
• C. Examine the chromatograms obtained in the test for related substances. The principal spot in the chromatogram obtained with 1 µl of the test solution is similar in position and size to the principal spot in the chromatogram obtained with reference solution (a).
• D. Dissolve about 10 mg in 1 ml of alcohol (50 per cent V/V) R, add 3 ml of a 10 g/l solution of calcium chloride R and 50 mg of zinc powder R and heat on a water-bath for 10 min. Filter the hot solution and allow to cool. Add 0.1 ml of benzoyl chloride R and shake for 1 min. Add 0.5 ml of ferric chloride solution R1 and 2 ml of chloroform R and shake. The aqueous layer is coloured light violet-red to purple.
• E. To 50 mg in a porcelain crucible add 0.5 g of anhydrous sodium carbonate R. Heat over an open flame for 10 min. Allow to cool. Take up the residue with 5 ml of dilute nitric acid R and filter. To 1 ml of the filtrate add 1 ml of water R. The solution gives reaction (a) of chlorides (2.3.1).

Chloramphenicol
TESTS
Acidity or alkalinity. To 0.1 g add 20 ml of carbon dioxide-free water R, shake and add 0.1 ml of bromothymol blue solution R1. Not more than 0.1 ml of 0.02M hydrochloric acid or 0.02M sodium hydroxide is required to change the colour of the indicator.
Specific optical rotation (2.2.7). Dissolve 1.50 g in ethanol R and dilute to 25.0 ml with the same solvent. The specific optical rotation is +18.5° to +20.5°.
Chlorides (2.4.4). To 1.00 g add 20 ml of water R and 10 ml of nitric acid R and shake for 5 min. Filter through a filter paper previously washed by filtering 5 ml portions of water R until 5 ml of filtrate no longer becomes opalescent on addition of 0.1 ml of nitric acid R and 0.1 ml of silver nitrate solution R1. 15 ml of the filtrate complies with the limit test for chlorides (100 ppm).
Loss on drying (2.2.32). Not more than 0.5 per cent, determined on 1.000 g by drying in an oven at 100°C to 105°C.
Sulphated ash (2.4.14). Not more than 0.1 per cent, determined on 2.0 g.

Chloramphenicol
ASSAY
• Dissolve 0.100 g in water R and dilute to 500.0 ml with the same solvent.
Dilute 10.0 ml of this solution to 100.0 ml with water R. Measure the
absorbance (2.2.25) at the maximum at 278 nm.
• Calculate the content of C11H12Cl2N2O5 taking the specific absorbance to be
297.

Chloramphenicol Capsules
Content of chloramphenicol, C11H12Cl2N2O5 95.0 to 105.0% of the stated amount.
Identification
Suspend a quantity of the contents of the capsules containing 0.1 g of Chloramphenicol in 60 ml of water and extract with two 20 ml quantities of petroleum spirit (boiling range, 120° to 160°). Wash the combined extracts with two 15-ml quantities of water, add the washings to the aqueous layer, extract with four 50-ml quantities of ether and evaporate the combined ether extracts. The residue complies with the following tests.
• A. Carry out the method for thin-layer chromatography, Appendix III A, using silica gel GF254 as the coating substance and a mixture of 90 volumes of chloroform, 10 volumes of methanol and 1 volume of water as the mobile phase. Apply separately to the plate 1 µl of each of two solutions in ethanol (96%) containing (1) 1% w/v of the residue and (2) 1% w/v of chloramphenicol EPCRS. After removal of the plate, allow it to dry in air and examine under ultraviolet light (254 nm). The principal spot in the chromatogram obtained with solution (1) corresponds to that in the chromatogram obtained with solution (2).

Chloramphenicol Capsules
• B. Dissolve 10 mg in 2 ml of ethanol (50%), add 4.5 ml of 1M sulphuric acid and 50 mg of zinc powder and allow to stand for 10 minutes. Decant the supernatant liquid or filter if necessary. Cool the resulting solution in ice and add 0.5 ml of sodium nitrite solution and, after 2 minutes, 1 g of urea followed by 1 ml of 2-naphthol solution and 2 ml of 10M sodium hydroxide; a red colour is produced. Repeat the test omitting the zinc powder; no red colour is produced.
Dissolution Comply with the dissolution test for tablets and capsules, Appendix XII D, using as the medium 900 ml of 0.1M hydrochloric acid and rotating the basket at 100 revolutions per minute. Withdraw a sample of 10 ml of the medium. Measure the absorbance of the filtered sample, suitably diluted if necessary, at the maximum at 278 nm, Appendix II B. Calculate the total content of chloramphenicol, C11H12Cl2N2O5, in the medium taking 297 as the value of A(1%, 1 cm) at the maximum at 278 nm.
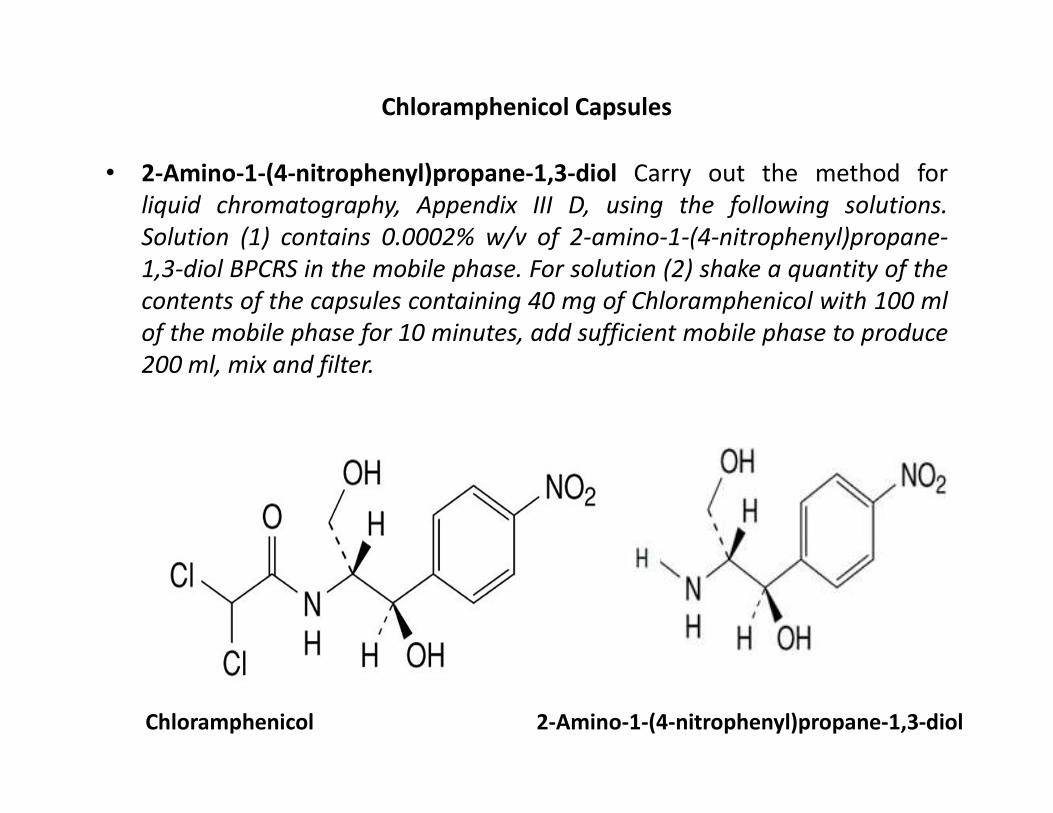
Chloramphenicol Capsules
• 2-Amino-1-(4-nitrophenyl)propane-1,3-diol Carry out the method for
liquid chromatography, Appendix III D, using the following solutions.
Solution (1) contains 0.0002% w/v of 2-amino-1-(4-nitrophenyl)propane-
1,3-diol BPCRS in the mobile phase. For solution (2) shake a quantity of the
contents of the capsules containing 40 mg of Chloramphenicol with 100 ml
of the mobile phase for 10 minutes, add sufficient mobile phase to produce
200 ml, mix and filter.
Chloramphenicol 2-Amino-1-(4-nitrophenyl)propane-1,3-diol

Chloramphenicol Capsules
• The chromatographic procedure may be carried out using (a) a stainless
steel column (10 cm × 4.6 mm), packed with stationary phase C (5 µm)
(Nucleosil C18 is suitable), (b) a mixture of 85 volumes of a 0.21% w/v
solution of sodium pentanesulphonate, 15 volumes of acetonitrile and 1
volume of glacial acetic acid as the mobile phase with a flow rate of 2 ml
per minute and (c) a detection wavelength of 272 nm.
• In the chromatogram obtained with solution (2) the area of any peak
corresponding to 2-amino-1-(4-nitrophenyl)-propane-1,3-diol is not
greater than the area of the peak in the chromatogram obtained with
solution (1).

Chloramphenicol Capsules
• Assay Dissolve a quantity of the mixed contents of 20 capsules containing
0.2 g of Chloramphenicol in 800 ml of water, warming if necessary to
effect solution, and add sufficient water to produce 1000 ml. Dilute 10 ml
to 100 ml with water and measure the absorbance of the resulting solution
at the maximum at 278 nm, Appendix II B. Calculate the content of
C11H12Cl2N2O5 taking 297 as the value of A(1%, 1 cm) at the maximum at
278 nm.
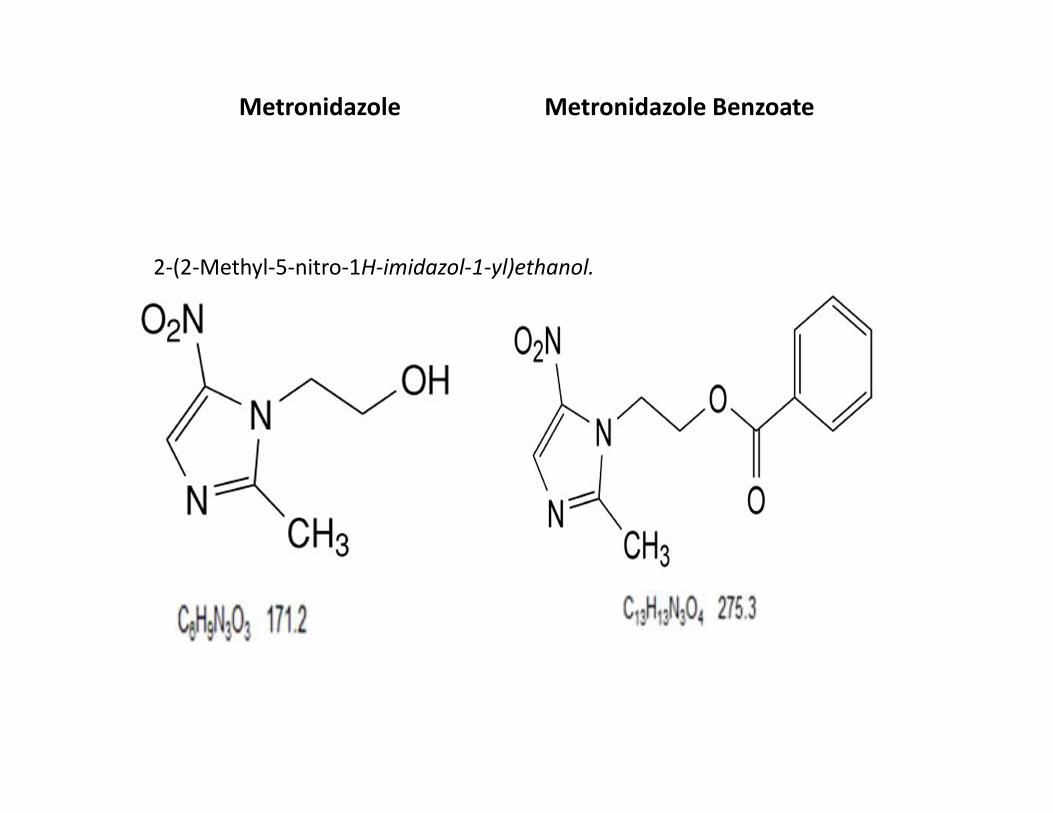
Metronidazole Metronidazole Benzoate
2-(2-Methyl-5-nitro-1H-imidazol-1-yl)ethanol.

Metronidazole
Preparations
• Metronidazole Gel
• Metronidazole Intravenous Infusion
• Metronidazole Suppositories
• Metronidazole Tablets
DEFINITION
• 2-(2-Methyl-5-nitro-1H-imidazol-1-yl)ethanol.
• Content: 99.0 per cent to 101.0 per cent (dried substance).

Metronidazole
IDENTIFICATION
• First identification: C.
• Second identification: A, B, D.
• A. Melting point (2.2.14): 159°C to 163°C.
• B. Dissolve 40.0 mg in 0.1M hydrochloric acid and dilute to 100.0 ml with the same acid. Dilute 5.0 ml of the solution to 100.0 ml with 0.1M hydrochloric acid. Examined between 230 nm and 350 nm (2.2.25), the solution shows an absorption maximum at 277 nm and a minimum at 240 nm. The specific absorbance at the maximum is 365 to 395.
• C. Infrared absorption spectrophotometry (2.2.24).
• Preparation: discs.
• Comparison: metronidazole CRS.
• D. To about 10 mg add about 10 mg of zinc powder R, 1 ml of water R and 0.25 ml of dilute hydrochloric acid R. Heat on a water-bath for 5 min. Cool. The solution gives the reaction of primary aromatic amines (2.3.1).

Metronidazole
TESTS
Heavy metals (2.4.8): maximum 20 ppm.
• 1.0 g complies with limit test C. Prepare the standard using 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying (2.2.32): maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 100-105°C for 3 h.
Sulphated ash (2.4.14): maximum 0.1 per cent, determined on 1.0 g.
ASSAY
• Dissolve 0.150 g in 50 ml of anhydrous acetic acid R. Titrate with 0.1M perchloric acid, determining the end-point potentiometrically (2.2.20).
• 1 ml of 0.1M perchloric acid is equivalent to 17.12 mg of C6H9N3O3.

Metronidazole
IMPURITIES
• A. R1 = R4 = H, R2 = CH3, R3 = NO2: 2-methyl-4-nitroimidazole,
• B. R1 = R2 = R4 = H, R3 = NO2: 4-nitroimidazole,
• C. R1 = CH2-CH2-OH, R2 = R4 = H, R3 = NO2: 2-(4-nitro-1H-imidazol-1-yl)ethanol,
• D. R1 = CH2-CH2-OH, R2 = R3 = H, R4 = NO2: 2-(5-nitro-1H-imidazol-1-yl)ethanol,
• E. R1 = CH2-CH2-OH, R2 = CH3, R3 = NO2, R4 = H: 2-(2-methyl-4-nitro-1H-imidazol-1-yl)ethanol,
• F. R1 = CH2-CH2-O-CH2-CH2-OH, R2 = CH3, R3 = H, R4 = NO2: 2-[2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethoxy]ethanol,
• G. R1 = CH2-CO2H, R2 = CH3, R3 = H, R4 = NO2: 2-(2-methyl-5-nitro-1H-imidazol-1-yl)acetic acid.

Metronidazole Tablets
Definition Metronidazole Tablets contain Metronidazole.
Content of metronidazole, C6H9N3O3 95.0 to 105.0% of the stated amount.
Identification
• A. Shake a quantity of the powdered tablets containing 0.1 g of Metronidazole with 40 ml of chloroform for 15 minutes, filter and evaporate the filtrate to dryness. The infrared absorption spectrum of the residue, Appendix II A, is concordant with the reference spectrum of metronidazole (RS 229).
• B. Shake a quantity of the powdered tablets containing 0.2 g of Metronidazole with 4 ml of 0.5M sulphuric acid and filter. To the filtrate add 10 ml of picric acid solution R1 and allow to stand. The melting point of the precipitate, after washing with water and drying at 105°, is about 150°, Appendix V A.
• C. Heat a quantity of the powdered tablets containing 10 mg of Metronidazole on a water bath with 10 mg of zinc powder, 1 ml of water and 0.25 ml of hydrochloric acid for 5 minutes, cool in ice, add 0.5 ml of sodium nitrite solution and remove the excess of nitrite with sulphamic acid. Add 0.5 ml of 2-naphthol solution and 2 ml of 5M sodium hydroxide. An orange-red colour is produced.

Metronidazole Tablets
• Related substances Carry out the method for liquid chromatography,
Appendix III D, using the following solutions. Solution (1) contains
0.00050% w/v of 2-methyl-5-nitroimidazole BPCRS in the mobile phase.
For solution (2) shake a quantity of the powdered tablets containing 0.2 g
of Metronidazole with 100 ml of the mobile phase for 5 minutes, dilute to
200 ml with the mobile phase, filter and use the filtrate. Solution (3)
contains 0.00050% w/v of 2-methyl-5-nitroimidazole BPCRS in solution (2).
2-methyl-5-nitroimidazole 2-(2-Methyl-5-nitro-1H-imidazol-1-yl)ethanol.

Metronidazole Tablets
• The chromatographic procedure may be carried out using (a) a stainless steel column (20 cm × 4.6 mm) packed with stationary phase C (10 µm) (Spherisorb ODS 1 is suitable), (b) a mixture of 30 volumes of methanol and 70 volumes of 0.01M potassium dihydrogen orthophosphate as the mobile phase with a flow rate of 1 ml per minute and (c) a detection wavelength of 315 nm. Record the chromatograms for 3 times the retention time of the principal peak in the chromatogram obtained with solution (2).
• Adjust the sensitivity so that the height of the peak due to 2-methyl-5-nitroimidazole in the chromatogram obtained with solution (3) is about 50% of full-scale deflection. Measure the height (a) of the peak due to 2-methyl-5-nitroimidazole and the height (b) of the lowest part of the curve separating this peak from the principal peak. The test is not valid unless a is greater than 10b.
• The area of any secondary peak in the chromatogram obtained with solution (2) is not greater than the area of the peak due to 2-methyl-5-nitroimidazole in the chromatogram obtained with solution (1).

Metronidazole Tablets
• Assay Weigh and powder 20 tablets. Transfer a quantity of the powder
containing 0.2 g of Metronidazole to a sintered-glass crucible and extract
with six 10 ml quantities of hot acetone. Cool, add to the combined
extracts 50 ml of acetic anhydride and 0.1 ml of a 1% w/v solution of
brilliant green in anhydrous acetic acid and titrate with 0.1M perchloric
acid VS to a yellowish green end point. Repeat the operation without the
powdered tablets. The difference between the titrations represents the
amount of perchloric acid required. Each ml of 0.1M perchloric acid VS is
equivalent to 17.12 mg of C6H9N3O3.












![Index [assets.cambridge.org] · associated leuconorite, 402 associated quartz mangerite, 402 coarse grain size, 401, 402 composition of plagioclase, 402 crystal size distribution](https://static.fdocuments.net/doc/165x107/606c9147757c7d7d903e2249/index-associated-leuconorite-402-associated-quartz-mangerite-402-coarse-grain.jpg)





