
PHASE TRANSFORMATIONS - Information and Library...
103
PHASE TRANSFORMATIONS Nucleation & Growth TTT and CCT Diagrams APPLICATIONS Transformations in Steel Precipitation Solidification & crystallization Glass transition Recovery, Recrystallization & Grain growth Phase Transformations in Metals and Alloys David Porter & Kenneth Esterling Van Nostrand Reinhold Co. Ltd., New York (1981)
Transcript of PHASE TRANSFORMATIONS - Information and Library...

PHASE TRANSFORMATIONS
Nucleation & Growth
TTT and CCT Diagrams
APPLICATIONS
Transformations in Steel
Precipitation
Solidification & crystallization
Glass transition
Recovery, Recrystallization & Grain growth
Phase Transformations in Metals and AlloysDavid Porter & Kenneth Esterling
Van Nostrand Reinhold Co. Ltd., New York (1981)

When one phase transforms to another phase it is called phase transformation.
Often the word phase transition is used to describe transformations where there is no
change in composition.
In a phase transformation we could be concerned about phases defined based on:
Structure → e.g. cubic to tetragonal phase
Property → e.g. ferromagnetic to paramagnetic phase
Phase transformations could be classified based on (pictorial view in next page):
Kinetic: Mass transport → Diffusional or Diffusionless
Thermodynamic: Order (of the transformation) → 1st order, 2nd order, higher order.
Often subtler aspects are considered under the preview of transformations.
E.g. (i) roughening transition of surfaces, (ii) coherent to semi-coherent transition of
interfaces.
Phase Transformations: an overview

Diffusional
PHASE TRANSFORMATIONS
Diffusionless
1nd order
nucleation & growth
PHASE TRANSFORMATIONS
2nd (& higher) order
Entire volume transforms
Based on
Mass
transport
Based on
order
E.g. MartensiticInvolves long range mass transport

Phases Defects Residual stress
Transformations in Materials
Defect structures can changePhases can transform Stress state can be altered
Phase
Transformation
Defect Structure
Transformation
Stress-State
Transformation
Geometrical Physical
MicrostructurePhases
Microstructural TransformationsPhases Transformations
Structural Property
Phase transformations are associated with change in one or more properties.
Hence for microstructure dependent properties we would like to additionally ‘worry about’
‘subtler’ transformations, which involve defect structure and stress state (apart from
phases).
Therefore the broader subject of interest is Microstructural Transformations.

What is a Phase?
What kind of phases exist?
What constitutes a transformation?
How can we cause a phase transformation to occur?
The stimuli: P, T, Magnetic field, Electric field etc.
What kind of phase transformations are there?
Why does a phase transformation occur?
Energy considerations of the system?
Thermodynamic potentials (G, A…)
Is melting point the same as the freezing point?Further: Does there exist a freezing point?
Some of the questions we would like to have an answer for…
Answers for some these questions may be found in other chapters

Energies involved
Bulk Gibbs free energy ↓
Interfacial energy ↑
Strain energy ↑
Important in solid to solid transformations
Revise concepts of surface and interface energy before starting on these topics
When a volume of material (V) transforms three energies have to be considered :
(i) reduction in G (assume we are working at constant T & P),
(ii) increase in (interface free-energy),
(iii) increase in strain energy.
In a liquid to solid phase transformation the strain energy term can be neglected (as the
liquid can flow and accommodate the volume/shape change involved in the transformation-
assume we are working at constant T & P).
Volume of transformed material
New interface created

Energies involved
Bulk Gibbs free energy ↓
Interfacial energy ↑
Strain energy ↑
The origin of the strain energy can be understood using the schematics as below. Eshelby
construction is used for this purpose.
In general a solid state phase transformation can involve a change in both volume and
shape. I.e. both dilatational and shear strains may be involved. For simplicity we consider
only change in volume of the material, leading to an increase in the strain energy of the
system (in future considerations).
Schematic of the Eshelby construction to understand the origin of the stresses due to phase transformation
of a volume (V): (a) region V before transformation, (b) the region V is cut out of the matrix and allowed to
transform (the transformation could involve both shape and volume changes), (c) the transformed volume
(V‘- shown to be larger in the figure) is inserted into the hole (here only volume change is shown for
simplicity), (c) the system is allowed to equilibrate. The continuity of the system is maintained during the
transformation. The system is strained as a larger volume V’ is inserted into the hole of volume V.
Only volume change
(a)
(b)(c) (d)
Considering only
volume change

Energies involved
Bulk Gibbs free energy ↓
Interfacial energy ↑
Strain energy ↑ Solid-solid transformation
Let us start understanding phase transformations using the example of the solidification of
a pure metal. (This process is a first order transformation*. First order transformations
involve nucleation and growth**).
There is no change in composition involved as we are considering a pure metal. If we
solidify an alloy this will involve long range diffusion.
Strain energy term can be neglected as the liquid melt can flow to accommodate the
volume change (assume we are working at constant T & P).
The process can start only below the melting point of the liquid (as only below the melting
point the GLiquid < GSolid). I.e. we need to Undercool the system. As we shall note, under
suitable conditions (e.g. container-less solidification in zero gravity conditions), melts can
be undercooled to a large extent without solidification taking place.
Click here to know more about order of a phase transformation
Nucleation
of
phase
Trasformation
→
+
Growth till
is
exhausted=
1nd order
nucleation & growth
*
**

↑ t
“For sufficient Undercooling”
Cru
de
sch
emat
ic!
Liq
uid
→ S
oli
dphas
e tr
ansf
orm
atio
n:
Soli
dif
icat
ion
3
21
4
5 6
Liquid
Solid
Growth of Crystal
Two crystal going to join to
form grain boundary
Growth of nucleated crystal
Solidification complete
Video snap shots of
solidification of stearic acid
Grain boundary
Caution: here we are seeing an
increase time experiment and
soon we will be ‘talking of’
increasing undercooling
experiments
See video here

Liquid → Solid phase transformation
Liquid (GL)
TmT →
G
→
T
Gv
Liquid (L) stableSolid (S) stable
T - Undercooling
On cooling just below Tm solid becomes stable, i.e. GLiquid < GSolid.
But even when we are just below Tm solidification does not ‘start’.
E.g. liquid Ni can be undercooled 250 K below Tm.
We will try to understand Why?
The figure below shows G vs T curves for melt and a crystal.
The undercooling is marked as T and the ‘G’ difference between the liquid and the solid
(which will be released on solidification) is marked as Gv (the subscript indicates that the
quantity G is per unit volume). Hence, Gv is a function of undercooling (T)
GL→S → ve
GL→S → +ve
Solid (GS) Assume for now that
we are at a fixed T
below the Tm
Note that Tm is the melting point
of the bulk solid

In Homogenous nucleation the probability of nucleation occurring at point in the parent
phase is same throughout the parent phase.
In heterogeneous nucleation there are some preferred sites in the parent phase where
nucleation can occur
Homogenous
Heterogeneous
Nucleation
NucleationSolidification + Growth=
Heterogenous nucleation sites
Liquid → solid walls of container, inclusions
Solid → solid inclusions, grain boundaries,
dislocations, stacking faults
As pointed out before solidification is a first order phase transformation involving
nucleation (of crystal from melt) and growth (of crystals such that the entire liquid is
exhausted).
Nucleation is a ‘technical term’ and we will try to understand that soon.
In solid solid phase transformation, which involve strain energy, heterogeneous
nucleation (defined below) is highly preferred. Even in liquid solid transformations
heterogeneous nucleation plays an very important role.
of crystals from melt of nucleated crystals till liquid is exhausted

Homogenous nucleation
ΔG (Volume).( ) (Interface).( )VG
).(4 ).(3
4 ΔG 23 rGr v
r2
r3
1
)( TfGv
Free energy change on nucleation
Reduction in bulk free energy increase in interface energy increase in strain energy
r
( )f r
Neglected in L → S transformations
Let us consider LS transformation taking place by homogenous nucleation. Let the
system be undercooled to a fixed temperature (T held constant). Let us consider the formation of a
spherical crystal of radius ‘r’ from the melt. We can neglect the strain energy contribution.
Let the change in ‘G’ during the process be G. This is equal to the decrease in bulk free
energy + the increase in interface free energy. This can be computed for a spherical
nucleus as below.
Note that below a value of ‘1’ the lower power of ‘r’ dominates;
while above ‘1’ the higher power of ‘r’ dominates.
In the above equation these powers are weighed with other
‘factors/parameters’, but the essential logic remains.
Note that GV is negative
Let us start with a ‘text-book’ description of nucleation before taking up an alternate
perspective
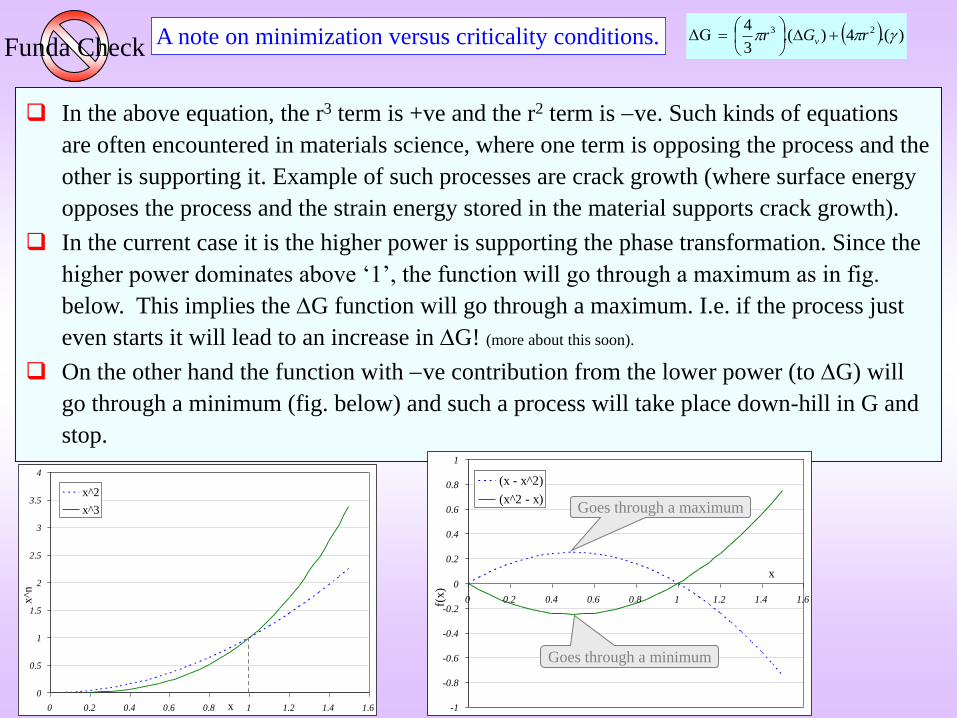
A note on minimization versus criticality conditions.Funda Check
In the above equation, the r3 term is +ve and the r2 term is ve. Such kinds of equations
are often encountered in materials science, where one term is opposing the process and the
other is supporting it. Example of such processes are crack growth (where surface energy
opposes the process and the strain energy stored in the material supports crack growth).
In the current case it is the higher power is supporting the phase transformation. Since the
higher power dominates above ‘1’, the function will go through a maximum as in fig.
below. This implies the G function will go through a maximum. I.e. if the process just
even starts it will lead to an increase in G! (more about this soon).
On the other hand the function with ve contribution from the lower power (to G) will
go through a minimum (fig. below) and such a process will take place down-hill in G and
stop.
).(4 ).(3
4 ΔG 23 rGr v
0
0.5
1
1.5
2
2.5
3
3.5
4
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6x
x^n
x^2
x^3
-1
-0.8
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
x
f(x)
(x - x^2)
(x^2 - x)Goes through a maximum
Goes through a minimum

).(4 ).(3
4 ΔG 23 rGr v
As we have noted previously G vs r plot will go through a maximum (implying that as a
small crystal forms ‘G’ will increase and hence it will tend to dissolve).
The maximum of G vs r plot is obtained by by setting dG/dr = 0. The maximum value of
G corresponds to a value of ‘r’ called the critical radius (denoted by superscript *).
If by some ‘accident’ (technically a ‘statistical random fluctuation’) a crystal (of ‘preferred’
crystal structure) size > r* (called supercritical nuclei) forms then it can grow down-hill in
‘G’. Crystals smaller than r* (called embryos) will tend to shrink to reduce ‘G’. The critical
value of G at r* is called G*.
Reduction in G (below the liquid state) is obtained only after r0 is obtained (which can be
obtained by setting G = 0).
0
dr
Gd0*
1 r
vGr
2*
2
Trivial solution
vGr
2*
2
3*
3
16
vGG
As Gv is ve, r*is +ve
r →
G
→
0G
0r
0G vGr
30
Supercritical nucleiEmbryos
Note that G is a function of T, r &
*r
0
dr
Gd
Note that we are
at a constant T
*G

r →
G
→
Decreasing r*
Dec
reas
ing
G*
Tm
23
2 2
16
3
mTG
T H
Using the Turnbull approximation (linearizing the G-T curve
close to Tm), we can get the value of G interms of the enthalpy
of solidification.
)( TfGv The bulk free energy reduction is a function of undercooling
What is the effect of undercooling (T) on r* and G*?
We have noted that GV is a fucntion of undercooling (T). At larger undercoolings GV
increases and hence r* and G* decrease. This is evident from the equations for r* and G*
as below (derived before).
At Tm GV is zero and r* is infinity!
That the melting point is not the same as the freezing point!!
This energy (G) barrier to nucleation is called the ‘nucleation barrier’.
vGr
2*
2
3*
3
16
vGG

T →
G →
Turnbull’s approximation
Tm
Solid (GS)
Liquid (GL)T
G
mf f
m m
T T TG H H
T T
fΔH heat of fusion
2
* 316
3
m
f
TG
H T

How are atoms assembled to form a nucleus of r* “Statistical Random Fluctuation”Quantum
Jump
To cause nucleation (or even to form an embryo) atoms of the liquid (which are randomly moving
about) have to come together in a order, which resembles the crystalline order, at a given instant of
time.
Typically, this crystalline order is very different from the order (local order), which exists in the liquid.
This ‘coming together’ is a random process, which is statistical in nature i.e. the liquid is exploring
‘locally’ many different possible configurations and randomly (by chance), in some location in the
liquid, this order may resemble the preferred crystalline order.
Since this process is random (& statistical) in nature, the probability that a larger sized crystalline
order is assembled is lower than that to assemble a smaller sized ‘crystal’.
Hence, at smaller undercoolings (where the value of r* is large) the chance of the formation of a
supercritical nucleus is smaller and so is the probability of solidification (as at least one nucleus is
needed which can grow to cause solidification). At larger undercoolings, where r* value is relatively
smaller, the chance of solidification is higher.
↑ T
r*
Tm
Ch
ance
s o
f n
ucl
eati
on
incr
ease
s

Here we try to understand: “What exactly is meant by the nucleation barrier?”.
It is sometime difficult to fathom out as to the surface energy can make freezing of a small
‘embryo’ energetically ‘infeasible’ (as we have already noted that unless the crystallite size is >
r0 the energy of the system is higher). Agreed that for the surface the energy lowering is not as
much as that for the bulk*, but even the surface (with some ‘unsaturated bonds’) is expected to
have a lower energy than the liquid state (where the crystal is energetically favoured). I.e. the
specific concern being: “can state-1 in figure below be above the zero level (now considered for
the liquid state)?” “Is the surface so bad that it even negates the effect of the bulk lowering?”
We will approach this mystery from a different angle by first asking the question: “what is
meant by melting point?” & “what is meant by undercooling?”.
What is meant by the ‘Nucleation Barrier’ an alternate perspective Funda Check
* refer to surface energy and surface tension slides.

The plot below shows melting point of Au nanoparticles, plotted as a function of the particle radius. It is to
be noted that the melting point of nanoparticles decreases below the ‘bulk melting point’ (a 5nm particle
melts more than 100C below Tmbulk). This is due to surface effects (surface is expected to have a lower
melting point than bulk!?*) actually, the current understanding is that the whole nanoparticle melts
simultaneously (not surface layer by layer).
Let us continue to use the example of Au. Suppose we are below Tmbulk (1337K=1064C, i.e. system is undercooled
w.r.t the bulk melting point) at T1 (=1300K T = 37K) and suppose a small crystal of r2 = 5nm forms in the liquid.
Now the melting point of this crystal is ~1200K this crystal will ‘melt-away’. Now we have to assemble a
crystal of size of about 15nm (= r1) for it ‘not to melt’. This needless to say is much less probable (and it is
better to undercool even further so that the value of r* decreases). Thus the mystery of ‘nucleation barrier’
vanishes and we can ‘think of’ melting point freezing point (for a given size of particle)!
Tm is in heating for the bulk material and in cooling if we take into account the size dependence of melting
point everything ‘sort-of’ falls into place .
Melting point, undercooling, freezing point (in the realm of homogenous nucleation)
Other materials like Pb, Cu, Bi,
Si show similar trend lines
* Surface atoms are loosely bound as compared to the bulk atoms.
T1
r1

Is the melting point same as the freezing point?Funda Check
Usually, as we heat a pure metal, it melts at single temperature called the melting point
(Tm). [Proviso, sufficient heat is available].
Somehow, ‘strangely’, the entire ‘lattice’ collapses at a single temperature.
However, in the cooling direction (i.e. on cooling the melt) freezing can occur at any
temperature below Tm.
At Tm itself (i.e. at zero undercooling) there is no tendency for solidification to start.
Though the solid state is energetically (in terms of G) favourable below the melting point,
freezing actually may not start ‘for long times’ if we are just below the melting point.
Heterogeneous nucleation sites may help the solidification process below melting point.
Hence, there is a fixed melting point, but there is no fixed freezing point (even for a pure
metal).

The process of nucleation (of a crystal from a liquid melt, below Tmbulk) we have described so
far is a dynamic one. Various atomic configurations are being explored in the liquid state some
of which resemble the stable crystalline order. Some of these ‘crystallites’ are of a critical size
r*T for a given undercooling (T). These crystallites can grow to transform the melt to a
solid by becoming supercritical. Crystallites smaller than r* (embryos) tend to ‘dissolve’.
As the whole process is dynamic, we need to describe the process in terms of ‘rate’ the
nucleation rate [dN/dt number of nucleation events/volume/time].
Also, true nucleation is the rate at which crystallites become supercritical. To find the
nucleation rate we have to find the number of critical sized crystallites (N*) and multiply it by
the frequency/rate at which they become supercritical.
If the total number of particles (which can act like potential nucleation sites in homogenous
nucleation for now) is Nt , then the number of critical sized particles given by an Arrhenius type
function with a activation barrier of G*.
Atomic perspective of nucleation: Nucleation Rate
kT
G
t eNN
*
*
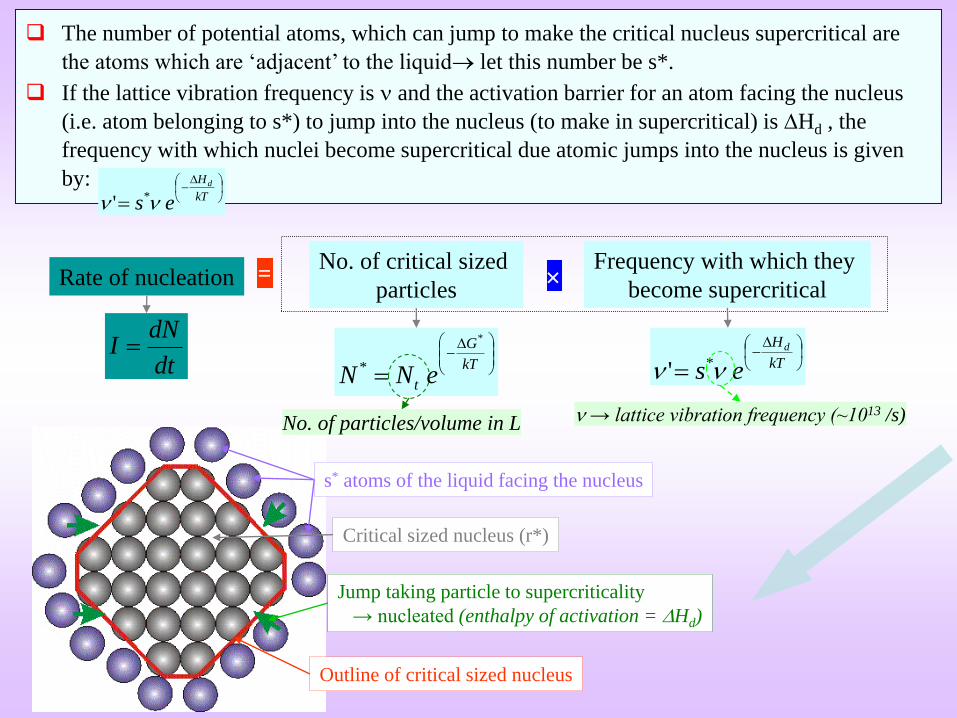
The number of potential atoms, which can jump to make the critical nucleus supercritical are
the atoms which are ‘adjacent’ to the liquid let this number be s*.
If the lattice vibration frequency is and the activation barrier for an atom facing the nucleus
(i.e. atom belonging to s*) to jump into the nucleus (to make in supercritical) is Hd , the
frequency with which nuclei become supercritical due atomic jumps into the nucleus is given
by:
No. of critical sized
particlesRate of nucleation
Frequency with which they
become supercritical=
dt
dNI
kT
G
t eNN
*
*
kT
Hd
es ' *
Critical sized nucleus (r*)
s* atoms of the liquid facing the nucleus
Outline of critical sized nucleus
Jump taking particle to supercriticality
→ nucleated (enthalpy of activation = Hd)
No. of particles/volume in L → lattice vibration frequency (~1013 /s)
kT
Hd
es ' *

T (
K)
→In
crea
sing
T
Tm
0 I →
T = Tm → G* = → I = 0
T = 0 → I = 0
kT
HG
t
d
esNI
*
*
G* ↑ I ↓
T ↑ I ↑
Note: G* is a function of T
The nucleation rate (I = dN/dt) can be written as a product of the two terms as in the equation
below.
How does the plot of this function look with temperature?
At Tm , G* is I = 0 (as expected if there is no undercooling there is no nucleation).
At T = 0K again I = 0
This implies that the function should reach a maximum between T = Tm and T = 0.
A schematic plot of I(T) (or I(T)) is given in the figure below.
An important point to note is that the nucleation rate is not a monotonic function of
undercooling.

Heterogenous nucleation
We have already talked about the ‘nucleation barrier’ and the difficulty in the nucleation
process. This is all the more so for fully solid state phase transformations, where the strain
energy term is also involved (which opposes the transformation).
The nucleation process is often made ‘easier’ by the presence of ‘defects’ in the system.
In the solidification of a liquid this could be the mold walls.
For solid state transformation suitable nucleation sites are: non-equilibrium defects such
as excess vacancies, dislocations, grain boundaries, stacking faults, inclusions and
surfaces.
One way to visualize the ease of heterogeneous nucleation
heterogeneous nucleation at a defect will lead to destruction/modification of the defect
(make it less “‘defective’”). This will lead to some free energy Gd being released → thus
reducing the activation barrier (equation below).
hetro,defectΔG (V) A ( )v s dG G G
Increasing Gd (i.e. decreasing G*)
Homogenous sites
Vacancies
Dislocations
Stacking Faults
Grain boundaries (triple junction…), Interphase boundaries
Free Surface

Heterogenous nucleation
Consider the nucleation of from on a planar surface of inclusion .
The nucleus will have the shape of a lens (as in the figure below).
Surface tension force balance equation can be written as in equation (1) below. The contact angle
can be calculated from this equation (as in equation (3)).
Keeping in view the interface areas created and lost we can write the G equation as below (2).
)( )()(A )(V ΔG lenslens circlecirclev AAG
Alens
Acircle
Acircle
Created
Created
Lost
CosSurface tension force balance
Interfacial Energies
Vlens = h2(3r-h)/3 Alens = 2rh h = (1-Cos)r rcircle = r Sin
Cos
(1)
(2)
is the contact angle
(3)
v
heteroG
r
2
*
3
2
3
* 323
4CosCos
GG
v
hetero
0
dr
Gd
3
homo
* 324
1CosCosGG *
hetero
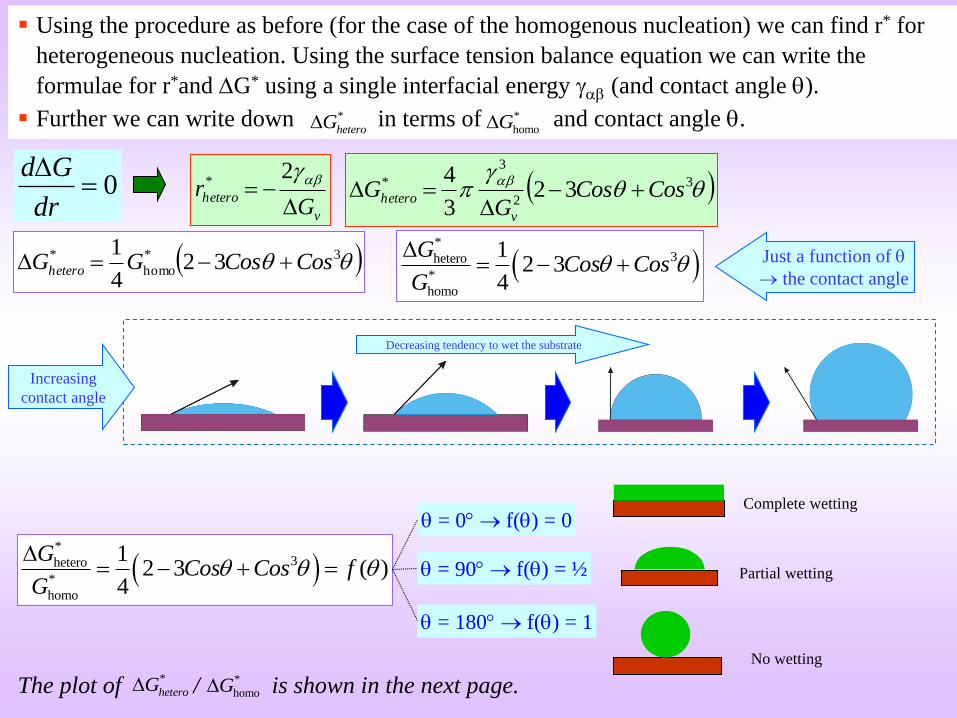
Using the procedure as before (for the case of the homogenous nucleation) we can find r* for
heterogeneous nucleation. Using the surface tension balance equation we can write the
formulae for r*and G* using a single interfacial energy (and contact angle ).
Further we can write down in terms of and contact angle . *
heteroG *
homoG
*
3hetero
homo
12 3
4*
GCos Cos
G
Just a function of
the contact angle
*
3hetero
homo
12 3 ( )
4*
GCos Cos f
G
= 0 f() = 0
= 90 f() = ½
= 180 f() = 1
The plot of / is shown in the next page.*
heteroG *
homoG
Increasing
contact angle
Complete wetting
No wetting
Partial wetting
Decreasing tendency to wet the substrate

0
0.25
0.5
0.75
1
0 30 60 90 120 150 180 (degrees) →
G
*het
ero
/
G*hom
o→
G*hetero (0o) = 0
no barrier to nucleation G*hetero (90o) = G*
homo/2
G*hetero (180o) = G*
homo
no benefit
Complete wetting No wettingPartial wetting
Cos
Plot of G*hetero/G*
homo is shown below. This brings out the benefit of heterogeneous nucleation vs homogenous nucleation.
If the phase nucleus (lens shaped) completely wets the substrate/inclusion (-phase) (i.e. = 0)
then G*hetero = 0 there is no barrier to nucleation.
On the other extreme if -phase does not we the substrate (i.e. = 180)
then G*hetero = G*
homo there is no benefit of the substrate.
In reality the wetting angle is somewhere between 0-180
Hence, we have to chose a heterogeneous nucleating agent with a minimum ‘’ value.

Choice of heterogeneous nucleating agent
How to get a small value of ? (so that ‘easy’ heterogeneous nucleation).
Choosing a nucleating agent with a low value of (low energy interface)
(Actually the value of ( ) will determine the effectiveness of the heterogeneous
nucleating agent → high or low )
Cos
Cos
How to get a low value of ?
We can get a low value of if:
(i) crystal structure of and are similar and
(ii) lattice parameters are as close as possible
Examples of such choices:
In seeding rain-bearing clouds → AgI or NaCl are used for nucleation of ice crystals
Ni (FCC, a = 3.52 Å) is used a heterogeneous nucleating agent in the production of
artificial diamonds (FCC, a = 3.57 Å) from graphite.
Heterogeneous nucleation has many practical applications.
During the solidification of a melt if only a few nuclei form and
these nuclei grow, we will have a coarse grained material (which
will have a lower strength as compared to a fine grained
material- due to Hall-Petch effect).
Hence, nucleating agents are added to the melt (e.g. Ti for Al
alloys, Zr for Mg alloys) for grain refinement.
kT
G
eII
*homo
0
homohomo
kT
G
eII
*hetero
0
heterohetero
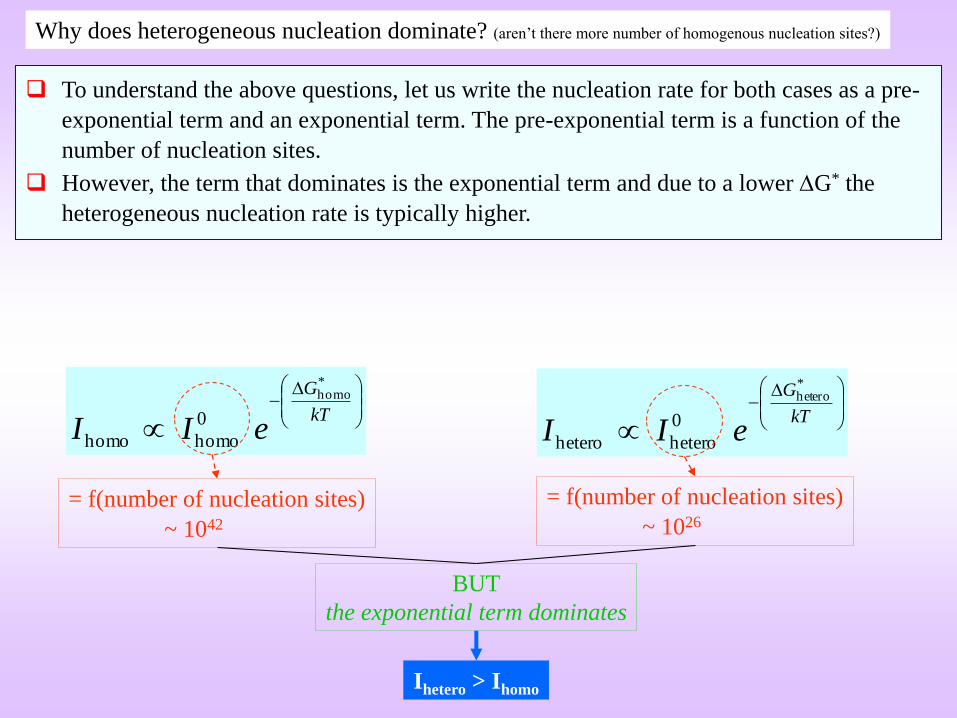
= f(number of nucleation sites)
~ 1042
= f(number of nucleation sites)
~ 1026
BUT
the exponential term dominates
Ihetero > Ihomo
To understand the above questions, let us write the nucleation rate for both cases as a pre-
exponential term and an exponential term. The pre-exponential term is a function of the
number of nucleation sites.
However, the term that dominates is the exponential term and due to a lower G* the
heterogeneous nucleation rate is typically higher.
Why does heterogeneous nucleation dominate? (aren’t there more number of homogenous nucleation sites?)

Heterogeneous nucleation in AlMgZn alloy

Nucleation of phaseTransformation
→ +
Growth of phase
till is exhausted*=
Diffusional transformations involve nucleation and growth. Nucleation involves the
formation of a different phase from a parent phase (e.g. crystal from melt). Growth
involves attachment of atoms belonging to the matrix to the new phase (e.g. atoms
‘belonging’ to the liquid phase attach to the crystal phase).
Nucleation we have noted is ‘uphill’ in ‘G’ process, while growth is ‘downhill’ in G.
Growth can proceed till all the ‘prescribed’ product phase forms (by consuming the parent
phase).
Growth

Hd – vatom Gv
Hd
phase
phase
At transformation temperature the probability of jump of atom from → (across the
interface) is same as the reverse jump
Growth proceeds below the transformation temperature, wherein the activation barrier for the
reverse jump is higher than that for the forward jump.
Growth
As expected transformation rate (Tr) is a function of nucleation rate (I) and growth rate
(U).
In a transformation, if X is the fraction of -phase formed, then dX/dt is the
transformation rate.
The derivation of Tr as a function of I & U is carried using some assumptions (e.g.
Johnson-Mehl and Avarami models).
Transformation rate

rate)Growth rate,on f(Nucleatiratetion Transforma
I, U, Tr →
T (
K)
→In
crea
sing
T
Tm
0
U
Tr
I
( , )r
dXT f I U
dt
Maximum of growth rate usually at higher
temperature than maximum of nucleation rate
We have already seen the curve for the nucleation rate (I) as a function of the
undercooling.
The growth rate (U) curve as a function of undercooling looks similar. The key difference
being that the maximum of U-T* curve is typically above the I-T curve*.
This fact that T(Umax) > T(Imax) give us an important ‘handle’ on the scale of the
transformed phases forming. We will see examples of the utility of this information later.
* The U-T curve is an alternate way of stating the U-T curve[rate sec1]

t →
X
→
0
1.0
0.5
3
t UI π
β
43
e 1X
Fraction of the product () phase forming with time the sigmoidal growth curve
Many processes in nature (etc.), e.g. growth of bacteria in a culture (number of bacteria
with time), marks obtained versus study time(!), etc. tend to follow a universal curve the
sigmoidal growth curve.
In the context of phase transformation, the fraction of the product phase (X) forming with
time follows a sigmoidal curve (function and curve as below).
Linear growth regime ~constant high growth rate
Incubation period slow growth (but with increasing growth rate with time)
Saturation phase decreasing growth rate with time
(region of law of diminishing returns)
Using ‘some’ model

From ‘Rate’ to ‘time’: the origin of Time – Temperature – Transformation (TTT) diagrams
A type of phase diagram
Tr (rate sec1) →
T (
K)
→
Tr
Tm
0
T (
K)
→
Tm
0
Time for transformation
Small driving
force for nucleation
Replot
( , )Rate f T t
Sluggish growth
The transformation rate curve (Tr-T plot) has hidden in it the I-T and U-T curves.
An alternate way of plotting the Transformation rate (Tr) curve is to plot Transformation
time (Tt) [i.e. go from frequency domain to time domain]. Such a plot is called the Time-
Temperature-Transformation diagram (TTT diagram).
High rates correspond to short times and vice-versa. Zero rate implies time (no transformation).
This Tt-T plot looks like the ‘C’ alphabet and is often called the ‘C-curve. The minimum
time part is called the nose of the curve.
Tt
Tt (time sec) →
Nose of the ‘C-curve’

Understanding the TTT diagram
Though we are labeling the transformation temperature Tm , it represents other transformations, in
addition to melting.
Clearly the Tt function is not monotonic in undercooling. At Tm it takes infinite time for
transformation.
Till T3 the time for transformation decreases (with undercooling) [i.e. T3 < T2 < T1] due to
small driving force for nucleation.
After T3 (the minimum) the time for transformation increases [i.e. T3 < T4 < T5] due to sluggish
growth.
This is a phase diagram where the blue region is the Liquid (parent) phase field and purplish region is
the transformed product (crystalline solid).
The diagram is called the TTT diagram because it plots the
time required for transformation if we hold the sample at
fixed temperature (say T1) or fixed undercooling (T1). The
time taken at T1 is t1.
To plot these diagrams we have to isothermally hold at
various undercoolings and note the transformation time.
I.e. instantaneous quench followed by isothermal hold.
Hence, these diagrams are also called Isothermal
Transformation Diagrams.
Similar curves can be drawn for (solid state)
transformation.

Clearly the picture of TTT diagram presented before is incomplete transformations may
start at a particular time, but will take time to be completed (i.e. between the L-phase field
and solid phase field there must be a two phase region L+S!).
This implies that we need two ‘C’ curves one for start of transformation and one for
completion. A practical problem in this regard is related to the issue of how to define start
and finish (is start the first nucleus which forms? Does finish correspond to 100%?) . Since practically it is
difficult to find ‘%’ and ‘100%’, we use practical measures of start and finish, which can
be measured experimentally. Typically this is done using optical metallography and a
reliable ‘resolution of the technique is about 1% for start and 99% for finish.
Another obvious point: as x-axis is time any ‘transformation paths’ have to be drawn such
that it is from left to right (i.e. in increasing time).
t (sec) →T (
K)
→
99% = finish
Increasing % transformation
TTT diagram → phase transformation
1% = start
Fraction
transformed
f volume fractionof
volume fractionof at tf
final volumeof
How do we define the fractions transformed?

f(t,T) determined by
Growth rate
Density and distribution of nucleation sites
Nucleation rate
Overlap of diffusion fields from adjacent transformed volumes
Impingement of transformed volumes
How can we compute Tt(T) (transformation time for each T)
The ‘C’ curve depends on various factors as listed in diagram below.
Some common assumptions used in the derivation are: (i) constant number of nuclei, (ii)
constant nucleation rate, (iii) constant growth rate.

( , )f F number of nucleationsites growthrate growthrate withtime
Constant number of nuclei (these form at the beginning of the transformation)
One assumption to simplify the derivation is to assume that the number of nucleation sites
remain constant and these form at the beginning of the transformation.
This situation may be approximately valid for example if a nucleating agent (inoculant) is
added to a melt (the number of inoculant particles remain constant).
In this case the transformation rate is a function of the number of nucleation sites (fixed)
and the growth rate (U).
Growth rate is expected to decrease with time.
In Avrami model the growth rate is assumed to be constant (till impingement).

Parent phase has a fixed number of nucleation sites Nn per unit volume (and these sites are
exhausted in a very short period of time
Growth rate (U = dr/dt) constant and isotropic (as spherical particles) till particles impinge
on one another
Derivation of f(T,t): Avrami Model
2 3 224 4 4n n nr Utf N N N U t dtdr Udt
At time t the particle that nucleated at t = 0 will have a radius r = Ut
Between time t = t and t = t + dt the radius increases by dr = Udt
The corresponding volume increase dV = 4r2 dr
1
dXf
X
This fraction (f) has to be corrected for impingement. The corrected transformed volume
fraction (X) is lower than f by a factor (1X) as contribution to transformed volume
fraction comes from untransformed regions only:
Without impingement, the transformed volume fraction (f) (the extended transformed
volume fraction) of particles that nucleated between t = t and t = t + dt is:
3 24
1n
dXN U t dt
X

3 2
0 0
41
X t t
n
t
dXN U t dt
X
3 3n4π N U t
3
βX 1 e
Based on the assumptions note that the growth rate is not part of the equation it is only the
number of nuclei.
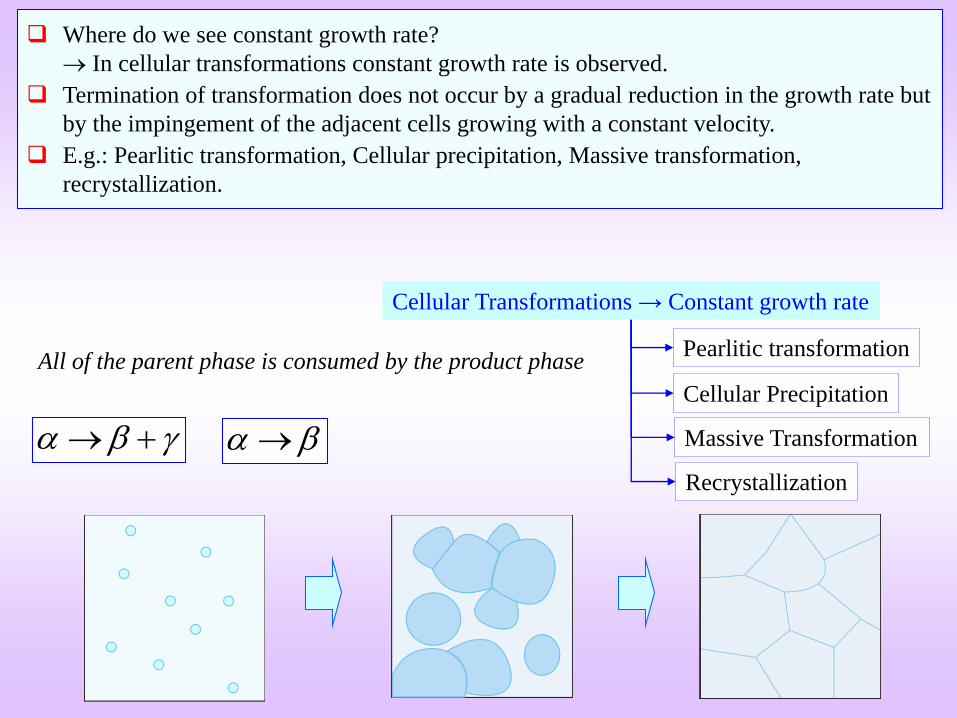
Cellular Transformations → Constant growth rate
Cellular Precipitation
Pearlitic transformation
Massive Transformation
Recrystallization
All of the parent phase is consumed by the product phase
Where do we see constant growth rate?
In cellular transformations constant growth rate is observed.
Termination of transformation does not occur by a gradual reduction in the growth rate but
by the impingement of the adjacent cells growing with a constant velocity.
E.g.: Pearlitic transformation, Cellular precipitation, Massive transformation,
recrystallization.
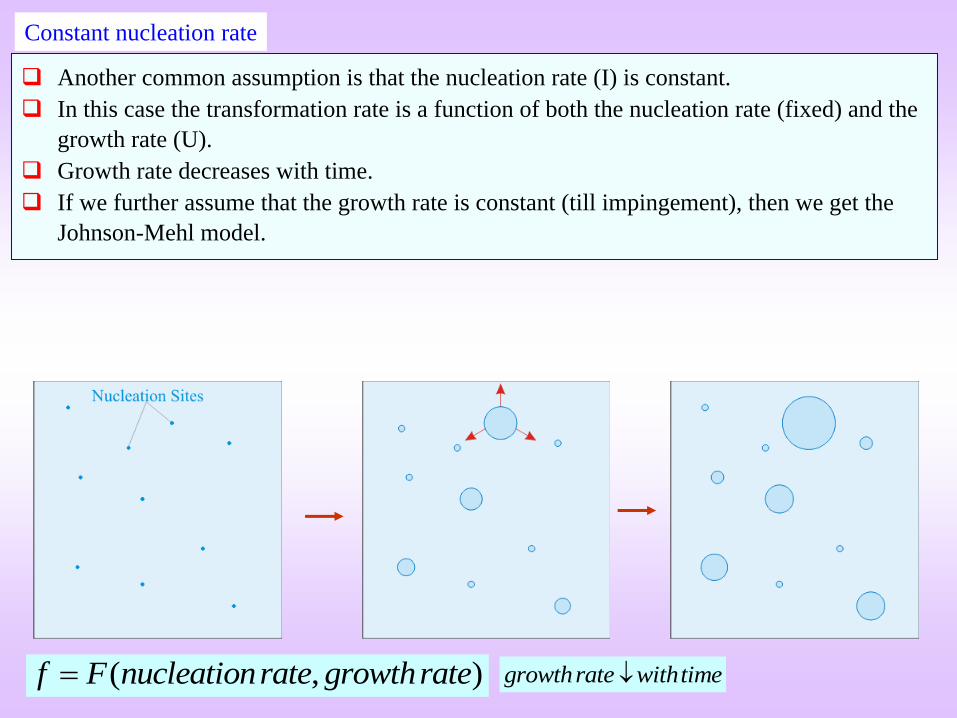
( , )f F nucleationrate growthrate
Constant nucleation rate
growthrate withtime
Another common assumption is that the nucleation rate (I) is constant.
In this case the transformation rate is a function of both the nucleation rate (fixed) and the
growth rate (U).
Growth rate decreases with time.
If we further assume that the growth rate is constant (till impingement), then we get the
Johnson-Mehl model.

Parent phase completely transforms to product phase ( → )
Homogenous Nucleation rate of in untransformed volume is constant (I)
Growth rate (U = dr/dt) constant and isotropic (as spherical particles) till particles impinge
on one another
Derivation of f(T,t): Johnson-Mehl Model
334 4
3 3( )r U t If Id d
At time t the particle that nucleated at t = 0 will have a radius r = Ut
The particle which nucleated at t = will have a radius r = U(t )
Number of nuclei formed between time t = and t = + d → Id
1
dXf
X
This fraction (f) has to be corrected for impingement. The corrected transformed volume
fraction (X) is lower than f by a factor (1X) as contribution to transformed volume
fraction comes from untransformed regions only:
Without impingement, the transformed volume fraction (f) (called the extended
transformed volume fraction) of particles that nucleated between t = and t = + d is:
334 4
1 3 3( )Idr U
dX
Xt Id

0 0
3(
4
1 3)
X t
U t IddX
X
3
t UI π
β
43
e 1X
t →
X
→
0
1.0
0.5
t →
X
→
0
1.0
0.5
3π I Uis a constant during isothermal transformation
3
For a isothermal transformation
Note that X is both a function of I and
U. I & U are assumed constant

APPLICATIONS
of the concepts of nucleation & growth
TTT/CCT diagrams
Phase Transformations in Steel
Precipitation
Solidification, Crystallization and Glass Transition
Recovery recrystallization & grain growth
As hyperlinks

Phase Transformations in Steel
Now we have the necessary wherewithal to understand phase transformations in steel
Phase diagram (Fe-Fe3C) and Concept of TTT diagrams
We shall specifically consider TTT and CCT diagrams for eutectoid, hypo- and hyper-
eutectoid steels.
Further we will consider the use of these diagrams to design heat treatments to get a
specific microstructure (each microstructure will give us a different set of properties).

%C →
T
→
Fe Fe3C
6.74.30.80.16
2.06
Peritectic
L + → Eutectic
L → + Fe3C
Eutectoid
→ + Fe3C
L
L +
+ Fe3C
1493ºC
1147ºC
723ºC
Fe-Cementite diagram
0.025 %C
0.1 %C
+ Fe3C
We have already seen the Fe-Fe3C phase diagram (please have a second look!)

Austenite
Pearlite
Pearlite + Bainite
Bainite
Martensite
100
200
300
400
600
500
800
723C
0.1 1 10 102 103 104 105
Eutectoid temperature
Ms
Mf
t (s) →
T
→
Eutectoid steel (0.8%C)
+ Fe3C
700
TTT diagram for
Eutectoid steel (0.8%C)
For every composition of steel we should draw a different TTT diagram.
To the left of the start C curve is the Austenite () phase field.
To the right of finish C curve is the ( + Fe3C) phase field.
Above Eutectoid
temperature there is no
transformation
Important points to be
noted:
The x-axis is log scale. ‘Nose’ of the ‘C’ curve is in
~sec and just below TE
transformation times may be
~day.
The starting phase has to
.
The ( + Fe3C) phase
field has more labels
included.
There are horizontal
lines labeled Ms and Mf.
‘Nose’ of ‘C’ curve

As pointed out before one of the important utilities of the TTT diagrams comes from the
overlay of microconstituents (microstructures) on the diagram.
Depending on the T, the ( + Fe3C) phase field is labeled with microconstituents like
Pearlite, Bainite.
We had seen that TTT diagrams are drawn by instantaneous quench to a temperature
followed by isothermal hold.
Suppose we quench below (~225C, below the temperature marked Ms), then Austenite
transforms via a diffusionless transformation (involving shear) to a (hard) phase known as
Martensite. Below a temperature marked Mf this transformation to Martensite is complete.
Once is exhausted it cannot transform to ( + Fe3C).
Hence, we have a new phase field for Martensite. The fraction of Martensite formed is not
a function of the time of hold, but the temperature to which we quench (between Ms and
Mf).
Austenite
Pearlite
Pearlite + Bainite
Bainite
Martensite
100
200
300
400
600
500
800
723C
0.1 1 10 102 103 104 105
Eutectoid temperature
Ms
Mf
t (s) →
T
→
Eutectoid steel (0.8%C)
+ Fe3C
700
How are these TTT diagrams drawn?
Samples are quenched into a salt bath maintained at
various temperatures (practical version of the
‘instantaneous quench’)
The samples are then quenched from this bath to room
temperature after various times.
Phase fraction of transformed phase is determined by
optical metallography.

Strictly speaking cooling curves (including finite quenching rates) should not be overlaid
on TTT diagrams (remember that TTT diagrams are drawn for isothermal holds!).
Isothermal hold at: (i) T1 gives us Pearlite, (ii) T2 gives Pearlite+Bainite, (iii) T3 gives
Bainite. Note that Pearlite and Bainite are both +Fe3C (but their morphologies are
different).
To produce Martensite we should quench at a rate such as to avoid the nose of the start ‘C’
curve. Called the critical cooling rate.
Austenite
Austenite
Pearlite
Pearlite + Bainite
Bainite
Martensite
100
200
300
400
600
500
800
723C
0.1 1 10 102 103 104 105
Eutectoid temperature
Not an isothermal
transformation
Ms
Mf
Coarse
Fine
t (s) →
T
→
Eutectoid steel (0.8%C)
700
T1
T2
T3
If we quench between Ms and Mf we
will get a mixture of Martensite and
(called retained Austenite).
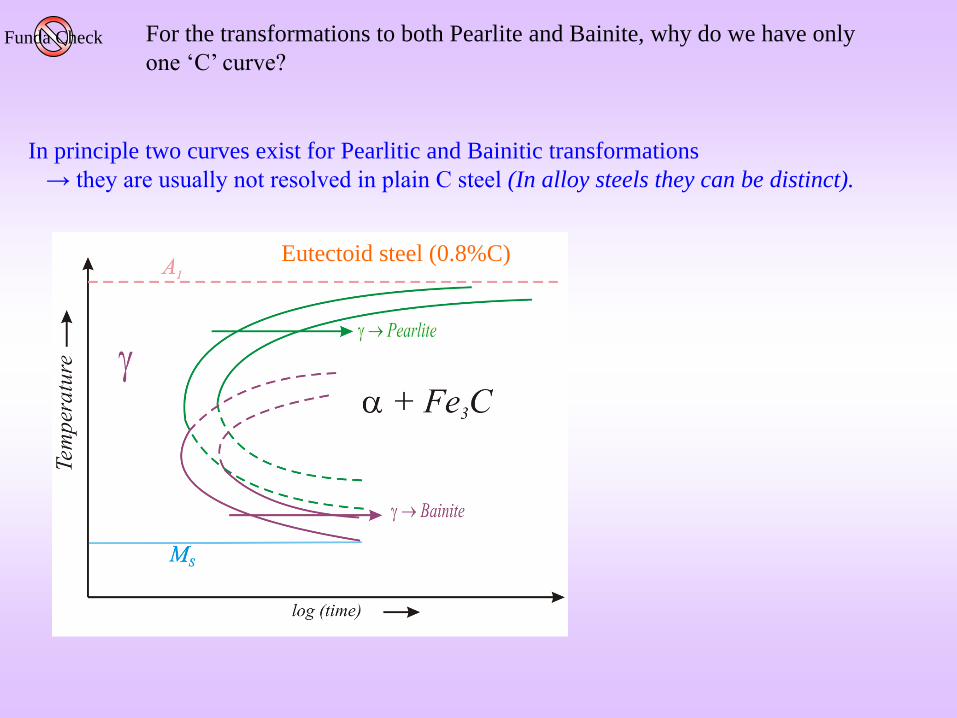
In principle two curves exist for Pearlitic and Bainitic transformations
→ they are usually not resolved in plain C steel (In alloy steels they can be distinct).
Eutectoid steel (0.8%C)
For the transformations to both Pearlite and Bainite, why do we have only
one ‘C’ curve?Funda Check
Atl
as
of
Iso
ther
ma
l T
ran
sfo
rma
tio
n a
nd
Co
oli
ng
Tra
nsf
orm
ati
on
Dia
gra
ms,
AS
M I
nte
rna
tio
na
l, M
eta
ls P
ark
, O
H,
19
77
.
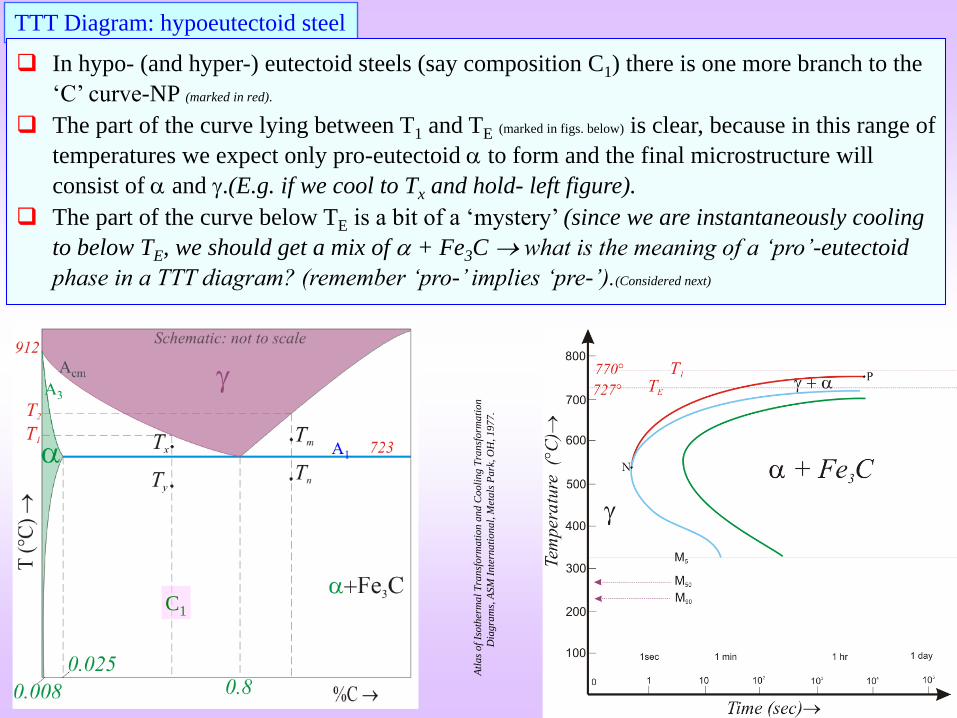
TTT Diagram: hypoeutectoid steel
Hypo-Eutectoid steel
In hypo- (and hyper-) eutectoid steels (say composition C1) there is one more branch to the
‘C’ curve-NP (marked in red).
The part of the curve lying between T1 and TE (marked in figs. below) is clear, because in this range of
temperatures we expect only pro-eutectoid to form and the final microstructure will
consist of and .(E.g. if we cool to Tx and hold- left figure).
The part of the curve below TE is a bit of a ‘mystery’ (since we are instantaneously cooling
to below TE, we should get a mix of + Fe3C what is the meaning of a ‘pro’-eutectoid
phase in a TTT diagram? (remember ‘pro-’ implies ‘pre-’).(Considered next)
C1

Why do we get pro-eutectoid phase below TE?
Suppose we quench instantaneously an hypo-eutectoid composition (C1) to Tx we should expect the
formation of +Fe3C (and not pro-eutectoid first).
The reason we see the formation of pro-eutectoid first is that the undercooling w.r.t to Acm is more
than the undercooling w.r.t to A1. Hence, there is a higher propensity for the formation of pro-eutectoid
.
Undercooling wrt Acm
(formation of pro-eutectoid )undercooling wrt A1 line
(formation of + Fe3C)
C1
Funda Check

Hyper-Eutectoid steel
T2
TE
Similar to the hypo-eutectoid case, hyper-eutectoid compositions (e.g. C2 in fig. below) have a
+Fe3C branch.
For a temperature between T2 and TE (say Tm (not melting point- just a label)) we land up with +Fe3C.
For a temperature below TE (but above the nose of the ‘C’ curve) (say Tn), first we have the
formation of pro-eutectoid Fe3C followed by the formation of eutectoid +Fe3C.
C2

Continuous Cooling Transformation (CCT) Curves
The TTT diagrams are also called Isothermal Transformation Diagrams, because the
transformation times are representative of isothermal hold treatment (following a instantaneous quench).
In practical situations we follow heat treatments (T-t procedures/cycles) in which (typically)
there are steps involving cooling of the sample. The cooling rate may or may not be
constant. The rate of cooling may be slow (as in a furnace which has been switch off) or
rapid (like quenching in water).
Hence, in terms of practical utility TTT curves have a limitation and we need to draw
separate diagrams called Continuous Cooling Transformation diagrams (CCT), wherein
transformation times (also: products & microstructure) are noted using constant rate cooling
treatments. A diagram drawn for a given cooling rate (dT/dt) is typically used for a range of
cooling rates (thus avoiding the need for a separate diagram for every cooling rate).
However, often TTT diagrams are also used for constant cooling rate experiments keeping
in view the assumptions & approximations involved.
The CCT diagram for eutectoid steel is considered next. Blue curve is the CCT curve and
TTT curve is overlaid for comparison.
Important difference between the CCT & TTT transformations is that in the CCT case
Bainite cannot form.

Eutectoid steel (0.8%C)
Martensite
100
200
300
400
600
500
800
723
100
200
300
400
600
500
800
723
0.1 1 10 102 103 104105
Eutectoid temperature
Ms
Mf
t (s) →
T
→
Original TTT lines
Cooling curves
Constant rate
Pearlite
1T 2T
Continuous Cooling Transformation (CCT) Curves
Important points to be
noted:
As before the x-axis is
log scale.
Bainite cannot form by
continuous cooling.
Constant rate cooling
curves look like curves
due to log scale in x-
axis. The higher cooling
rate curve has a higher
(negative) slope.
As time is one of the
axes, no treatment curve
can be drawn where time
decreases or remains
constant.
dTT
dtConstant Cooling rate
1T 2T>
Start
Finish
CR1 CR2
CR1 CR2
A
B
C
D
E
F
G
CCT curvesCCT curves

The CCT curves are to the right of the corresponding TTT curves. Why?Funda Check
As the cooled sample has spent more time at higher temperature, before it intersects the
TTT curve (virtually superimposed) and the transformation time is longer at higher T
(above the nose) CCT curves should be to the right of TTT curves.
Eutectoid steel (0.8%C)
Martensite
100
200
300
400
600
500
800
723
100
200
300
400
600
500
800
723
0.1 1 10 102 103 104105
Eutectoid temperature
Ms
Mf
t (s) →T
→
Original TTT lines
Cooling curves
Constant rate
Pearlite
1T 2T
Further points to be noted:
Using CR1: the phase begins to transform to pearlite, but the transformation is not completed. The
remaining transforms to Martensite on crossing the Mf line (point C).
Using CR2: the phase completely transforms to pearlite (after point E). Hence, there is no significance
of the crossing of the CR2 line of the Ms (point F) and Mf lines (point G).

Common heat treatments involving cooling
Common cooling heat treatment labels (with increasing cooling rate) are:
Full anneal < Normalizing < Oil quench < Water quench
The microstructures produced for these treatments are:
Full Anneal (furnace cooling) Coarse Pearlite
Normalizing (Air cooling) Fine Pearlite
Oil Quench Matensite (M) + Pearlite (P)
Water Quench Matensite
To produce full martensite we have to avoid the ‘nose’ of the TTT diagram (i.e. the
quenching rate should be fast enough).
Within water or oil quench further parameters determine the actual quench rate (e.g. was the
sample shaken?).

Different cooling treatments
M = Martensite
P = Pearlite
M = Martensite
P = Pearlite
Eutectoid steel (0.8%C)
100
200
300
400
600
500
800
100
200
300
400
600
500
800
723
0.1 1 10 102 103 104 105
t (s) →
T
→
Water q
uen
ch
Oil quench
Norm
alizing
Full anneal
Coarse P
P M M + Fine P
It is important to note that for a single composition, different cooling treatments give
different microstructures these give rise to a varied set of properties.
After even water quench to produce Martensite, further heat treatment (tempering) can be
given to optimize properties like strength and ductility.
Note: this is ‘Microstructure
Engineering’ (changing properties
without changing the composition)

What are the typical cooling rates of various processes?
Process Cooling rate (K/s) Comments
Furnace cooling (Annealing) 105 – 103 Typically for solid samples
Air Cooling 1 – 10 “
Oil Quenching* ~100 “
Water Quenching* ~500 “
Splat Quenching 105 For molten material
Melt-Spinning 106 – 108 “
Evaporation, sputtering 109 (expected) Gaseous state involved
* Depends on conditions discussed later

Pearlite
Nucleation and growth
Heterogeneous nucleation at grain boundaries
Interlamellar spacing is a function of the temperature of transformation
Lower temperature → finer spacing → higher hardness
→ + Fe3C
Lamellae of Pearlite in ~0.8% carbon steel

(100) || (111)C
Branching mechanismOrientation Relation:
Kurdyumov-Sachs (010) || (110)C
(001) || (112)C
1 Let us consider the heterogeneous nucleation of one of the phases of the pearlitic
microconstituent (say Fe3C), at a grain boundary of Austenite (). Further let this
precipitate be bound by a coherent interface on one side and a incoherent interface on the
other side. The incoherent interface will be glissile (mobile) and will grow into the
corresponding grain (2).
The orientation relation (OR) between and Fe3C is refered to as the Kurdyumov-
Sachs OR (as in fig. below).
2,3 The region surrounding this Fe3C precipitate will be depleted in Carbon and the
conditions will be right for the nucleation of adjacent to it.
4 The process is repeated to give rise to a pearlitic colony. Branching of an advancing
plate may also be observed.
Mechanism of Pearlitic transformation: arising of the lamellar microstructure
321 4

Bainite
Bainite formed at high temperature (~ 350C) has a feathery appearance and is called
‘Feathery Bainite’.
Bainite formed at lower temperature (~ 275C) has a needle-like appearance and is called
‘acicular Bainite’.
The process of formation of bainite involves nucleation and growth
Acicular, accompanied by surface distortions
** Lower temperature → carbide could be ε carbide (hexagonal structure, 8.4% C)
Bainite plates have irrational habit planes
Ferrite in Bainite plates possess different orientation relationship relative to the parent
Austenite than does the Ferrite in Pearlite
→ + Fe3C**
Micrograph courtesy: Prof. Sandeep Sangal

0.8% C steel, the sample was quenched
in a salt bath having 400°C temperature
and then it was held for 2 hours.
More images of Bainite
Micrograph courtesy: Prof. Sandeep Sangal, Swati Sharma
AFM image
Micrograph courtesy: Prof. Sandeep Sangal, Swati Sharma

Shape of the Martensite formed → Lenticular (or thin parallel plates)
Associated with shape change (shear)
But: Invariant plane strain (observed experimentally) → Interface plane between Martensite and
Parent remains undistorted and unrotated
This condition requires:
1) Bain distortion → Expansion or contraction of the lattice along certain crystallographic
directions leading to homogenous pure dilation
2) Secondary Shear Distortion → Slip or twinning
3) Rigid Body rotation
Characteristic of Martensitic transformations
Surface deformations caused by the Martensitic plate

MartensiteC
BCT
C
FCCQuench
% 8.0
)( '
% 8.0
)(
Martensitic transformation can be understood by first considering an alternate unit cell for the
Austenite phase as shown in the figure below.
If there is no carbon in the Austenite (as in the schematic below), then the Martensitic
transformation can be understood as a ~20% contraction along the c-axis and a ~12% expansion of
the a-axis → accompanied by no volume change and the resultant structure has a BCC lattice (the
usual BCC-Fe) → c/a ratio of 1.0.
Change in Crystal Structure
~20% contraction of c-axis
~12% expansion of a-axis
FCC → BCC
In Pure Fe after
the Matensitic transformation
c = a
FCC Austenite alternate choice of Cell

Martensite
In the presence of Carbon in the octahedral voids of CCP (FCC) -Fe (as in the schematic below) →
the contraction along the c-axis is impeded by the carbon atoms. (Note that only a fraction of the
octahedral voids are filled with carbon as the percentage of C in Fe is small).
However the a1 and a2 axis can expand freely. This leads to a product with c/a ratio (c’/a’) >1
→ 1-1.1.
In this case there is an overall increase in volume of ~4.3% (depends on the carbon content) → the
Bain distortion*.
C along the c-axis
obstructs the contraction
Tetragonal
MartensiteAustenite to Martensite → ~4.3 % volume increase
* Homogenous dilation of the lattice (expansion/contraction along crystallographic axis) leading to the formation of a new lattice is called
Bain distortion. This involves minimum atomic movements.

Martensite in 0.6%C steel

But shear will distort the lattice!
Slip Twinning
Average shape
remains undistorted

The martensitic transformation occurs without composition change.
The transformation occurs by shear without need for diffusion.
The atomic movements required are only a fraction of the interatomic spacing.
The shear changes the shape of the transforming region
→ results in considerable amount of shear energy
→ plate-like shape of Martensite.
The amount of martensite formed is a function of the temperature to which the sample is
quenched and not of time.
Hardness of martensite is a function of the carbon content
→ but high hardness steel is very brittle as martensite is brittle.
Steel is reheated to increase its ductility
→ this process is called TEMPERING.
Summary of characteristics of Martensitic transformation

% Carbon →
Har
dn
ess
(R
c) →
20
40
60
0.2 0.4 0.6
Harness of Martensite as a
function of Carbon content
Properties of 0.8% C steel
Constituent Hardness (Rc) Tensile strength (MN/m2)
Coarse pearlite 16 710
Fine pearlite 30 990
Bainite 45 1470
Martensite 65 -
Martensite tempered at 250C 55 1990

ROLE OF ALLOYING ELEMENTS
• + Simplicity of heat treatment and lower cost
• Low hardenability
• Loss of hardness on tempering
• Low corrosion and oxidation resistance
• Low strength at high temperatures
Plain Carbon Steel
Element Added
Segregation / phase separationSolid solution
Compound (new crystal structure)
• ↑ hardenability
• Provide a fine distribution of alloy carbides during tempering
• ↑ resistance to softening on tempering
• ↑ corrosion and oxidation resistance
• ↑ strength at high temperatures
• Strengthen steels that cannot be quenched
• Make easier to obtain the properties throughout a larger section
• ↑ Elastic limit (no increase in toughness)
Alloying elements
• Alter temperature at which the transformation occurs
• Alter solubility of C in or Iron
• Alter the rate of various reactions
Interstitial
Substitutional

P ►Dissolves in ferrite, larger quantities form iron phosphide → brittle (cold-shortness)
S►Forms iron sulphide, locates at grain boundaries of ferrite and pearlite poor ductility
at forging temperatures (hot-shortness)
Si ► (0.2-0.4%) increases elastic modulus and UTS
Cu►0.8 % soluble in ferrite, can be used for precipitation hardening
Pb►Insoluble in steel
Cr►Corrosion resistance, Ferrite stabilizer, ↑ hardness/strength, > 11% forms passive
films, carbide former
Ni► Austenite stabilizer, ↑ strength ductility and toughness,
Mo► Dissolves in & , forms carbide, ↑ high temperature strength,
↓ temper embrittlement, ↑ strength, hardenability
Sample elements and their role

Alloying Element (%) →
Bri
nel
l H
ard
nes
s→
v
0 2 4 6 8 1060
100
140
180
Cr
Cr + 0.1%C
Mn
Mn +0.1% C
Alloying elements increase hardenability but the major contribution to hardness comes from
Carbon

Mn, Ni are Austenite stabilizers
Cr is Ferrite stabilizer
Shrinking phase field with ↑ Cr
C (%) →
Tem
per
ature
→
0 0.4 0.8 1.61.2
5% Cr
12% Cr15% Cr
0% Cr
C (%) →
Tem
per
atu
re→
0 0.4 0.8 1.61.2
0.35% Mn
6.5% Mn
Outline of the phase field

Austenite Pearlite
Bainite
Martensite100
200
300
400
600
500
800
Ms
Mf
t →
T
→
TTT diagram for Ni-Cr-Mo low alloy steel
~1 min

TTT diagram of low alloy steel (0.42% C, 0.78% Mn,
1.79% Ni, 0.80% Cr, 0.33% Mo)U.S.S. Carilloy Steels, United States Steel Corporation, Pittsburgh, 1948)

0
100
200
300
400
500
600
700
800
900
1000
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35
Engineering Strain (e)
En
gin
eeri
ng
Str
ess
(s)
[MP
a]
0.4% C - Slow cooled
0.8% C - Slow cooled
0.8% C - quenched
Effect of carbon content and heat treatment on properties of steel
150
200
250
300
350
400
450
0.5 0.6 0.7 0.8 0.9 1 1.1C %
Vik
ers
Ha
rdn
ess
Slowly cooled- 0.6%C
Quenched- 0.8% C
Slowly cooled- 0.8% C
Slowly cooled- 1.0% C
Hardness
Tensile Test

Precipitation

The presence of dislocations weakens the crystal → leading to easy plastic deformation.
Putting ‘hindrance’ to dislocation motion increases the strength of the crystal.
Fine precipitates dispersed in the matrix provide such an impediment.
Strength of Al → 100 MPa
Strength of Duralumin with proper heat treatment (Al + 4% Cu + other alloying elements)
→ 500 MPa.
Precipitation Hardening
If a high temperature solid solution is slowly cooled, then coarse (large sized) equilibrium
precipitates are produced. These precipitates have a large distance between them. These
precipitates have incoherent boundaries with the matrix (incoherent precipitates).
Such (coarse) precipitates, which have a large inter-precipitate distance, are ‘not the best’ in
terms of the increase in the hardness.
Hence, we device a 3 step process to obtain a fine distribution of precipitates, which have a
low inter-precipitate distance, to obtain a good increase in hardness.
Philosophy behind the process steps in Precipitation Hardening
Coarse incoherent
precipitates, with large inter-
precipitate distance
Click here to know more about: “how fine
precipitates can give rise to hardening”Multi-step process used to
obtain a fine distribution of
precipitates (with small
inter-precipitate distance)

Al-Cu phase diagram: the sloping solvus line and the design of heat treatments
The Al-Cu system is a model system to understand precipitation hardening (typical
composition chosen is Al-4 wt.% Cu).
Primary requirement (for precipitation hardening) is the presence of a sloping solvus line
(i.e. high solubility at high temperatures and decreasing solubility with decreasing
temperature). In the Al rich end, compositions marked with a shaded box can only be used
for precipitation hardening.
Sloping Solvus line:
high T → high solubility
low T → low solubility of Cu in Al
Al
Cu

4 % Cu
+
→ +
Slow equilibrium cooling gives rise to coarse
precipitates which is not good in impeding
dislocation motion.*
RT
Cu
TetragonalCuAl
RT
Cu
FCC
C
Cu
FCCcoolslow
o
% 52
)(
% 5.0
)(
550
% 4
)( 2
*Also refer section on Double Ended Frank-Read Source in the chapter on plasticity: max = Gb/L

C
A
B
Heat (to 550oC) → solid solution
Quench (to RT) →
Age (reheat to 200oC) → fine precipitates
4 % Cu
+
CA
B
To obtain a fine distribution of precipitates the cycle A → B → C is used
Note: Treatments A, B, C are for the same composition
Supersaturated solution
Increased vacancy concentration
Heat treatment steps to obtain a fine distribution of precipitates
Assume that we start with a material having coarse
equilibrium precipitates (which has been obtained by prior slow cooling of the
sample).
A: We heat the sample to the single phase region () in
the phase diagram (550C).
B: We quench (fast cooling) the sample in water to
obtain a metastable supersaturated solid solution (the amount
of Cu in the sample is more than that allowed at room temperature according to the
phase diagram).
C: We reheat the sample to relatively low temperature
(~180C/200C) get a fine distribution of precipitates. We
have noted before that at ‘low’ temperatures nucleation is dominant over growth.

Log(t) →
Har
dn
ess
→ 180oC
100oC
20oC
Higher temperature less time of aging to obtain peak hardness
Lower temperature increased peak hardness
optimization between time and hardness required
Schematic curves →
Real experimental curves
are in later slides
Note: Schematic curves shown- real curves considered later

Log(t) →
Har
dn
ess
→
180oC
T
OveragedUnderaged
Peak-aged
Region of solid solution
strengthening→ Hardness is higher than that of Al
(no precipitation hardening)
Region of precipitation
hardening
(but little/some solid solution
strengthening)
Dispersion of
fine precipitates
(closely spaced)
Coarsening
of precipitates
with increased
inter-precipitate spacing
Not zero of
hardness scale

Log(t) →
Har
dn
ess
→
180oC Peak-aged
Particle radius (r) →
CR
SS
Incr
ease
→
2
1
r r
1
Particle
shearingParticle
By-pass)(tfr
Section of GP zone parallel to (200) plane

Log(t) →
Har
dn
ess
→
T
Increasing size of precipitates with increasing interparticle (inter-precipitate) spacing
A complex set of events are happening in parallel/sequentially during the aging process
→ These are shown schematically in the figure below
Interface goes from coherent to semi-coherent to incoherent
Precipitate goes from GP zone → ’’ → ’ →

Cu rich zones fully coherent with the matrix → low interfacial energy
(Equilibrium phase has a complex tetragonal crystal structure which has incoherent
interfaces)
Zones minimize their strain energy by choosing disc-shape to the elastically soft <100>
directions in the FCC matrix
The driving force (Gv Gs) is less but the barrier to nucleation is much less (G*)
2 atomic layers thick, 10nm in diameter with a spacing of ~10nm
The zones seem to be homogenously nucleated (excess vacancies seem to play an
important role in their nucleation)
GP Zones

Atomic image of Cu layers in Al matrix
Bright field TEM micrograph of an Al-4% Cu alloy
(solutionized and aged) GP zones.
5nm
5nm
Selected area diffraction (SAD) pattern, showing
streaks arising from the zones.

Due to large surface to volume ratio the fine precipitates have a tendency to coarsen
→ small precipitates dissolve and large precipitates grow
Coarsening
↓ in number of precipitate
↑ in interparticle (inter-precipitate) spacing
reduced hindrance to dislocation motion (max = Gb/L)

Phase Transformations in Metals and Alloys, D.A. Porter and K.E. Easterling, Chapman & Hall, London, 1992.
''(001) || (001)
''[100] || [100]
'(001) || (001)
'[100] || [100]
10 ,100nmthick nmdiameter
Distorted FCC
Tetragonal
UC composition Al4Cu2 = Al2Cu
Becomes incoherent
as ppt. grows
BCT, I4/mcm (140),
a = 6.06Å, c = 4.87Å, tI12
''
UC composition Al6Cu2 = Al3Cu
UC composition Al8Cu4 = Al2Cu
'

Phase Transformations in Metals and Alloys, D.A. Porter and K.E. Easterling,Chapman & Hall, London, 1992.
Schematic diagram showing the lowering of the Gibbs free energy of the system on sequential transformation:
GP zones → ’’ → ’ →
Successive lowering if free
energy of the system

The activation barrier for
precipitation of equilibrium ()
phase is large
But, the free energy benefit in each step is small compared to the
overall single step process
Single step
(‘equilibrium’) process
Schematic plot

Precipitation processes in solids, K.C. Russell, H.I. Aaronson (Eds.), The Metallurgical Society of AMIE, 1978, p.87
In this diagram additionally information has
been superposed onto the phase diagram (which
strictly do not belong there- hence this diagram
should be interpreted with care)
The diagram shows that on aging at various
temperatures in the + region of the phase
diagram various precipitates are obtained first
At higher temperatures the stable phase is
produced directly
At slightly lower temperatures ’ is produced
first
At even lower temperatures ’’ is produced first
The normal artificial aging is usually done in
this temperature range to give rise to GP zones
first


Base Alloy Precipitation Sequence
Al Al-Ag GPZ (Spheres) ' (plates) (Ag2Al)
Al-Cu GPZ (Discs) '' (Discs) ' (Plates) (CuAl2)
Al-Cu-Mg GPZ (Rods) S' (Laths) S (Laths, CuMgAl2)
Al-Zn-Mg GPZ (Spheres) ' (Plates) (Plates/Rods, Zn2Mg)
Cu Cu-Be GPZ (Discs) ' (CuBe)
Cu-Co GPZ (Spheres) (Plates, Co)
Fe Fe-C -carbide (Discs) Fe3C (Plates)
Fe-N '' (Discs) Fe4N (Plates)
Ni Ni-Cr-Ti-Al ' (Cubes/Spheres)
Precipitation Sequence in some precipitation hardening systems
(Morphology and compound stoichiometry are given in brackets)
[1] J.M. Silcock, T.J. Heal and H.K. Hardy, J. Inst. Metal. 82 (1953-54) 239.
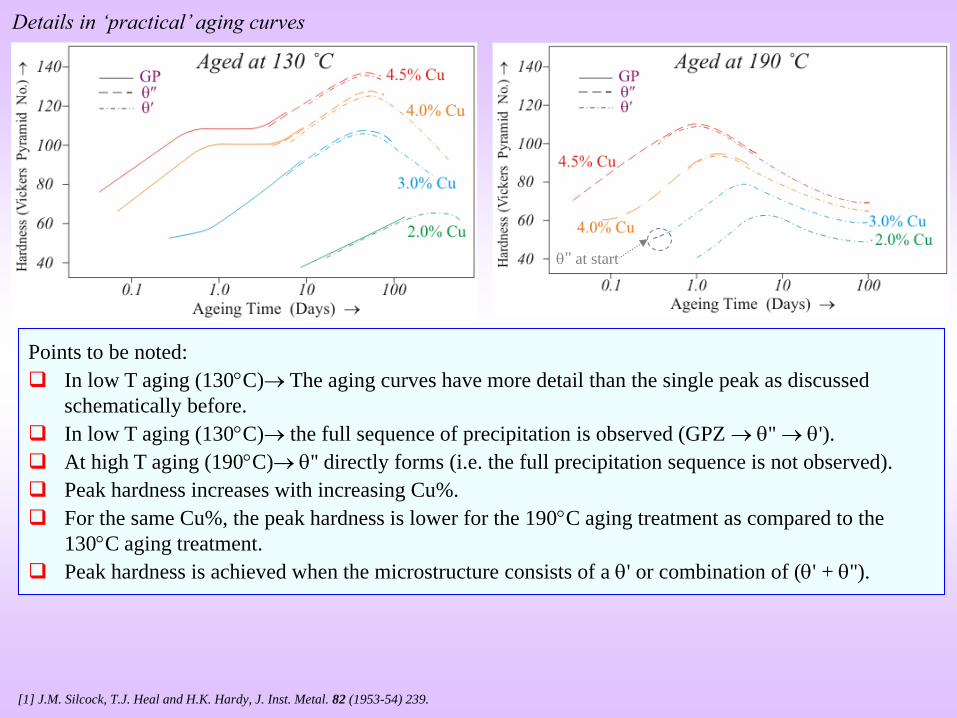
Details in ‘practical’ aging curves
Points to be noted:
In low T aging (130C) The aging curves have more detail than the single peak as discussed
schematically before.
In low T aging (130C) the full sequence of precipitation is observed (GPZ '' ').
At high T aging (190C) '' directly forms (i.e. the full precipitation sequence is not observed).
Peak hardness increases with increasing Cu%.
For the same Cu%, the peak hardness is lower for the 190C aging treatment as compared to the
130C aging treatment.
Peak hardness is achieved when the microstructure consists of a ' or combination of (' + '').
’’ at start

There will be a range of particle sizes due to time of nucleation and rate of
growth
As the curvature increases the solute concentration (XB) in the matrix adjacent to
the particle increases
Concentration gradients are setup in the matrix → solute diffuses from near the
small particles towards the large particles
small particles shrink and large particles grow
with increasing time * Total number of particles decrease
* Mean radius (ravg) increases with time
Particle/precipitate Coarsening
Gibbs-Thomson effect

Gibbs-Thomson effect

Rate controlling
factor
Interface diffusion rate
Volume diffusion rate
3 3
0avgr r kt
ek D X
r0 → ravg at t = 0
D → Diffusivity
Xe → XB (r = )
D is a exponential function of temperature
coarsening increases rapidly with T
2
avg
avg
dr k
dt r
small ppts coarsen more
rapidly
0r
avgr
t
3Linear versus relation maybreak down due todiffusionshort-circuits
or if theprocessisinterface controlled
avgr t

Rateof coarsening depends on (diffusion controlled)eD X
Precipitation hardening systems employed for high-temperature applications must
avoid coarsening by having low: , Xe or D
Nimonic alloys (Ni-Cr + Al + Ti)
Strength obtained by fine dispersion of ’ [ordered FCC Ni3(TiAl)] precipitate in FCC Ni
rich matrix
Matrix (Ni SS)/ ’ matrix is fully coherent [low interfacial energy = 30 mJ/m2]
Misfit = f(composition) → varies between 0% and 0.2%
Creep rupture life increases when the misfit is 0% rather than 0.2%
Low
Nimonic 90: Ni 54%, Cr 18-21%, Co 15-21%, Ti 2-3%, Al 1-2%

ThO2 dispersion in W (or Ni) (Fine oxide dispersion in a metal matrix)
Oxides are insoluble in metals
Stability of these microstructures at high temperatures due to low value of Xe
The term DXe has a low value
Low Xe
ThO2 dispersion in W (or Ni) (Fine oxide dispersion in a metal matrix)
Cementite dispersions in tempered steel coarsen due to high D of interstitial C
If a substitutional alloying element is added which segregates to the carbide → rate of
coarsening ↓ due to low D for the substitutional element
Low D


















