
PCR large byrapidwalkingthrough cDNA - pnas.org filePCR(library...
5
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 8563-8567, October 1991 Biochemistry A PCR procedure to determine the sequence of large polypeptides by rapid walking through a cDNA library (dynein/leapfrog PCR/cilia/flagelia/sea urchin) I. R. GIBBONS*, DAVID J. ASAIt, NATHAN S. CHING*, GREGORY J. DOLECKI*, GABOR MOCZ*, CHERYL A. PHILLIPSON*, HENING REN*, WEN-JING Y. TANG*, AND BARBARA H. GIBBONS* *Pacific Biomedical Research Center, University of Hawaii, Honolulu, HI 96822; and tDepartment of Biological Science, Purdue University, West Lafayette, IN 47907 Communicated by Howard C. Berg, July 15, 1991 ABSTRACT A procedure that uses the PCR to make rapid successive steps through a random-primed cDNA library has been developed to provide a method for sequencing very long genes that are difficult to obtain as a single clone. In each successive step, the portions of partial clones that extend out from the region of known DNA sequence are amplified by two stages of PCR with nested, outward-directed primers designed -50 bases in from the end of the known sequence, together with a general primer based on the sequence of the vector. This procedure has been used to determine the coding sequence of the cDNA for the (3 heavy chain of axonemal dynein from embryos of the sea urchin Tripneustesgratifla. By starting from a single parent clone, whose translated amino acid sequence overlapped the microsequence of a tryptic peptide of the a3 heavy chain, and making 3 such walk steps downstream and 14 walk steps upstream, we obtained a sequence of 13,799 base pairs that had an open reading frame of 13,398 base pairs. This sequence encodes a polypeptide with 4466 residues of Mr 511,804 that is believed to correspond to the complete /3 heavy chain of ciliary outer arm dynein. amplify selectively the regions of clones that extend the known sequence in a defined direction. The speed with which these walk steps can be taken, and the suitability of the products for direct sequencing, make this a relativey fast and efficient procedure for determining the sequence of very large polypeptides. Since successive walk steps can tie taken without necessarily completing determination of the inter- vening sequence, we designate this procedure "leapfrog" PCR (library extension and amplification by PCR for rapidly obtaining the gene). As an example, we describe the use of this technique to determine the complete nucleotide sequence [13,398 base pairs (bp)] encoding the p heavy chain polypeptide of axone- mal dynein, an energy-transducing ATPase involved in mi- crotubule-based cell motility (3) whose study has been hin- dered by the lack of information regarding its primary struc- tureJ Analysis of the deduced amino acid sequence of the dynein ,B heavy chain and its relationship to the function of dynein in microtubule-based motility is presented elsewhere (4). The transcripts that encode very high molecular weight polypeptides are difficult to sequence in their entirety be- cause they are rarely obtained as single clones. In a cDNA library prepared by priming the poly(A)+ RNA with oligo- (dT), the 3' region of the desired coding segment is well represented, but when the message is very long [>5 kilobases (kb)], clones of the 5' region are relatively sparse and some regions are often completely absent from the library. cDNA libraries prepared by random-priming the poly(A)+ RNA have the advantage that all regions of the desired sequence are nearly evenly represented in the library, but the average insert size is usually relatively small so that a large number of overlapping clones need to be obtained to cover the full sequence. Standard protocols for walking progressively through a library by screening with a labeled probe based on the terminal region of the previously known sequence are impractical for retrieving an entire large coding sequence from such a library, partly because of the labor involved in isolating new clones for each step of the large number of walk steps involved and partly because until each clone is se- quenced there is no information regarding the extent to which it provides new sequence, as opposed to extensive overlap of the region of already known sequence. We report here a rapid technique for using the PCR to extend a single parent clone by making successive steps through a random-primed cDNA library. This procedure, which combines aspects of those of Ohara et al. (1) and Friedman et al. (2), uses two stages of PCR amplification to MATERIALS AND METHODS Preparation of cDNA Library. Eggs from four female sea urchins (Tripneustes gratilla) were fertilized with sperm from a single male and allowed to develop for 16 hr at 23°C. Poly(A)+ RNA was isolated from mesenchyme blastula em- bryos that had been deciliated with hypertonic seawater (5) and allowed to reciliate for 60 min. The poly(A)+ RNA was used to prepare a size-selected, random-primed cDNA li- brary in the bacteriophage Agtll and yielded a total of 1.4 x 106 independent recombinants. The library was divided into 57 aliquots that were amplified individually in a suitable host bacterium (Escherichia coli, strain Y1088). DNA to be used as PCR template was prepared by the plate lysate method (6) from each of the separate amplified library aliquots. The resulting 57 batches of DNA were pooled into 24 template samples before use. PCR Amplification. Routine PCR amplifications were per- formed with 30 cycles in a Perkin-Elmer/Cetus DNA Ther- mal Cycler, using a cycle of denaturation for 1.0 min at 94°C, annealing for 2.0 min at 60°C, and an extension at 72°C for 1.5 min that was incremented by 5 sec per cycle. The 100-,ul reaction mixture contained 10 pmol of each primer and 2.0 units of Taq DNA polymerase (Promega) with the manufac- turer's buffer system, together with 0.3-1 ,ug of pooled library DNA (which is 3-10 ng of insert DNA) in the first stage amplification, and 1.0 Al of the first stage product in the second amplification (see Results). DNA Sequencing. Nucleotide sequencing was performed on template purified directly from the product of PCR am- tThe sequence reported in this paper has been deposited in the GenBank data base (accession no. X59603). 8563 The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Transcript of PCR large byrapidwalkingthrough cDNA - pnas.org filePCR(library...

Proc. Natl. Acad. Sci. USAVol. 88, pp. 8563-8567, October 1991Biochemistry
A PCR procedure to determine the sequence of large polypeptidesby rapid walking through a cDNA library
(dynein/leapfrog PCR/cilia/flagelia/sea urchin)
I. R. GIBBONS*, DAVID J. ASAIt, NATHAN S. CHING*, GREGORY J. DOLECKI*, GABOR MOCZ*,CHERYL A. PHILLIPSON*, HENING REN*, WEN-JING Y. TANG*, AND BARBARA H. GIBBONS**Pacific Biomedical Research Center, University of Hawaii, Honolulu, HI 96822; and tDepartment of Biological Science, Purdue University,West Lafayette, IN 47907
Communicated by Howard C. Berg, July 15, 1991
ABSTRACT A procedure that uses the PCR to make rapidsuccessive steps through a random-primed cDNA library hasbeen developed to provide a method for sequencing very longgenes that are difficult to obtain as a single clone. In eachsuccessive step, the portions of partial clones that extend outfrom the region of known DNA sequence are amplified by twostages of PCR with nested, outward-directed primers designed-50 bases in from the end ofthe known sequence, together witha general primer based on the sequence of the vector. Thisprocedure has been used to determine the coding sequence ofthe cDNA for the (3 heavy chain of axonemal dynein fromembryos of the sea urchin Tripneustesgratifla. By starting froma single parent clone, whose translated amino acid sequenceoverlapped the microsequence of a tryptic peptide of the a3heavy chain, and making 3 such walk steps downstream and 14walk steps upstream, we obtained a sequence of 13,799 basepairs that had an open reading frame of 13,398 base pairs. Thissequence encodes a polypeptide with 4466 residues of Mr511,804 that is believed to correspond to the complete /3 heavychain of ciliary outer arm dynein.
amplify selectively the regions of clones that extend theknown sequence in a defined direction. The speed with whichthese walk steps can be taken, and the suitability of theproducts for direct sequencing, make this a relativey fast andefficient procedure for determining the sequence of very largepolypeptides. Since successive walk steps can tie takenwithout necessarily completing determination of the inter-vening sequence, we designate this procedure "leapfrog"PCR (library extension and amplification by PCR for rapidlyobtaining the gene).As an example, we describe the use of this technique to
determine the complete nucleotide sequence [13,398 basepairs (bp)] encoding the p heavy chain polypeptide of axone-mal dynein, an energy-transducing ATPase involved in mi-crotubule-based cell motility (3) whose study has been hin-dered by the lack of information regarding its primary struc-tureJ Analysis of the deduced amino acid sequence of thedynein ,B heavy chain and its relationship to the function ofdynein in microtubule-based motility is presented elsewhere(4).
The transcripts that encode very high molecular weightpolypeptides are difficult to sequence in their entirety be-cause they are rarely obtained as single clones. In a cDNAlibrary prepared by priming the poly(A)+ RNA with oligo-(dT), the 3' region of the desired coding segment is wellrepresented, but when the message is very long [>5 kilobases(kb)], clones of the 5' region are relatively sparse and someregions are often completely absent from the library. cDNAlibraries prepared by random-priming the poly(A)+ RNAhave the advantage that all regions of the desired sequenceare nearly evenly represented in the library, but the averageinsert size is usually relatively small so that a large numberof overlapping clones need to be obtained to cover the fullsequence. Standard protocols for walking progressivelythrough a library by screening with a labeled probe based onthe terminal region of the previously known sequence areimpractical for retrieving an entire large coding sequencefrom such a library, partly because of the labor involved inisolating new clones for each step of the large number ofwalksteps involved and partly because until each clone is se-quenced there is no information regarding the extent to whichit provides new sequence, as opposed to extensive overlap ofthe region of already known sequence.We report here a rapid technique for using the PCR to
extend a single parent clone by making successive stepsthrough a random-primed cDNA library. This procedure,which combines aspects of those of Ohara et al. (1) andFriedman et al. (2), uses two stages of PCR amplification to
MATERIALS AND METHODSPreparation of cDNA Library. Eggs from four female sea
urchins (Tripneustes gratilla) were fertilized with sperm froma single male and allowed to develop for 16 hr at 23°C.Poly(A)+ RNA was isolated from mesenchyme blastula em-bryos that had been deciliated with hypertonic seawater (5)and allowed to reciliate for 60 min. The poly(A)+ RNA wasused to prepare a size-selected, random-primed cDNA li-brary in the bacteriophage Agtll and yielded a total of 1.4 x106 independent recombinants. The library was divided into57 aliquots that were amplified individually in a suitable hostbacterium (Escherichia coli, strain Y1088). DNA to be usedas PCR template was prepared by the plate lysate method (6)from each of the separate amplified library aliquots. Theresulting 57 batches of DNA were pooled into 24 templatesamples before use.PCR Amplification. Routine PCR amplifications were per-
formed with 30 cycles in a Perkin-Elmer/Cetus DNA Ther-mal Cycler, using a cycle of denaturation for 1.0 min at 94°C,annealing for 2.0 min at 60°C, and an extension at 72°C for 1.5min that was incremented by 5 sec per cycle. The 100-,ulreaction mixture contained 10 pmol of each primer and 2.0units of Taq DNA polymerase (Promega) with the manufac-turer's buffer system, together with 0.3-1 ,ug of pooledlibrary DNA (which is 3-10 ng of insert DNA) in the firststage amplification, and 1.0 Al ofthe first stage product in thesecond amplification (see Results).DNA Sequencing. Nucleotide sequencing was performed
on template purified directly from the product of PCR am-
tThe sequence reported in this paper has been deposited in theGenBank data base (accession no. X59603).
8563
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

8564 Biochemistry: Gibbons et al.
plification by agarose gel electrophoresis, followed by ad-sorption on glass milk (Bio 101, La Jolla, CA), and elution.Double-strand sequencing was performed by the dideoxynu-cleotide chain termination procedure using Sequenase 2(United States Biochemical) as recommended by the manu-facturer, except for the inclusion of0.5% Nonidet P-40 in thereaction mixture (7) and the replacement of Mg2+ by Mn2 ,as these modifications reduced the incidence of compressionlines. We obtained a median of 240 readable bases persequencing reaction.
Primers. Oligonucleotides were synthesized on a PCRMate model 391 synthesizer (Applied Biosystems). In mostcases, 24-mers were used to prime walk steps into the libraryand 16-mers were used for sequencing primers. Two generalprimers based on the sequence of the A arms were synthe-sized and used: 1222, 5'-d(TTGACACCAGACCAACTGG-TAATG)-3'; 1218A, 5'-d(GCTACAGTCAACAGCAACT-GATGG)-3'.Amino Acid Sequencing. To confirm the identity of the
sequence determined, four major tryptic peptides (215, 130,124, and 110 kDa) of the dynein ( heavy chain from spermflagella (8) were separated by electrophoresis on polyacryl-amide gels in the presence of SDS, electroblotted ontoImmobilon poly(vinylidene difluoride) membrane, and se-quenced at their N termini (9).
RESULTSLibrary Screening. Initial screening for clones whose ex-
pressed fusion protein reacted with afflinity-purified polyclo-nal antiserum against dynein heavy chains yielded 64 uniquepositives. Fifteen of these clones passed a second screen inwhich their radiolabeled DNA hybridized with a band of >12kb on a Northern blot of embryo poly(A)+ RNA. The fusionproteins from these clones were used to adsorb selectivelyantibodies from the purified antiserum, and the resultantepitope-selected antibodies were tested to determine whetherthey stained a rational pattern of peptides on blots of pho-tocleaved and trypsin-digested (3 heavy chain (10).
Validation of Parent Clone. The sequence from the cloneL13-14, which passed all screens and purified antibodiesspecific for the 130-kDa tryptic peptide of the 83 heavy chain,
was found to possess an open reading frame whose deducedamino acid sequence partially overlapped the microsequenceoftheN terminus ofthe 124-kDa tryptic peptide that is knownto be derived from the 130-kDa peptide (G.M., unpublisheddata). Extension of L13-14 in the upstream direction yieldedamplified portions of clones whose deduced amino acidsequence completed the microsequence of the 124-kDa pep-tide and also contained the microsequence of the 130-kDatryptic peptide close to its expected distance 85 residuesupstream of the 124-kDa peptide. On the basis of thisevidence, L13-14 was accepted as an authentic dynein .3heavy chain clone.Walk Steps Through the Library. Because of the paucity of
valid immunopositive clones to cover such a long cDNAsequence and the potential problem of immunologically re-lated isoforms, we devised a strategy for rapidly and effi-ciently walking through the library by utilizing PCR to stepoutward from the region of known sequence without the timeand labor involved in subcloning. To obtain sufficient spec-ificity (1), the procedure uses two successive stages of PCRamplification with a nested pair of outward-directed oligo-nucleotide primers designed in the region ofknown sequencetogether with a general primer based on the sequence of oneof the two A arms. In addition, prior adjustment of thecomplexity of the template DNA by separating the libraryinto aliquots before amplifying it in the host bacterium isimportant, for it allows one to avoid an inconvenient super-imposition ofproduct bands upon electrophoresis after PCR,while retaining a sufficient density to keep the number ofsamples manageable.To obtain as many PCR amplification products as possible,
we ran each of the 24 template samples with each of thegeneral A primers (1218A and 1222) plus the first ofthe nestedpair of specific primers (Fig. 1). Thus, there were 48 PCRtubes in each stage of a walk step (24 template samples vs.1218A and the same 24 vs. 1222). The second stage of PCRamplification was then performed with specific primer 2, thesame general A primer as was used in the first stage, and 1 ,lIfrom each of the product tubes of the first stage. Uponelectrophoresis of the products after this second amplifica-tion, many of the gel lanes showed one to three bands that
known sequence from walk stop n..............
........I
/1222general (orX primer \1218A/
1r.a.....................arm. . . . . . ....................... . .
lambda arm
Walk step n + 1
1st stage PCR30 cycles
2nd stage PCR30 cycles primer 2
II
Purification and sequencing
FIG. 1. Schematic of PCR amplification for walk step n + 1. Known sequence from walk step n is used to design new specific primers 1and 2 on the outward-directed strand of the DNA. Validation that the n + 1 sequence is a continuation of the n sequence is given by the 50-bpregion by which specific primer 2 is inset from the end of step n.
I.Speci f icprimer 1
lambda arm
. . .
Proc. Natl. Acad. Sci. USA 88 (1991)
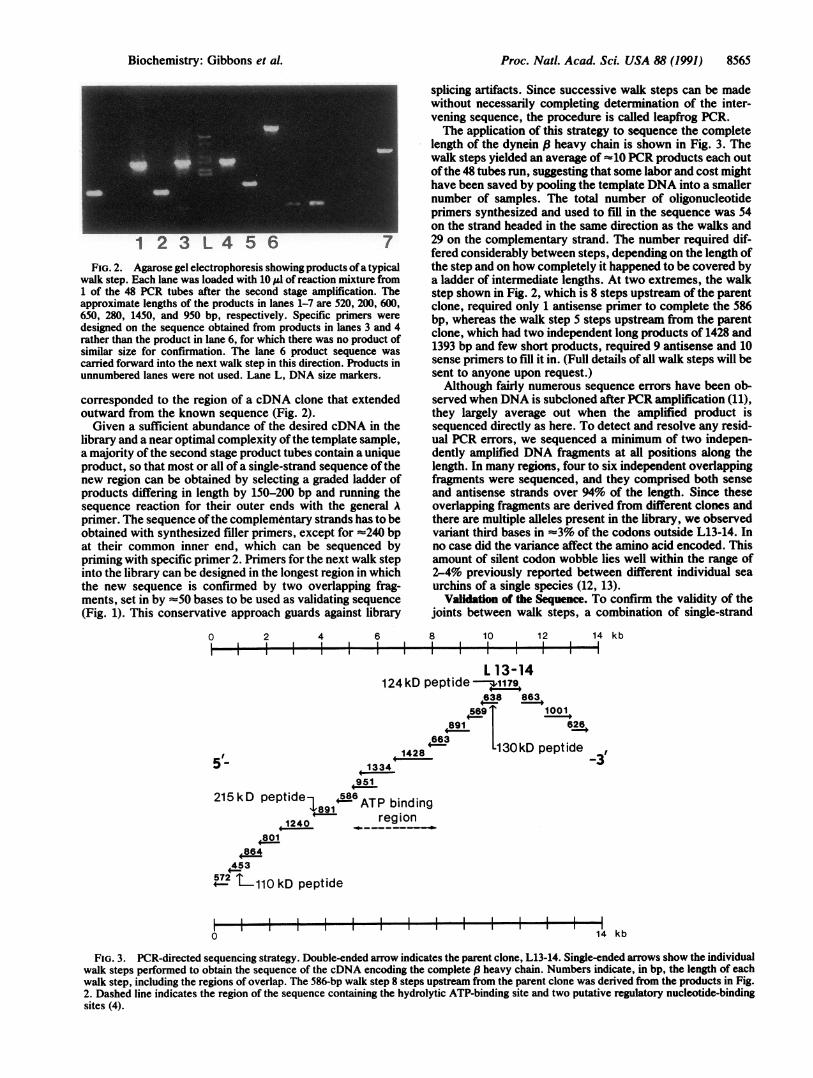
Proc. Natl. Acad. Sci. USA 88 (1991) 8565
FIG. 2. Agarose gel electrophoresis showing products ofa typicalwalk step. Each lane was loaded with 10 1l of reaction mixture from1 of the 48 PCR tubes after the second stage amplification. Theapproximate lengths of the products in lanes 1-7 are 520, 200, 600,650, 280, 1450, and 950 bp, respectively. Specific primers weredesigned on the sequence obtained from products in lanes 3 and 4rather than the product in lane 6, for which there was no product ofsimilar size for confirmation. The lane 6 product sequence wascarried forward into the next walk step in this direction. Products inunnumbered lanes were not used. Lane L, DNA size markers.
corresponded to the region of a cDNA clone that extendedoutward from the known sequence (Fig. 2).Given a sufficient abundance of the desired cDNA in the
library and a near optimal complexity ofthe template sample,a majority of the second stage product tubes contain a uniqueproduct, so that most or all ofa single-strand sequence of thenew region can be obtained by selecting a graded ladder ofproducts differing in length by 150-200 bp and running thesequence reaction for their outer ends with the general Aprimer. The sequence ofthe complementary strands has to beobtained with synthesized filler primers, except for =240 bpat their common inner end, which can be sequenced bypriming with specific primer 2. Primers for the next walk stepinto the library can be designed in the longest region in whichthe new sequence is confirmed by two overlapping frag-ments, set in by =50 bases to be used as validating sequence(Fig. 1). This conservative approach guards against library
0 2 4 6l
splicing artifacts. Since successive walk steps can be madewithout necessarily completing determination of the inter-vening sequence, the procedure is called leapfrog PCR.The application of this strategy to sequence the complete
length of the dynein .8 heavy chain is shown in Fig. 3. Thewalk steps yielded an average of 10 PCR products each outofthe 48 tubes run, suggesting that some labor and cost mighthave been saved by pooling the template DNA into a smallernumber of samples. The total number of oligonucleotideprimers synthesized and used to fill in the sequence was 54on the strand headed in the same direction as the walks and29 on the complementary strand. The number required dif-fered considerably between steps, depending on the length ofthe step and on how completely it happened to be covered bya ladder of intermediate lengths. At two extremes, the walkstep shown in Fig. 2, which is 8 steps upstream of the parentclone, required only 1 antisense primer to complete the 586bp, whereas the walk step 5 steps upstream from the parentclone, which had two independent long products of 1428 and1393 bp and few short products, required 9 antisense and 10sense primers to fill it in. (Full details of all walk steps will besent to anyone upon request.)Although fairly numerous sequence errors have been ob-
served when DNA is subcloned after PCR amplification (11),they largely average out when the amplified product issequenced directly as here. To detect and resolve any resid-ual PCR errors, we sequenced a minimum of two indepen-dently amplified DNA fragments at all positions along thelength. In many regions, four to six independent overlappingfragments were sequenced, and they comprised both senseand antisense strands over 94% of the length. Since theseoverlapping fragments are derived from different clones andthere are multiple alleles present in the library, we observedvariant third bases in -3% of the codons outside L13-14. Inno case did the variance affect the amino acid encoded. Thisamount of silent codon wobble lies well within the range of2-4% previously reported between different individual seaurchins of a single species (12, 13).
Validation of the Sequence. To confirm the validity of thejoints between walk steps, a combination of single-strand
8 10 12 14 kb
L 13-14124 kD peptide I
:8914663
if1428
1001,626p
pept ide0:>- ( ~~~1334
+951
215 kD peptide 586 ATP bindingreg ion
453
7 -110 kD peptide
0I I I II I 1o 14 kb
FIG. 3. PCR-directed sequencing strategy. Double-ended arrow indicates the parent clone, L13-14. Single-ended arrows show the individualwalk steps performed to obtain the sequence of the cDNA encoding the complete s heavy chain. Numbers indicate, in bp, the length of eachwalk step, including the regions of overlap. The 586-bp walk step 8 steps upstream from the parent clone was derived from the products in Fig.2. Dashed line indicates the region of the sequence containing the hydrolytic ATP-binding site and two putative regulatory nucleotide-bindingsites (4).
Biochemistry: Gibbons et al.
Ic -

8566 Biochemistry: Gibbons et al.
14.5 kb _
1 2 3 4 5 6
FIG. 4. Northern analysis of sea urchin embryonic poly(A)+RNA probed with L13-14 and with cDNA from various regions alongthe ,8 heavy chain. Stringency of final wash was 0.5x SET (0.075 MNaCI/15 mM Tris-HCl/1 mM EDTA)/0.1% SDS/0.1% sodium pyro-phosphate at 60°C. Lane 1, probed with L13-14 (positions 9942-11221). Lanes 2-6, probed with cDNA from the following positions:2, 773-1143; 3, 5746-6096; 4, 6504-7744; 5, 7805-8665; 6, 12157-12787. Numbers correspond to positions in the nucleotide sequence.Lines indicate the origin for each lane of the gel. Smear below the14.5-kb band in lanes 2-6 is of unknown source.
cDNA synthesis by reverse transcriptase using poly(A)+RNA primed with a selection of the oligonucleotide primersused for walking, followed by PCR amplification with anoppositely directed second primer was used to amplify longerpieces ofcDNA that bridged one to three joints. In all cases,the joints were validated by the presence of an electropho-retic band of the predicted length in the product DNA.
Further confirmation was obtained by Northern blotting,which showed that L13-14 and probes from five other posi-tions along the length of the sequence all hybridized againstan 414.5-kb band (Fig. 4).
Features of the Sequence. Starting from L13-14, the first ofa series of in-frame stop codons occurred after three walksteps in the 3' direction. That this stop codon represents thetrue 3' end of the coding sequence is supported by thepresence of nearby stop codons in the other reading framesand by the occurrence of significant sequence differencebetween alleles, including insertions and deletions, in theregion beyond it.
In the 5' direction, the first walk step contained theremainder of the N-terminal sequence of the 124-kDa pep-tide. The N-terminal microsequence of the 130-kDa peptidewas in approximately the anticipated position 85 residuesfurther upstream. The N-terminal microsequence of the215-kDa peptide was located 8 walk steps upstream from theN terminus of the 130-kDa peptide. The 215-kDa peptidecontains the ATP-binding consensus sequence GPAGTGKTlocated 660 residues from its N terminus, close to thepredicted position of the Vl photocleavage site (4, 8).The sequence corresponding to the N terminus of the
110-kDa tryptic peptide was located 13 walk steps upstream
of L13-14. The N terminus of the complete /3 heavy chain ispredicted to be =20 kDa beyond the N terminus of the110-kDa peptide (8). At position 205 in this region, 14 walksteps from L13-14, occurs the nucleotide sequence ACT-ATGG that has an in-frame methionine and conforms to theconsensus of eukaryotic initiation sites (14). The first ofseveral in-frame stop codons occurs at nucleotide 1%, onlyslightly further 5'. The next ATG codon downstream is atposition 294; it has much less favorable flanking bases andalso is located too close to the start of the 110-kDa peptide.Therefore, we believe that the ATG at position 208 is theprobable translation initiation codon for the dynein /3 heavychain.
Starting from nucleotide 208, a continuous open readingframe runs for 13,398 nucleotides. The deduced amino acidsequence contains 4466 residues and has a calculated Mr of511,804 (4). In the region of overlap, this sequence shows-90%o identity at the nucleotide level and 98% identity at theamino acid level to two partial clones of dynein ,/ chain froma different species of sea urchin examined by Ogawa (15, 16).Two regions have been identified in which the deduced
amino acid sequence of certain clones varies from thatreported. Starting at nucleotide 2037, four of the clonessequenced were 15 bases shorter and lacked the five aminoacids TGVRR (Fig. SA). The second variation starts atnucleotide 10271, where two of the five clones examinedcontain a 33-base out-of-frame insertion that changed theamino acid sequence beginning at amino acid 3356 from LPGto LLTGNFFCCFMTAG (Fig. 5B). These variations maycorrespond to alternative splicing that produces differentversions of the dynein ,8 heavy chain for different cells in theembryo, although the possibility of their being cloning arti-facts remains open until the genomic sequence has beenexamined.
DISCUSSIONThe major advantages of the procedure described are that itenables the complete screening of a cDNA library for theportions of clones that extend a region of known sequencewithin 48 hr and that the products of this screening constitutea suitable template for direct sequencing. Combined with theuse of an oligonucleotide synthesizer as part of the project,this procedure makes it possible to take successive stepsthrough a library at a rate of better than 1 per week. In thepresent instance, once the parent clone had been identified,the sequencing of the P heavy chain of dynein took -3months for a team of four people. We believe that the cost iscompetitive with that of other manual procedures.The rapidity with which walk steps can be taken is partic-
ularly important in determining the sequence of a messagewhose length substantially exceeds the average insert size inthe library, so that many steps are required to obtain a
A 2029 GAA CAT CCG ACT GGT GTC AGG AGA ATC TTA GAG 2061608 E H P T G V R R I L E 618
2029 GAA CAT CCG --- --- --- --- --- ATC TTA GAG 2046608 E H P I L E 613
B 10270 ACT TTG C-- --- --- --- --- --- --- --- --- --- --- -CT GGT GAT GTC 102873355 T L P G D V 3360
10270 ACT TTG CTC ACT GGC MAC TTT TTT TGT TGT TTT ATG ACA GCT GGT GAT GTC 103203355 T L L T G N F F C C F N T A G D V 3371
FIG. 5. Sequence variations found in cDNA encoding dynein (3 chain. In each case, the upper line represents the major sequence reported(4). (A) A 15-base in-frame deletion found in four clones. (B) A 33-base out-of-frame insertion found in two clones.
Proc. Natl. Acad Sci. USA 88 (1991)

Proc. Natl. Acad. Sci. USA 88 (1991) 8567
complete sequence. Since the procedure amplifies only theportions of clones that extend out from the known sequence,along with an overlap of -50 bases required for validation,the size of the product band indicates directly the amount ofnew sequence that it contains.The principal disadvantage of this procedure is that it does
not directly yield clones that can be used for protein expres-sion. The PCR-amplified DNA is not well suited for thispurpose because the accumulated Taq polymerase errors inindividual DNA molecules, although irrelevant for directsequencing, introduce defects into the expressed polypeptidemolecules. However, this is not a severe disadvantage, foronce the sequence is known it is relatively simple to preparean expression library in an appropriate vector by use ofspecific primers.The procedure described may be useful in sequencing the
cDNA for other high molecular weight polypeptides that aretoo long for a full-length clone to be obtained.
We thank Dr. Kazuo Ogawa for kindly sending us copies of refs.15 and 16 prior to publication and Pat Harwood for assistance withthe immunoscreening. This work was supported in part by NationalInstitutes of Health Grants GM30401 to I.R.G. and HD06565 toB.H.G., and by a National Science Foundation grant to D.J.A.
1. Ohara, O., Dorit, R. L. & Gilbert, W. (1989) Proc. Nati. Acad.Sci. USA 86, 5673-5617.
2. Friedman, K. D., Rosen, N. L., Newman, P. J. & Montgom-ery, R. R. (1990) in PCR Protocols: A Guide to Methods andApplications, eds. Innis, M. A., Gelfand, D. H., Sninsky, J. J.& White, T. H. (Academic, San Diego), pp. 253-258.
3. Gibbons, I. R. (1988) J. Biol. Chem. 263, 15837-15840.4. Gibbons, I. R., Gibbons, B. H., Mocz, G. & Asai, D. J. (1991)
Nature (London) 352, 640-643.5. Stephens, R. E. (1978) Biochemistry 17, 2882-2891.6. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular
Cloning:A Laboratory Manual (Cold Spring Harbor Lab., ColdSpring Harbor, NY), pp. 2.118-2.120.
7. Bachmann, B., Luke, W. & Hunsmann, G. (1990) NucleicAcids Res. 18, 1309.
8. Mocz, G., Tang, W.-J. Y. & Gibbons, I. R. (1988) J. Cell Biol.106, 1607-1614.
9. Matsudaira, P. (1987) J. Biol. Chem. 262, 10035-10038.10. Gooderham, K. (1984) in Methods in Molecular Biology, ed.
Walker, J. M. (Humana, Clifton, NJ), Vol. 1, pp. 193-202.11. Keohavong, P. & Thilly, W. G. (1989) Proc. Natl. Acad. Sci.
USA 86, 9253-9257.12. Britten, R. J., Cetta, A. & Davidson, E. H. (1978) Cell 15,
1175-1186.13. Palumbi, S. R. & Metz, E. C. (1991) Mol. Biol. Evol. 8,
227-239.14. Kozak, M. (1986) Cell 44, 283-292.15. Ogawa, K. (1991) Proc. Jpn. Acad. B 67, 27-31.16. Ogawa, K. (1991) in Biology of Echinodermata, eds. Yanagi-
sawa, T., Yasumasu, I., Oguro, C., Suzuki, N. & Motokawa,T. (Balkema, Rotterdam), in press.
Biochemistry: Gibbons et al.


















