
PART - McGraw-Hill...
12
7 PART Neoplasia
Transcript of PART - McGraw-Hill...

7
P A R T
Neoplasia


CHAPTER 109
■
GENOME INSTABILITY, DNA REPAIR, AND CANCER
977
S E C T I O N
20
CARCINOGENESIS
C H A P T E R 1 0 9
Genome Instability, DNA Repair, and Cancer
Kenneth H. KraemerThomas M. Rünger
INTRODUCTION
The integrity of the genome of all livingorganisms is constantly threatened byexogenous and endogenous DNA-damag-ing agents. Exogenous DNA-damagingagents include physical agents, such asultraviolet (UV) or ionizing radiation,and a wide variety of chemical agents,such as components of cigarette smoke.Endogenous DNA damage arises fromregular metabolic processes within thecell, mediated, for example, by reactiveoxygen species. Maintaining the stabil-ity of the genome is of utmost impor-tance to all living organisms. Therefore,since early evolution, all organismsranging from prokaryotes to eukaryoteshave been equipped with mechanismsthat react to and repair DNA damageand thereby maintain genomic stability.The types of damage produced includealterations in the structure of nucleo-tides, DNA strand breaks, DNA cross-links, and DNA adducts. Different types
of DNA-damaging agents induce differ-ent types of DNA damage (Table 109-1),which in turn require different re-sponses and repair pathways for pro-cessing (Table 109-2).
1
If DNA damage is not repaired ade-quately, it may lead to altered cell func-tion, cell death, or the formation of muta-tions (alterations of the DNA sequence)in the damaged cells. These DNA dam-age–induced mutations will persist aslong as the affected cell survives. At a cel-lular level, mutations in vital genes canlead to alterations of cell functions or ma-lignant transformation. Accumulation ofmutations may lead to organ dysfunc-tion, aging, and cancer. Although mostDNA damage is adequately repaired,none of the cellular responses is 100 per-cent effective in repairing all DNA dam-age under all circumstances.
A hereditary or acquired impairmentin the way cells respond to DNA dam-age may result in genome instabilitywith an increased rate of mutation for-mation. Numerous hereditary disordersare characterized by such genome insta-bility (reviewed in Chap. 140). Many,but not all, of those are associated withan increased cancer risk and/or acceler-ated aging.
Exposure of the skin to UV light hasmultiple cellular and clinical effects, in-cluding an increase in skin cancer risk.The photocarcinogenesis cascade ofevents (Fig. 109-1) exemplifies the linkbetween genome instability, DNA re-pair, and cancer. UV light produces atype of DNA damage involving the gen-eration of photoproducts, which are al-terations in the structure of nucleotides.The major DNA photoproducts are cy-clobutane pyrimidine dimers (CPDs;Fig. 109-2) and 6,4-pyrimidine–pyrimi-done dimers. Unrepaired CPDs or 6,4-pyrimidine–pyrimidone dimers may re-sult in characteristic mutations: C to Tsingle base and CC to TT tandem base
substitution mutations.
2–4
Such muta-tions are typical for UV light exposuresand only rarely are induced by othermutagens. They have therefore beentermed
UV-signature mutations
(see Fig.109-2).
GENOME INSTABILITY, DNA REPAIR, AND CANCER
AT A GLANCE
■
DNA can be damaged by physical agents (ultraviolet or ionizing radiation) or chemi-cal agents in the environment.
■
DNA damage may lead to mutations (changes in DNA sequence).
■
The ability of cells to repair DNA damage and to maintain genome stability is of utmost importance to prevent malignant transformation.
■
Different agents induce different types of DNA damage, which in turn require dif-ferent responses and repair pathways.
■
A number of hereditary disorders are characterized by genome instability due to defects in genes involved in DNA repair or DNA damage signaling.
■
Many different laboratory tests can be used to diagnose genome instability and/or DNA repair defects.
■
Inherited or acquired genome instability is associated with an increased cancer risk.
�
Lentigo maligna melanoma. A large lentigomaligna on the left cheek with the typical varie-gation in color. The lesion is flat and macular andrepresents in situ melanoma. In the center of theirregular lesion there is a pitch-black nodule thatindicates a switch from the radial to the verticalgrowth phase and thus invasiveness: The lesionis now called lentigo maligna melanoma.

SECTION 20
■
CARCINOGENESIS
978
DNA repair is an important cellulardefense mechanism that prevents muta-tion formation at sites of DNA damageafter UV exposure. However, it is notthe only defense mechanism (see Fig.109-1). Most mutations are generatedduring replication of damaged DNA.Therefore, a damage-induced arrest incell cycling, which allows more time forrepair, is another important cellulardamage response that prevents muta-tion formation.
5,6
Furthermore, pro-grammed cell death (apoptosis) preventsthe survival of cells with overwhelmingDNA damage, and through that mecha-nism the frequency of cells with UV-induced mutations is also reduced.
7
Other inherent defense mechanismsagainst the ultimately carcinogenic prop-erties of UV light include increased mel-anogenesis and thickening of the epider-mis and stratum corneum, which protectfrom future DNA damage, as well as re-moval of mutated cells through host im-mune responses (see Fig. 109-1).
DNA DAMAGE AND REPAIR
More than 100 DNA repair genes havebeen identified (http://www.cgal.icnet.uk/DNA_Repair_Genes.html#NER). The nu-cleotide excision repair (NER) pathway
acts on DNA damaged by UV radiation,repairing CPDs and other photoproducts,as well as on DNA damaged by certaincarcinogens (such as benzo-[a]-pyrene)(see Table 109-2). In NER, the damagednucleotide is removed and replaced with
undamaged DNA. Multiple proteins actin concert to repair UV-induced DNAdamage (Fig. 109-3). Defects in these re-pair genes can cause human diseases (Ta-ble 109-3), including xeroderma pigmen-tosum (XP), Cockayne syndrome, andtrichothiodystrophy (for details on thesedisorders, see Chap. 140). For instance,XP can be caused by a defect in any oneof several genes involved in NER. Basedon cell fusion experiments, cells/patientswith defects in the same gene are consid-ered to be in the same complementationgroup, and in different complementationgroups if different genes are affected. IfDNA repair in the fused cell is normal-ized, with the wild-type gene from eachcell giving rise to a functional protein thatis absent in the other cell, the cells “com-plement” each other and are in differentcomplementation groups. Seven suchcomplementation groups have beenidentified (XPA to XPG), which impliesthat mutations in any of seven distinctgenes can cause XP.
Transcribed genes are repaired fasterthan the rest of the genome. In the NERpathway, the first steps involving DNAdamage recognition are different in non-transcribed and transcribed genes (glo-bal genome NER and transcription-coupled NER). In non-transcribed genesand noncoding areas, which representmost of the genome, the
XPE
and
XPC
gene products bind to UV-damagedDNA, marking it for further processing.In contrast, DNA damage in transcribedgenes is probably sensed by a stalled
TABLE 109-1
Cellular Damage Induced by Physical and Chemical Agents
A
GENT
D
AMAGE
Ultraviolet radiation Dipyrimidine cyclobutane dimers (TT, TC, CT, or CC), pyrimidine-pyrimidone (6-4) photoproducts (mostly TC), DNA-protein cross-links
X-irradiation DNA single- and double-strand breaks, oxidative base damage (see below) Psoralens plus ultraviolet A
DNA-psoralen monoadducts, DNA interstrand cross-links (binds to T at TA sequences)
Mitomycin C DNA interstrand cross-linksBenzo-[a]-pyrene Bulky adductsReactive oxygen species
Oxidative base damage (8-oxo-deoxyguanine, thymine glycol), cyclopurines (A or G) making bulky lesions
TABLE 109-2
Types of DNA Damage and Associated DNA Repair Pathways
T
YPE
OF
DNA D
AMAGE
DNA R
EPAIR
P
ATHWAY
DNA photoproducts (CPDs, 6,4-PP) Nucleotide excision repairOxidative base modifications (e.g., 8-oxoG) Base excision repairIncorrect DNA base pairing Mismatch repairDNA double-strand breaks Non-homologous end joining, homologous
recombination (i.e., recombination repair)DNA adducts Nucleotide excision repairDNA cross-links (interstrand and intrastrand) Recombination repairPersistent DNA lesions Translesion (bypass) DNA synthesis
CPDs = cyclobutane pyrimidine dimers; 6,4-PP = 6,4-pyrimidine–pyrimidone photoproducts; 8-oxoG = 8-oxo-deoxyguanine.
�
FIGURE 109-1
Photocarcinogenesis cascade of events. Exposure to ultraviolet (UV) light inducestypical types of DNA damage, namely, cyclobutane pyrimidine and 6,4-pyrimidine–pyrimidone photo-products. These often generate single and tandem base substitution mutations (C
→
T and CC
→
TT) thatare typical for UV light exposure and are therefore termed
UV-signature mutations
. With sufficient num-bers of inactivating mutations in crucial genes (tumor suppressor genes), individual cells may undergomalignant transformation, clonally expand, and form skin cancers. Several inherent defense mechanismscounteract this chain of events (see text).
Inherent defense mechanisms
Pigmentationskin thickening
UV-exposure
DNA damageDNA photoproducts
Mutationincl. C→T transitions
Skin cancermonoclonal expansion of mutated
keratinocytes or melanocytes
DNA repaircell cycle arrestapoptosis
Removal of mutated cells(immune-surveillance)
CHAPTER 109
■
GENOME INSTABILITY, DNA REPAIR, AND CANCER
979
�
FIGURE 109-2
Example of ultraviolet (UV) light–inducedgeneration of DNA damage and a subsequent mutation. On di-rect excitation of the DNA molecule by UV light, adjacent pyrim-idine bases (cytosine or thymine) may form covalent bonds be-tween them, which leads to creation of pyrimidine dimers. Theillustration shows an example in which two covalent bindingsgenerate a tri-cyclic cyclobutane ring between two pyrimidines.Hence, this type of UV light–induced DNA damage is called a
cyclobutane pyrimidine dimer
, a common type of DNA photo-product. The nucleotide excision repair DNA repair system func-tions to remove this damage, which results in the normal DNAsequence (
upper box
). If the damage is not repaired, this typeof DNA photoproduct can lead to formation of a typical C
→
Tsingle base substitution mutation (
lower box
). This is most likelyto occur on replication of damaged DNA and misincorporationof adenine opposite the cytosine-containing photoproduct. Anexample of such a UV-signature mutation is shown on theright.
4
Note that the mutation is located within a run of sevenpyrimidines, a common location for UV-signature mutations.
�
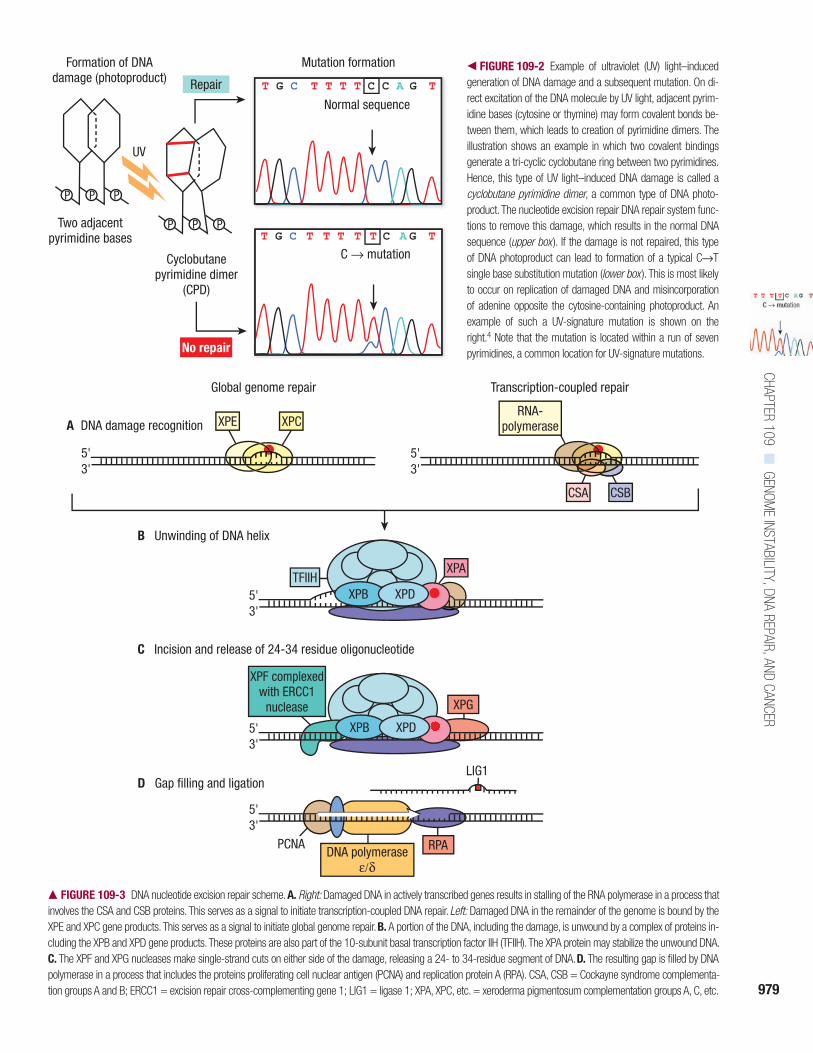
FIGURE 109-3
DNA nucleotide excision repair scheme.
A.
Right:
Damaged DNA in actively transcribed genes results in stalling of the RNA polymerase in a process thatinvolves the CSA and CSB proteins. This serves as a signal to initiate transcription-coupled DNA repair.
Left:
Damaged DNA in the remainder of the genome is bound by theXPE and XPC gene products. This serves as a signal to initiate global genome repair.
B.
A portion of the DNA, including the damage, is unwound by a complex of proteins in-cluding the XPB and XPD gene products. These proteins are also part of the 10-subunit basal transcription factor IIH (TFIIH). The XPA protein may stabilize the unwound DNA.
C.
The XPF and XPG nucleases make single-strand cuts on either side of the damage, releasing a 24- to 34-residue segment of DNA.
D.
The resulting gap is filled by DNApolymerase in a process that includes the proteins proliferating cell nuclear antigen (PCNA) and replication protein A (RPA). CSA, CSB = Cockayne syndrome complementa-tion groups A and B; ERCC1 = excision repair cross-complementing gene 1; LIG1 = ligase 1; XPA, XPC, etc. = xeroderma pigmentosum complementation groups A, C, etc.
Repair
No repair
Normal sequence
T G C T T T T C C A G T
C → mutationT G C T T T T T C A G T
P P P
UV
Two adjacentpyrimidine bases
Cyclobutanepyrimidine dimer
(CPD)
P P P
Formation of DNAdamage (photoproduct)
Mutation formation
B Unwinding of DNA helix
PCNADNA polymerase
ε/δ
5'3'
5'3'
5'3'
LIG1
XPB XPD
DNA damage recognition
Global genome repair
5'3'
A
Transcription-coupled repair
XPE XPC
CSA CSB
TFIIHXPA
C Incision and release of 24-34 residue oligonucleotide
XPB XPD
XPF complexedwith ERCC1
nuclease XPG
RPA
D Gap filling and ligation
5'3'
RNA-polymerase

SECTION 20
■
CARCINOGENESIS
980
RNA polymerase acting in conjunctionwith the CSA and CSB (Cockayne syn-drome complementation groups A andB) gene products. After the DNA dam-age recognition steps, global genomeNER and transcription-coupled NER fol-low the same pathway. The
XPA
geneproduct probably functions in conjunc-tion with replication protein A, thetranscription factor IIH (TFIIH), XPF,and excision repair cross-complement-ing gene 1 (ERCC1). The followingsteps occur in both non-transcribed andtranscribed gene repair:
• The
XPB
and
XPD
gene products par-tially unwind the DNA in the region ofthe damage, thereby exposing thelesion for further processing. Theseproteins are part of the TFIIH basaltranscription factor (see Fig. 109-3B).
• The
XPF
gene product, in a complexwith ERCC1, makes a single-strandnick at the 5
′
side of the lesion,whereas the
XPG
gene product makesa similar nick on the 3
′
side, whichresults in the release of a region of
approximately 30 nucleotides contain-ing the damage (see Fig. 109-3C).
• The resulting gap is filled by DNApolymerase using the other (undam-aged) strand as a template in a processinvolving proliferating cell nuclearantigen. DNA ligase 1 seals theregion, restoring the original undam-aged sequence (see Fig. 109-3D).
Other DNA repair pathways includebase excision repair, recombination re-pair, and mismatch repair. Defects inmismatch repair are also associatedwith human diseases—Muir-Torre syn-drome and human non-polyposis coloncancer. More extensive descriptions ofthese pathways may be found in refs. 1,8, 9, and 10.
REGULATION OF CELLULAR RESPONSES TO DNA DAMAGE
As outlined in the introduction, severalcellular responses to DNA damage con-tribute to the maintenance of genomeintegrity. These include cell cycle arrest,
apoptosis (programmed cell death), andDNA repair.
5,11–18
Because these re-sponses need to be carefully orches-trated, there are many proteins involvedin the signaling of DNA damage and theregulation of DNA damage responses(Fig. 109-4). Different types of DNA-damaging agents and different types ofDNA damage require different DNAdamage responses. Fig. 109-4 presentssome of the major players in signalingDNA damage and regulating DNA dam-age responses. As with defects in DNArepair genes (see Table 109-3), defects inmany of these DNA damage–signalinggenes (boxed in Fig. 109-4) are also im-plicated in hereditary disorders of ge-nome instability (Table 109-4; for fur-ther details see Chap. 140).
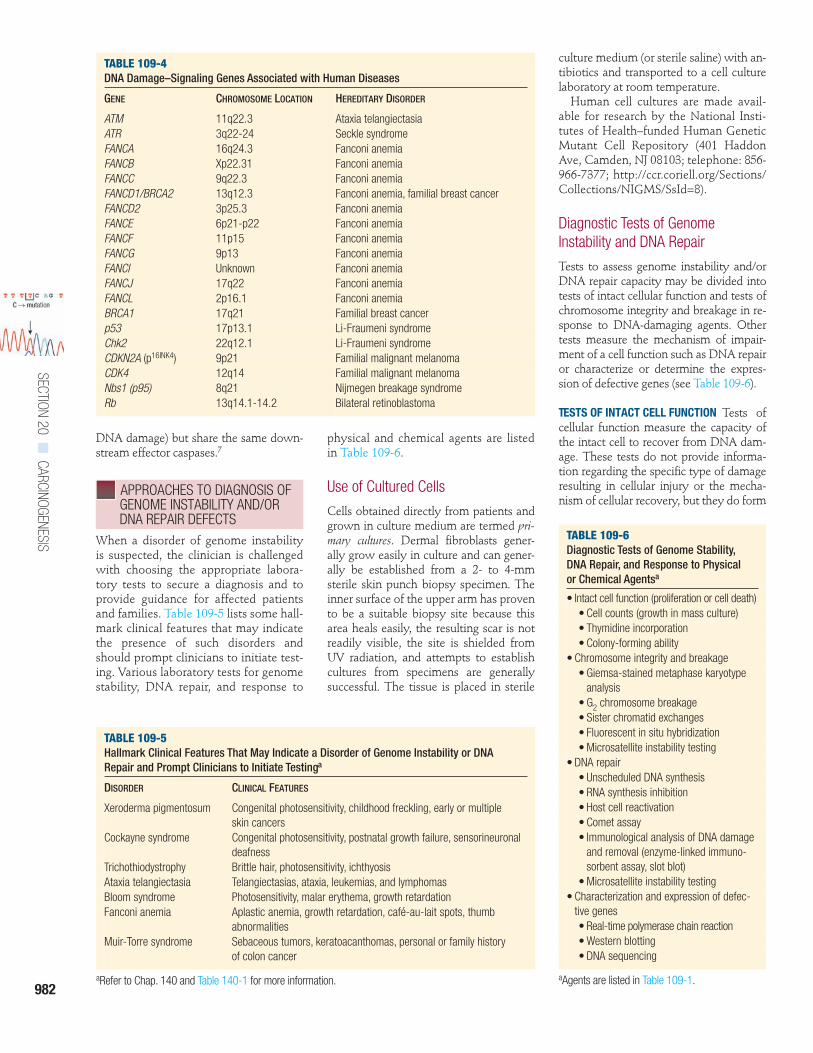
The tumor suppressor gene p53,termed the
guardian of the genome
,
19
playsa pivotal role in regulating and orches-trating these responses and is mutated inmany cancers, including cutaneous squa-mous cell carcinomas. Upstream regula-tors of p53 in the cellular DNA damageresponse pathway are ATM (ataxia tel-angiectasia mutated) and ATR (ataxiatelangiectasia- and Rad3-related) genes.One of p53’s several functions is theregulation of the cell cycle in responseto DNA damage. After cell division (mi-tosis), cells have 23 pairs of chromo-somes and are in the G
1
phase of the cellcycle. The chromosomes then replicateduring DNA synthesis, or S phase, andas a result have twice the number ofchromosomes (G
2
phase) just before mi-tosis (M phase). The cell stops cycling(arrests) in specific cell cycle phasescalled
cell cycle checkpoints
. An importantdownstream effector in preventing cellsfrom entering S phase (G
1
/S checkpoint)is p21.
5,11
p53 also induces NER by tran-scriptionally inducing XPC, XPE/p48,and GADD45.
20,21
If cells enter S phase with unrepairedDNA damage, or if cells are UV exposedduring S phase, regular DNA poly-merases stall at DNA photoproducts andfall off the DNA strand. For these in-stances, cells are equipped with severalspecialized DNA polymerases for trans-lesional DNA synthesis.
22
DNA polym-erase eta is one of these; it is specializedto bypass DNA photoproducts but mayintroduce mutations while doing so.
23
Itis mutated in XP variant, which is clini-cally indistinguishable from XP (see Ta-ble 109-3, Chap. 140).
24,25
This demon-strates the importance of this second lineof defense against the mutagenic and car-cinogenic consequences of DNA photo-products. It was reported that p53 and
TABLE 109-3
DNA Repair Genes Associated with Human Diseases
G
ENE
a
C
HROMOSOME
L
OCATION
R
EPAIR
P
ATHWAY
F
UNCTION
XPA
9q22.3 NER Binding of damaged DNA
XPB (ERCC3)
2q21 NER DNA helicase, part of TFIIH
XPC
3p25 NER Binding of damaged DNA, global genome repair
XPD (ERCC2)
19q13.3 NER DNA helicase, part of TFIIH
XPE (DDB2)
11p12-p11 NER Binding of damaged DNA, global genome repair
XPF (ERCC4)
16p13.3-p13.11
NER DNA endonuclease
XPG (ERCC5)
13q22 NER DNA endonuclease
TTDA (GTF2H5)
6p25.3 NER Part of TFIIH
CSA (ERCC8)
5q12 NER Transcription-coupled repair
CSB (ERCC6)
10q11 NER Transcription-coupled repair
XPV (polymer-ase eta)
6p21.1-p12 Bypass DNA damage bypass polymerase
MSH2
2p22-p21 MMR Mismatch repair (Muir-Torre syndrome, HNPCC)
MLH1
3p21.3 MMR Mismatch repair (Muir-Torre syndrome, HNPCC)
PMS1
2q31-q33 MMR Mismatch repair (HNPCC)
PMS2
7p22 MMR Mismatch repair (HNPCC)
MSH6
2p16 MMR Mismatch repair (HNPCC)
TFGBR
23p22 MMR Mismatch repair (HNPCC)
MLH3
14q24.3 MMR Mismatch repair (HNPCC)
ERCC = human excision repair cross-complementing genes that correct defects in cultured hamster cells; HNPCC = hereditary non-polyposis colon cancer; NER = nucleotide excision repair; MMR = mismatch repair; TFIIH = transcription factor IIH.
aThe genes designated XP are defective in the corresponding xeroderma pigmentosum complementation group, and those designated CS are defective in the corresponding Cockayne syndrome complementation group.

CHAPTER 109■
GENOME INSTABILITY, DNA REPAIR, AND CANCER
981
p21 also downregulate the activity ofthis translesional DNA synthesis tomaintain a low mutagenic activity at theprice of reduced damage bypass.26
If this translesional DNA synthesisfails, cells can use recombination repairto resolve stalled replication forks.27
When invoked in response to damagefrom UV light, this third line of defenseis mediated by activation of the Fanconianemia/BRCA DNA damage response
pathway.28 The exact mechanisms thatinitiate these DNA damage responsesignaling cascades is under investiga-tion. Telomeres, which are repeats ofTTAGGG that cap the ends of chromo-somes and whose ends form a loopstructure, are suggested to be importantplayers in sensing DNA damage, for ex-ample, through opening of the telomereloop.29 Other sensors may be stalledDNA or RNA polymerases, or proteins
that detect bending of the DNA helix atsites of DNA damage.
Apoptosis is a regulated physiologicprocess leading to cell death characterizedby cell shrinkage, membrane blebbing,and DNA fragmentation. A group of cys-teine proteases called caspases are centralregulators of apoptosis. Triggers may beextrinsic or intrinsic to the cell (e.g., DNAdamage) and involve separate initiatorcaspases (e.g., caspase 2 in response to
� FIGURE 109-4 Signaling cascades that regulate apoptosis, DNA repair, translesional DNA synthesis, and activation of cell cycle checkpoints in response toDNA damage. This is a highly simplified diagram that depicts only the most important players in the intricate and interwoven DNA damage–signaling networks.The traditional thinking was that ATM (ataxia telangiectasia mutated gene) is activated (phosphorylated) in response to ionizing radiation (IR) and ATR (ataxia tel-angiectasia- and Rad3-related gene) is activated in response to ultraviolet (UV) radiation, but newer data indicate that both are activated by IR and UV. ATM/ATRcan activate (i.e., phosphorylate) p53 either directly or indirectly through activation (phosphorylation) of Chk2 (an ATM target) or Chk1 (an ATR target). Throughtranscriptional activation of p21 and subsequent inhibition of cyclin-dependent kinases, which usually drive cells from the G1 into the S phase of the cell cycle,activated p53 activates the G1/S checkpoint (i.e., arrests cells in G1). The G1/S checkpoint is also activated by Chk1/Chk2-induced phosphorylation and thendegradation of Cdc25A and subsequent failure to activate cyclin-dependent kinases. Phosphorylation and subsequent degradation of Cdc25A, Cdc25B, andCdc25C also mediate the G2/M arrest, as does p21. Intra–S phase arrest is mediated through activation (phosphorylation) of p95/Nbs1. p53 also induces globalgenome nucleotide excision repair (GG-NER) through transcriptional activation of XPC, XPE/p48, and GADD45. Translesional DNA synthesis has been shown tobe downregulated by p53 via p21. Recombination repair is mediated through the Fanconi anemia (FA)/BRCA pathway, which in turn is dependent on ATR acti-vation. The mitochondrial pathway of apoptosis is activated through activation of Bax by caspase 2 and p53, the initiator caspase 9, and the effector caspases3, 6, and 7 (for references, see text). Gene products that are defective in hereditary human diseases are boxed. DDB2 = DNA damage binding protein 2; XPC,XPE = xeroderma pigmentosum complementation groups C and E.
DNA damage Caspase 2
Caspase 9
Caspase 3
Bax
Apoptosis
ATM ATR
FA/BRCApathway
Recombination repair
Chk2Chk1
p95/Nbs1
Cdc25A, B, Cdegradation
A, B, C A
Error-prone translesionalDNA synthesis
p53
XPE/DDB2
XPC
GADD45
GG-NER
G2/M G1/SCell cyclecheckpoints:
intra S
p21
SECTION 20■
CARCINOGENESIS
982
DNA damage) but share the same down-stream effector caspases.7
APPROACHES TO DIAGNOSIS OF GENOME INSTABILITY AND/OR DNA REPAIR DEFECTS
When a disorder of genome instabilityis suspected, the clinician is challengedwith choosing the appropriate labora-tory tests to secure a diagnosis and toprovide guidance for affected patientsand families. Table 109-5 lists some hall-mark clinical features that may indicatethe presence of such disorders andshould prompt clinicians to initiate test-ing. Various laboratory tests for genomestability, DNA repair, and response to
physical and chemical agents are listedin Table 109-6.
Use of Cultured Cells
Cells obtained directly from patients andgrown in culture medium are termed pri-mary cultures. Dermal fibroblasts gener-ally grow easily in culture and can gener-ally be established from a 2- to 4-mmsterile skin punch biopsy specimen. Theinner surface of the upper arm has provento be a suitable biopsy site because thisarea heals easily, the resulting scar is notreadily visible, the site is shielded fromUV radiation, and attempts to establishcultures from specimens are generallysuccessful. The tissue is placed in sterile
culture medium (or sterile saline) with an-tibiotics and transported to a cell culturelaboratory at room temperature.
Human cell cultures are made avail-able for research by the National Insti-tutes of Health–funded Human GeneticMutant Cell Repository (401 HaddonAve, Camden, NJ 08103; telephone: 856-966-7377; http://ccr.coriell.org/Sections/Collections/NIGMS/SsId=8).
Diagnostic Tests of Genome Instability and DNA Repair
Tests to assess genome instability and/orDNA repair capacity may be divided intotests of intact cellular function and tests ofchromosome integrity and breakage in re-sponse to DNA-damaging agents. Othertests measure the mechanism of impair-ment of a cell function such as DNA repairor characterize or determine the expres-sion of defective genes (see Table 109-6).
TESTS OF INTACT CELL FUNCTION Tests ofcellular function measure the capacity ofthe intact cell to recover from DNA dam-age. These tests do not provide informa-tion regarding the specific type of damageresulting in cellular injury or the mecha-nism of cellular recovery, but they do form
TABLE 109-6Diagnostic Tests of Genome Stability, DNA Repair, and Response to Physical or Chemical Agentsa
• Intact cell function (proliferation or cell death)• Cell counts (growth in mass culture)• Thymidine incorporation• Colony-forming ability
• Chromosome integrity and breakage• Giemsa-stained metaphase karyotype
analysis• G2 chromosome breakage• Sister chromatid exchanges• Fluorescent in situ hybridization• Microsatellite instability testing
• DNA repair• Unscheduled DNA synthesis• RNA synthesis inhibition• Host cell reactivation• Comet assay• Immunological analysis of DNA damage
and removal (enzyme-linked immuno-sorbent assay, slot blot)
• Microsatellite instability testing• Characterization and expression of defec-
tive genes• Real-time polymerase chain reaction• Western blotting• DNA sequencing
aAgents are listed in Table 109-1.
TABLE 109-4DNA Damage–Signaling Genes Associated with Human Diseases
GENE CHROMOSOME LOCATION HEREDITARY DISORDER
ATM 11q22.3 Ataxia telangiectasiaATR 3q22-24 Seckle syndromeFANCA 16q24.3 Fanconi anemiaFANCB Xp22.31 Fanconi anemiaFANCC 9q22.3 Fanconi anemiaFANCD1/BRCA2 13q12.3 Fanconi anemia, familial breast cancerFANCD2 3p25.3 Fanconi anemiaFANCE 6p21-p22 Fanconi anemiaFANCF 11p15 Fanconi anemiaFANCG 9p13 Fanconi anemiaFANCI Unknown Fanconi anemiaFANCJ 17q22 Fanconi anemiaFANCL 2p16.1 Fanconi anemiaBRCA1 17q21 Familial breast cancerp53 17p13.1 Li-Fraumeni syndromeChk2 22q12.1 Li-Fraumeni syndromeCDKN2A (p16INK4) 9p21 Familial malignant melanomaCDK4 12q14 Familial malignant melanomaNbs1 (p95) 8q21 Nijmegen breakage syndromeRb 13q14.1-14.2 Bilateral retinoblastoma
TABLE 109-5Hallmark Clinical Features That May Indicate a Disorder of Genome Instability or DNA Repair and Prompt Clinicians to Initiate Testinga
DISORDER CLINICAL FEATURES
Xeroderma pigmentosum Congenital photosensitivity, childhood freckling, early or multiple skin cancers
Cockayne syndrome Congenital photosensitivity, postnatal growth failure, sensorineuronal deafness
Trichothiodystrophy Brittle hair, photosensitivity, ichthyosisAtaxia telangiectasia Telangiectasias, ataxia, leukemias, and lymphomasBloom syndrome Photosensitivity, malar erythema, growth retardationFanconi anemia Aplastic anemia, growth retardation, café-au-lait spots, thumb
abnormalitiesMuir-Torre syndrome Sebaceous tumors, keratoacanthomas, personal or family history
of colon cancer
aRefer to Chap. 140 and Table 140-1 for more information.
CHAPTER 109■
GENOME INSTABILITY, DNA REPAIR, AND CANCER
983
the basis for identifying cells as hypersen-sitive to DNA-damaging agents and areoften used as simple screening tests.
Cell Counts or Thymidine Incorporation.One of simplest tests after exposure toUV radiation or x-rays is the assessmentof the growth rate in mass culture by
using a microscope or an automated cellcounter to count the number of cells orby measuring incorporation of radioac-tive thymidine into newly synthesizedDNA (see Tables 109-6 and 109-7).
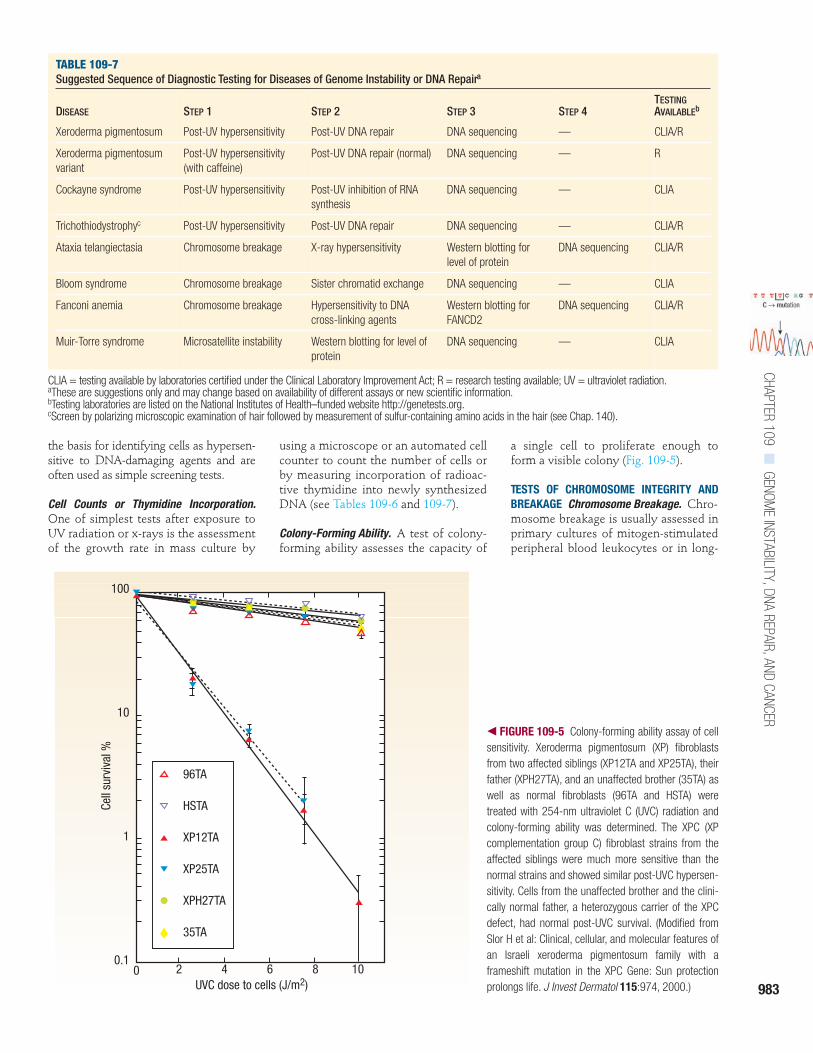
Colony-Forming Ability. A test of colony-forming ability assesses the capacity of
a single cell to proliferate enough toform a visible colony (Fig. 109-5).
TESTS OF CHROMOSOME INTEGRITY ANDBREAKAGE Chromosome Breakage. Chro-mosome breakage is usually assessed inprimary cultures of mitogen-stimulatedperipheral blood leukocytes or in long-
TABLE 109-7Suggested Sequence of Diagnostic Testing for Diseases of Genome Instability or DNA Repaira
DISEASE STEP 1 STEP 2 STEP 3 STEP 4TESTING AVAILABLEb
Xeroderma pigmentosum Post-UV hypersensitivity Post-UV DNA repair DNA sequencing — CLIA/R
Xeroderma pigmentosum variant
Post-UV hypersensitivity (with caffeine)
Post-UV DNA repair (normal) DNA sequencing — R
Cockayne syndrome Post-UV hypersensitivity Post-UV inhibition of RNA synthesis
DNA sequencing — CLIA
Trichothiodystrophyc Post-UV hypersensitivity Post-UV DNA repair DNA sequencing — CLIA/R
Ataxia telangiectasia Chromosome breakage X-ray hypersensitivity Western blotting for level of protein
DNA sequencing CLIA/R
Bloom syndrome Chromosome breakage Sister chromatid exchange DNA sequencing — CLIA
Fanconi anemia Chromosome breakage Hypersensitivity to DNA cross-linking agents
Western blotting for FANCD2
DNA sequencing CLIA/R
Muir-Torre syndrome Microsatellite instability Western blotting for level of protein
DNA sequencing — CLIA
CLIA = testing available by laboratories certified under the Clinical Laboratory Improvement Act; R = research testing available; UV = ultraviolet radiation.aThese are suggestions only and may change based on availability of different assays or new scientific information.bTesting laboratories are listed on the National Institutes of Health–funded website http://genetests.org.cScreen by polarizing microscopic examination of hair followed by measurement of sulfur-containing amino acids in the hair (see Chap. 140).
� FIGURE 109-5 Colony-forming ability assay of cellsensitivity. Xeroderma pigmentosum (XP) fibroblastsfrom two affected siblings (XP12TA and XP25TA), theirfather (XPH27TA), and an unaffected brother (35TA) aswell as normal fibroblasts (96TA and HSTA) weretreated with 254-nm ultraviolet C (UVC) radiation andcolony-forming ability was determined. The XPC (XPcomplementation group C) fibroblast strains from theaffected siblings were much more sensitive than thenormal strains and showed similar post-UVC hypersen-sitivity. Cells from the unaffected brother and the clini-cally normal father, a heterozygous carrier of the XPCdefect, had normal post-UVC survival. (Modified fromSlor H et al: Clinical, cellular, and molecular features ofan Israeli xeroderma pigmentosum family with aframeshift mutation in the XPC Gene: Sun protectionprolongs life. J Invest Dermatol 115:974, 2000.)
96TA
HSTA
XP12TA
XP25TA
XPH27TA
35TA
UVC dose to cells (J/m2)
Cell
surv
ival
%
100
10
1
0.10 2 4 6 8 10

SECTION 20■
CARCINOGENESIS
984
term cultures of fibroblasts or lympho-blastoid cell lines. Cell cycle progressionis stopped at metaphase by treatment ofthe cells with an inhibitor of mitosissuch as colchicine. In this procedure, the23 pairs of metaphase chromosomesfrom a single cell are spread over a dis-crete area of the slide and stained (usu-ally with Giemsa stain). Preparationsmay be analyzed for the number ofchromosomes per metaphase, the mor-phology of the individual chromo-somes, and the attachments or rear-rangements of chromosomes in relationto each other.
Sister Chromatid Exchange. During DNAreplication, chromatids occasionally ex-change positions along the arms of achromosome. This sister chromatid ex-change may be detected by permittingthe cells to grow through two cycles ofreplication in medium containing thenucleic acid analog bromodeoxyuridine(Fig.109-6). Sister chromatid exchangesare thought to be related to DNA re-combination repair, although their pre-cise significance is not understood.
TESTS OF DNA REPAIR Unscheduled DNASynthesis. One of the most commonlyused tests of NER is unscheduled DNAsynthesis. This test has been used tomeasure DNA repair in intact humanskin, in cultured epidermal or dermalcells, and in blood cells, and for prenataldiagnosis using amniotic fluid cells. Un-scheduled DNA synthesis testing mea-sures the repair-associated DNA synthe-sis in G1 or G2 cells (which usually donot synthesize DNA). Cells are treatedwith UV radiation or another DNA-damaging agent and then incubated inmedium containing radioactive thymi-dine. During the process of NER, thedamage is removed and the radioactivethymidine is incorporated into the re-paired region. The cells are treated withfixative, coated with autoradiographic(photographic) emulsion, and kept inthe dark for an appropriate interval, andthen the emulsion is developed. UV ra-diation of normal fibroblasts results in alarge increase in the number of grainsseen over all the nuclei. In marked con-trast, irradiation of the XP fibroblaststhat cannot repair DNA damage resultsin very few grains over the nuclei.
RNA Synthesis Inhibition. After exposureto UV radiation, normal cells tempo-rarily reduce RNA synthesis from activegenes. Synthesis resumes when thedamage is repaired. This resumption of
RNA synthesis is delayed in cells frompatients with Cockayne syndrome andsome forms of XP.
Host Cell Reactivation. The host cell reac-tivation assay relies on the fact that plas-mids do not have the ability to repair
� FIGURE 109-6 A. Sister chromatid exchange (SCE) assay of chromosome integrity. After the first cycleof replication, the DNA of the newly synthesized strand is labeled with bromodeoxyuridine (BrUdR), whereasthe older strand is unlabeled. Such chromosomes appear uniformly dark with Giemsa stain. After a secondcycle of replication in BrUdR-containing medium, one arm of a chromosome will contain two labeled chro-matids, whereas the other will contain one labeled and one unlabeled chromatid. The doubly substitutedarm will stain lightly, whereas the singly substituted arm will stain darkly. If an SCE occurred during replica-tion, a portion of each chromosome arm will be doubly substituted and the remainder singly substituted withBrUdR. B. Undamaged normal cultured peripheral blood lymphocytes have approximately 10 SCEs permetaphase. C. Cultured peripheral blood lymphocytes from a patient with Bloom syndrome have a manifoldincrease in SCEs. (From Chaganti RS, Schonberg S, German J: A manyfold increase in sister chromatid ex-changes in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A 71:4508, 1974, with permission.)
Stage of cell cycle
G1
S•G2
Firstmetaphase
G1
S•G2
Secondmetaphase
BrUdR
Uniformly darkstained chromosome
BrUdR
No SCE SCE
Differentialstaining
A
B C

CHAPTER 109■
GENOME INSTABILITY, DNA REPAIR, AND CANCER
985
damage to their DNA but depend on cel-lular repair systems. Thus, damaged plas-mids would be expected to be expressedat a higher level in cells with normal re-pair capacity. A non-replicating plasmidthat contains the gene for the firefly en-zyme luciferase, constructed to permitexpression in human cells,30 is widelyused; generation of light provides a quan-titative endpoint for its repair (Fig. 109-7).
Comet Assay. The comet assay is a sin-gle cell–based technique that allows de-tection and quantitation of DNA dam-age. Damaged nuclei are embedded inagarose, lysed, and exposed to an elec-tric field. The DNA migrates out of thenucleus forming a “comet” whenstained. The length of the comet is pro-portional to the fragmentation of theDNA. This assay can be modified fordetection of single-strand or double-strand DNA breaks, UV damage, or oxi-dative DNA damage. Cells from pa-tients with XP have defective repair inthe post-UV comet assay.
Microsatellite Instability. Normal DNA hastens of thousands of regions with re-peats of the dinucleotide CA or othershort motifs up to five nucleotides long.
In normal individuals each of these mi-crosatellites (also called simple sequencerepeats or short tandem repeats) has a uni-form size. However, these sizes arehighly variable among different individ-uals and are often used for DNA “finger-printing.” The appearance of abnor-mally longer or shorter simple sequencerepeats in different tissues or tumorsfrom a patient is called microsatellite in-stability. This can be associated with adefect in mismatch repair genes.
CHARACTERIZATION AND EXPRESSION OFDEFECTIVE GENES Real-Time PolymeraseChain Reaction. In some genome insta-bility genes, mutations create prematurestop codons for protein synthesis. Thesemutations result in low levels of mes-senger RNA (mRNA) for the gene prod-uct through a process called nonsense-mediated message decay. The mRNA levelscan be accurately measured by use ofquantitative reverse transcriptase real-time polymerase chain reaction. For ex-ample, low XPC mRNA levels havebeen found in cells from most XP pa-tients with defects in this gene.31
Western Blotting. Some mutations resultin reduced levels or size of encoded
proteins, often through creation of pre-mature stop codons.31 Reduced proteinlevels are most commonly detected byWestern blotting. Cells are lysed andthe proteins are extracted and separatedby gel electrophoresis. The separatedproteins are transferred to a membraneand probed with an antibody that isspecific for the protein of interest. Theintensity of the antibody staining re-flects the amount of protein in the cells,and its location on the membrane is anindication of the size of the proteinmolecules.
DNA Sequencing. Direct sequencing ofdefective genes is the gold standard fordetermination of the presence of a mu-tation. The final step in confirmation ofa diagnosis may be DNA sequencing todetermine the disease-causing muta-tion. By definition, disease-causing mu-tations alter the function of genes. Ofnote, however, not every alteration inthe sequence of a gene alters the func-tion of the encoded protein. In the hu-man genome there are millions of singlenucleotide polymorphisms, or changesin one nucleotide, that are not associ-ated with disease and may not evenchange the amino acid composition of
� FIGURE 109-7 Plasmid host cell reactivation assay with assignment to xeroderma pigmentosum (XP) complementation group. A. The plasmid is damagedby ultraviolet (UV) radiation and introduced into cultured human cells by a transfection technique. The cells’ DNA repair enzymes repair the damage in a mannersimilar to the repair of cellular DNA. Repaired DNA will then function to transcribe the plasmid-encoded luciferase gene in the human cell. The amount of lu-ciferase activity within the host cells therefore reflects the efficiency of the cellular DNA repair system. This assay can also be used to determine the comple-mentation group by co-transfecting UV-treated plasmid plus plasmids expressing wild-type XP complementary DNA (cDNA). B. The results of plasmid host cellreactivation experiments and complementation group assignment with DNA excision repair–deficient XP46DC cells. UV-treated plasmid showed low expressionin the XP65BE cells that was increased only by co-transfection with a plasmid expressing the wild-type XPC cDNA. This result indicates that XP65BE cells are inXP complementation group C (XPC). rLU = relative light units; pCMVluc and pLUC = plasmids containing gene for luciferase; pXPA–pXPG = plasmids express-ing xeroderma pigmentosum complementation groups A–G.
100
10
1
0.1
0
Dose of UVC to plasmid (J/m2)
Host
cel
l rea
ctiv
atio
nm
ean
rela
tive
luci
fera
se a
ctiv
ity(rL
U; %
of u
nirr
adia
ted
cont
rol)
500 10000
Post UV luciferace expression in complemented XPC cell line
B
pLUC + pXPApLUC + pXPBpLUC + pXPCpLUC + pXPDpLUC + pXPFpLUC + pXPG
1. Irradiation of plasmid with UV
Formation of DNA damage (DNA photoproducts)
2. Transfection of host cells
DNA repair and expression of luciferase
3. Determine luciferase activity in cell extract
Luciferase activity reflects repair efficiencyof host cells
A Host cell reactivation assay with the plasmid pCMVluc
2 d

SECTION 20■
CARCINOGENESIS
986
the encoded protein. In recessive disor-ders, each clinically unaffected parenthas one normal allele and one poten-tially disease-causing mutation in theother allele. The affected child receivesan allele with a disease-causing muta-tion from each parent. The two muta-tions must be in the same gene, al-though they need not necessarily beidentical to each other.
Considerations in Diagnostic Testing
Diagnosis of disorders of genome insta-bility or DNA repair is often a multi-step process. A suggested sequence ofsteps is listed in Table 109-7. As newtests are developed and new informa-tion is obtained about these disorders,testing procedures may change accord-ingly. In addition, decisions regardingthe extent of testing performed may bemade with consideration of how muchthe tests cost and whether additionalinformation would alter treatment. Forexample, DNA sequencing is rarely re-quired for establishment of a diagnosisof XP in a child with classical clinical
features and cells that are hypersensi-tive to killing by UV and have defectiveDNA repair. But if a family has onechild affected with XP and is consider-ing having additional children, DNA se-quencing might offer them the possibil-ity of prenatal diagnosis through DNAsequencing of a trophoblast biopsyspecimen.
Genetic counseling is an importantcomponent of patient management forthese genetic diseases. This functionmay be performed by the treating phy-sician or by a trained genetic counselor.
In the United States, only laboratoriescertified in accordance with the ClinicalLaboratory Improvement Act (CLIA) areallowed to perform these specializedtests in the context of patient care. Testsperformed in research laboratories gen-erally have limited use in clinical prac-tice. However, a research laboratorymay identify a disease mutation in cellsfrom a patient that could then be con-firmed in a CLIA-certified laboratoryand used in clinical practice. A listing oflaboratory testing facilities for these dis-eases may be found at the website
http://genetests.org, funded by the Na-tional Institutes of Health.
KEY REFERENCES
The full reference list for all chapters is available at www.digm7.com.
1. Friedberg EC et al: DNA Repair andMutagenesis. Washington, DC, ASMPress, 2006
2. Wikondahl NM, Brash DE: Ultravioletradiation induced signature mutationsin photocarcinogenesis. J Invest DermatolSymp Proc 4:6, 1999
8. Bootsma D et al: Nucleotide excisionrepair syndromes: Xeroderma pigmen-tosum, Cockayne syndrome, and tri-chothiodystrophy, in The Genetic Basis ofHuman Cancer, 2nd ed, edited by Vogel-stein B, Kinzler KW. New York,McGraw-Hill, 2002, p 211
21. Hanawalt PC: Subpathways of nucleo-tide excision repair and their regulation.Oncogene 21:8949, 2002
22. Lehmann AR: Replication of damagedDNA by translesion synthesis in humancells. FEBS Lett 579:873, 2005
31. Khan SG et al: Reduced XPC DNArepair gene mRNA levels in clinicallynormal parents of xeroderma pigmento-sum patients. Carcinogenesis 27:84, 2006
C H A P T E R 1 1 0
Carcinogenesis: ChemicalAndrzej A. DlugoszStuart H. Yuspa
CUTANEOUS CANCER AND PUBLIC HEALTH
Cutaneous cancers account for morethan one-half of all malignancies diag-nosed in the United States, totaling morethan 1 million non-melanoma skin can-cers (NMSCs) [800,000 to 900,000 basalcell carcinomas (BCCs) and 200,000 to300,000 squamous cell carcinomas(SCCs)] and over 62,000 melanomas, ac-cording to estimates for 2006 by theAmerican Cancer Society. The precisenumber of NMSCs is unknown, becauseunlike other malignancies, they are notroutinely reported to registries. Al-though only a small fraction of patientswith NMSC will die from their disease,which is nearly always SCC, the fre-quency of these cancers nonetheless re-
sults in an estimated death rate between1000 and 2000 per year. In contrast, al-though much less common than NMSC,melanoma has a death rate estimated atnearly 8000 per year,1 and a staggering 1 in59 individuals born today is predicted todevelop melanoma during his or her life-time, based on 2001–2003 data compiledby the National Cancer Institute’s Surveil-lance Epidemiology and End Results pro-gram (http://www.seer.cancer.gov). Be-cause they are so common, cutaneouscancers have a major impact on healthcare costs: in addition to the mortalityburden, treatment is associated withconsiderable morbidity and cosmetic de-fects. For these reasons, understandingthe etiology and pathogenesis of thesemalignancies is a significant publichealth goal, and development of ratio-nal, non-deforming therapies to reducemorbidity and mortality is urgentlyneeded. The high prevalence of skin can-cer, the external location of the tumors,and well-defined pre-neoplastic lesionsall provide an excellent opportunity forstudying the factors regulating cutane-ous cancer induction in humans. Thosequalities that facilitate the study of cuta-neous neoplasms in human populationshave also been useful in establishing rel-
evant animal models. Advances in mo-lecular genetics, keratinocyte cell cul-ture, and development of geneticallyaltered mice and reconstructed humanskin models have greatly facilitated theanalysis of basic mechanisms of cutane-ous carcinogenesis. The main focus ofthis chapter is on NMSC: the reader isreferred to Chap. 124 for further discus-sion of melanoma.
CHEMICALS ASSOCIATED WITH SKIN CANCER INDUCTION IN HUMANS
The ultraviolet radiation (UVR) in sun-light is the primary etiologic agent for allskin cancers, and, thus, UVR is the majorcarcinogen in the human environment.The powerful carcinogenic activity ofUVR is attributable to its ability to dam-age DNA and cause mutations, its ca-pacity to clonally expand incipient neo-plastic cells whose altered signalingpathways provide a survival advantagein the face of ultraviolet-induced cyto-toxicity, its ability to induce reactive oxy-gen species, and its activity as an im-mune suppressant (see Chap. 112). Theassociation of UVR with skin cancer is sostrongly supported by clinical, epidemio-


















