
Part 2 - UAH · Appropriate values: 0.2 – 0.3 CASPT2 IMAGINARY: imaginary shift SHIFT: real shift...
17
The 7 th MOLCAS Workshop Part 2 Cris, Alessio, Felipe and Marco
Transcript of Part 2 - UAH · Appropriate values: 0.2 – 0.3 CASPT2 IMAGINARY: imaginary shift SHIFT: real shift...

The 7th MOLCAS Workshop Part 2
Cris, Alessio, Felipe and Marco

Outline
• Density Fitting and Cholesky Decomposition
• Selecting the Active Space
• CASSCF and CASPT2 parameters
• RASSCF
• Solvent Effects: Polarized Continuum Model (PCM)
• Some Practical Hints

Density Fitting
Fitting of a “density” with an external predifined auxiliary basis
Two-electron integrals
Fitting coefficients optimized by minimizing the error
Electron-electron repulsion
• Auxiliary basis have to be adapted for each new method and valence basis set.
• Standard error in total energy around 10-2 kcal/mol.
• Analytic gradients and hessians available.

Cholesky Decomposition to approximate the two-electron integrals
On-the-fly generation of auxiliary basis
Cholesky Decomposition
• The accuracy of the decomposition depends on a single parameter.
• The level of accuracy is independent of the method.
• Standard error in total energy between 10-4 and 10-6 kcal/mol.
• Analytic gradients and hessians not yet available.
Lower triangular matrix
Calibration of Cholesky Auxiliary Basis Sets for Multiconfigurational Perturbation Theory Calculations of Excitation Energies, J. Boström, M. G. Delcey, F. Aquilante, Luis Serrano-Andrés, T. B. Pedersen, R. Lindh, J. Chem. Theor. Comput., 6:747-754 (2010).

Density Fitting or Cholesky Decomposition
&GATEWAY Title = CH4 molecule coord = CH4.xyz basis = STO-3G group = c1 RIJ / RIJK / RIC / RICD &SEWARD MEDIUM Cholesky / HIGH Cholesky &SCF
Decomposition threshold: 10-6 Decomposition threshold: 10-8
Use Cholesky Decomposition Resolution of Identity to treat the two-electron integrals (default threshold for on-the-fly generation of auxiliary basis: 10-4)
Density Fitting options ε
Cholesky-vectors rank
Full matrix rank
10-4
10-6
10-8
10-2

Selecting the Active Space
generates localized orbitals
&Gateway Coord=butadiene.xyz Basis=ANO-RCC-MB Group=noSym RICD &SEWARD &SCF >> LINK butadiene.ScfOrb INPORB &Localisation; Occu >> LINK butadiene.LocOrb INPORB &Localisation; Virt &Grid_it; All; Ascii
Calculations of molecular orbitals and density in a set of cartesian grid points.

Selecting the Active Space
OUTPUT
• $Project.localisation.molden • $Project.grid
>>> LINK butadiene.GvOrb INPORB &RASSCF; LumOrb
Orbital localisation makes use of the fact that a Hartree-Fock wave function is invariant under unitary transformations of the occupied orbitals:
where U is unitary (i.e. orthogonal for real orbitals). The same is true for the inactive or active orbitals in a CASSCF wave function.
Default method: Pipek-Mezey
The Localisation program defines U through an iterative maximization of a localisation functional.

CASSCF and CASPT2 parameters
Increase LEVShift parameter in case of slow or difficult convergence
CASSCF
Define a level shift value for the super-CI Hamiltonian. Appropriate values: 0.2 – 0.3
CASPT2
IMAGINARY: imaginary shift SHIFT: real shift
to avoid singularities created by weakly coupled intruders (e.g. in a potential curve)
First-order perturbation
Level shift
Second-order energy
The net effect is that the energy is almost unaffected except in the vicinity of the weak singularity, which is removed

CASSCF and CASPT2 parameters
IPEA shift: by default, H0 includes a 0.25 shift that modifies the energies of active orbitals, to reduce the systematic error which leads to an overestimation of the correlation energy for open shell systems.
It also reduces intruder problems.
In some cases IPEA = 0 gives better correlation with the experiments.
CASPT2

RASSCF
Módulo para llevar a cabo cálculos SCF multiconfiguracionales, tanto RASSCF como CASSCF.
Tratamiento de los MOs:
Espacio orbital completo: Espacio orbital RASSCF:
RASSCF CASSCF SD-CI …

RASSCF
Nactel = 8 1 1
Número total de electrones activos Número máximo de huecos permitidos en RAS1
Número máximo de electrones en el espacio RAS3
*RASSCF energy for CH4 at a fixed nuclear geometry *File: RASSCF.energy.CH4 * &GATEWAY coord = CH4.xyz basis = STO-3G group = C1 &SEWARD &RASSCF Title = CH4 molecule Spin = 1
Inactive = 1 Ras1 = 1; Ras2 = 6; Ras3 = 1 CIRoot= 2 1 1
Número total de raíces Dimensión de la matriz CI de partida
StateAverage ( 1= mismos peso para cada raíz)

Polarized Continuum Model (PCM)
•Efectos del disolvente en Molcas a través de dos modelos: - Modelo de Kirkwood - PCM (es el más común y realista de los dos)
•Se basa en cavidades construidas como una envolvente de esferas centradas en los átomos.
•El campo de fuerzas se describe en términos de cargas aparentes repartidas en pequeños elementos de la superficie de la cavidad. Así, se intenta reproducir el potencial electrostático generado por el disolvente.
•Para trabajar con este modelo debemos modificar el input con el fin de: - Indicar los parámetros de reacción en GATEWAY o SEWARD. - Añadir una serie de Keywords indispensables en algunos módulos.
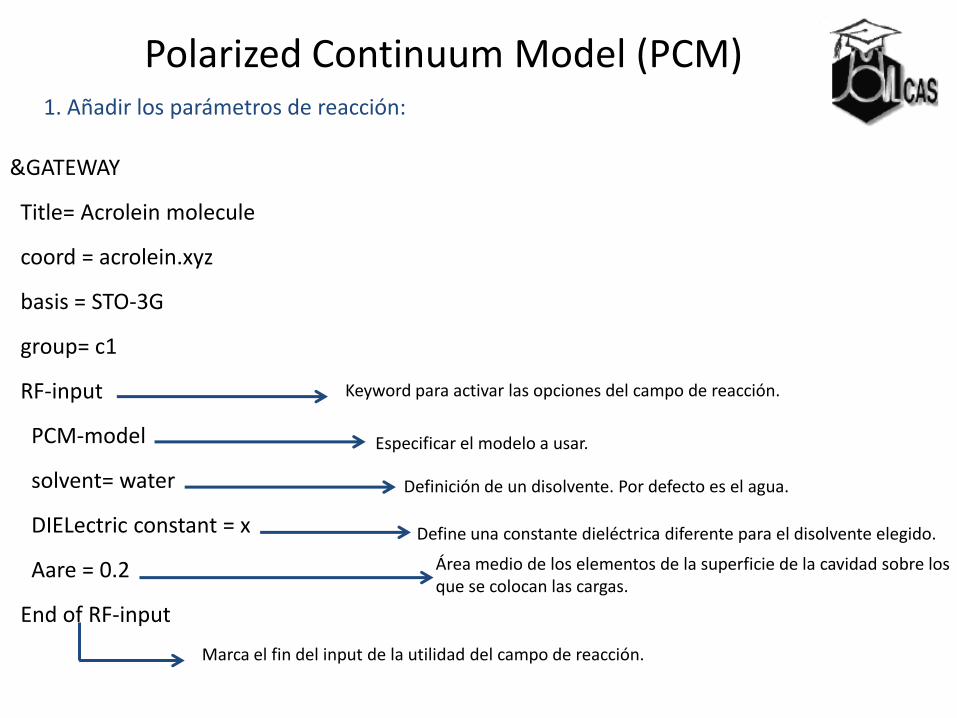
Polarized Continuum Model (PCM) 1. Añadir los parámetros de reacción:
&GATEWAY
Title= Acrolein molecule
coord = acrolein.xyz
basis = STO-3G
group= c1
RF-input
PCM-model
solvent= water
DIELectric constant = x
Aare = 0.2
End of RF-input
Keyword para activar las opciones del campo de reacción.
Especificar el modelo a usar.
Definición de un disolvente. Por defecto es el agua.
Define una constante dieléctrica diferente para el disolvente elegido.
Área medio de los elementos de la superficie de la cavidad sobre los que se colocan las cargas.
Marca el fin del input de la utilidad del campo de reacción.

Polarized Continuum Model (PCM) 1. Cálculos en estado fundamental:
&SCF
&RASSCF Spin= 1
Nactel= 6 0 0
Inactive= 12
Ras2= 5
CiRoot= 5 5 1
RFRoot= 5
&CASPT2
Multistate= 1 5
RFPert
No requiere ninguna Keyword adicional.
No requiere ninguna Keyword adicional excepto en cálculos StateAverage . Indica la raíz para la cual el campo de reacción es generado.
Para estos módulos se requiere que el campo de reacción sea incluido como una perturbación. Esta Keyword añade los efectos del campo de reacción al Hamiltoniano de un electrón como una perturbación constante.

Polarized Continuum Model (PCM) 2. Cálculos en estado excitado:
Durante una transición electrónica se observa
-una respuesta lenta: campo de fuerzas de los núcleos fijos tras excitación/emisión.
-una respuesta rápida: campo de fuerzas de la carga electrónica modificada tras la excitación/emisión.
Estado fundamental (equilibrio)
Estado excitado (no-equilibrio)
&GATEWAY Title= Acrolein molecule coord = acrolein.xyz; basis = STO-3G; group= c1 RF-input PCM-model; solvent= water End of RF-input &SEWARD &RASSCF Spin= 1; Nactel= 6 0 0; Inactive= 12; Ras2= 5 CiRoot= 1 1 1 &CASPT2 Multistate= 1 1 RFPert &RASSCF Spin= 1; Nactel= 6 0 0; Inactive= 12; Ras2= 5 CiRoot= 5 5 1 RFRoot= 5 NONEquilibrium &CASPT2 Multistate= 1 5 RFPert
Cálculo de equilibrio ( en el estado inicial)
Cálculo de no-equilibrio (en el estado final)
Congela la parte lenta (estado inicial) y calcula la parte rápida (estado final)

Some Practical Hints
Before using IMAGINARY, SHIFT or changing IPEA, try to increase the active space (also by including RAS1 and RAS3).
Multiconfigurational approach
Hardware/software issues
Memory can be more important than CPU speed: do not overload it (MOLCASMEM = 2000, as maximum).
By default, coordinates given explicitly in the input file are in Bohr, while coordinates given by an external file are in Ångström. Only cartesian coordinates can be inserted manually.
GATEWAY

Some Practical Hints
Do not feel that theory is always inferior to experiment.
In many cases experimentalists need us more than we need them, because they don’t know at all what they have measured.

















![Imaginary Phone: Learning Imaginary Interfaces by ... · Imaginary Phone: Learning Imaginary Interfaces by ... Our depth camera is a PMD[vision] CamCube that provides frames at 40Hz](https://static.fdocuments.net/doc/165x107/5ae222be7f8b9a097a8c8939/imaginary-phone-learning-imaginary-interfaces-by-phone-learning-imaginary.jpg)
