
PacBio® SMRT Sequencing Technology, Applications &...
86
FIND MEANING IN COMPLEXITY © Copyright 2013 by Pacific Biosciences of California, Inc. All rights reserved. 10/28/2013 PacBio® SMRT Sequencing Technology, Applications & Roadmap
Transcript of PacBio® SMRT Sequencing Technology, Applications &...

FIND MEANING IN COMPLEXITY
© Copyright 2013 by Pacific Biosciences of California, Inc. All rights reserved.
10/28/2013
PacBio® SMRT Sequencing Technology,
Applications & Roadmap

Outline
2
Technology RoadMap
Q&A
Applications
General Technology Overview/Current Performance

Single Molecule, Real-Time (SMRT®) DNA Sequencing
3
PacBio® RS Trace

The PacBio® Difference
Observes single molecules in real time to provide high-throughput
SMRT® Sequencing of DNA and base modifications simultaneously
• Generate finished genomes
• Discover a broad spectrum of
base modifications
• Characterize complex variations
• Extraordinarily long read lengths
• Extremely high accuracy
• Exquisite sensitivity
• Direct detection of a broad spectrum of DNA base modifications
• Shortest run time
• Least GC bias
• No amplification bias

Typical PacBio® RS II Results

SMRT® Sequencing Accuracy
Data generated with P4-C2 chemistry on PacBio® RS II;
Analyzed using Quiver with 2.0.1 SMRT® Analysis
Perspective: Understanding SMRT Sequencing Accuracy

Detection of DNA Base Modifications by SMRT®
Sequencing
Flusberg et al. (2010) Nature Methods 7: 461-465
7

Detectable by other Sequencing Methods
Signatures of Different DNA Base Modifications
8
Prokaryotic Eukaryotic DNA Damage

Integrated End-to-End Solutions
Easy, user-friendly,
web-based solutions
Streamlined data analysis
and viewing
Support for novice and
expert users

The PacBio® RS Helps Resolve Genetically Complex Problems
10
Generate finished
assemblies
De Novo Assembly
Comprehensively
characterize
genomic variation
Targeted
Sequencing
Automatically detect
DNA base
modifications
Base Modification
Detection

Microbiology on the PacBio RS

100K FoodBorne
Pathogen genome
project
Increase food safety using microbe systems biology
Discover the genetic constituents that are robust to be predictive biomarkers for specific traits
Rapid ID and tracking
Understand evolution to build more robust detection systems
New isolate emergence and persistence http://100kgenome.vetmed.ucdavis.edu
2012 HHSInnovate Secretary’s Choice Awardee

The Value of De Novo, Finished Microbial Genomes
• Less than 1% of the Earth’s
microbiome is known
• Horizontal gene transfer is
wide-spread and frequent
• High-quality, finished genomes are
the starting point for:
– Functional genomic studies
– Comparative genomics
– Forensics
– Metagenomics
Chain et al. (2009) Science 326: 236-237
Fraser et al. (2002) J Bacteriology 184: 6403-6405
Read Primer: The Value of Finished Bacterial Genomes

New Bioinformatics Solution:
Finishing Genomes Using Only PacBio® Reads
Full push-button solution from beginning to end
• Longest reads for continuity
• All reads for high consensus accuracy
Hierarchical Genome Assembly Process (HGAP)
HGAP Nature Methods (2013)

PacBio® Advantage of Even Coverage
Rubrivivax gelatinosus Gemmatimonadetes species
(*not drawn to scale*)
S. Noble, J. Yu, P. Maness, J. Chen, K. Wawrousek, C.
Eckert (NREL, U Wyoming)
S. Polson, R. Marine, D. Nasko, M. Radosevich, J. DeBruyn, E. Wommack
(U Delaware, U Tennessee)
5,075,070 bp
71.3% GC
5,312,117 bp
72.6% GC
20,957 bp
72.0% GC ~10x
1,106,726 bp
72.7% GC
1,040,999 bp
72.8% GC 235,512 bp
64.9% GC
47,366 bp
73.5% GC ~5x

Improving the Plasmodium Genome (23.3 MB)
Malaria:
– 350-500 million infections per year
– 1 million deaths per year
– 20% average GC content Plasmodium falciparum
454® pyrosequencing Sanger sequencing Illumina® sequencing SMRT® sequencing
Progeny Parents Reference genome 30 SMRT Cells
7C126 SC05 Dd2 HB3 NP-3D7-S NP-3D7-L 3D7
Number of Contigs 9,452 9,597 4,511 2,971 26,920 22,839 98
N50 Contig Size (kb) 3.3 3.3 11.6 20.6 1.5 1.6 1,242
Largest Contig (kb) 36.7 34.4 79.2 111.9 29.1 24.0 2,534
Number of assembled bases (Mb) 20.8 21.1 19.5 23.4 19.0 21.1 23.5
Average Coverage 33× 36× 7.8× 7.1× 43× 64× 155×
Sample provided by the Broad Institute & Sarah Volkmann (Harvard School of Public Health)
Samarakoon et al. (2011) BMC Genomics 12: 116.

Understanding Virulence in Infectious Diseases
Whooping cough
– Highly contagious
– Especially severe in children
– 48.5 million infections per year
– 295,000 deaths per year, 90% in developing countries
– Vaccines ~80% effective, but emergence of vaccine escape strains
– Infections & deaths on the rise in recent years, declared epidemic in California
(2010), Washington & Vermont (2012), UK (2012)
Bordetella pertussis

The Pertussis Genome is Extremely Repetitive
Bordetella pertussis E. coli
>1 kb
>2 kb
>5 kb
Repeats
>99% identical with length:
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands

Year Strain Sequencing Genome size Reference
2003 Tohama Sanger:
• 87,500 paired-end reads (1-4kb shotgun libraries)
• 2,560 paired-end reads (10-20kb pBAC library)
• 41,700 sequencing reads during finishing
4,086,186 bp Parkhill et al.
Nature Genetics
35: 32-40
Complete Pertussis Genomes

Year Strain Sequencing Genome size Reference
2003 Tohama Sanger 4,086,186 bp Parkhill et al.
2011 CS 454 & Sanger:
• 329,480 454 reads yielding 287 contigs
• 11,444 paired-end ABI3730 reads
• Filled gaps through sequencing of PCR products
4,124,236 bp Zhang et al.
J Bacteriology
193: 4017-4018
Year Strain Sequencing Genome size Reference
2003 Tohama Sanger:
• 87,500 paired-end reads (1-4kb shotgun libraries)
• 2,560 paired-end reads (10-20kb pBAC library)
• 41,700 sequencing reads during finishing
4,086,186 bp Parkhill et al.
Nature Genetics
35: 32-40
Complete Pertussis Genomes

Year Strain Sequencing Genome size
2013 B1917 6 SMRT Cells 4,102,176 bp
2013 B1920 8 SMRT Cells 4,114,613 bp
2013 B3405 6 SMRT Cells 4,109,986 bp
2013 B3582 8 SMRT Cells 4,104,315 bp
2013 B3585 8 SMRT Cells 4,106,397 bp
2013 B3640 8 SMRT Cells 4,110,999 bp
2013 B3658 6 SMRT Cells 4,103,245 bp
2013 B3913 6 SMRT Cells 4,109,515 bp
2013 B3921 4 SMRT Cells 4,111,519 bp
Finish Challenging Genomes with a Few SMRT® Cells
Complexity of Genome
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands
Strains of Bordetella pertussis genomes
>1 kb
>2 kb
>5 kb
Repeats
>99% identical
with length:
Watch Jonas Korlach present the complete story (AGBT 2013)

Compare Genome Organization Between Strains
1917
3582
1920
3585
3640
3405
3658
3913
3921
CS
Tohama
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands

Organization of Virulence Genes Differs Between Strains
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands

Phylogeny of Sequenced Pertussis Strains
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands

High Mobile-Element Diversity
using PHAST (http://phast.wishartlab.com)
Phage Element
To
ham
a
B19
17
B19
20
B34
05
B35
82
B35
85
B3
64
0
B36
58
B39
13
B39
21
Prophage Brucella suis 1330
Prochlorococcus phage P-SSM2
Burkholderia_phage_BcepGomr
Prophage Brucella suis 1330
Feldmannia_species_virus
Lactococcus_phage_bIL312
Cronobacter phage phiES15
Pseudoalteromonas phage H105/1
Spiroplasma_kunkelii_virus_SkV1_CR2_3x
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands
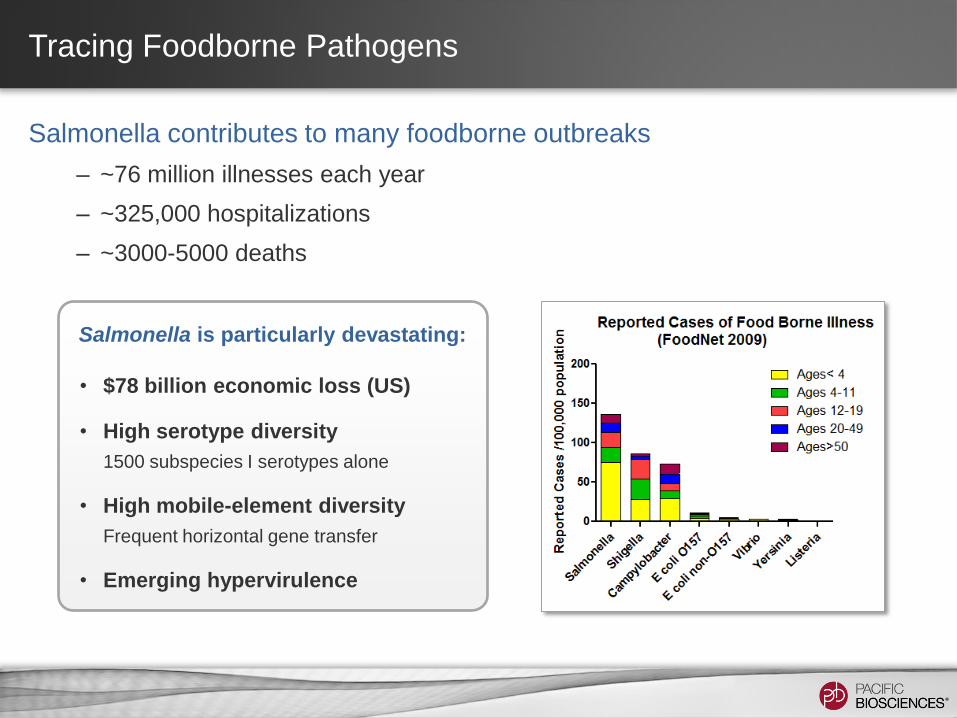
Tracing Foodborne Pathogens
Salmonella contributes to many foodborne outbreaks
– ~76 million illnesses each year
– ~325,000 hospitalizations
– ~3000-5000 deaths
Salmonella is particularly devastating:
• $78 billion economic loss (US)
• High serotype diversity
1500 subspecies I serotypes alone
• High mobile-element diversity
Frequent horizontal gene transfer
• Emerging hypervirulence

Assemblies of Salmonella Genomes
• HGAP assemblies of the complete Salmonella genome were
constructed in only a few weeks and revealed additional novel
genetic elements
Strain
Sequencing
(PacBio® RS) Genome size (bp) Additional genomic elements
S. Bareilly (SAL2881) 8 SMRT® Cells 4,730,611 78,193 bp
S. Heidelberg (318_04) 8 SMRT Cells 4,793,478 117,929 bp; 35,296 bp; 3969 bp
S. Heidelberg (2069) 8 SMRT Cells 4,783,941 110,345 bp; 37,704 bp
S. Typhimurium (2048) 8 SMRT Cells 4,967,892 142,804 bp; 48,532 bp
S. Javiana (1992_73) 8 SMRT Cells 4,629,444 24,013 bp; 17,094 bp
S. Cubana (2050) 12 SMRT Cells 4,977,480 166,668 bp; 122,863 bp
S. St.Paul SP3 8 SMRT Cells 4,730,130 none
S. St.Paul SP48 8 SMRT Cells 4,940,224 44,606 bp; 40,801 bp
Collaboration with M. Allard, E. Brown, E. Strain, M. Hoffman, T. Muruvanda, S. Musser (FDA),
R. Roberts (NEB), B. Weimer (UC Davis)
Read about the Genome

Diagnosing Active Salmonella Outbreaks with Finished Genomes
Clinical Arizona isolate from October
2012 produce-related outbreak
Complete process from isolate to finished genomic
sequence in <1 week on PacBio® RS
Finished Assembly Results:
• 1 chromosome
• 2 plasmids containing never-before-seen sequence
• Observed 4 active 6-mA methyltransferases
Collaboration with M. Allard, E. Brown, E. Strain, M. Hoffman, T. Muruvanda, S. Musser (FDA), R. Roberts (NEB), B. Weimer (UC Davis)
Genome Announc. March/April (2013) 1:doi:10.1128/genomeA.00081-13

Methylome of the German E. coli outbreak strain. The
inner and outer red circles show the kinetic signals. The
colored internal tracks show the different methylation motif
distributions.
Genome-wide detection of methylation for the German E. coli outbreak strain.
Characterization of Methylation Profiles
• Methyltransferases bind specifically to DNA motifs in a genome and methylate bases
• PacBio® software locates modified sites and motifs
Case Study: Beyond Four Bases - Epigenetic Modifications Prove Critical to Understanding E. coli Outbreak

CTGCAG Motif is Unique to Outbreak Strain
30
Methyltransferase Motif (weblogo) C227
outbreak 55989 782-09 17-2 734-09 760-09 35-10 042 1010
M.EcoGI Non-specific
M. EcoGII Non-specific
M. EcoGV
M. EcoGVIII
M. EcoGVI
M. EcoGIV
M. EcoGIII
M. EcoGVII
M. EcoGIX Non-specific
M. EcoGDam

CTGCAG Methylation Affects Gene Expression
31
Up-regulated
Down-regulated

New Bacterial Modification Systems Identified
Methyltransferase specificities for bacteria
sequenced recently on the PacBio® RS System
Type of Methyltransferase
(sequenced) I II III
Biochemically / Genetically
Characterized 69/20 854/722 22/21
Putatives from GenBank 3,480 15,226 1,600
Previously known
Previously unknown

Listeria monocytogenes Epigenomes
• Characterize strain methyltransferase diversity
• Identify novel methyltransferases
Serogroup 1/2a 1/2b 4b
Methyltransferase
Specificity
Modified
Base 861
878
899
1846
2074
2625
2626
2676
859
867
911
2624
G4599
1493
1494
1495
5'-GATC-3'
3'-CTAG-5' m6A
5’-GATC-3’
3’-CTAG-5’ X
5'-GACN5GGT-3'
3'-CTGN5CCA-5' m6A
5'-GAN6TGCG-3'
3'-CTN6ACGC-5' m6A
5'-TACN7GTNG-3'
3'-ATGN7CANC-5' m6A
5'-TAGRAG-3'
3'-ATCYTC-5' m6A
5'-GTATCC-3'
3'-CATAGG-5' m6A
complete methylation
partial methylation
5’-GAxTC-3’
5’-Gm6
ATC-3’
Collaboration with C. Tarr (CDC), H. den Bakker (Cornell U),
R. Roberts (NEB), B. Weimer (UC Davis)

Requirements for Achieving High-Quality Finished Genomes
1. High Consensus Accuracy
– Lack of systematic bias
2. Lack of sequence context bias
– GC content
– Low complexity sequence
3. Long sequence reads
– Resolve repeats, plasmids

Finished Microbial Genomes & Epigenomes on the
PacBio® RS II
35

PacBio® Benefits for Microbial Genomes
• Highest consensus accuracy (>99.999%)
• Complete bacterial chromosomes with minimal or no gaps
• Capture associated phage or plasmid elements
• Epigenomic characterization
• Simple sample preparation
• Rapid results in one week
• Full push-button bioinformatics solutions
• Cost effective
PacBio® Microbial Applications

Solutions for Plant & Animal Genomes

Discover Biology with Extraordinary Read Lengths
Complete microbial genomes and improve assemblies of larger organisms
• Highest N50
• Fewest fragments
• Detect structural variation
• 99.999% consensus accuracy
Read lengths up to 20 kb, unbiased genome coverage, and high accuracy
Finished bacterial genome
www.pacb.com/denovo

Improve and Finish Genomes with the PacBio® System
De novo Assembly
Complete genomes with
PacBio reads alone
Combine technologies
for best of both worlds
2 3
2 3
1
1
Scaffold
Establish framework for genome
and resolve ambiguities
Span Gaps
Polish genomic regions with up to
10x improvement
• HGAP
• PacBio2CA /
Celera® Assembler
• Others (Mira,
Cerulean, etc)
• AHA
• PB Jelly 2
• PB Jelly

Long Reads Span Difficult Genomic Regions
Address genomic challenges
with longer read lengths
• Resolve long palindromes
• Identify structural variants
• Obtain accurate microsatellite lengths
• Span homopolymeric, low-complexity, and
highly repetitive regions
• Delineate tandem repeats
Loomis et al. (2013) Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene.
Genome Research, 23(1):121-8
Fragile X gene with >2 kb of repeat regions
PacBio® reads span extreme
CGG repeats and AT-rich regions

Improving Atlantic Cod (G. morhua )
Genome with PacBio® Data
http://www.slideshare.net/flxlex/combining-pacbio-with-short-read-technology-for-improved-de-novo-genome-assembly
http://www.slideshare.net/flxlex/a-different-kettle-of-fish-entirely-bioinformatic-challenges-and-solutions-for-whole-de-novo-
genome-assembly-of-atlantic-cod-and-atlantic-salmon#btnNext
“When we looked at these PacBio
reads mapping to the assembly, we
saw them crossing large gaps of
even multiple kilobases. I could
see that the problem of STRs
and heterozygosity could be
addressed by this technology.”
Lex Nederbragt, Ph.D.
Research Fellow
University of Oslo
Case Study: Long Reads Offer Unique Insight

English et al. (2012) Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read
Sequencing Technology. PLoS One.
Towards Gap-Free Reference Genomes
D. melanogaster (139.5 Mb)
D. pseudoobscura (176.04 Mb)
M. undulatus (1.23 Gb)
C. atys (2.82 Gb)
Original PacBio Original PacBio Original PacBio Original PacBio
Gap Count 4651 311 6026 1852 49,376 39,204 186,841 66,211
Total Gap Size (Mb) 3.19 0.54 6.67 3.61 154.9 134.6 197.5 79.3
Contig N50 (kb) 64 723.6 53 224.4 134.4 233.27 34.92 128.38
Contig N50
Improvement
1030.6%
(11.3x) 323.4%
(4.2x) 73.6%
(1.74x) 267.6%
(3.68x)

Improve Assemblies with Low PacBio® Coverage
“With the RS, the contigs from
our de novo assembly of the 400
Mbp rice genome are several fold
better than the state-of-the-art
ALLPATHS-LG assembly using
short reads”
Michael C. Schatz, Ph.D.
Assistant Professor of Quantitative Biology
Cold Spring Harbor Laboratory
Rice Genome Assembly (Oryza sativa pv Nipponbare: 400 MB)
Contig N50
HiSeq® Fragments 50x 2x100bp @ 180
3,925
MiSeq® Fragments 23x 459bp
8x 2x251bp @ 450
6,332
Illumina® Mates 50x 2x100bp @ 180
36x 2x50bp @ 2100
51x 2x50bp @ 4800
18,248
PBeCR + Illumina reads 7x 3500bp ** MiSeq reads for
correction
50,995
PBeCR + Illumina reads 19x ** MiSeq reads for correction
155 kb
http://schatzlab.cshl.edu/presentations/2013-04-10.UVA.De%20novo%20assembly%20of%20complex%20genomes.pdf
http://schatzlab.cshl.edu/presentations/2013-06-18.PBUserMeeting.pdf
Case Study: The Next Frontier in Assembly – Long Reads Offer Finished Genomes

Hybrid Assembly for Improving Larger Genomes
Melopsittacus
undulatus
(parrot)
vs.
Taeniopygia
guttata
(Zebra finch) ®
®
More Gene Content in
Assemblies with PacBio®
Long Reads
Koren et al. (2012) Hybrid error correction and
de novo assembly of single-molecule
sequencing reads.
Nature Biotechnology 30, 693–700.

Tackle De Novo Assembly of Large Genomes
Novel Rumen Fungal Genome (100.95 MB) (Orpinomyces sp. Strain C1A)
• Anaerobic fungal cultures from Angus steer
• Motivation to understand biomass degradation
• 10x PacBio® sequencing improved Illumina® assembly
Illumina platform
(29 Gb) Illumina + PacBio platforms
(1 Gb)
Genome Assembly Size 105.1 MB 100.85 MB
# ambiguous bp 91,688 bp 0
# Contigs 82,325 32,574
N50 1 KB+ Contigs 2,226 bp 3,373 bp
N90 1 KB+ Contigs 1,072 bp 1,829 bp
Avg. Gene Model Length 903 1,623
# introns 2,458 35,697
# gene models 14,594 16,437
% GC Content 15.8 17
Youssef NH et al. (2013) The Genome of the Anaerobic Fungus Orpinomyces sp. Strain C1A Reveals the Unique Evolutionary History of a
Remarkable Plant Biomass Degrader. Appl Environ Microbiol, 79(15):4620-34
Significant improvements in gene models and transcript alignment.

Detect Transposable Elements
Improved genome assemblies
allows for transposable
element analysis.
Transposable element breakdown
for fungus Orpinomyces sp. Strain
C1A.
46
TE Class Detected occurance
Total Length (bp) % Genome Coverage
Cla
ss I
LTR
Copia 2,482 1,197,759 1.19
Gypsy 2,752 1,533,722 1.58
Pao 49 5,972 0.01
no
n-L
TR :
LIN
ES L1 77 38,441 0.04
L2 76 28,505 0.03
CR1 3 492 0.00
Rex 8 3,937 0.00
RTE 157 40,204 0.04
RTE-X 545 214,720 0.21 C
lass
II hA 9 1,926 0.00
MuDR 495 269,488 0.27
Pdinton 11 1,552 0.00
Total 6,664 3,336,718 3.31%
Youssef NH et al. (2013) The Genome of the Anaerobic Fungus Orpinomyces sp. Strain C1A Reveals the Unique Evolutionary History of a
Remarkable Plant Biomass Degrader. Appl Environ Microbiol, 79(15):4620-34

Resolve Difficult BACs
Aluminum tolerance in maize is important for drought resistance and
protecting against nutrient deficiencies
• Segregating population localized a QTL on a BAC, but unable to genotype
with short-read sequencing because of high repeat content and GC skew
• BAC assembly with PacBio® long reads revealed a triplication of the
ZnMATE1 membrane transporter
Genomic organization of the MATE1 locus
Maron, LG et al. (2012) A rare gene copy-number variant that contributes to maize aluminum tolerance and adaptation to acid soils. PNAS

PacBio® Long Reads Span Full Length Transcript
• Recover missing exons
• Gene structure annotation
• Identify gene isoforms and splicing events
• New gene identification in absence of reference genome
Koren et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads.
Nature Biotechnology 30, 693–700.
Example Annotation with Corn Transcriptome

Arabidopsis Assembly Offers Glimpse of De Novo SMRT
Sequencing for Larger Genomes
Sample Ler-0 Ler-0 Short Read
Assembly (2011)
Assembl
y Total
Size
124,572,7
84
110,357,1
64
Missing
significan
t chunk
# contigs 540 4,662 8.6X
more
Contig
N50
6,190,353 66,600 ~1/90th
PacBio’s
assembly
Max
Contig
Length
12,982,39
0
462,490
~ 1/30th of
PacBio’s
assembly
General Conclusion:
PacBio’s data provides a more complete assembly.

Pacific Biosciences’ Customers are Improving Large
Genome Assemblies
Large genomes assemblies being improved using
SMRT® Sequencing:
– Cotton
– Rice
– Wheat
– Salmon
– Sea Bass
– Medaka
– Pig
– …and more

PacBio® Benefits for Large Genomes
• PacBio complements short reads to improve
new and existing de novo assemblies
• Improve N50 contig length with modest
5x coverage
• Scaffold PacBio® long reads to set framework
for genome completion
• Resolve troublesome gaps with low-complexity
and repetitive genomic regions
• Increase annotations of gene structure with
transcripts
• Catalog transposable elements
PacBio® De Novo Assembly Homepage

PacBio® Targeted Biomedical Research
Applications

Targeted Sequencing: High-Resolution Insights
Exquisite sensitivity and specificity to fully
characterize genetic complexity
– Multi-kilobase reads
– 99.999% consensus accuracy
– Linear variant detection to <0.1% frequency
– Access to the entire genome
SNP Detection and Validation Repeat Expansions
Full-Length Transcripts and Splice Variants
Compound Mutations and Haplotype Phasing Minor Variants Detection
www.pacb.com/target

HLA Complex
54

PacBio® SMRT ® Full-Length Gene HLA Typing
55

Long-Read SNP Phasing
• Long reads provide haplotype directly
• Example: heterozygous SNPs across 5 kb amplicon at 20x coverage
maternal:
paternal:
maternal:
paternal:
vs.
~5 kb
Phasing:
A--C
T--A
A--A
T--C
…AGACACGACATGCG… …TCTGCACCGGCCT…
…GACTTGTCCGCGTT… …CAGCTTGAGGATA…
…AGACACGACATGCG…
…GACTTGTCCGCGTT…
…CAGCTTGAGGATA…
…TCTGCACCGGCCT…
56

Long-Read SNP Phasing
• Long reads provide haplotype directly
• Example: heterozygous SNPs across 5 kb amplicon at 20x coverage
• Phasing:
9 A--A
7 T--C
0 A--C
0 T--A
1 A--del
1 del--C
1 T--del
1 C--C
~5 kb 57

Full Phasing Information for HLA Haplotyping
• Amplified HLA-A region via long-range PCR (~3,000 bases)
• SMRT® sequencing of the full gene (all exons & introns) with long reads
• Phase and compare to HLA database:
Sample 1 HLA-A Type Comment
Best Match A*02:05:01
2nd Best
Match
A*02:06:01 5 SNPs from
the best match
Sample 2 HLA-A Type Comment
Best Match A*02:01:01:01
2nd Best
Match
A*02:07:01 only one SNP
from the best
match
Differ by 7 SNPs over
3 kb region
58

FLT3 Compound Mutations and Haplotype Phasing
• FLT3 mutations impact acute myeloid leukemia treatment
• Activating internal tandem duplication (ITD) mutations in FLT3 detected in ~
20% of AML patients and associated with a poor prognosis
• Potential resistance mutations located > 800 bp away from ITD region
F691 D835 Y842 E608
ITD
(20-100 bp repeat) > 800 bp
One PacBio® Read Spans Region
Smith et al. (2012) Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukemia. Nature
485, 260–263.
Case Study: A New Hope in Acute Myeloid Leukemia Treatment

Secondary and Rare Polyclonal Mutations for Resistance Identified
Pre-Treatment Relapse Normal Control #1
Subject
Number Mutation
Native
Codon
Alternative
Codon
Observed
Alternative
Codon
Frequency in
ITD+
Sequences
Total Number
of ITD+
Sequences
Sampled
Observed
Alternative
Codon
Frequency
In ITD+
Sequences
Total Number
of ITD+
Sequences
Sampled
Observed
Alternative
Codon
Frequency
Total Number
of Sequences
Sampled
1009-003 D835Y GAT TAT 0.21% 482 8.4% 332 0.00% 768
D835V GAT GTT 0.00% 482 3.3% 332 0.13% 768
D835F GAT TTT 0.00% 482 10.2% 332 0.00% 768
1011-006 D835Y GAT TAT 0.00% 196 41.0% 402 0.00% 768
1011-007 F691L TTT TTG 0.18% 561 6.2% 341 0.22% 450
D835Y GAT TAT 0.00% 930 3.0% 436 0.00% 768
D835V GAT GTT 0.43% 930 29.6% 436 0.13% 768
1005-004 F691L TTT TTG 0.00% 496 29.6% 513 0.22% 450
1005-006 D835Y GAT TAT 0.00% 171 39.5% 261 0.00% 768
D835F GAT TTT 0.00% 171 2.7% 261 0.00% 768
1005-007 D835Y GAT TAT 0.00% 57 4.0% 378 0.00% 768
D835V GAT GTT 0.00% 57 47.4% 378 0.13% 768
1005-009 D835Y GAT TAT 0.00% 19 50.6% 445 0.00% 768
1005-010 F691L TTT TTG 0.00% 387 25.3% 150 0.22% 450
Coupled Secondary Mutations Rare Polyclonal Mutations (<5%)
Smith et al. (2012) Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukemia. Nature
485, 260–263.

Trinucleotide Repeat Disorders
• Set of genetic disorders caused by trinucleotide repeat expansion
• A mutation where trinucleotide repeats in certain genes exceed the normal,
stable, threshold, which differs per gene
• Disease Examples:
– Autism
– Mental retardation (especially males)
– Huntington’s disease
– Fragile X syndrome
AGGTAT CGGCGGCGGCGGCGGCGGCGGCGGCGG AGATC …
AGGTAT CGGCGGCGGCGGCGGCGGCGGCGGCGG AGATC …
120
600+

A First Look into a Repeat Expansion Disorder
• Repeat regions sequenced accurately
• Sequence through up to 750 CGG repeats
Loomis et al. (2013) Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene. Genome Research, 23(1):121-8
Case Study: SMRT® Sequencing Provides a First Look at Repeat Expansion Disorder Sequence

Filling the Genomic Gap in MUC5AC Gene using PacBio Long
Reads and De Novo Assembly
63

SMRT® Sequencing of Intact, Full-Length HIV-1 Genomes From
Single Molecules to Study HIV Transmission
• Complete HIV-1 genomes from single
molecules
– Sanger-quality, fully phased
– Samples of complex mixtures
• Eliminate need for Single Genome
Amplification
• Full genomic characterization of clinical
transmission events
Donor (chronic infection)
Recipient (acute infection)
Full HIV Genome – 9, 084 bp
Collaboration with CFAR site at Emory University
Poster: Rapid Sequencing of HIV-1 Genomes as Single Molecules from Simple and Complex Samples

Reliably Detect Variant Mutations Below 0.1% Frequency
All minor variants
reliably detected
down to 0.08%
L180M
254 C A
S202G
320 A G
M204V
326 A G
Single SMRT® Cell provides enough data to detect HBV minor variants:
Poster: Sensitive Detection of Minor Variants and Viral Haplotypes

Complete Capture of Full-Length, No Assembly Transcripts
• Recover missing exons
• Gene-structure annotation
• Identify gene isoforms and splicing events
• New gene identification
Single PacBio read covers 32-exon mRNA:
Poster: Full-Length cDNA Sequencing on the PacBio® RS
• Entire length of human glutamyl-prolyl-tRNA synthetase mRNA
• cDNA primers observed at both ends
• PolyA tail observed at the 3’ end indicates correct end capture

A Strength of PacBio Long Read Technology is the
Unambiguous Detection of Splice Isoforms
CCS alignments uncover multiple isoforms of the CDK4 transcript. Exons 2, 3, 4, 5, 6, and 7 are
skipped in various combinations. The 5’ end also shows variable transcription start sites.

Survey of the human transcriptome using PacBio
68

Validation of Illumina® SNPs with SMRT® Sequencing
• Whole‐exome hybrid capture and deep sequencing to identify somatic
mutations in 92 primary medulloblastoma‐normal pairs
• All SNPs studied were validated (including 2 bp deletion in CTDNEP1)
69
PacBio®
Illumina®
PacBio®
Illumina®
Pugh et al. (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106-110.

Accurate SNP Detection
Discordant PacBio® data confirmed by
PCR-Sanger to be 100% correct
Genomic Regions Associated
with Mental Retardation
• Comparison of a BAC libraries thought to
be highly similar to HG19 reference
genome
• Discordant bases (~30 sites) between
PacBio data and HG19 (Sanger) data
were identified
• PacBio calls 100% confirmed by PCR-
Sanger
Clone HG19 Ref PacBio Sanger
BAC 1
T G G
T -- --
T A A
G T T
C T T
C T T
G C C
C G G
BAC 2
C T T
C G G
A G G
G A A
T C C
T C C
C T T
BAC 3
T -- --
T -- --
T C C
T C C
G T T
A C C
BAC 4
A G G
T C C
A T T
T -- --
T G G
In collaboration with Evan Eichler (HHMI, University of Washington)

Implementation of cancer sequencing in the clinic

Using PacBio sequencing in CLIA setting
72

Pathogenic Repeat Panels Carrier Screening
73

TNR
74

Beyond Targeted Sequencing: SMRT® Sequencing of Whole Human Genome Reveals Undetected Variations
Mt. Sinai human-genome sequencing (NA12878 from CEPH)
• Detect clinically significant variants not detected with short-read technologies
• Identify unexplored structural variants across the genome
• Develop new, clinically relevant gene panels only identifiable using PacBio® technology
454® Reads
Number of reads ~100M
Mapped coverage ~15X
Single and Paired-End Illumina® Reads
Number of reads ~100s of M
Mapped coverage ~30X
PacBio® Reads
Number of Reads ~12M
Mapped coverage 10X+
Mean subread length 2,766
Mean unrolled read length 4,066
95th Percentile 11,630
Accuracy (error-corrected reads) >99%
Watch Eric Schadt present the complete story

PacBio® Benefits for Targeted Biomedical Research
• Achieve >99.999% consensus accuracy (QV 50)
• Direct strand-specific haplotype phasing with multi-kilobase reads
• Resolve troublesome gaps with low-complexity and repetitive genomic
regions
• Improve gene-structure annotation with intact, full-length transcripts
• High-resolution detection of low-frequency minor variants
• Finish human genomes
PacBio® Targeted Sequencing Applications Page

Recent Publications
1) Efficient and accurate whole genome assembly and methylome profiling of E. coli
Authors:Jason G Powers, Victor J Weigman, Jenny Shu, John M Pufky, Donald Cox and Patrick Hurban
2) An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome
Authors:Marco Ferrarini, Marco Moretto, Judson A Ward, Nada Surbanovski, Vladimir Stevanovic, Lara Giongo, Roberto Viola,
Duccio Cavalieri, Riccardo Velasco, Alessandro Cestaro and Daniel J Sargent
3) Reducing assembly complexity of microbial genomes with single-molecule sequencing
Authors: Sergey Koren, Gregory P Harhay, Timothy PL Smith, James L Bono, Dayna M Harhay, Scott D Mcvey, Diana Radune,
Nicholas H Bergman and Adam M Phillippy
4) Genome Reference and Sequence Variation in the Large Repetitive Central Exon of Human MUC5AC
Authors: Xueliang Guo, Shuo Zheng, Hong Dang, Rhonda G Pace, Jaclyn R Stonebraker, Corbin D Jones, Frank Boellmann, George
Yuan, Prashamsha Haridass, Olivier Fedrigo, David L Corcoran, Max A Seibold, Swati S Ranade, Michael R Knowles, Wanda K
O'Neal, and Judith A Voynow
5) Comparing the genomes of Helicobacter pylori clinical strain UM032 and Mice-adapted derivatives
Authors: Yalda Khosravi, Vellaya Rehvathy, Wei Yee Wee, Susana Wang, Primo Baybayan, Siddarth Singh, Meredith Ashby, Junxian
Ong, Arlaine Anne Amoyo5, Shih Wee Seow, Siew Woh Choo, Tim Perkins, Eng Guan Chua, Alfred Tay, Barry James Marshall, Mun
Fai Loke, Khean Lee Goh, Sven Pettersson and Jamuna Vadivelu
6) The advantages of SMRT sequencing
Authors: Richard J Roberts, Mauricio O Carneiro and Michael C Schatz
7) McGill University Team Develops Rapid Genome Sequencing Technique for Outbreak Monitoring 77

FIND MEANING IN COMPLEXITY
© Copyright 2013 by Pacific Biosciences of California, Inc. All rights reserved.
PacBio® Roadmap

PacBio® Advances in Chemistry and Software
79
Early PacBio chemistries
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
q108 q208 - 453
q308 q408 q109 q209 -
1012
q309 -
1734
q409 q110 q210- lpr
q310 q410 q111 q211 - fcr
q311 q411 - ecr2
q112 q212 - c2
q312 q412 - xl
q113 q213 q313 q413 -
p5c3
453 1012
1734 LPR
FCR
ECR2
C2–C2
P4–C2
P5–C3
8,500 bp
Read L
ength
(b
p)
2008 2009 2010 2011 2012 2013

BluePippin™ System for Size Selection

Pol Protecting
Scaffold
Dye
Pol
Polymerase
surface that
dye can
access
Dye cannot
access
polymerase
surface
Photodamage Mitigation: Photo-Protected Analogs
Large macromolecule scaffold can prevent the dye from touching the polymerase
Dye
81

BluePippin™ Size-Selection Protocol
P5-C3, E coli 20 kb BluePippin™ size-selected library, 3-hour movie
P5-C3, E. coli 10 kb library, 3-hour movie
Avg Subread lengths: 3,427 bp
Subread N50: 5,607 bp
Mapped Reads: 47,321
Avg Subread lengths: 7,537 bp
Subread N50: 10,725 bp
Mapped Reads: 41,301
Official Procedure for 20 kb Template Preparation

Impact of Improvements for Microbial Assembly
0
4
8
12
16
20
24
28
32
Jan 2012
C2 Release Sept 2012
MagBead
Dec 2012
XL Release
32 SMRT Cells
Two SMRTbell™ libraries
DevNet tools - hybrid
Degree of Completeness & Quality
SM
RT
® C
ells
<35 contigs,
Q40
<5-10 contigs, Q50,
plus methylome
16 SMRT Cells, 2 libraries
Hybrid - Celera® Assembly
Identify base modifications
Quiver for consensus (DevNet)
4 - 8 SMRT Cells
Single SMRTbell library
HGAP assembly pipeline w/ Quiver
Automated methylation detection
Q2 2013
150K, Size
Selection
1 SMRT Cell
Size-selected library
HGAP w/ Quiver
Single contig per
chromosome, Q50,
plus methylome
Examples based on E. coli

Upcoming Improvements Will Make Generation of 10X
PacBio® Coverage of 3 GB Genomes More Economical
84
Estimated per
SMRT® Cell
Throughput (MB)*
Estimated Number
of SMRT® Cells
per 10X
Instrument
Run Time
(days)
Beginning of 2013 ~100 300 38
Today’s
Throughput
-150K
~200 150 19
Optimally loaded
size-selected
libraries
~400 75 7
Photo-protected
Analogs ~800 38 4
one SMRT Cell =
one microbial
de novo genome
& epigenome

FIND MEANING IN COMPLEXITY
© Copyright 2013 by Pacific Biosciences of California, Inc. All rights reserved.

Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, and SMRTbell are trademarks of Pacific Biosciences in
the United States and/or other countries. All other trademarks are the sole property of their respective owners.


















