
Oxygen Toxicity - hboorcca.com 6 Oxygen Toxicity.pdf · Oxygen Toxicity K.K.Jain I,~~, Prolonged...
12
Oxygen Toxicity K.K.Jain I,~ ~, Prolongedexposure to oxygen at high pressure can have toxic effects, particularly on the central nervousystem,but at pressures used clinically it does not pose a problem. The main topics discussed here are: Pathophysiology of CNS Oxygen Toxicity 48 Pathologyof Oxygen Toxicity 49 PulmonaryOxygen Toxicity 51 Oxygen-Induced Retinopathy 52 Factorsthat Enhance Oxygen Toxicity 52 CentralNervous System Oxygen Toxicity 53 ClinicalMonitoring for Oxygen Toxicity 56 ProtectionAgainst Oxygen Toxicity 56 Extensionof Oxygen Tolerance 57 Conclusionand Directions for Future Research 58 - i -
Transcript of Oxygen Toxicity - hboorcca.com 6 Oxygen Toxicity.pdf · Oxygen Toxicity K.K.Jain I,~~, Prolonged...

Oxygen Toxicity
K.K.Jain
I,~~,
Prolongedexposure to oxygen at high pressure can have toxic effects, particularly on the centralnervoussystem,but at pressures used clinically it does not pose a problem. The main topics discussedhereare:
Pathophysiology of CNS Oxygen Toxicity 48Pathologyof Oxygen Toxicity 49PulmonaryOxygen Toxicity 51Oxygen-Induced Retinopathy 52Factorsthat Enhance Oxygen Toxicity 52CentralNervous System Oxygen Toxicity 53ClinicalMonitoring for Oxygen Toxicity 56ProtectionAgainst Oxygen Toxicity 56Extensionof Oxygen Tolerance 57Conclusionand Directions for Future Research 58
-
i
-

48 Chapter 6
-- ~----- --_.- - - -
Introduction
Priestley (1775, see Chapter 1), the discoverer of oxygen,theorized about the effects of hyperoxia in this charmingpassage:
We may also infer from these experiments, that though puredephlogisticated air might be very useful as a medicine, it
might 110tbe so properfor use in the healthy state of the body:
for, as a cal1dle bums out much faster in dephlogisticated than
in COlllmOI1air, so we might, as might be said, live out toofast,and the al1imal powers be too SOOI1exhausted il1 this pure kindof air. A moralist, at least, may say that the air which l1aturehas provided for us is as good as we deserve.
Paul Bert ( 1878, see Chapter 1) was the first to actually document the toxicity of oxygen. He conducted experiments totest the effects of hyperbaric oxygen (HBO), not only onhimself but also on other life forms. Indeed, seizures result
ing from the toxicity of oxygen to the central nervous systemare still referred to as the "Paul Bert effect." Although hiswork is a classic, Bert com pletely missed pulmonary toxicityas an effect of normobaric oxygen. This was later discoveredby Lorraine Smith (1899) and is fittingly referred to as the"Lorraine Smith effect." Bean (1945) studied the toxic effects
of continuous e:x.'P0sure to HBO beyond the point of seizures, to irreversible neurological damage and eventualdeath; this problem is now widely known as the "John Beaneffect. "
Behnke et al (1936) carried out a variety of experimentsin human subjects to show the effects of oxygen toxicity. Asa result of these earlier studies it became generally acceptedthat a 3-h exposure at 3 ATA and a 30- to 40-min exposureat 4 ATA were the limits of safe tolerance by healthy humanadults. It is now generally accepted that HBO at 3 ATA affects primarily the nervous system, while the respiratorysystem is affected independently at 2 ATA. There is a vastamount of literature on basic mechanism of oxygen toxicity (Bean 1945; Balentine 1982).
This chapter describes mainly the toxic effects of HBO.
Normobaric hyperoxia, which usually leads to pulmonaryoxygen poisoning, has been dealt with in detail elsewhere(Jain 1989a).
Pathophysiology of Oxygen Toxicity
The molecular basis of CNS as well as pulmonary oxygenpoisoning, involves generation of reactive oxygen species(ROS). This has been known as the free radical theory ofoxygen poisoning. The basis of this theory, for the CNSo:x.l'gentoxicity, is that an increased generation ofROS during HBO may ultimately lead to alterations in cerebral energy metabolism and electrical activity due to membraneslipid peroxidation, enzyme inhibition, and/or enzymemodulation. Although HBO-induced generation of ROScould directly alter the functions of various SH-containingenzymes, membrane-bound enzymes and structures as
well as the nucleus, the physiological effects of HBO mayalso indirectly cause hypoxic-ischemia, acidosis, anemia,and hyperbilirubinemia.
At higher pressures of oxygen, events in the brain are aprelude to a distinct lung pathology. The experimental observation that CNS-mediated component of lung injurycan be attenuated by selective inhibition of neuronal nitricoxide synthase (nNOS) or by unilateral transection of the
vagus nerve has led to the hypothesis that extrapulmonary,neurogenic events predominate in the pathogenesis ofacute pulmonary oxygen toxicity in HBO, as nNOS activitydrives lung injury by modulating the output of centralautonomic pathways (Demchenko et al 2007).
Free Radical Mechanisms
Oxygen free radicals are products of normal cellular oxidation- reduction processes. Under conditions of hyperoxia,their production increases markedly. The nature of the ox-
Table 6.1
Animal Experimental Studies of the Effect of HBO on Brain lipoperoxide Levels
EffectsAuthors and year Pressure
Zirkle et al (1965)
4ATA
Galvin (1962)
2ATA
Jerrett et al (1973)
5ATA
Yusaet al (1987)
3ATA
Torbati et al (1992)
5ATA
Clinical level of CNS toxicity correlated with elevated lipoperoxide content; convulsionswere considered to be due to raised AChE (acetylcholinesterase) activity
Cerebral peroxide levelnot elevated;no difference in the peroxide levelbetween convulsing and nonconvulsing animals
H202levels were elevated except in those animals given supplemental a-tocopherol
Rise ofH202level in brain by 300% when symptoms ofCNS toxicity became apparent
Direct demonstration of reactive oxygen species before onset of CNS convulsions

Oxygen Toxicity
Metabolism of oxygenvia free radicals
1 Unknown mechanism
~ Lipoperoxides ~
Disruptionof membranes 1 Oxidation of glutathione
Oxidation of SH groups 1Enzymes inhibited
~Xidation of pyridine
nucleotides
1Impaired energy
~ production
Cell injury
(Alteredmembranes, impaired mitochondria, impaired neurotransmission,inhibition of nuclear function and/or protein synthesis)
1Cell death
49
Figure 6.1Summary of the hypothesis of oxygentoxicity. SH = sulfhydryl. (Reproducedfrom Chance and Boveris 1978, by permission.)
ygenmoleculemakes it susceptible to univalent reductionreactionsin the cells to form superoxide anion (02-) a highlyreactivc,cytotoxic free radical. In turn, other reactionproductsof oxygen metabolism, including hydrogen peroxide(H20Z), hydroA")'1radicals (OH·), and singlet oxygen(IO~), can he formed. These short-lived forms are capableofoxidizingthe sulfhydryl (SH) groups of enzymes, interactwithDNA, and promote lipoperoxidation of cellularmembrancs.Animal studies showing the effect of HBO inraisingthccerebral peroxide content and correlating it withC;-':S toxicityare listed in Table 6.1.
Boverisand Chance presented an excellent unifying concrptof thcmechanism of oxygen toxicity in 1978, which isaclassicnow.H20Z generation as a physiological event hasorendocumented in a variety of isolated mitochondria andisrapidlyenhanced by hyperoxia. Superoxide ions, generatedsubmitochondrial fractions, are the source of H202
(Bo\'eris& Chance 1978). This hypothesis is shown in Figurr6.1.Asa primary event, the free radical chain reactionsproducelipoperoxidation. Lipoperoxides, in turn, will havedisruptivceffects on the structure of the biomembranes,inhibitenzymes with SH groups, and shift the cellular redmstateof glutathione toward oxidation. This will be tranImittedthrough the secondary events to pyridine nucleotides,withthe mitochondrial NAD H oxidation resulting in
impaired energy production. Enzyme inhibition, alteredenergy production, and decrease or loss of function may beconsequences of either increased peroxides or a decline inthe antioxidant defence.
Although increased generation of ROS before the onsetof HBO-induced convulsions has been demonstrated in
conscious rats, their production in association with oxygentoxicity has not been demonstrated satisfactorily in humansubjects. There are increased electron spin resonance signals from blood of persons exposed to HBO but these return to normal within 10 min of cessation of exposure toHBO.
Pathology of Oxygen Toxicity
The pathology of oxygen toxicity has been documentedcomprehensively in a classical work on this topic (Balentine 1982). The various manifestations of O:A")'genpoisoning are summarized in Figure 6.2. It is well known thatthe development of pulmonary and CNS toxicity dependsupon the partial pressure and the duration of exposure,as shown in Figure 6.3. Fortunately, the early effects ofpoisoning are completely reversible, but prolonged expo~
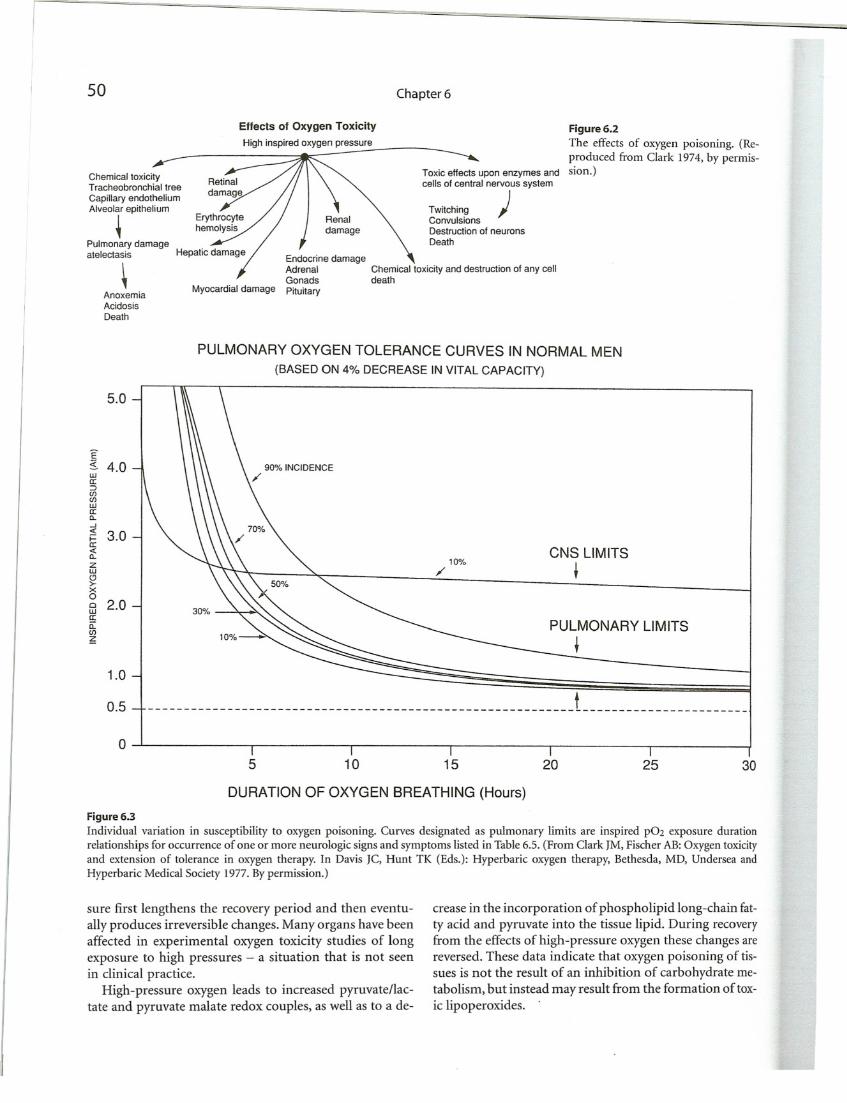
Chemical toxicity and destruction of any celldeath
TwitchingConvulsionsDestruction of neuronsDeath
50
Chemical toxicityTracheobronchial tree
Capillary endotheliumAlveolar epithelium
~Pulmonary damageatelectasis
AnoxemiaAcidosisDeath
Effects of Oxygen Toxicity
High inspired oxygen pressure
Myocardial damage
Chapter 6
Toxic effects upon enzymes andcells of central nervous system
)
Figure 6.2
The effects of oxygen poisoning. (Reproduced from Clark 1974, by permission.)
PULMONARY OXYGEN TOLERANCE CURVES IN NORMAL MEN
(BASED ON 4% DECREASE IN VITAL CAPACITY)
5.0
E5. 4.0wa::::>U)U)wa:a..-'i:§ 3.0a:...:a..ZwCJ>xofiJ 2.0a:a:U)~
1.0
30%
90% INCIDENCE/
10%/CNS LIMITS
t
PULMONARY LIMITS
t
0.5
o
---------------- t _
5 10 15 20 25 30
DURATION OF OXYGEN BREATHING (Hours)
Figure 6.3
Individual variation in susceptibility to oxygen poisoning. Curves designated as pulmonary limits are inspired pOz exposure durationrelationships for occurrence of one or more neurologic signs and symptoms listed in Table 6.5. (From Clark 1M, Fischer AB: Oxygen toxicityand extension of tolerance in oxygen therapy. In Davis IC, Hunt TK (Eds.): Hyperbaric oxygen therapy, Bethesda, MD, Undersea andHyperbaric Medical Society 1977. By permission.)
sure first lengthens the recovery period and then eventually produces irreversible changes. Many organs have beenaffected in experimental oxygen toxicity studies of longexposure to high pressures - a situation that is not seenin clinical practice.
High-pressure oxygen leads to increased pyruvate/lactate and pyruvate malate redox couples, as well as to a de-
crease in the incorporation of phospholipid long-chain fatty acid and pyruvate into the tissue lipid. During recoveryfrom the effects of high-pressure oxygen these changes arereversed. These data indicate that m,"ygenpoisoning of tissues is not the result of an inhibition of carbohydrate metabolism, but instead may result from the formation of toxic lipoperoxides.

II
Oxygen Toxicity 51
Thepowerexpression for cumulative oxygen toxicity andtheexponential recovery have been successfully applied torariousfeaturesof oxygen toxicity at the Israel Naval MedicalInstitutein Israel (Arieli et a/2002). From the basic equation,
theauthorsderived expressions for a protocol in which P02
changeswith time. The parameters of the power equationweresolvedby using nonlinear regression for the reductioninritalcapacity (DeltaVC) in humans:
OeltaVC = 0.0082 x tz (POz/101.3)(4.57)
wheret is the time in hours and P02 is expressed in kPa.
Therecovery of lung volume is:
IJdtaVC(t) = DeltaVC(e) x e(-(-Q.42 + 0.00379 POz)t)
whereDdtaVC(t) is the value at time t of the recovery, Del
taVC(e)is the value at the end of the hyperoxic exposure,andPO! is the prerecovery oxygen pressure.
Datafrom different experiments on CNS oxygen toxicityinhumansin the hyperbaric chamber were analyzed alongwithdatafrom actual closed-circuit oxygen diving by usinga maximum likelihood method. The parameters of themodelwere solved for the combined data, yielding thepowerequation for active diving:
K = tz (P02/101.3)(6.8),
I,"heret is in minutes.
Itwassuggested that the risk of CNS oxygen toxicity indiringcan be derived from the calculated parameter of thenormaldistribution:
Z = [In(t) - 9.63 + 3.38 x In(POz/101.3) ]/2.02
Therecoverytime constant for CNS oxygen toxicity wascalculatedfrom the value obtained for the rat, taking intoaccountthe effect of body mass, and yielded the recoverycquation:
K(t) = K(e) x e(-0.079t)
\·;hereK(t) and K(e) are the values of K at time t of the
rccorcrrprocess and at the end of the hyperbaric oxygencxposure,respectively, and t is in minutes.
Pulmonary Oxygen Toxicity
Thisis usually a manifestation of prolonged exposureImurethan 24 h) to normobaric 100% oxygen, as well asduringexposure to HBO from 2 to 3 ATA O2 in humanandexperimentalanimals. The pathology and pathophysiologrof pulmonary oxygen toxicity are described inmoredetail elsewhere (Jain 1989b). The major mechanismbr which HBO produces lung injury in rabbits is bystimUlatingthromboxane synthesis. Lung injury inducedbyireeradicalshas been demonstrated in an animal moddoismokeinhalation, and the free radicals clear up after
about an hour. Normobaric 100% oxygen given for onehour does not increase the level of free radicals in this
model, but HBO at 2.5 ATA does so.
Acute changes in the lungs resulting from oxygen toxicity consist of alveolar and interstitial edema, alveolarhemorrhages, and proteinaceous exudates. This is followed by an inflammatory reaction. Further prolongedexposure to oxygen leads to a proliferative phase, whichincludes proliferation of type II epithelial cells and fibroblasts, followed by collagen deposits. Healing may occurafter discontinuance of oxygen exposure, but areas of fibrosis and emphysema may remain. In patients with heartfailure or in patients with reduced cardiac ejection fractions, HBO may contribute to pulmonary edema by increasing left ventricular afterload, increasing oxidativemyocardial stress, decreasing left ventricular complianceby oxygen radical-mediated reduction in nitric oxide, increasing pulmonary capillary permeability, or by causingpulmonary oxygen toxicity (Weaver & Churchill 2001).
Repeated exposure to HBO at intervals insufficient toallow total recovery from pulmonary oxygen toxicity maylead to cumulative effects. The progression of toxicity canbe monitored by serial pulmonary function studies. Theconcept of a "unit pulmonary toxic dose" (UPTD) has beendeveloped (Bardin & Lambertsen 1970), and this allowscomparison of the pulmonary effects of various treatmentschedules of HBO (Table 6.2). The UPTD is designed toexpress any pulmonary toxic dose in terms of an equivalentexposure to m:ygen at 1 ATA. It is only an arbitrary measureand does not allow for the recovery between HBO expo
sures. For example, 10 HBO treatments at 2.4 ATA for90 min each would give the patient a UPTD of more than200 and would indicate significant pulmonary toxicity witha 20% reduction in vital capacity. In practice, however, noclinical evidence of pulmonary toxicity is seen with thisschedule. There is no significant impairment of pulmonarydiffusing capacity in divers who have been intermittentlyexposed to HBO at 4 ATA for years.
Prolonged exposure to elevated oxygen levels is a frequent and important clinical problem. Superoxide dismutase (SOD) and catalase, the major intracellular antioxidant enzymes, cooperate in the detoxification of freeoxygen radicals produced during normal aerobic respiration. Therapeutic approaches designed to deliver SOD orcatalase to these intracellular sites would be useful in mit
igating the pulmonary oxygen toxicity. A number of approaches to deliver these enzymes have not been successful. Adenovirus-mediated transfer to lungs of both catalase and SOD cDNA has been shown to protect againstpulmonary oxygen toxicity. Distal airway epithelial cells,including type II alveolar and nonciliated bronchiolarepithelial cells, are important targets for oxygen radicalsunder the hyperoxic condition. The accessibility of thesedistal airway epithelial cells to in vivo gene transferthrough the tracheal route of administration, suggests the

52 Chapter 6
Table 6.2
Cumulative Pulmonary Oxygen Toxicity Indices for CommonlyUsed Oxygen Therapy Tables (Bardin & Lambertsen 1970)
Therapy table
• Chronic osteomyelitis/radionecrosis120 min oxygen at 33 fsw90 min oxygen at 45 fsw
• Anaerobic infection
45 min oxygen/15 min air/45 min oxygenat 60 fsw
45 min oxygen at 60-0 fsw with 8 minat 20 fsw and 27 min at 10 fsw
• CO intoxication
45 min oxygen at 60 fsw30 min oxygen at 60-30 fswIS min air/60 min oxygen at 30 fsw30 min oxygen at 30-0 fsw
'USN6
• USN 6 extended
20 min oxygen/5 min air at 60 fsw15 min air/60 min oxygen at 30 fsw20 min oxygen/5 min air at 60 fswandIS min air/60 min oxygen at 30 fsw
• USN 6A
• USN 6A extended
20 min oxygen/5 min air at 60 fsw15 min air/60 min oxygen at 60 fsw20 min oxygen/5 min air at 60 fswandIS min air/60 min m...ygen at 30 fsw
• IFEM 7A (air and oxygen)
• IFEM 7A alternating 50/50 Nitrox with air30 min on/30 min off from 100-70 fsw
UPTDa
300270
401
445
645
718787
860
690
763
833
906
1813
2061
rity (Gleissner et a12003). HBO (2.8 ATA,80% oxygen) given to premature rats does not result in retinopathy, whereascontrol animals given normobaric 80% oxygen developedretinopathy. This topic is discussed further in Chapter 32.
Factors that Enhance Oxygen Toxicity
Various factors which enhance oxygen toxicity are listed inTable 6.3. Combining HBO with the substances listed, together with morbid conditions such as fever, should definitely be avoided.
Mild hyperthermia (38.5 DC)has been used therapeutically for a number of conditions. An increase of temperatureincreases oxygen uptake by body tissues. Hyperthermia maythus be expected to enhance oxygen toxicity.Transient biochemical side effectsof mild hypothermia such as hyperammonemia can be inhibited by HBO, but this combinationshould be used cautiously to avoid oxygen toxicity.
It is generally believed that high humidity enhances oxygen toxicity as manifested by lung damage and convulsions. This has been experimentally verified in rodents exposed to HBO (515 to 585 kPa) under conditions of lowhumidity as well as 60% relative humidity.
Physical exercise definitely lowers the threshold for CNSoxygen toxicity in the rat over the entire range of pressuresfrom 2 to 6 ATA.This observation should be kept in mindin planning physical exercise in hyperbaric environments(see Chapter 4). Various enzymes inhibited by hyperoxiaare shown in Table 6.4. This may explain how hyperoxialeads to oxygen toxity.
GlutdChione reductase is an integra! component of theantioxidant defence mechanism. Inhibition of brain glutathione reductase by carmustine lowers the threshold forseizures in rats exposed to HBO.
UPTD value indicates duration (min) of oxygen breathing at 1.0ATA that would cause equivalent degree of pulmonary intoxication (measured as decrease in vital capacity)
potential for in vivo transfer of MnSOD and extracellularSOD genes as a future approach in the prevention of pulmonary oxygen toxicity (Tsan 2001).
Oxygen-Induced Retinopathy
Retrolental fibroplasia is considered to be an oxygen-induced obliteration of the immature retinal vessels when
100% oxygen is given to premature infants. A recent studyshowed that oxygen therapy for more than 3 days, in infantsdelivered following 32-36 weeks of gestation, was not associated with an increased risk of retinopathy of prematu-
Table 6.3
Enhancers of Oxygen Toxicity
• GasesCarbon dioxideNitrous oxideHormonesInsulin
Thyroid hormonesAdrenocortical hormonesNeurotransmitters
Epinephrine and norepinephrineDrugs and chemicalsAcetazolamide
DextroamphetamineNH4C1
ParaquatAspirin
DisulfiramGuanethidine
• Trace metalsIron
Copper• Morbid conditions
Fever
Vitamin E deficiency convulsions
Congenital spherocytosisPhysiological states of increased metabolism
Physical exerciseHyperthermiaDiving

Table 6.4
EnzymesInhibited by Hyperoxia at 1-5 ATA
Oxygen Toxicity
Central Nervous System OxygenToxicity
53
I. Emhdcn-Mcyerhof pathway
Ph.)sph()glllCokinase
Phosphoglucomutase
(;Iyccraldchydc-phosphate-dehydrogenase*
2. Conversion of p)TUvate to acetyl-CoA
Pyruvatc oxidasc
3. Tricarboxylic acid cycle
Succinatc dchydrogenase*
[I.-ketoglutarate dehydrogenase*
:'Ialate dehrdrogenase*
.1.Ehtron transport
Succinatc dehydrogenase*
.\Ialatc dehydrogenase*
(;Iyccraldehyde-phosphate dehydrogenase*
IJl'NII dehydrogenase*
Lactate dehydrogenase'Xanthinc oxidase
D-Amino acid oxidase
5. :\curolransmitter synthetic enz)Tnes
(;Iutamic acid dccarboxylase
Cholinc acetylase
I)opa dccarboxylase
5-IITI' decarboxylase
Phenylalanine hydroxylase
Tyrosine hydroxylase
o. Proteolysis and hydralysis
Cathcpsin
Papain
Unspccified pro teases and peptidases
Unspccified in autolysis
ArginaseUrcasc
Ribonuclease
/. :'kmbrane transportNA!, Ki-ATPase+
~. :'Iolccular oxygen reduction pathwayCatalasc
9.0thcrs
Acctatc ki nase
Ccrcbrosedase
Cholinc oxidase
Fatty acid dehydrogenase
Formic acid dehydrogenase
(;Iulamic dehydrogenase
(;Iutamic synthetase
(;Iyoxylase
Ilydrogcnase
Isocitrate lyase
Malate syntase
:'!yo kinase (adenylate kinase)
Phosphate transacetylaseTransaminase
Zymohcxilse (aldolase)
.Astcrisks indicate enzymes containing essential sulfhydryl (SH)groups, emphasized as being inactivated by oxidation of these
gnHips.
Effect on Cerebral Metabolism
Disturbances of cerebral metabolism resulting from hyperoxia have been described in Chapter 2. HBO at 2 ATAhasbeen shown to stimulate rCMRGI slightly, but does not result in any toxic manifestations. Oxidative metabolism ofthe brain is usually not affected by pressures up to 6 ATA.In the primary rat cortical culture, HBO exposure to 6 ATAfor 30, 60, and 90 min increased the lactate dehydrogenase(LDH) activity in the culture medium in a time-dependentmanner (Huang et al 2000). Accordingly, the cell survivalwas decreased after HBO exposure. Pretreatment with theNMDA antagonist MK-801 protected the cells against theHBO-induced damage. The protective effectwas also notedin the cells pretreated with L-N(G)-nitro-arginine methylester, an NO synthase inhibitor. These results suggest thatactivation of NMDA receptors and production of NO playa role in the neurotoxicity produced by HBO exposure.
There is no evidence that seizures are related to oxidative
metabolic changes. However, increase of glucose utilizationprecede the onset of electrophysiological manifestations ofCNS oxygen toxicity). Increased nitric oxide (NO) production during prolonged HBO exposure is responsible for escape from hyperoxic vasoconstriction in cerebral blood arterioles. The finding suggests that NO overproduction initiates CNS oxygen toxicity by increasing rCBF,which allowsexcessive oxygen to be delivered to the brain (Demchenkoet al 2001). The hypothetical pathophysiologic pathwaysleading to acute and chronic CNS oxygen toxicity are illustrated in Figure 6.4.
Effect on Neurotransmitters
Neurotransmitters have been shown to be downregulatedunder hyperbaric hyperoxia (Courtiere et al1991). Withthe recognition of nitric oxide (NO) as a neurotransmitter,its relationship to hyperia has been studied. Experimentalstudies of Zhang et al (1993) showed that rats can be protected against oxygen toxicity by a combination of a monoamine oxidase inhibitor and a nitric oxide (NO) synthaseinhibitor. Their data showed that protection against oxygentoxicity by these agents is not related to the preservation ofthe GABA pool. They found that oxygen-dependent noradrenaline metabolism and NO synthesis appear to be inactive during oxygen neurotoxicity. Oury et al (1992) consider NO to be an important mediator in oxygen neurotoxicity and suggest that extracellular superoxide dismutaseincreases oxygen neurotoxicity by inactivation of NO.
Exposures to HBO at 2 and 2.8 ATAstimulated neuronalNO synthase (nNOS) and significantly increased steady-state
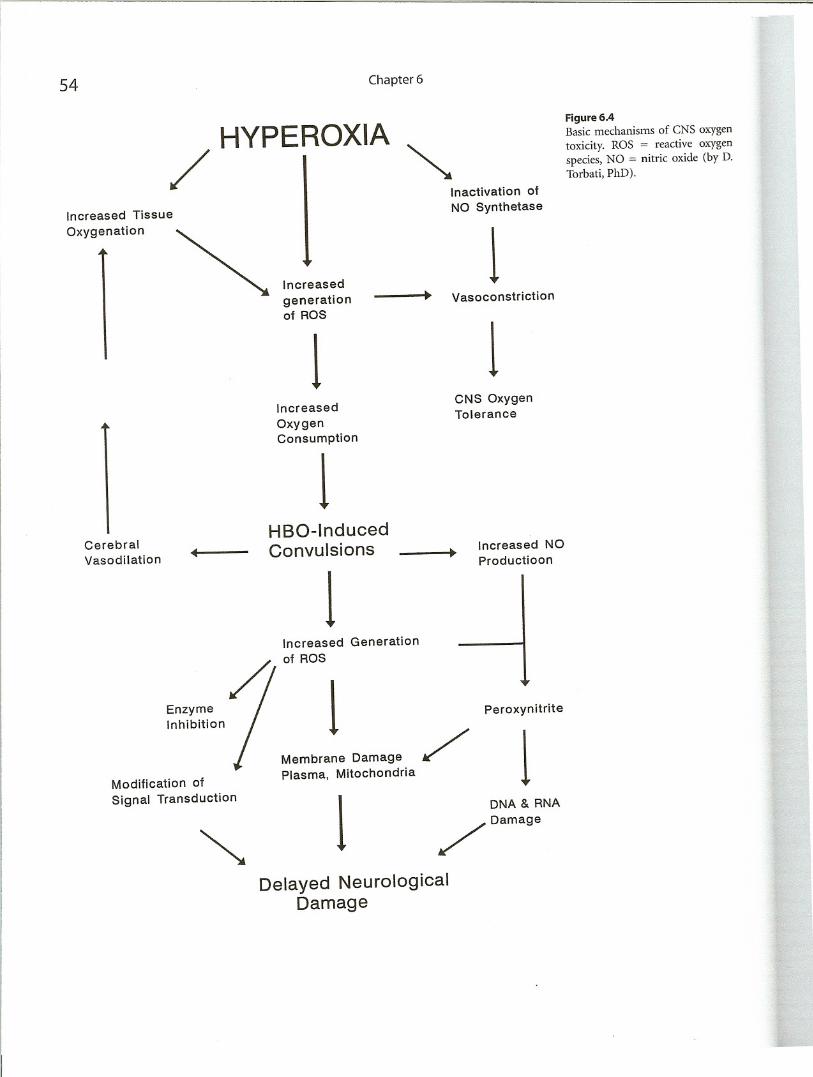
HYPEROXIA
/ I ~
54
Increased Tissue
Oxygenation
Increased
generationof ROS
1Increased
OxygenConsumption
1
Chapter 6
Inactivation of
NO Synthetase
1Vasoconstriction
1CNS OxygenTolerance
Figure 6.4Basic mechanisms of CNS oxygen
toxicity. ROS := reactive oxygen
species, NO = nitric oxide (by D.Torbati, PhD).
Cerebral
HBO-Induced
Vasodilation
4Convulsions Increased NO•
1
Productioon
Increased Generation
of ROS
Enzyme
/1Inhibition
Peroxynitrite
Membrane Damage
/1Modification of
Plasma, Mitochondria
Signal Transduction
1DNA & RNA
~
/ Damage
Delayed Neurological
Damage

Oxygen Toxicity 55
(Thomrl IlJ 2(03). At both pressures, elevations in NO conctntrationwere inhibited by the nNOS inhibitor 7-nitroindazoleand the calcium channel blocker nimodipine. Infu
sionof superoxide dismutase inhibited NO elevation at 2.8,hutnot 2 ATAHBO. Hyperoxia increased the concentrationoft\O associatedwith hemoglobin. These findings highlighttht complexityof oxidative stress responses and may helptxplainsomeof the dose responses associated with therapeuticapplicationsof hyperbaric oxygen.
Ammoniaand Amino Acids
Singleseizures induced in rats subjected to HBO at 6 ATAhaw heen shown to be associated with accumulation of
ammoniaand alterations in amino acids in the brain, with
lht greatestchanges taking place in the striatum (Mialon,·t IlJ 1992). These changes were considered to be caused byanincreasein oxidative deamination or possibly the resultofgliallililureto capture released amino acids. The subseIlutnlimbalancebetween the excitatory and inhibitory mediatorsin the striatum was offered as an explanation of thertcurrenceof seizures in animals maintained on HBO.
Changesin the Electrical Activity of the BrainandSeizures
Ctlnsciousrats and rabbits exposed to HBO usually demon,Iratean increased EEG slow wave activity which eventuallydmlops into bursts of paroxysmal electrical discharges.Theseelectricalevents precede the onset of visible HBO-inducedconvulsions,and therefore were suggested as an early,igmof CNS oxygen toxicity in experimental animals. In\'itw studieswith HBO also show changes in neuronal electricalactivity,which may be associated with seizures.
Theseizureassociated with HBO usually occurs towardlheend of the o},l'gen exposure while the patient is beingdWlIllpressed.It is a violent motor discharge with a briefrtriod of breathholding. In such cases, therefore, decomprmioll should be temporarily halted until the seizure isom; otherwise there could be rupture of lung alveoli.
(Jx~'gen-inducedseizures are not a contraindication for
furtherHBOtherapy. Further HBO treatments may be carriedout at lower pressures and shorter exposures. Anticonvulsantmedications are usually not indicated, but maybeuStd.ln animal experiments Carbamazepine (Reshef etrlllL)91) and vigabatrin (Tzuk et a11991) have been foundiii he dTcctivc in preventing HBO-induced convulsions.:\cupuncturehas been claimed to protect against oxygeninducedcon\'ulsions by increasing GABA in the brain levelsI\\'u rt oJ 1992).
Epileps)'has been listed as a contraindication for usingHBO therapy.This is based on the assumption that oxy-
gen is liable to precipitate a seizure in an epileptic patientand such an event in a chamber might be detrimental to
the patient. Seizures in epileptic patients are rare duringHBO therapy where pressures less than 2 ATA are used.There is no published study that reexamines this issue.
The question therefore still arises: is HBO really dangerous for an epileptic? If epilepsy is included in the contraindications for HBO, patients with head injuries andstrokes who happen to have seizures would be deprivedof the benefit of HBO therapy. The mechanism of epilepsy
in such patients is different from that of an m."ygen-induced convulsion. It has even been shown that EEG ab
normalities in stroke patients improve with HBO treatment (Wassmann 1980). It is possible that HBO mayabort a seizure from a focus with circulatory and meta
bolic disturbances by correcting these abnormalities. Seizures are extremely rare and no more than a chance occurrence during HBO sessions at pressures between 1.5and 2 ATA even in patients with a history of epilepsy.
Neuropathology
In experimental studies, there is no damage to the CNSof rats exposed to HBO until the pressure exceeded 4 ATA.The brain damage is increased by CNS-depressant drugs,increase of pCOl, acetazolamide, and NH4Cl. Permanentspastic limb paralysis has been observed in rats (the JohnBean effect) after repeated exposure to high oxygen pressures (over 5 ATA). There is selective necrosis of white
matter both in the spinal cord and the brain, and this isconsidered to be the effect ofhyperoxia. HBO-induced ratbrain lesions, examined by electron microscopy, show twotypes of nerve cell alterations: (I) type A lesions characterized by pyknosis and hyperchromatosis of the nervecells, vacuolization of the cytoplasm, and simultaneousswelling of the perineural glial processes; (2)type B lesionsare characterized by lysis in the cytoplasm and karyorrhexis.
Manifestations of CNS Oxygen Toxicity
Signs and symptoms of CNS m.]'gen toxicity are listed inTable 6.5.
Clinical Monitoring for Oxygen Toxicity
The most important factor in early detection of oxygen toxicity is the observation of signs and symptoms. For monitoring pulmonary function, determination of vital capacityis the easiest and most reliable parameter, as it is reducedbefore any irreversible changes occur in the lungs. EEG

56
Table 6.5Signs and Symptoms of CNSToxicity
Chapter 6
Table 6.6Factors Protecting Against Generalized Oxygen Toxicity
tracings do not show any consistent alterations before theonset of seizures and are not a reliable method of early detection of oxygen toxicity.
Decrease in [9,10-3H] oleic acid incorporation by human erythrocytes detected ill vitro after HBO exposure illvivo may reflect an early event in the pathogenesis of m.:ygen-induced cellular injury and may be a useful monitoring procedure.
An increase in CBF velocity (BCFV) precedes onset ofsymptoms of oxygen toxicity during exposure to 280 kPaoxygen, which may be followed by seizure (Koch et al2008). At rest a delay of approximately 20 min precedes theonset of CNS oxygen toxicity and seizure can be abortedwith timely oxygen reduction.
Facial pallorSweatingBradycardiaChoking sensationPalpitationsEpigastric tensionsSleepinessDepressionEuphoriaApprehensionChanges of behaviorFidgetingDisinterestClumsiness
\fisualsymptomsLoss of acuityDazzleLateral movement
Decrease of intensityConstriction of visual field
Acoustic symptomsMusic
Bell ringingKnockingUnpleasant olfactory sensationsUnpleasant gustatory sensationsRespiratory changesPantingGruntingHiccoughsInspiratory predominanceDiaphragmatic spasmsSevere nausea
Spasmodic vomiting\fertigoFibrillation ofIipsLip twitchingTwitching of cheek and noseSyncopeConvulsions
Antioxidants, free radical scavengers, and trace mineralsallopurinolascorbic acid
glycinemagnesiumselenium
superoxide dismutase, SODtyloxapolvitamin E
Chemicals and enzymes modifying cerebral metabolismargininecoenzyme QlO and carnitinegamma-aminobutyric acid, GABAglutathionehemocarnisineinterleukin-6
leukotriene B4 antagonist SC-41930paraglycine and succinic acidsodium succinate and glutamate
Drugsadrenergic-blocking and ganglion-blocking drugsbarbituratesBCNU
chlorazepatediazepamergot derivatives: lisuride and quinpiroleisonicotinic acid hydrazidelevodopalithiummilecide
MK-801 (a competitive NMDA receptor antagonist)neuroleptics: chlorpromazine, thorazinepropranolol
Intermittent exposure to HBOacclimatization to hypoxiainterposition of air-breathing periods
Endocrine factors
adrenalectomyhypophysectomythyroidectomy
Gene therapy
Protection Against Oxygen Toxicity
Various agents and measures for prevention or treatmentof oxygen toxicity are listed in Table 6.6; these are mostlyexperimental. The most promising agents are the antioxidants. The use of vitamin E (tocopherol) is based on thefree-radical theory of oxygen toxicity. It has been used toprotect premature infants (who lack vitamin E) against oxygen toxicity. Dietary suppJementation with selenium andvitamin E, which increase the cerebral as well as extracerebral GSH content, does not protect rats against the effectof HBO by delaying the onset of first electrical discharge(Boadi et al1991). However, such diets may still be advantageous in promoting recovery and reversal of toxic process,as occurs between consecutive HBO exposures or during intermittent oxygen exposure (Bleiberg &Kerem 1988).
Not all of the dietary free-radical scavengers are effectiveincounteracting oxygen toxicity. In animal experiments, nocorrelation was found between ill vitro inhibition of lipidperoxidation and ill vivo protection against oxygen toxicity.
Hypothermia has been considered to be a protectoragainst o},.}'gentoxicity, but HBO at 5 ATAinduces hypothermia in mice, and this has little protective effect againstconvulsions.
Every clinician who treats patients should be aware ofoxygen toxicity, although it is rare. At pressures of1.5 ATA,even prolonged use in patients with cerebrovascular disease has not led to any reported case of oxygentoxicity. It should not be assumed that experimental observations regarding oxygen toxicity under hyperbaricconditions are· applicable to normobaric conditions.

Oxygen Toxicity 57
\\'hereasdisulfiram protects against hyperbaric oxygen, itpotentiatesthe toxicity of normobaric oxygen in rats. AsCorhicacid is also a free radical scavenger and protectsagainstoxygen toxicity, but large doses of this vitaminmayprovecounterproductive in treating oxygen toxicityif the reducing enzymes are overloaded. An oxidized ascorbatemight actually potentiate oxygen toxicity throughlipoperoxideformation. Mg2+ has a double action againsttheundesirable effects of oxygen. It is a vasodilator andailoa calciumblocker and protects against cellular injury.\Iagnesium sulfate suppresses the electroencephalographicmanifestations of CNS oxygen toxicity and an antiConvulsanteffect has been demonstrated in rats exposedto HBO at 6ATA. A prophylactic regimen of 10 mmol\Ig" 3h bd(m a session of HBO and 400 mg of vitaminEJaily,starting a couple of days before the HBO treatment,is useful in preventing oxygen toxicity, but no controlledstudy has been done to verify the efficacy of thisregime.
Thedetoxit)'ing function of cytochrome c to scavengeROS in mitochondria has been confirmed experimentally1,\lin& Jian-xing2007). A concept of mitochondrial radicalmetabolism is suggested based on the two electronleakpathwaysmediated by cytochrome c that are metabolicroutes of oxygen free radicals. The main portion ofoxygenconsumed in the electron transfer of respiratorychainis used in ATP synthesis, while a subordinate partlit' oxygenconsumed by the leaked electrons contributes10 ROS generation. The models of respiratory chain opmting with two cytochrome c-mediated electron-leakpathwaysand a radical metabolism of mitochondria ac
cnmpaniedwith energy metabolism are helpful in underItandingthe pathological problems caused by oxygen toxicity
Distalairway epithelial cells, including type II alveolarJndnonciliatedbronchiolar epithelial cells, are importanttargebI()r0~radicals under the hyperoxic condition. TheJccessibilityof these distal airway epithelial cells to in vivogenetransfer through the tracheal route of administration,suggeststhe potential for in vivo transfer of MnSODandextracellularSOD genes as a future approach in thepreventionof pulmonary O2 toxicity (Tsan 2001).
Extension of Oxygen Tolerance
"!ll!eranceto oxygenprimarily means tolerance to the toxicefli:cts,becausethe physiological effects have no prolongedCOIMjUeI1Ces.This subject has been discussed in detail byLJmbertsen(1988). He considers a positive emphasis oneX!endingoxygen tolerance as desirable, as opposed to areltrictirefear of oxygen poisoning. The following arecompikdfrom his comments regarding extension of oxygcntolerance.
Tolerance Extension by Adaptation
At low levels of atmospheric hyperoxia, some forms of true
protective adaptation appear to occur, such as that relatedto changing antioxidant defenses in some tissues. At higher
oxygen pressures, some adaptation could conceivably occur in some cells of the intact human being with progressive and severe poisoning in other cells. At very high oxygen
pressure, rapid onset of poisoning would make adaptationinadequate and too late.
Tolerance Extension by Drugs
A pharmacological approach, such as that of providing freeradical scavengers, will attain broad usefulness only if thedrug can attain the free permeability of the oxygen molecule. The drug should reach the right location at the righttime, and remain effective there in the face of continuous
hyperoxia, without itself inducing any toxic effects. Thereis no such ideal drug available at present.
Tolerance Extension by Interrupted Exposureto Oxygen
Interruption of exposure to HBO is known to extend thesafe exposure time. In experimental animals, intermittentexposure to HBO postpones the gross symptoms of oxygentoxicity along with changes in enzymes, such as superoxidedismutase, in the lungs (Harabin et aI1990). Species differences were noted in this study; biochemical variables weremore pronounced in guinea pigs than in rats.
There is no accepted procedure for quantifying the recovery during normoxia. A cumulative oxygen toxicity index - K, when K reaches a critical value (Kc) and the toxic
effect is manifested, can be calculated using the followingequation:
where t(e) is hyperoxic exposure time and POz is oxygenpressure and c is a power parameter.
Recovery during normoxia (reducing K) is calculated bythe following equation
K2 = K. x e[-rt(r)J
where t(r) is recovery time, r being the recovery time constant.
A combination of accumulation of oxygen toxicity andits recovery can be used to calculate central nervous systemoxygen toxicity. Predicted latency to the appearance of thefirst electrical discharge in the electroencephalogram,which precedes clinical convulsions, was compared to mea-

58 Chapter 6
sured latency for seven different exposures to HBO, followed by a period of normoxia and further HBO exposure(Arieli & Gutterman 1997). Recovery followed an exponential path, with r = 0.31 (SD 0.12) min (-1). Calculation ofthe recovery of the CNS oxygen toxicity agreed with thepreviously suggested exponential recovery of the hypoxicventilatory response and was probably a general recoveryprocess. The authors concluded that recovery can be applied to the design of various hyperoxic exposures.
Inclusion of air breaks in prolonged HBO treatmentschedules is a recognized practice. The return to normobaric air between HBO sessions may lead to low pOz seizures, which are also described as a "switch off' phenomenon. However, much research still needs to be done to find
the ideal schedules to extend oxygen tolerance.
Effect of HBO on the CNS of Newborn Mammals
Newborn mammals are extremely resistant to the CNS effects of HBO compared to adults. Indirect evidence indicates that HBO in newborn rats induces a persistent cerebral vasoconstriction concurrently with a severe and maintained reduction in ventilation. The outcome of these
exposures may be as follows:
• Extension of tolerance to both CNS and pulmonary oxygen toxicity,
• Creation of a hypoxic-ischemic condition in vulnerableneuronal structures, and
• Impairment of circulatory and ventilatory responses tohypoxic stimuli on return to air breathing, with subsequent development of a hypoxic-ischemic condition.
These events may set the stage for development of delayedneurological disorders.
Conclusion and Directions for FutureResearch
The exact mechanism underlying oxygen toxicity to theCNS is not known, but the free radical theory appears to
be the most likely explanation. The role of nitric oxide inthe effect of HBO has also been established. Fortunately,
CNS oxygen toxicity is rare because most HBO treatments
are carried out at pressures below 2.5 ATA, and the duration of treatment does not exceed 90 min. Nevertheless, a
physician treating patients with HBO must be aware of oxygen toxicity. There is no rational prevention or treatment,but free radical scavengers are used in practice to preventthe toxic effects of oxygen. Until a better understanding ofthe mechanism of oxygen toxicity and better methods oftreatment are available, use of the free radical scavengersthat are available appears to be a reasonable practice, particularly when these are relatively nontoxic. In situationswhere prolonged exposures to HBO are required, the benefits of treatment versus the risks of oxygen toxicity shouldbe carefully weighed.
The chemiluminescence index, which is a measure of tis
sue lipid peroxidation indicates individual sensitivity of thebody to HBO. Such a technique would enable the predictionof the effectiveness of HBO treatment as well as control its
duration. Oxygen toxicity can also be exploited for therapeutic purposes. One example of this is the use of HBO as anantibiotic. Induced oxygen toxicity by HBO with protectionof the patient by free radical scavengers should be investigated as an adjunctive treatment for AIDS, because the virusresponsible for this condition has no protective mechanismsagainst free radicals. Since induction of antioxidative defencemechanisms has been determined after HBO exposure, amodified treatment regimen of HBO therapy may avoidgenotoxic effects (Speit et a12002).
The methods for estimating free radicals are still cumbersome and not in routine use. More practical methods shouldbe developed as a guide to the safe limits ofHBO therapy.
The molecular basis of oxygen toxicity should be sought
at the cellular and organelle levels. Simultaneous monitoringof cerebral, electrical, circulatory, and energy-producingfunctions is a useful tool for determining the safety marginsof HBO, as well as for tracing the primary mechanisms ofoxygen toxicity in the CNS.
Mammalian cell lines have been shown to develop toler
ance to oxygen by repetitive exposure to HBO at 6 to 10ATAfor periods up to 3 h. Repeated screening of various cell linesmay lead to the discovery of oxygen-resistant cell types,which might provide an insight into the factors inherent inthe development of oxygen tolerance.
The latest approach to counteract pulmonary oxygen
toxicity is gene therapy by viral-mediated transfer SODand catalase to the pulmonary epithelium. This appearsto be the most promising method of delivery of these en
zymes.


















