
Popular Science Background: How the optical microscope became ...

1
SBPG programme
December 2010
Optical microscopy basics
Advanced Technology Development Group
B. VojnovicGray Institute for Radiation Oncology and Biology, Oxford
The Randall Centre, King’s College, London

2
Light Microscopy basics….
We’ve probably all learnt about
what a microscope does, but there
is no harm in being reminded…
A microscope does not ‘make small
objects larger’,
it shows us how small objects interact
with light…

3
Optical microscopy
Optical microscopes have been used in biomedical science for several hundred years
Although the basic principles have been understood for a long time, increasingly sophisticated methods are being continuously developed
Although microscopes are used to magnify objects, this is only part of the story………….. they principally give a representation of the way light interacts with the structures of the sample under investigation. Interference, diffraction effects, the limitations of the finite wavelength of light play a crucial role
Contrast is generated by a wide range of interactions
In the last 10-20 years there has been a tremendous resurgence of optical methods
Modern instruments have become so sophisticated that most users are not familiar with the basic principles – and often will not obtain the best performance
Although resolution is crucial, in biology other considerations are also important, such as speed, minimisation of damage to the sample, ability to ‘see’ transparent samples etc

4

5
Particles and energies
Electron volt = 1.602 176 462 x 10-19 Joules
Electron mass = 9.10938215(45) × 10−31 kg
Elementary charge = 1.602 176 462 x 10-19 Coulombs
Photon = zero mass and zero electric charge
100 nm 3 x 1015 Hz 12.4 eV200 nm 1.5 x 1015 Hz 6.20 eV500 nm 6 x 1014 Hz 2.48 eV1000 nm 3 x 1014 Hz 1.24 eV
E = h = h c/
Energy
Planck’s constant
frequency
Speed of light
wavelength
Planck’s constant = 6.626 068 76 x 10-34 Joule seconds

6
Where it all started….
Antonie van Leeuwenhoek
(1632-1723)
Ball lens (=shortest focal length) microscope
Van Leeuwenhoek maintained throughout his life that there were aspects of microscope construction "which I only keep for myself", including in particular his most critical secret: how did he create the lens……
Not much has changed!.....try obtaining a lens prescription from Zeiss, Nikon, others…..

7
Microscopes can be beautiful…
From http://www.antique-microscopes.com/

8
http://micro.magnet.fsu.eduAn excellent web site
Where to learn all about microscopy….
Also: https://support.svi.nl/wiki/index.php

9
Lens basics
u v
ff
V
U
u v f
1 1 1+ =
V v U u
=
Biconvex Plano-convex
Positive meniscus
Negative meniscus
Plano-concave
Biconcave
8 f
Point object at infinite
distance
Lens forms using
spherical surfaces
10
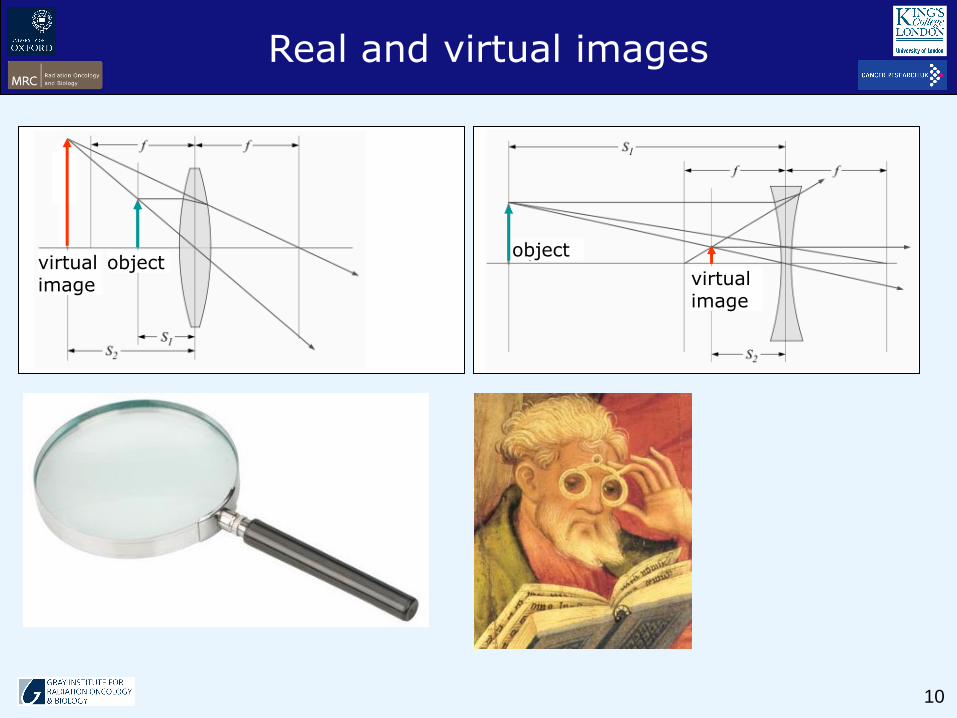
Real and virtual images
object
virtual image
objectvirtual image

11
Spherical aberration
Coma
image
Lens aberrations
Parallel rays (from object at infinity) do not converge to a single point (focus)
Aberrations reduced by utilising smaller portion of lens input area: aperture reduction

12
Chromatic aberration
Achromatic doublet
Crown glass
Flint glass
Astigmatism
on-axis
off-axis
Lens aberrations

13
Compound lenses
Multiple elements used to correct for individual aberrations: examples of astigmats and achromatic astigmats

14
Wave diffraction
Google earth:Panama canal entrance
Francesco Grimaldi coined the term diffraction (Latin diffringere, 'to break into pieces‘)Thomas Young, Augustin-Jean Fresnel, Christiaan Huygens, Joseph von Fraunhofer

15
The ability of an imaging system to resolve detail is ultimately limited by diffraction
A plane wave incident on a circular lens (or spherical mirror) is diffracted
Light is not focused to a point but forms an Airy disk having a central spot in the focal plane with radius to first null of:
where λ is the wavelength of the light and N is the f-number (focal
length divided by diameter) of the imaging optics.
In object space, the corresponding angular resolution is:
Diffraction
D is the diameter of the entrance pupil of the imaging lens

16
objective
first objective lens
= 30o
NA = n sin()
NA = 0.5 NA = 0.75
= 48.7o
NA = 0.95
= 72.1o
n = refractive index (air=1, water = 1.3, oil =1.4)
Numerical aperture
Resolution = 1.22 / 2NA
Resolution = 1.22 / NA objective + NA condenser
= half-angle of light cone collected

17
half-angle subtended and
% collected; air objective
0
10
20
30
40
50
60
0 0.2 0.4 0.6 0.8 1
numerical aperture
an
gle
(d
eg
) o
r p
erc
en
tag
e
θ/2
% collected
Objective numerical aperture
0.3 na 660 nm 1.342 m
Resolution = 1.22 / 2NA
500 nm
450 nm
1.021 m
0.813 m
0.75 na 660 nm 0.537 m
500 nm
450 nm
0.408 m
0.325 m

18
Objects in the optical microscope that are either self-luminous or illuminated by a large-angle cone of light form Airy patterns at the intermediate image plane that are incoherent and do not interfere with each other
This allows the determination of the minimum separation distance between adjacent Airy patterns by examination of the total intensity distribution (the sum of intensities) when these patterns are closely spaced or overlapping
When the separation distance between adjacent Airy patterns is greater than the central disc radius, the sum of the intensities yields two individual peaks. As the discs approach each other, the separation distance will reach a value equal to the central disc radius, a condition known as the Rayleigh criterion
At even closer approach, the separation distance is less than the central disc radius and the sum of the two peaks merges into a single peak. In the latter instance, the two Airy patterns are said not to be resolved
Microscope resolution

19
Airy pattern from point sources
Amplitude (EM field)
Intensity (brightness)
Airy disc

20
Köhler illumination –parfocality in a microscope
21
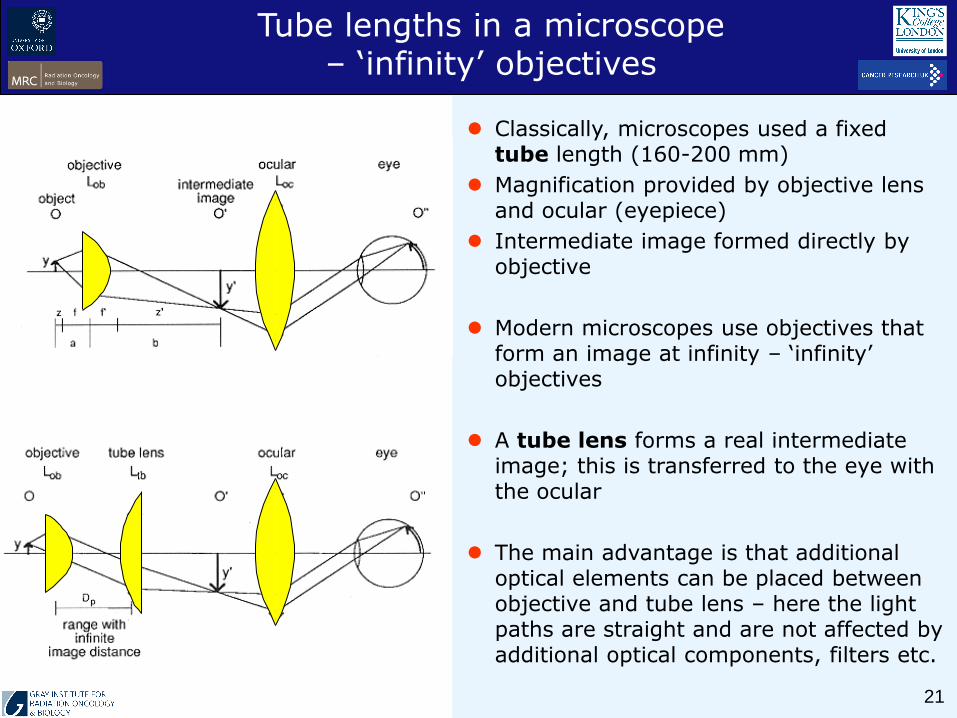
Classically, microscopes used a fixed tube length (160-200 mm)
Magnification provided by objective lens and ocular (eyepiece)
Intermediate image formed directly by objective
Modern microscopes use objectives that form an image at infinity – ‘infinity’ objectives
A tube lens forms a real intermediate image; this is transferred to the eye with the ocular
The main advantage is that additional optical elements can be placed between objective and tube lens – here the light paths are straight and are not affected by additional optical components, filters etc.
Tube lengths in a microscope – ‘infinity’ objectives

22IMAGING LIGHT PATHS ILLUMINATION LIGHT PATHS
Eye
Eyepiece
Intermediate
image plane
Object
Condenser
.
Collector Lens
Lamp
Objective Objective back focal plane
Tube
lens
Light paths in
transmitted-light
microscope using
Köhler illumination
Condenser diaphragm - determines
condenser numerical aperture
Field diaphragm - determines
illuminated sample area

23
Johann Ploem
(1927 -
Georges Nomarski
(1919 - 1997)
Frits Zernike
(1888-1966)
Phase Contrast
microscopy
Robert Day Allen
(1927-1986)
Fluorescence
microscopy
Differential
Interference
Contrast
Video microscopy
Ernst Abbe
(1840-1905)
Microscopy developments in modern times

24
Microscope resolution
focal plane focal plane
focal plane
intensity
intensity
intensity
Airy discAiry disc
Airy disc
0.2 na 0.94 na
1.4 na
Point source
Spherical wavefront
objective
Spherical wavefront

25
Coverglass correction
Modern objectives are precision optical components designed to maximise the numerical apertures and the working distances.
Most optical defects (chromatic aberration, spherical aberration, astigmatism, coma, flatness of field etc. are highly corrected.
Design is very specialised activity …….cost can be high
Microscope objectives

26
Objective immersion
Air (dry) objective water immersion objective oil objective
air gap
water
interface
oil
interface
coverslipcoverslip

27
Objective classes and numerical aperture
Achromatic Fluorite Apochromatic
Plan Plan-achromat
Plan Fluorite
Plan Apochromat
Improved colour correction
Impro
ved fie
ld f
latn
ess
28
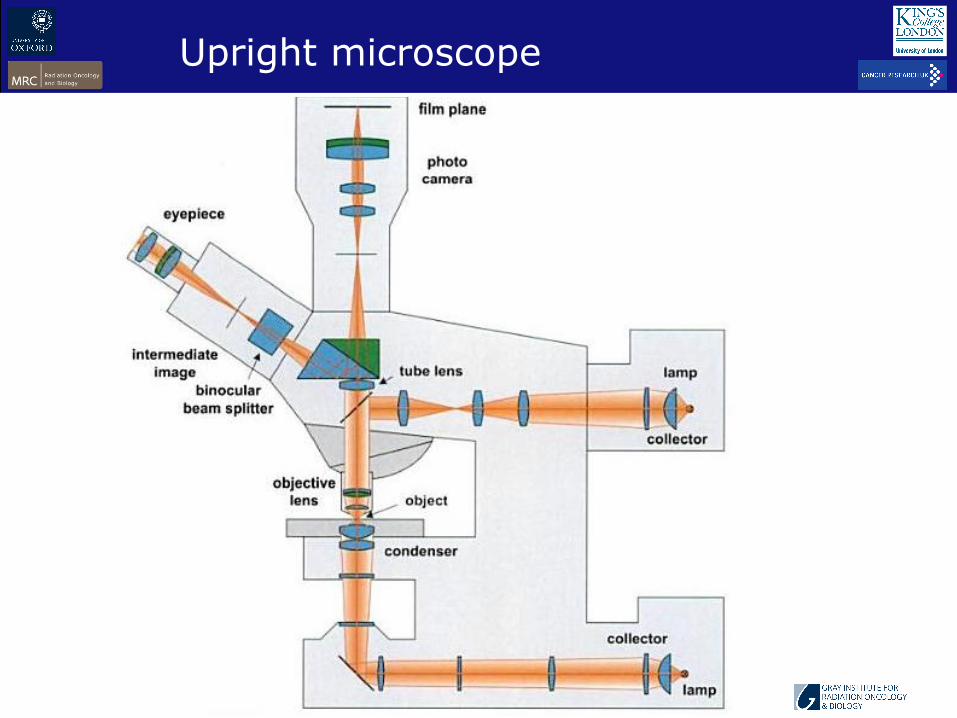
Upright microscope

29
Inverted microscope

30
Simplest of all the light microscopy techniques
Sample illumination is via transmitted white light, i.e. sample is illuminated from below and observed from above (or vice-versa)
Limitations include low contrast of most biological samples and low apparent resolution due to the blur of out of focus material
Simplicity of the technique and the minimal sample preparation required are significant advantages
Bright field microscopy
from: http://iheartguts.com/tag/brightfield-microscopy

31
Improves (and reverses) the contrast of unstained, transparent specimens
Uses a light source to minimize the amount of directly-transmitted (unscattered) light entering the imaging plane, collecting only the light scattered by the sample
Darkfield can dramatically improve image contrast—especially of transparent objects – while requiring little equipment setup or sample preparation
Suffers from low light intensity in final image of many biological samples, and provides low apparent resolution
Rheinberg illumination is a variant of dark field illumination transparent, coloured filters are inserted just before
the condenser light rays at high aperture are differently coloured
than those at low aperture (e.g. specimen background is red while the object appears yellow)
Dark field microscopy
annulusdisc condenser
objective
condenser
objective
sample

32
Gives the image a 3-dimensional appearance and can highlight otherwise invisible features
Technique based on this method is Hoffmann's modulation contrast, often used on inverted microscopes applied to cell culture work
Oblique illumination suffers poor contrast of many biological samples; low apparent resolution due to out of focus regions), but nevertheless can highlight otherwise invisible structures
Oblique illumination
light from condenser
sample
objective
objective
light ‘wedge’
http://www.microscopy-uk.org.uk
http://www.modulationoptics.com/

33
Widely used technique that shows differences in refractive index as difference in contrast
Developed by Frits Zernike in late 1930s (awarded the Nobel Prize in 1953)
Contrast is excellent but limited to imaging of thin objects
Halo is formed even around small objects, which obscures detail
Uses circular annulus in the condenser which produces a cone of light
This cone is superimposed on a similar sized ring within the phase-objective
Every objective has a different size ring: every objective requires appropriate condenser setting
Ring in the objective has special optical properties
Reduces the direct light in intensity
Creates an artificial phase difference of ~ /4
Since properties of this direct light have changed, interference with the diffracted light takes place: the result is a phase contrast image.
Phase contrast microscopy
phase
plate
objective
sample
condenser
annulus
complementary
rings
diffracted
rays

34
Developed by Georges Nomarski
Differences in optical phase seen as differences in relief
Apparent relief is an optical effect – not ‘real’
Contrast is excellent; condenser aperture can be used fully open, thus reducing depth of field and maximizing resolution
Requires a polarized light source to function
The system consists of a Wollaston prism in the condenser that splits light into an ordinary and an extraordinary beam; the spatial difference between the two beams is minimal (<< resolution of the objective)
After passage through the specimen, the beams are reunited by a similar prism in the objective
In a homogeneous specimen, there is no difference between the two beams, and no contrast is being generated
Near a refractive boundary, the difference between the ordinary and the extraordinary beam will generate a relief in the image
Differential interference contrast
analyser
Wollaston prism
objective
sample
condenser
Wollaston prism
analyser

35
DIC vs. phase contrast
analyser
Wollaston prism
objective
sample
condenser
Wollaston prism
analyser
image planeimage plane
Phase-
contrast
objective
objective
sample
condenser
undiffracted
light
diffracted
light

36
Modern microscopes provide ‘easy’ solutions for microphotography and electronic image recording.
Experienced microscopists often prefer a hand drawn image rather than a photograph
subject knowledge allows accurate conversion of an image into a precise drawing: a camera provides a single in-focus plane
The creation micrographs requires a monocular microscope
both eyes are open
the eye not observing down the microscope is concentrated on a sheet of paper besides the microscope
Without moving the head or eyes, record the observed details by tracing round the observed shapes by simultaneously ‘seeing’ the pencil point in the microscope image
Image recording

37
Time-lapse microscopy
Inverted microscope configuration
Incubator around the microscope
Control of temperature, humidity and pH (CO2)
Challenges: preserving focus, power outages (!)

38
When certain compounds are illuminated with high energy (blue) light, they emit light of a different, lower frequency (redder light)
This effect is referred to as fluorescence, or more generally luminescence
Almost all biological specimens have their own characteristic autofluorescence, based on their chemical makeup
Fluorescence microscopy is of critical importance in modern life sciences
Extremely sensitive, allows detection of single molecules
Many different fluorescent dyes can be used to stain different structures
A particularly powerful method is the combination of antibodies coupled to a fluorochrome as in immunostaining
Antibodies can be tailored specifically for a chemical compound
e.g. artificial production of proteins, based on the genetic code (DNA): used to immunize e.g. rabbits, form antibodies which bind to the protein. The antibodies are then coupled chemically to a fluorochrome and can be used to trace the proteins in the cells under study
Highly-efficient fluorescent proteins such as the green fluorescent protein(GFP) have been developed using gene fusion (process which links the expression of the fluorescent compound to that of the target protein)
Fluorescence microscopy

39
Fluorescence emission differs in wavelength from the excitation light: a fluorescence image ideally only shows labelled structure of interest
High specificity led to the widespread use of fluorescence light microscopy in biomedical research
Fluorescence dyes of different spectral characteristics used to stain different biological structures: simultaneous detection while still exhibiting specificity but differentiated from individual dye colour
Most fluorescence microscopes are operated in the epi-illumination mode (illumination and detection from one side of the sample) to decrease the amount of excitation light entering the imaging device
Fluorescence microscopy

40
Sequence of events leading to fluorescence
From: http://micro.magnet.fsu.edu

41
arc lamp
Johann Ploem
Dichromatic cube
Epi-illuminated fluorescence microscope
‘cube’
objective

42
Modern inverted fluorescence microscope

43
Objective
Specimen
Objective back
focal Plane
Eye
Eyepiece
Tube Lens
Intermediate
Image plane
or camera plane
Emission Filter
Dichromatic
Mirror
Excitation
Filter
Lamp
Arc
arc images
Condenser
Diaphragm
Field
Diaphragm
arc image
MICROSCOPE EPI-FLUORESCENCE
KÖHLER ILLUMINATION LIGHT PATHS
Collector Lens
Ftube‘4f’ telescope
lenses
determines area
of sample
illuminateddetermines
condenser
excitation
intensity
F1
F2F1
SCAN MIRROR HERE IN
BEAM SCANNING SYSTEM
Filter Cube

44
Under some circumstances, e.g. photobleaching or the presence of salts of heavy metals, etc., emitted light may be significantly reduced or stopped altogether
Fading - There are conditions that may affect the re-radiation of light and thus reduce the intensity of fluorescence. This reduction of emission intensity is generally called fading.
Fading is subdivided into quenching and bleaching
Bleaching is irreversible decomposition of the fluorescent molecules because of light intensity in the presence of molecular oxygen
Fluorescent probe molecule reports
‘classified’ information on behaviour of
surrounding molecules
Quenching also results in reduced fluorescence intensity and frequently comes about as a result of oxidizing agents or the presence of salts of heavy metals or halogen compounds
Sometimes quenching results from the transfer of energy to other so-called acceptor molecules physically close to the excited fluorophores, a phenomenon known as resonance energy transfer.
Fluorescence limitations

45
Fluorophore molecule can emit typ. 103 - 104 quanta
signal-to-noise
ratio
spatial
resolution
speedThe three Ss or the
‘eternal fluorescence triangle’
Fluorescence photon budgets
Any of the three can be enhanced at the expense of the
other two
Available quanta can be resolved in time or in wavelength,
but only up to maximum available

46
106 molecules @ 1 M cell ≈ 1 pl
1010 photons per cell maximum
At max. excitation ≈ 1013 photons per sec
Fluorescence lifetimes ≈ 10-9 - 10-8 sec typical
Collection, QE limitations ≈ 1-10% photons ‘used’
108 -109 detected quanta per cell practical
At max. excitation ≈ 1011 -1012 photons per sec
s/n ratio ≈ 104 -105 for steady-state signal
Distributed over time, space
Fluorescence photon budgets

47
Photon (optical) shot noise (1)
Photon arrival times into a camera, or detector, are statistically independent, or uncorrelated events
When the physical signal that we observe is based on light, then the quantum nature of light plays a significant role!
A single photon at = 500 nm carries an energy of E = h = hc/ = 3.97 x 10-19 Joules

48
The intensity of a source will yield the average number of photons collected, but knowing the average number of photons which will be collected will not give the actual number collected
The actual number collected will be more than, equal to, or less than the average, and the distribution about that average will be a Poisson distribution
Cannot assume that, in a given pixel for two consecutive but independent observation intervals of length T, the same number of photons will be counted
The probability distribution for p photons in an observation window of length T seconds
where is the rate or intensity measured in photons per second
Photon (optical) shot noise (2)

49
Siméon Denis Poisson 1781-1840
1 photon4 photons10 photons
Poisson distribution
For sufficiently large numbers of photons, (typ. >>15-20) the normal (Gaussian) distribution with mean p and variance p (standard deviation p1/2), is a good approximation to the Poisson distribution.
FPoisson (x, p ) Fnormal (x; = p, 2 = p)
Johann Karl Friedrich Gauss 1777–1855
0 20 40 60 80 1000
0.02
0.04
0.06
0.08
0.1
0.1
0
P m n( )
1000 n
0 20 40 60 80 1000
0.02
0.04
0.06
0.08
0.1
0.1
0
G m n( )
1000 n
50
output
sensor
photons
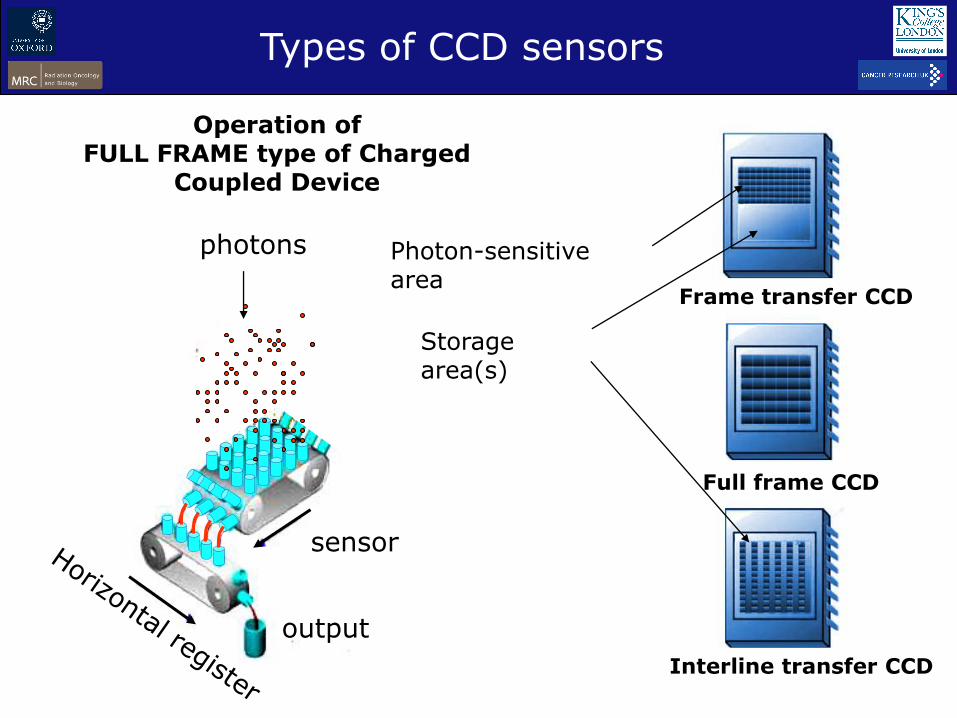
Operation of FULL FRAME type of Charged
Coupled Device
Frame transfer CCD
Full frame CCD
Interline transfer CCD
Types of CCD sensors
Photon-sensitive area
Storage area(s)

51
channels
channelsGlass
structure
input
photoelectronsecondary
electrons
glass
channel
wall
output
electrons
Semiconducting layer
Electrode
- High voltage +
Electrode plating (on each face)
Microchannel plate image intensifier

52
Electron multiplying CCD imager
EMCCD technology is a digital scientific detector innovation first introduced to the imaging community in early 2000, followed by spectroscopy versions in early 2005
As charge is transferred through each stage the phenomenon of impact ionization is utilized to produce secondary electrons, and hence EM gain
When this is done over several hundred stages, the resultant gain can be controlled from unity to hundreds or even thousands of times.
storage
section
imaging
section
o/p
charge-
voltage
conversion
Multiplication
register
readout
register
Unlike a conventional CCD, an EMCCD is not limited by the readout noise of the output amplifier.
a solid state Electron Multiplying (EM) register is added to the end of the normal serial register; it allows weak signals to be multiplied before any readout noise is added by the output amplifier
The EM register has several hundred stages that use higher than normal clock voltages

53
10.4 electrons pixel-1
0.8 electrons pixel-1
Electron multiplication – typ. x1000
EMCCD: the ‘ultimate’ camera?

54
Image generated in a completely different way to normal (wide-field) microscopy
Uses a scanning point (or points) of light instead of full sample illumination
Confocal microscopy provides significant improvements in optical sectioning by blocking the influence of out-of-focus light which would otherwise degrade the image
Confocal microscopy is commonly used where 3D structure is important or where out-of-focus light is to be rejected
Confocal microscopy

55
Rotating Nipkow disc
Fast image acquisition
Alternative and more commonly used arrangement uses scanning laser beams (LSM)
The rotating disc confocal microscope
Petran and Hadravsky: original dual-port optical system

56
Confocal imaging can offer a the resolution x1.4 wrt resolution obtained withconventional microscopes
A confocal microscope can be used in reflection mode and still exhibit the sameout-of-focus rejection performance – sometimes used for detecting goldparticles used in immuno-gold labeling
In a confocal imaging system a single point ofexcitation light (or sometimes a group of points or aslit) is scanned across the specimen. This is adiffraction-limited spot, produced either by imagingan illuminated aperture situated in a conjugate focalplane to the specimen or, more usually, by focusing aparallel laser beam
With only a single point illuminated, the illuminationrapidly falls off above and below the plane of focus asthe beam converges and diverges, thus reducingexcitation of fluorescence for interfering objectssituated out of the focal plane
Fluorescence light passes through a pinhole situatedin a focal plane conjugate to that of the specimen.Light passing through the image pinhole is detectedby a photodetector
Confocal microscopy

57
The laser-scanning confocal microscope
objective
dichromatic
mirror
photodetector
cell
pinhole 2
pinhole 1
laser beam
x
yscanning
spot moves in
x,y directions
light from only one
focal plane passes
through pinhole

58
Two-photon excitation - principle
two-photon excitation
single-photon excitation
300 400 500 600 700 800 900

59
Two-photon excitation – how it works
Only a single voxel is
excited at any one time
This excitation point is
scanned in x,y and z
and an image is built
up in a few seconds
An ultrashort laser
pulse is used for
excitation – very high
peak power, very low
average power

60
emission filter
xy scanner
objective
tube lens
mirror
dichroic mirror
emission filterPMT detectors
collector lens
dichroic mirror
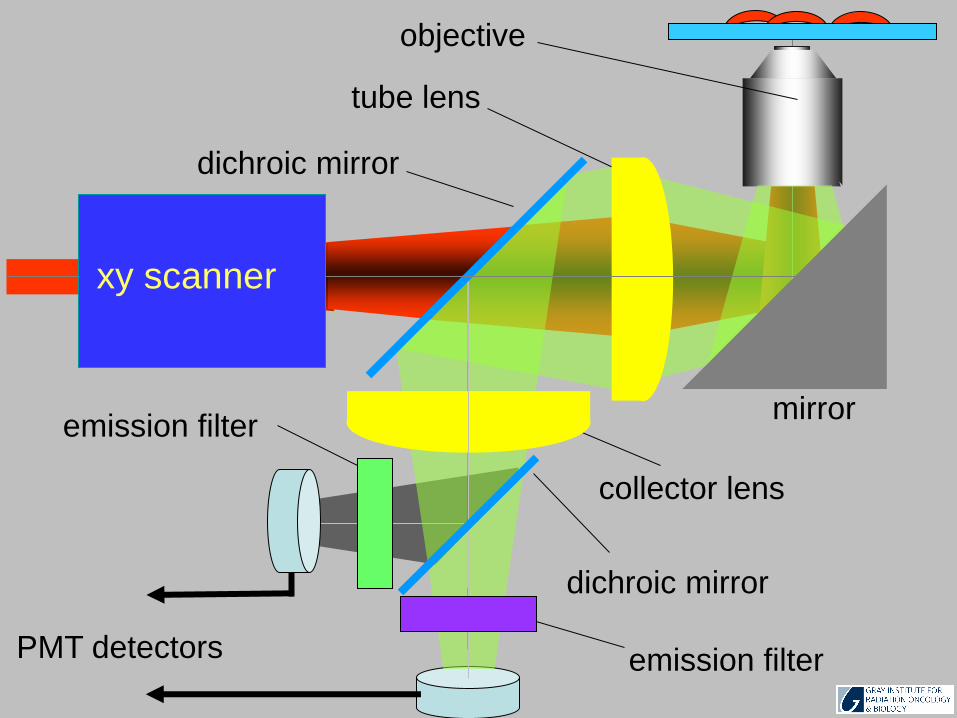
61
emission filter
objective
tube lens
mirror
dichroic mirror
emission filterPMT detectors
collector lens
dichroic mirror
xy scanner

62
Ex vivo 2P imaging - rat gut
With 2P excitation, it is
possible to image
deeply into specimens,
(>>100 microns)
Image sequence of successive optically-sectioned layers
63
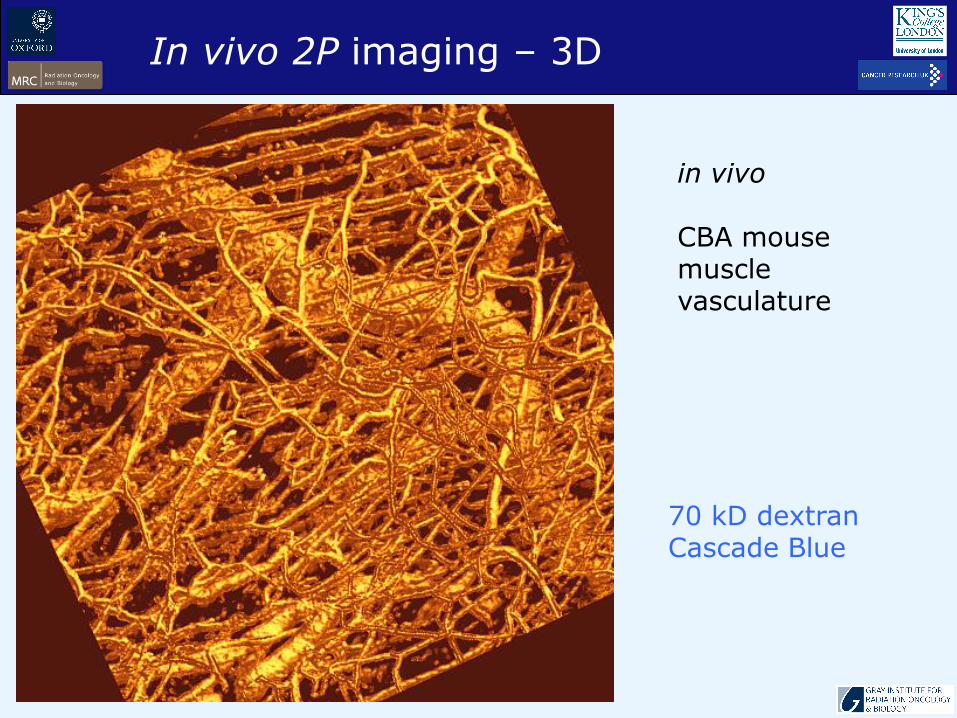
in vivo
CBA mouse muscle vasculature
70 kD dextranCascade Blue
In vivo 2P imaging – 3D
64
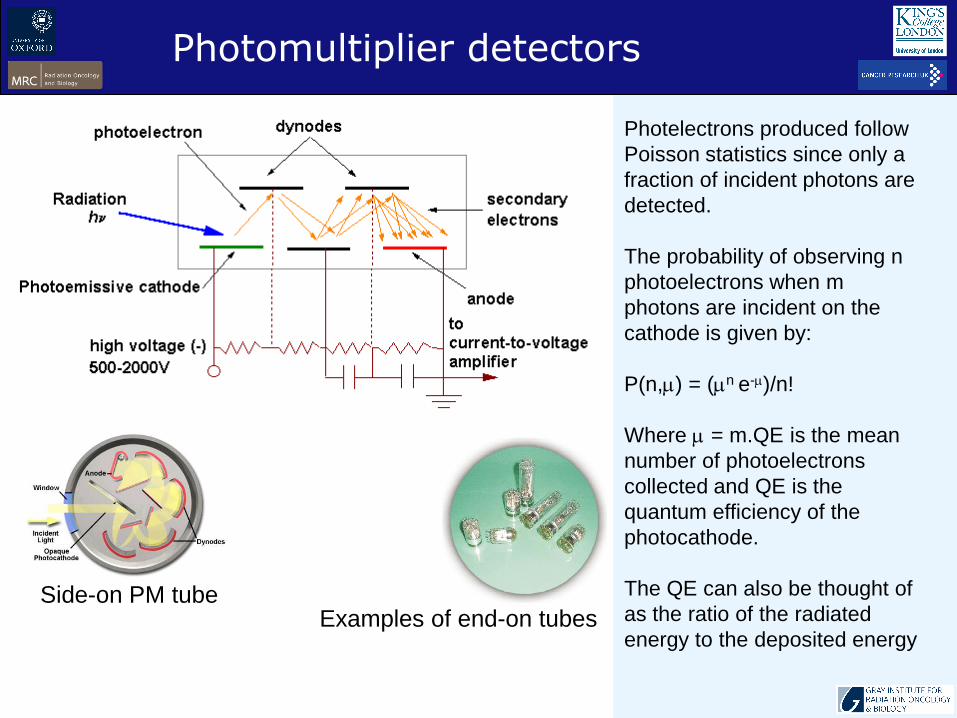
Photelectrons produced follow
Poisson statistics since only a
fraction of incident photons are
detected.
The probability of observing n
photoelectrons when m
photons are incident on the
cathode is given by:
P(n,) = (n e-)/n!
Where = m.QE is the mean
number of photoelectrons
collected and QE is the
quantum efficiency of the
photocathode.
The QE can also be thought of
as the ratio of the radiated
energy to the deposited energy
Photomultiplier detectors
Side-on PM tubeExamples of end-on tubes

65
The quantum efficiency (in %) at a given wavelength λ is related to the
radiant sensitivity R by:
QE = (124/λ) x R (measured in mA/W)
Photomultipliers

66
High quantum efficiency, high gainSmall active area
Avalanche photodiodes
Operation modes:
Linear: current to light intensityGeiger mode: pulse output

67
P22 fibrosarcoma
BD9 rat
Vasculature
(70 kD dextran / FITC)
+ autofluorescence
In vivo 2P imaging – 3D

68
Ex vivo 2P imaging
Mouse earMouse skin

69
P22 fibrosarcoma
BD9 ratHT29 human colon carcinoma
SCID mouse

70
High resolution, microvessel imaging
Liver
metastasis
formation
GFP tumour
cells

71
Fluorescence lifetime imaging
…..able to image molecular
interactions – and applied to
far-field methods with near-field
performance

72Intensity image Lifetime image
Kinetic trace at every pixel acquired (time resolution 130 ps)Kinetics analysed at every pixel to derive lifetime ()Lifetime mapped in (false colour) x (intensity)
Fluorescence lifetime imaging - FLIM
Analysis of the excited state lifetime of a
population of fluorescent probe molecules
Spatially resolved
acquisition of data
Informs on
molecular
environment
+

73
Donor emission
quenched
Protein
Donor
GFP
Protein
Acceptor-
labelled protein
Donor
GFP
Donor
emission
Főrster Resonance Energy Transfer (FRET)
FRET
Sensitized
emission
When donor emission spectrum overlaps with acceptor absorption spectrum
When dipole donor and acceptor moments are aligned Probability of FRET [separation]-6 (0.1-10 nm range)

74
Donor emission
quenched
Protein
Donor
GFPFRET
Sensitized
emission
Population of FRET species
= A1 / (A2+A1)
A1 = amplitude of quenched donor lifetime
A2 = amplitude of control donor lifetime
FRET efficiency
= (D - ) / D
D = control donor lifetime
= quenched donor lifetime
Unquenched
donor lifetime
D
Fully quenched
donor lifetime
Partial FRET
population
+ D
Főrster Resonance Energy Transfer (FRET)

75
FRET vs. Co-localisation
~ 0.25 - 0.5 m
vs FRETCo-localisation
Microscopic resolution Nanoscopic resolution
LOCATION INTERACTION
Far-field method with near-field res’n
Resolved
pixel/voxel

76
TACdiscriminator
excitation
arrival time
photodetectors
discriminator ADC
reset
time address
memory
event
xy address
add
Time-correlated single photon counting
signal
reference

77
1
10
100
0 10 ns5.02.5 7.5
distribution
Intensity
imageLifetime
image
In vivo 2P lifetime imaging

78
Location of site of PAK1-Cdc42 interaction.
MP FLIM used to determine the extent of
FRET between the GFP-PAK1 donor and
anti-HA-Cy3 acceptor in cells co-expressing
GFP-PAK1 and HA-tagged Cdc42 (WT and
N17 variants).
FLIM/FRET example: GFP donor, Cy3 acceptor


















