Nuclear Magnetic Resonance Parametersiupab/madhu1.pdf · 2010. 1. 1. · • Structure...
116
Nuclear Magnetic Resonance Parameters P. K. Madhu Department of Chemical Sciences Tata Institute of Fundamental Research Homi Bhabha Road Colaba Mumbai 400 005, India
Transcript of Nuclear Magnetic Resonance Parametersiupab/madhu1.pdf · 2010. 1. 1. · • Structure...

Nuclear Magnetic Resonance Parameters
P. K. MadhuDepartment of Chemical Sciences
Tata Institute of Fundamental ResearchHomi Bhabha Road
ColabaMumbai 400 005, India

Understanding the nuclear spin systemSpin choreography
Characterisation of various materials
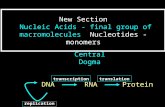
Structure of compounds, biomoleculesProteins, nucleic acids……….
Magnetic resonance imaging, MRI
NMR: What are the Potentials?

• Structure elucidation• Natural product chemistry• Synthetic organic chemistry
• Study of dynamic processes• Reaction kinetics• Study of equilibrium (chemical or structural)
• Structure determination of macromolecules• Proteins• Nucleotides, protein/DNA complexes• Polysaccharides
• Drug Design• Structure-Activity Relationship (SAR) by NMR
• Medicine• Magnetic Resonance Imaging• Metabonomics: combined use of spectroscopy & multivariate statistical approaches
to studies of biofluids, cells & tissues. Gives a unique metabolic fingerprint for eachcomplex biological mixture, sensitive to change
NMR: What are the Potentials?

Magnetic Resonance
Nuclear spins areour spies to probestructure
Superconducting magnetsgive us the medium andmechanism to manipulatethe spins
Talking to the spins ina language that they understand, which meanswe have to resonate withthem
Nuclear
NMR

Nuclear Spins
ElectronProtonNeutron
Nuclear spin is a fundamental property just like charge and mass
Deuterium atom:1 electron, 1 proton, and 1 neutron
Electronic spin = 1/2Nuclear spin = 1
NMR uses the nuclear properties and nuclear spins to eavesdrop on others
Paired nuclear spins are or of no use to work as nuclear spies
NMR needs unpaired, socially non-committed, nuclear spins to act as spies
Helium atomNet nuclear spin = 0NMR non-observabe

H1
Li2
Be3
Na11
Mg12
K19
Ca20
Rb37
Sr38
Cs55
Ba56
Fr87
Ra88
Sc21
Ti22
Y39
Zr40
La57
Hf72
Ac89
V23
Cr24
Mn25
Fe26
Co27
Ni28
Cu29
Zn30
Ga31
Ge32
As33
Se34
Br35
Kr36
Nb41
Ta73
Mo42
W74
Tc43
Re75
Ru44
Os76
Rh45
Ir77
Pd46
Pt78
Ag47
Au79
Cd48
Hg80
In49
C6
Si14
B5
Al13
N7
P15
O8
S16
F9
Cl17
Ar18
Ne10
He2
Sn50
Sb51
Te52
I53
Xe54
Rn86
At85
Po84
Bi83
Pb82
Tl81
Xn element with one I=1/2 isotope
Xn element with two (three) I=1/2 isotopes
NMR Periodic Table: Nuclei with Spins-1/2
Common nuclei probed are 13C, 15N, 29Si, and 31P

H1
Li2
Be3
Na11
Mg12
K19
Ca20
Rb37
Sr38
Cs55
Ba56
Fr87
Ra88
Sc21
Ti22
Y39
Zr40
La57
Hf72
Ac89
V23
Cr24
Mn25
Fe26
Co27
Ni28
Cu29
Zn30
Ga31
Ge32
As33
Se34
Br35
Kr36
Nb41
Ta73
Mo42
W74
Tc43
Re75
Ru44
Os76
Rh45
Ir77
Pd46
Pt78
Ag47
Au79
Cd48
Hg80
In49
C6
Si14
B5
Al13
N7
P15
O8
S16
F9
Cl17
Ar18
Ne10
He2
Sn50
Sb51
Te52
I53
Xe54
Rn86
At85
Po84
Bi83
Pb82
Tl81
Xn
Xn
Xn
I = 3/2 I = 5/2 I = 7/2
Xn
I = 9/2
Xn
5/2 + 7/2 etc.
NMR Periodic Table: Nuclei with Half-Integer Spins>1/2
Many of the nuclei of industrial importance are quadrupolar spin nucleimanifesting in ceramics, glasses, zeolites and catalysts

Nuclear Spins
Charge
Spin
Nuclear spin
Iμ γ=Gyromagnetic ratio, depends on the nucleus
Magnetic moment Spin quantum number

A spinning gyroscopein a gravity field A spinning charge
in a magnetic field
Nuclear Spins & Magnetic Field

Nuclear Spin

NMR Spies – Inside a Magnetic Field

NMR Spies – In Action

NMR Spies – The Tools
How do the spins probe the medium?
Chemical shift anisotropy
Dipole-dipolecouplings
Through-bondcouplings
Quadrupolarcouplings

Nuclear Spin Interactions
Spin Interactions
External Internal
Zeeman,HZ RF, HRF(t)
CSA,HCS Dipole,HDD Scalar,HJ Quad,HQ
The isotropic parts (manifest in solution-state) are time independentAnisotropic parts cause line broadening

Internal Nuclear Spin Interactions
Spin Interactions
Chemical shift Spin-spin couplings
Isotropicchemical shift
Chemical shiftanisotropy, CSA
Scalar, J-couplings Dipolar
Heteronuclear Homonuclear
Quadrupolar
Isotropic quad.shift
1st, 2nd order quad. Interaction, anisotropic
Electric Magnetic
Spin ½, 1H, 13C…..Spin>½, 23Na, 17O…..

NMR: Nuclear Spins, Magnetic Moments, and Resonance

The Electromagnetic Spectrum

RF waves in
Nuclear magnetic momentsin the sample
B0, External magnetic field
RF waves out
The frequency of emitted RF waves reveals information about the magnetic environment of atomic nuclei

Populations and Nuclear Magnetisation

Nuclear Magnetic Resonance

Typical 1H NMR Spectrum
CH3-CH2-OH

Sensitivity of NMR Spectroscopy

Isotope Net Spin γ / MHz T-1 Abundance %
1H 1/2 42.58 99.982H 1 6.54 0.0153H 1/2 45.41 0.031P 1/2 17.25 100.023Na 3/2 11.27 100.014N 1 3.08 99.6315N 1/2 4.31 0.3713C 1/2 10.71 1.10819F 1/2 40.08 100.0
Sensitivity of NMR Spectroscopy
Relative sensitivity of nuclei depends onGyromagneticGyromagnetic ratio (ratio (γγ))Natural abundance of the isotope Natural abundance of the isotope

1H NMR spectra of organic compound
8 scans ~12 secs
13C NMR spectra8 scans ~12 secs
13C NMR spectra10,000 scans ~4.2 hours
Sensitivity of NMR Spectroscopy

Energy Levels: Why Big Magnets Are Needed?

30 MHz
7 kGauss
500 MHz117 kGauss
Do We Need Bigger/Higher Magnets?
Highest superconducting magnet currently available is 23 T yieldingproton Larmor frequency of 1000 MHz

Chemical Shift- Usefulness of NMR
CH3CH2OH
NMR used for structural characterisationShould be able to distinguish functional groups unambiguously

Chemical Shift- Usefulness of NMR
Hb
Hc
C
Ha
Cl
νa νb νc
frequency

Pioneer of NMR in India
Prof. Dharmatti was a student of Felix Bloch in Stanford and discoveredchemical-shift phenomenon

Fourier-Transform NMR

Typical Experiment in NMR: RF Pulse
z
x
y
Mo
z
x
y
z
x
yMo
RF off
RF on
RF off
Effect of a 90o x pulse

z
x
yMo
z
x
y
Moω
z
x
y
Time
x
y
RF receivers pick up the signals I
After the Pulse: Nuclear Spin Evolution
The spins precess in the xy plane and relax to the equilibrium value, free induction decay

Fourier Transformation

Radio-Frequency Pulse
Pulse
Amplitude
Phase
Frequency
90x
Music
Volume
Timing
Pitch

Spin Relaxtion
There are two primary causes of spin relaxation:
Spin - lattice relaxation, T1, longitudinal relaxation
Spin - spin relaxation, T2, transverse relaxation
lattice

Spin Relaxation, T1
T1 determines the repetition rate of an experiment
90x
n
Signal-to-noise enhancement
For an optimum signal, there is a need to wait for a few T1 times before which an experiment can be repeated for signal averaging
The time scale with which the z-component of the magnetisation has felaxed back to the equilibrium magnetisation, M0

T1 or T2relaxation time
Correlation time
Short
Long
T1 minimum
T1 and T2 at short correlation times
optimal frequency for T1 relaxation (MHz frequencies)
T1
T2
Fast motionShort τc
Slow motionLong τc
Gases
Small molecules
Medium sizedmolecules
Large molecules
0 0.2 0.4 0.6 0.8 1.0 ns
Behaviour of T1 and T2 Relaxation Times

180x 90x
τ
z
y
x
z
y
x
90x
z
y
x
z
y
x
90x
z
y
x
z
y
x
90x
τ=0
some τ
large τ
T1 Measurement- Inversion Recovery Experiment

T1 Measurement- Inversion Recovery Experiment
180x 90x
τ
τ
0
Measure the NMR signal as a function of τ
Bloch equation for longitudinal magnetisation: 0
1
zz M MdMdt T
−= −
10 (1 2 )T
zM M eτ
−
= −
t

Mz = Mo (1- 2e -τ/T1 )
Mz
τ
Inversion Recovery - Measure NMR Intensity as a function of the delay time τ and fit to an exponential function
τ
0
0
T1 Measurement- Inversion Recovery Experiment

time
Currentamplitude
frequency
The faster the dephasing, the faster the decay of the time domain signal, the broader the line
Line widths are related to T2 relaxation. LW ~ 1/ T2 (not always true due to inhomogeneous broadening)
T2 is always faster (shorter) than or equal to T1
Transverse Relaxation Time, T2

T2 Measurement- Spin-Echo Experiment
90x 180x
τ/2 τ/2 t
Bloch equation for transverse magnetisation: , ,
1
x y x ydM Mdt T
= −
2, 0
Tx yM M e
τ−
=Mx(t)
τ
Plot of the peak amplitude as a functionof τ in the spin-echo experiment

Spin-Echo Experiment
Physics Today, 1953
E. Hahn, Physical Reivew, 80, 1950

90x 180x
τ/2 τ/2 t
Spin-Echo Experiment
y
z
y
x
y
x
y
x
y
x
90x
180x
evolution
evolutionMagnetisation is refocusedFormation of an echoDecay of echo only due to T2

T2 Relaxation, Line Width and Correlation Times
4pηwr3H
τc = 3kBT
ηw = viscosity of the solvent
r3H = hydrated radius
τc (ns)
0
10
20
15
5
25
2 4 6 8 10 12 14
Δν FWHM(Hz)
See Cavanagh et al. Protein NMR spectroscopy, pages 16-19.

1H
NH
NH
NH
15N
9.0 8.0
NH
NH
mobile, flexible chain has narrowerline widths than globular protein
T2 Relaxation, Line Width and Correlation Times

NH
NH
NH N
HNH
Mobility is also expressed in T1relaxation times.
t = 10 us
t = 100 us
t = 1000 us
t = 5000 us
T1 Relaxation and Mobility

Intensity and Resolution
To summarise:
The first point of the FID determines the intensity of the resonance signal
The duration of the FID determines the resolution of the resonance signal

Information in a NMR SpectraInformation in a NMR Spectra
ObservableObservable NameName QuantitativeQuantitative InformationInformation
Peak position Chemical shifts (δ) δ(ppm) = uobs –uref/uref (Hz) chemical (electronic) environment of nucleus
Peak Splitting Coupling Constant (J) Hz peak separation neighboring nuclei(intensity ratios) (torsion angles)
Peak Intensity Integral unit less (ratio) nuclear count (ratio)
relative height of integral curve quantifying
Peak Shape Line width Δυ = 1/πT2 molecular motionpeak half-height chemical exchange
1) Energy E = hυ
h is Planck constantυ is NMR resonance frequency 10-10 10-8 10-6 10-4 10-2 100 102
wavelength (cm)
γ-rays x-rays UV VIS IR μ-wave radio

a) Small local magnetic fields (Bloc) are generated by electrons as they circulate around nuclei.
b) These local magnetic fields can either oppose or augment the external magnetic field1) Typically oppose external magnetic field2) Nuclei “see” an effective magnetic field (Beff) smaller then
the external field3) σ – magnetic shielding or screening constant
i. depends on electron densityii. depends on the structure of the compound
Beff = Bo - Bloc Beff = Bo( 1 - σ )HO-CH2-CH3
de-shielding high shieldingShielding – local field opposes Bo
ν = γBo/2π
Chemical ShiftChemical Shift
σ – reason why we observe three distinct NMR peaks instead of one based on strength of B0

Proton Peaks are Referenced Against TMSProton Peaks are Referenced Against TMS
TMSshift in Hz
0
Si CH3CH3
CH3
CH3
tetramethylsilane“TMS”
n
Rather than measure the exact resonance position of a peak, we measure how far downfield it is shifted from TMS.
Highly shieldedprotons appearway upfield.
downfield

TMSshift in Hz
0ndownfield
The shift observed for a given protonin Hz also depends on the frequencyof the instrument used.
Higher frequencies= larger shifts in Hz
Higher Fields (Frequencies) Give Larger ShiftsHigher Fields (Frequencies) Give Larger Shifts

ppmppm and Hzand Hz
1H operating frequency Hz equivalent of ppm
500 MHz 500 Hz
600 MHz 600 Hz
800 MHz 800 Hz

chemicalshift = δ =
shift in Hzspectrometer frequency in MHz
= ppm
parts permillion
Chemical ShiftChemical Shift
0
10
10
10 0
0
PPM
PPM
PPM
500 MHzHigher frequency leads to more dispersion
800 MHz
600 MHz

H3C Si CH3
CH3
CH3
•For protons, ~ 15 ppm:
•For carbon, ~ 220 ppm:0
TMS
ppm
210 715 5
Aliphatic
Alcohols, protons ato ketones
Olefins
AromaticsAmides
AcidsAldehydes
ppm
50150 100 80210
Aliphatic CH3,CH2, CH
Carbons adjacent toalcohols, ketones
Olefins
Aromatics,conjugated alkenes
C=O of Acids,aldehydes, esters
0TMS
C=O inketones
Chemical-Shift Scales

Chemical-Shift Scales
aliphaticC-H
CH on Cnext to pi bonds
C-H where C is attached to anelectronega-tive atom
alkene=C-H
benzeneCH
aldehydeCHO
acidCOOH
23467910 0X-C-H X=C-C-H
Interpretation of most 1D spectra is possible with this information

Factors Determining Resonance Positions of 1H
• Deshielding by electronegative elements
•Anisotropic fields usually due to pi-bonded electrons in the molecule
•Deshielding due to hydrogen bonding

Deshielding by an Electronegative Element
C HClelectronegativeelement
δ- δ+
δ- δ+
Chlorine “deshields” the proton,that is, it takes valence electron density away from carbon, whichin turn takes more density fromhydrogen deshielding the proton.
0 ppm
highly shieldedprotons appearat high field
“deshielded“protons appear at low field
deshielding moves protonresonance to lower field

Electronegativity Dependence of Chemical Shift
Compound CH3X
Element X
Electronegativity of X
Chemical shift δ
CH3F CH3OH CH3Cl CH3Br CH3I CH4 (CH3)4Si
F O Cl Br I H Si
4.0 3.5 3.1 2.8 2.5 2.1 1.8
4.26 3.40 3.05 2.68 2.16 0.23 0
Dependence of the Chemical Shift of CH3X on the element X
deshielding increases with theelectronegativity of atom X
TMSmostdeshielded

Substitution Effects on Chemical Shift
CHCl3 CH2Cl2 CH3Cl 7.27 5.30 3.05 ppm
-CH2-Br -CH2-CH2Br -CH2-CH2CH2Br 3.30 1.69 1.25 ppm
mostdeshielded
mostdeshielded
The effect decreaseswith increasing distance
The effect increases withgreater numbersof electronegativeatoms

Aromatic Ring Current Effects
The presence of pi-bonds leads to anisotropic fields and affect chemical shift
Aromatic ring currents are observed in molecules like benzene and naphthalene
The applied magnetic field gives rise to a ring current in the delocalisedpi-electrons of the aromatic ring
Benzene rings have the greatest effect
Ring protons can get deshielded (induced and static fields add)while inner protons get shielded (induced and static fields oppose)
Ring current
Induced field

Aromatic Ring Current Effects
Secondary magnetic fieldgenerated by circulating πelectrons deshields aromaticprotons
Circulating π electrons
Ring Current in BenzeneRing Current in Benzene
Bo
Deshielded
H H fields add together
Ring protons come at 7.3 ppminstead of the 5.6 ppm of the vinylicprotons in cyclohexane

Influence of Hydrogen Bonding Influence of Hydrogen Bonding
OC
OR
H
HCO
OR
Carboxylic acids have stronghydrogen bonding - theyform dimers.
With carboxylic acids the O-Habsorptions are found between10 and 12 ppm very far downfield.
O
OO
HCH3
In methyl salicylate, which has stronginternal hydrogen bonding, the NMRabsortion for O-H is at about 14 ppm,way, way downfield.

O HR
O R
HHO
RThe chemical shift dependson how much hydrogen bondingis taking place.
Alcohols vary in chemical shiftfrom 0.5 ppm (free OH) to about5.0 ppm (lots of H bonding).
Hydrogen bonding lengthens theO-H bond and reduces the valence electron density around the proton- it is deshielded and shifted
downfield in the NMR spectrum.
Hydrogen Bond and Chemical Shift

Influence of Hydrogen Bonding Influence of Hydrogen Bonding
free H-bonded
OH upfield shifted,No H-bonding
OH downfieldfieldshifted, H-bonding

SPIN-SPIN SPLITTING

Multiplets in Spectra
Splitting of the resonances, multiplets, are due to spin-spin coupling andCan be predicted by the n+1 rule

Scalar Coupling
I1
I2e-
e-
Fermi contact
J-coupling is facilitated by the electrons in the bonds that connect the nuclei
Scalar coupling, through-bond coupling
The coupling constants can be related to a number of physical properties: Hybridisation, dihedral bond angles, and electronegativity of substituents
Geminal and vicinal couplings may be used to determine the bond angles and torsion angles

Geminal Scalar Coupling: 2J Coupling
Smaller the J value, larger the bond angle

The magnitude of the J coupling is dictated by the torsionangle between the two coupling nuclei according to the Karplus equation.
Vicinal Scalar Coupling: 3J Coupling
CC
H
HH
H θ
J = A + B cos(θ) + C cos2(θ)

right-handed alpha helix 3JNHα = 3.9antiparallel beta sheet 3JNHα = 8.9parallel beta sheet 3JNHα = 9.7
Scalar Coupling

Spin System Classification
Chemical equivalence among the spins:
• If the spins of the same isotopic species
• There exists a molecular symmetry operation that exchanges the two spins
Magnetic equivalence among the spins:
• This is a stronger form of the chemical equivalence
• The spins should have the same chemical shifts
• Either the spins have identical couplings to all other spins in themolecule or there are no other spins in the molecule
Ha
Hb Fb
Fa
1,1-difluoroethene
Ha
OR
HbH1
H2H3
Ha
OR
HbH3
H1H2
Ha
OR
HbH2
H3H1
CH3-CH2-OR

Spin System Classification
Ha
Hb Fb
FaNot magnetically equivalent
δHa= δHb, and δFa= δFb butJHaFb≠JHaFa and JHbFb≠JHbFa
Also here JHa JHb ≠0
It is an AA’XX’ spin system
H
HF
F
In this case, difluoromethane, 1Hs and 19Fs are magnetically equivalent not due to rotation, but to symmetry around the carbon. It is an A2X2 system
Ha
OR
HbH1
H2H3
Ha
OR
HbH3
H1H2
Ha
OR
HbH2
H3H1
Magnetically equivalent
It is an A2X3 spin system
The most shielded spin isnotated as A

C C
H H
H
C C
H H
H
two neighboursn+1 = 3triplet
one neighbourn+1 = 2doublet
this hydrogen peakis split by its two neighbours
these hydrogens aresplit by their singleneighbour
Scalar Coupling and Splitting of Resonances
n+1 rule

Exception To The n+1 RuleException To The n+1 Rule
Protons that are equivalent by symmetryusually do not split one another
CH CHX Y CH2 CH2X Y
no splitting if x=y no splitting if x=y
1)
2) Protons in the same groupusually do not split one another
CH
HH or C
H
H

Some Splitting PatternsSome Splitting Patterns
CH2 CH2X Y
CH CHX Y
CH2 CH
CH3 CH
CH3 CH2
CH3
CHCH3
( x = y )
( x = y )

J J
J
J J
The coupling constant is the distance J (measured in Hz) between the peaks in a multiplet.
J is a measure of the amount of interaction between the two sets of hydrogens creating the multiplet.
C
H
H
C H
H
H
J
Coupling ConstantCoupling Constant

C C
H H
C CH
H
C CHH
CH
H
6 to 8 Hz
11 to 18 Hz
6 to 15 Hz
0 to 5 Hz
three bond 3J
two bond 2J
three bond 3J
three bond 3J
trans
cis
geminal
vicinal
Representative Coupling ConstantsRepresentative Coupling Constants

I S
J ≠ 0
I S
J = 0
Scalar Coupling- Splitting of Resonances
A splitting of a signal means that we have more energies involved in the transition of a certain nuclei. So why do we have more energies?
Coupling constants do not depend on the applied magnetic field, unlike the CSA

Scalar Coupling- Splitting of Resonances
Bo
19F
19F
1H
1H
Nucleus
Electron
The nuclear magnetic moment of 19F polarises the F bonding electron (up), which, since we are following quantum mechanics rules, makes the other electron point down (the electron spins have to be antiparallel).
Now, since we have different states for the 1H electrons depending on the state of the 19F nucleus, we will have slightly different energies for the 1H nuclear magnetic moment (remember that the 1s electron of the 1H generates an induced field…).
This difference in energies for the 1H result in a splitting of the 1H resonance line.

Scalar Coupling- Splitting of Resonances
Bo
1H 1H
C
Similar analysis for a CH2 system as well.
The state of one of the 1H spins gets transmitted to the other 1H spins via the electrons in the bond (things get a bit complicated here due to the sp3 hybridiation etc. in general).

C CH H
C CH HA A
upfielddownfield
Bo
50 % ofmolecules
50 % ofmolecules
aligned with Bo opposed to Bo
neighbour aligned neighbour opposed
At any given time about half of the molecules in solution willhave spin +1/2 and the other half will have spin -1/2.
+1/2 -1/2
Shift of HA is Affected by the Spin of its Neighbour

C CH H
C CH H
one neighbourn+1 = 2doublet
one neighbourn+1 = 2doublet
yellow spinsblue spins
The resonance positions (splitting) of a given hydrogen is affected by the possible spins of its neighbor.
Spin Arrangements

C C
H H
H
C C
H H
H
two neighborsn+1 = 3triplet
one neighborn+1 = 2doublet
methylene spinsmethine spins
Spin Arrangements

A XA X
A1
A2
A1
A2
Bo E
J = 0J > 0
E4
E3E2
E1
A X A X
A1
A2A2
A1
Bo E
J = 0 J < 0E4
E3E2
E1
A1
A2
ν A1 = ν A2
J = 0
A1 A2
ν A1 ν A2
J > 0
A2 A1
ν A2 ν A1
J < 0
Scalar Coupling- Energy Level Picture

Scalar Coupling: First-Order Spectra
In weakly coupled spin systems: Consider ethyl acetate, CH2 at 4.5 ppm and CH3 at 1.5 ppm
J is typically 7 Hz, hence weakly coupled, also calledfirst-order spin system
ββαβ βααα
βββααβ αβα βαααββ βαβ ββα
αααCH2 CH3
Each 1H in CH3 will see three possible states of 1H in CH2Each 1H in CH2 will see four possible states of 1H in CH3Note the 1H in CH3 and that in CH2 are equivalent

11stst order systems (continued)order systems (continued)
•A coupled to n identical nuclei X (of spin 1/2) yields n + 1 lines in the spectrum of A.
•Therefore, the CH2 in EtOAc will show up as four lines, or a quartet. •Analogously, the CH3 in EtOAc will show up as three lines, or a triplet.
• The separation of the lines will be equal to the coupling constant between the two types of nuclei (CH2’s and CH3’s in EtOAc, approximately 7 Hz).
• Intensities can also be derived from the diagram of the possible states:
• Since we have the same probability of finding the system in any of the states,and states in the same rows have equal energy, the intensity will havea ratio 1:2:1 for the CH3, and a ratio of 1:3:3:1 for the CH2.
CH3
CH2
J (Hz)
4.5 ppm 1.5 ppm
Scalar Coupling: First-Order Spectra

The splitting of the resonance of a nuclei A by a nuclei X with spin number I will be 2I + 1.
Scalar Coupling: First-Order Spectra
8
4 4
2 22 2
1 111 1111
2
1
Coupling to the first 1H(2 * 1/2 + 1 = 2)
Coupling to the second 1H
Coupling to the third 1H
In general, the number of lines in these cases will be a binomial expansion, known as the Pascal Triangle:
Start with one 1H
1 : n / 1 : n ( n - 1 ) / 2 : n ( n - 1 ) ( n - 2 ) / 6 : ...

Scalar Coupling: First-Order Spectra
11 1
1 2 11 3 3 1
1 4 6 4 1
Here n is the number of equivalentspins 1/2 we are coupled to: Theresults for several n’s is
• In a spin system in which we have a certain nuclei coupled to more than one nuclei, all first order, the splitting will be basically an extension of what we saw before.
• Say that we have a CH (A) coupled to a CH3 (M) with a JAM of 7 Hz, and to a CH2 (X) with a JAX of5 Hz. We basically go in steps. First the big coupling, which will give a quartet:
• Then the small coupling, which will split each linein the quartet into a triplet:
• This is called a triplets of quartet (big effect is the last…).
7 Hz
5 Hz

CCH3 CH3
N
H
O O+
-
1:6:15:20:16:6:1 in higher multiplets the outer peaksare often nearly lost in the baseline
2-Nitropropane

Scalar Coupling: Strong Coupling
SecondSecond--order spectra, AB spin systemorder spectra, AB spin system
B2
B1
J > 0
A2
A1
23
αα
αβcosθ+βαsinθ
ββ
0 0| | | |
1
4
A X AXJω ω π− ∼
−αβsinθ+βαcosθ
E4 = 1/2 ω0A + 1/2 ω0BX + 1/4 πJAB
E3 = 1/2 D- 1/4 πJAB
E2 = - 1/2D - 1/4 πJAB
E1 = -1/2 ω0A - 1/2 ω0B + 1/4 πJAB
E4 = 1/2 ω0A + 1/2 ω0BX + 1/4 πJAB
E3 = 1/2 D- 1/4 πJAB
E2 = - 1/2D - 1/4 πJAB
E1 = -1/2 ω0A - 1/2 ω0B + 1/4 πJAB
sin 2θ=J/Dsos2θ= (ω0A - 1/2 ω0BX)/DD=[(ω0A - ω0B)2+πJ2]1/2
sin 2θ=J/Dsos2θ= (ω0A - 1/2 ω0BX)/DD=[(ω0A - ω0B)2+πJ2]1/2
ω0A ω0X
JAB JAB1-sin 2θ 1-sin 2θ
1+sin 2θ1+sin 2θ

Δν >> J
Δν = 0
Scalar Coupling: Second-Order Spectra
••As As ΔνΔν approaches J, more and more transition of similar energy occurapproaches J, more and more transition of similar energy occur
•Our system is now a second-order system. We have effects that are not predicted by the simple multiplicity rules that were described earlier

1 2 3 4A BνZνA νB
| JAB | = | f1 - f2 | = | f3 - f4 || JAB | = | f1 - f2 | = | f3 - f4 |Δν2 = | ( f1 - f4 ) ( f2 - f3 ) |
νA = νZ - Δν / 2νB = νZ + Δν / 2
Δν2 = | ( f1 - f4 ) ( f2 - f3 ) |
νA = νZ - Δν / 2νB = νZ + Δν / 2
I2 I3 | f1 - f4 |= =
I1 I4 | f2 - f3 |
I2 I3 | f1 - f4 |= =
I1 I4 | f2 - f3 |•Peak intensities can be computed similarly:
•AB system has the roofing effect: coupled pairs will lean towards each other, making a little roof:
•The chemical shifts of nuclei A and B are not at the center of the doublets. They will be at the center of mass of both lines. Δν the νA - νB chemical shift difference
Scalar Coupling: Second-Order Spectra

Cl
Cl
Cl
HX Cl
ClHA HA
Cl
Cl
HA
HB
HA
Cl
Transition from A2X to A2B
Scalar Coupling: Second-Order Spectra

100 MHz
200 MHz
123456
123
200 Hz
400 Hz
J = 7.5 Hz 7.5 Hz
J = 7.5 Hz 7.5 Hz
Coupling constants areconstant - they do not change at differentfield strengths
The shift isdependanton the field
ppm
Separationis larger
J and Magnetic Field: Comparison
100 Hz
200 Hz

100 MHz
200 MHz
123456
123
100 Hz
200 Hz
J = 7.5 Hz
J =7.5 Hz
ppm4
200 Hz
400 Hz
56
J = 7.5 Hz
Note the compression ofmultiplets in the 200 MHzspectrum when it is plotted on the same scale as the 100 MHz spectruminstead of on a chart whichis twice as wide.
Separationis larger
J and Magnetic Field: Comparison

123
123
100 MHz
200 MHz
Why buy a higherfield instrument?
Spectra aresimplified!
Overlapping multiplets areseparated.
123
50 MHzJ = 7.5 Hz
J = 7.5 Hz
J = 7.5 HzSecond-ordereffects are minimized.
J and Magnetic Field: Comparison

INTEGRATION

Integration of a Peak
Besides identifying the type of hydrogen, we can also obtain the relative Numbers of each type of hydrogen by integration
Integration is determining the area under a peak
The are under a peak is proportional to the number of hydrogens that generate the peak

Integration of a Peak
Benzyl AcetateThe integral line rises an amount proportional to the number of H in each peak
integralline
55: 22: 33 = 5: 2: 3 simplest ratio of the heights

Summary So Far!
• Each different type of hydrogen gives a peak or group of peaks(multiplet)
•The chemical shift, in ppm, is suggestive of the type of hydrogen generating the peak
•The integral gives the relative numbers of each type of hydrogen
•Spin-spin splitting gives the number of hydrogens on adjacent carbons
•The coupling constant J also gives information about the arrangement of the atoms involved, dihedral angles for instance

13C NMR
12C is not NMR active, I=0
13C is NMR active, I=1/2
However, 13C is only 1.08% abundant:
•Low gyromagnetic ratio•Signals about 6000 times weaker than 1H
The chemical-shift range of 13C is larger than that of 1H, about 0-200 ppm

13C NMR
For a given field strength 13C has its resonance at adifferent (lower) frequency than 1H.
1H11.7 T 500 MHz14.1 T 600 MHz18.89 T 800 MHz 13C
11.7 T 125 MHz14.1 T 150 MHz18.89 T 200 MHz
Divide the hydrogenfrequency by 4 (approximately)for carbon-13

13C NMR
Due to the low natural abundance of 13C spins, the probability of finding two 13C atoms next to each other in a single molecule is very small
13C-!3C coupling NO! Not probable
13C spectra are determined by many molecules contributing to the spectrum, eachhaving only one 13C atom
However, 13C does couple to 1H (spin ½)
13C-1H coupling YES! Very common

13C-1H J Coupling, Heteronuclear Coupling
The effect of attached protons on 13C resonances
n+1 = 4 n+1 = 3 n+1 = 2 n+1 = 1
C13
3 protons 2 protons 1 proton 0 protons
H
H
H
C13 H
H
C13 H C13
Methylcarbon
Methylenecarbon
Methinecarbon
Quaternarycarbon
( n+1 rule applies ) (J’s are large ~ 100 - 200 Hz)

13C-1H J Coupling, Heteronuclear Coupling
13C coupledto the hydrogens
Ethyl phenylacetate

Decoupling: Removing J Couplings
13C NMR spectrum of quinoline13C NMR spectrum of quinoline
1H decoupled 13C spectrum
13C spectrum scalar coupled to 1H
In cases of many nuclear spins, J couplings can reduce the sensitivitysnd crowd the spectra, hence, the need to remove them-Heteronuclear/homonuclear J decoupling

Heteronuclear Decoupling in Solution-State NMR
Decoupling
(π/2)y
S
I1H
13C
RF
Decoupling
S spin detection

Heteronuclear Decoupling in Solution-State NMR

Double-Resonance Techniques
Heteronuclear decoupling- A double-resonance scheme
Others being Nuclear Overhauser Effect, Spin Tickling and othersophisticated schemes
NMR Spectroscopy, A Physicochemical View- Robin HarrisModern NMR Techniques for Chemistry Research- Andrew DeromeNMR: The Toolkit- P. J. Hore, J. Jones, and S. WimperisSpin Dynamics, Basics of NMR- Malcolm H. Levitt

Decoupling: Spectral Simplification
Ethyl phenylacetate

13C Chemical Shift Chart
AldehydesKetones
Acids AmidesEsters Anhydrides
Aromatic ringcarbons
Unsaturated carbon - sp2
Alkynecarbons - sp
Saturated carbon - sp3
electronegativity effects
Saturated carbon - sp3
no electronegativity effects
C=O
C=O
C=CC C
200 150 100 50 0
8 - 30
15 - 55
20 - 60
40 - 80
35 - 80
25 - 65
65 - 90
100 - 150
110 - 175
155 - 185
185 - 220
C-O
C-Cl
C-Br
R3CH R4C
R-CH2-R
R-CH3
RANGE
/

nitriles
acid anhydridesacid chlorides
amides
esters
carboxylic acidsaldehydesα,β-unsaturated ketones
ketones
220 200 180 160 140 120 100 ppm
13C PPM Chart for Carbonyl and Nitrile Functional Groups
13C Chemical Shift Chart

Structural Determination with 1D NMR
•Structure determination of small molecules possible with 1D NMR
•Chemical shift, J coupling, geometry ………..
•Other sophisticated experiments
•INEPT•DEPT•Nuclear Overhauser Effect, NOE•Relaxation measurements•Exchange experiments•Diffusion experiments•Variable temperature experiments

Homodecoupling
• Simple & quick means of determining if two spins are coupled• Effective on molecules with simple, well-dispersed spectra• Involves irradiation of selected resonance with low power, thus
eliminating any coupling to this spin• Comparison of homodecoupled spectrum with the normal one,
coupling partners of the irradiated peak are determined
Homodec. spectrum of ethyl benzene
Normal spectrum
Irr. at quartet
Irr. at triplet
CH2-CH3
2D counterpart COSY

•Conclusions
•NMR is a very powerful method to probe geometry, dynamics, and other structural information parameters
•Chemical shift, scalar coupling, relaxation rate constants, peak integrals are some of the methods used in one-dimensional NMR
•1D NMR can yield a host of information regarding a wide range of molecules
•1H and 13C are the normal probes in 1D NMR
•Other probes, such as, 31P, 29Si, 11B, and many other spin ½ or spin higher nucleimay be used to characterise materials
•NMR also is a thorough test bed for many quantum mechanics principles
•A versatile tool with far reaching implications in Physics, Chemistry, Biology, and Medicine