
Novel MSH6 MutationsinTreatment...
11
Biology of Human Tumors Novel MSH6 Mutations in Treatment-Naïve Glioblastoma and Anaplastic Oligodendroglioma Contribute to Temozolomide Resistance Independently of MGMT Promoter Methylation Stephanie A. Nguyen 1,2 , Owen D.M. Stechishin 1,2 , H. Artee Luchman 1,2 , Xueqing Q. Lun 3,4 , Donna L. Senger 3,4 , Stephen M. Robbins 3,4 , J. Gregory Cairncross 1,3,5 , and Samuel Weiss 1,2,3 Abstract Purpose: The current standard of care for glioblastoma (GBM) involves a combination of surgery, radiotherapy, and temozolomide chemotherapy, but this regimen fails to achieve long-term tumor control. Resistance to temozolomide is largely mediated by expression of the DNA repair enzyme MGMT; however, emerging evidence suggests that inactivation of MSH6 and other mismatch repair proteins plays an important role in temozolomide resistance. Here, we investigate endogenous MSH6 mutations in GBM, anaplastic oligodendroglial tumor tissue, and corresponding brain tumor–initiating cell lines (BTIC). Experimental Design: MSH6 sequence and MGMT promoter methylation were determined in human tumor samples and BTICs. Sensitivity to temozolomide was evaluated in vitro using BTICs in the absence and presence of O 6 -benzylguanine to deplete MGMT. The influence of MGMT and MSH6 status on in vivo sensitivity to temozolomide was evaluated using intracranial BTIC xenografts. Results: We identified 11 previously unreported mutations in MSH6 in nine different glioma samples and six paired BTIC lines from adult patients. In addition, MSH6 mutations were documented in three oligodendrogliomas and two treatment-na € ve gliomas, both previously unreported findings. These muta- tions were found to influence the sensitivity of BTICs to temozolomide both in vitro and in vivo, independent of MGMT promoter methylation status. Conclusions: These data demonstrate that endogenous MSH6 mutations may be present before alkylator therapy and occur in at least two histologic subtypes of adult glial neoplasms, with this report serving as the first to note these mutations in oligodendroglioma. These findings broaden our understanding of the clinical response to temozolomide in gliomas. Clin Cancer Res; 20(18); 4894–903. Ó2014 AACR. Introduction Malignant glial neoplasms are the most frequent primary intra-axial brain tumors in adults. The current standard of care for glioblastoma (GBM), the most aggressive of glio- mas, involves a combination of surgery, radiotherapy, and temozolomide chemotherapy (reviewed in ref. 1). Despite aggressive treatment, long-term tumor stabilization of GBM is uncommon and the median survival time of good per- formance patients of ages 70 years or less is only 14.6 months (2). The most complete and most durable responses to temozolomide and the longest overall survival times are seen in the subgroup of tumors with epigenetic silencing of the MGMT enzyme that normally repairs the cytotoxic O 6 -methylguanine (O 6 -MeG) DNA adducts induced by temozolomide (3). However, MGMT methyla- tion status does not correlate with temozolomide sensitivity in all patients, and thus MGMT expression is insufficient to explain temozolomide resistance in all tumors (1, 3, 4). Recent findings from The Cancer Genome Atlas (TCGA) implicate defects in the DNA mismatch repair (MMR) system, primarily involving mutations of MSH6, as addi- tional mediators of temozolomide resistance (5). MMR proteins recognize O 6 -MeG adducts not removed by MGMT and initiate an apoptotic response that leads to cell death (6). Hence, sensitivity to temozolomide likely requires both 1 Hotchkiss Brain Institute, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada. 2 Department of Cell Biology and Anatomy, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada. 3 Clark Smith Brain Tumor Research Centre, Southern Alberta Cancer Research Institute, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada. 4 Department of Oncology, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada. 5 Department of Clinical Neurosciences, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada. Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). S.A. Nguyen and O.D.M. Stechishin contributed equally to this article. Corresponding Author: Samuel Weiss, Hotchkiss Brain Institute, Faculty of Medicine, University of Calgary, HRIC 1A10, 3330 Hospital Drive NW, University of Calgary, Calgary, AB T2N 4N1, Canada. Phone: 403-220- 6953; Fax: 403-210-9382; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-13-1856 Ó2014 American Association for Cancer Research. Clinical Cancer Research Clin Cancer Res; 20(18) September 15, 2014 4894 on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856
Transcript of Novel MSH6 MutationsinTreatment...

Biology of Human Tumors
NovelMSH6Mutations in Treatment-Naïve Glioblastoma andAnaplastic Oligodendroglioma Contribute to TemozolomideResistance Independently of MGMT Promoter Methylation
Stephanie A.Nguyen1,2, OwenD.M. Stechishin1,2, H. Artee Luchman1,2, XueqingQ. Lun3,4, Donna L. Senger3,4,Stephen M. Robbins3,4, J. Gregory Cairncross1,3,5, and Samuel Weiss1,2,3
AbstractPurpose: The current standard of care for glioblastoma (GBM) involves a combination of surgery,
radiotherapy, and temozolomide chemotherapy, but this regimen fails to achieve long-term tumor
control. Resistance to temozolomide is largely mediated by expression of the DNA repair enzyme
MGMT; however, emerging evidence suggests that inactivation of MSH6 and other mismatch repair
proteins plays an important role in temozolomide resistance. Here, we investigate endogenous MSH6
mutations in GBM, anaplastic oligodendroglial tumor tissue, and corresponding brain tumor–initiating
cell lines (BTIC).
Experimental Design: MSH6 sequence and MGMT promoter methylation were determined in human
tumor samples andBTICs. Sensitivity to temozolomidewas evaluated in vitrousing BTICs in the absence and
presence of O6-benzylguanine to deplete MGMT. The influence of MGMT and MSH6 status on in vivo
sensitivity to temozolomide was evaluated using intracranial BTIC xenografts.
Results: We identified 11 previously unreported mutations in MSH6 in nine different glioma samples
and six paired BTIC lines from adult patients. In addition, MSH6 mutations were documented in three
oligodendrogliomas and two treatment-na€�ve gliomas, both previously unreported findings. These muta-
tionswere found to influence the sensitivity of BTICs to temozolomide both in vitro and in vivo, independent
of MGMT promoter methylation status.
Conclusions: These data demonstrate that endogenousMSH6mutationsmaybe present before alkylator
therapy and occur in at least two histologic subtypes of adult glial neoplasms, with this report serving as the
first to note these mutations in oligodendroglioma. These findings broaden our understanding of the
clinical response to temozolomide in gliomas. Clin Cancer Res; 20(18); 4894–903. �2014 AACR.
IntroductionMalignant glial neoplasms are themost frequent primary
intra-axial brain tumors in adults. The current standard ofcare for glioblastoma (GBM), the most aggressive of glio-
mas, involves a combination of surgery, radiotherapy, andtemozolomide chemotherapy (reviewed in ref. 1). Despiteaggressive treatment, long-term tumor stabilization of GBMis uncommon and the median survival time of good per-formance patients of ages 70 years or less is only 14.6months (2). The most complete and most durableresponses to temozolomide and the longest overall survivaltimes are seen in the subgroup of tumors with epigeneticsilencing of the MGMT enzyme that normally repairs thecytotoxic O6-methylguanine (O6-MeG) DNA adductsinduced by temozolomide (3). However, MGMT methyla-tion status does not correlatewith temozolomide sensitivityin all patients, and thus MGMT expression is insufficient toexplain temozolomide resistance in all tumors (1, 3, 4).
Recent findings from The Cancer Genome Atlas (TCGA)implicate defects in the DNA mismatch repair (MMR)system, primarily involving mutations of MSH6, as addi-tional mediators of temozolomide resistance (5). MMRproteins recognizeO6-MeGadducts not removed byMGMTand initiate an apoptotic response that leads to cell death(6).Hence, sensitivity to temozolomide likely requires both
1Hotchkiss Brain Institute, Faculty of Medicine, University of Calgary,Calgary, Alberta, Canada. 2Department of Cell Biology and Anatomy,Faculty ofMedicine,University ofCalgary,Calgary, Alberta, Canada. 3ClarkSmith Brain Tumor Research Centre, Southern Alberta Cancer ResearchInstitute, Faculty of Medicine, University of Calgary, Calgary, Alberta,Canada. 4Department of Oncology, Faculty of Medicine, University ofCalgary, Calgary, Alberta, Canada. 5Department of Clinical Neurosciences,Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
S.A. Nguyen and O.D.M. Stechishin contributed equally to this article.
Corresponding Author: Samuel Weiss, Hotchkiss Brain Institute, Facultyof Medicine, University of Calgary, HRIC 1A10, 3330 Hospital Drive NW,University of Calgary, Calgary, AB T2N 4N1, Canada. Phone: 403-220-6953; Fax: 403-210-9382; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-13-1856
�2014 American Association for Cancer Research.
ClinicalCancer
Research
Clin Cancer Res; 20(18) September 15, 20144894
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

the absence of MGMT and the presence of functional MMR.Inactivation of MMR has been previously reported to be aconsequence of temozolomide exposure. All four MSH6mutations reported in the TCGA dataset were identified inMGMT-methylated, postchemoradiotherapy samples (5),and subsequently shown to apparently be de novomutationsabsent in the corresponding pretreatment biopsies (7).However, at the early stages of tumor evolution, MSH6mutations may be only present in a small fraction of tumorcells and thus below the level of detection with randombiopsy. Mutations in other MMR genes, including MSH2andMLH1, were also present in the TCGA dataset, but weremutually exclusive from mutations of MSH6 in all but oneGBM (5). The combination of MGMT promoter methyla-tion andMSH6 inactivation has also been associated with ahypermutator phenotype characterized by an aberrantlyhigh frequency of G�C to A�T transitions at non-CpG sites(5, 8). However, the precise detail concerning the incidenceofMSH6mutations in acquired temozolomide resistance ofGBM and other glial tumors remains unclear. Some studieshave concluded that MSH6 mutations are not associatedwith MGMT promoter methylation status (9), and fewstudies have investigated MSH6 in glial tumors, other thanGBM, that are also routinely treated with temozolomide(10).Here, we report for the first time, the presence of endog-
enous MSH6 mutations in GBM and anaplastic oligoden-droglioma and their impact on chemotherapeutic responsein brain tumor–initiating cell (BTIC) lines derived fromthese tumors. We have identified 11 previously unreported
nonsynonymous MSH6 mutations in nine different tumorsamples and six paired BTIC lines. Moreover, in five tumorsamples for which normal DNA from paired patient bloodwas available for comparison, we found that the MSH6mutations were somatic, not germline, in nature. Further-more, in previously treated recurrent cases, the presence ofMSH6 mutations influenced response to temozolomide inboth in vitro culture and in vivo xenografts of BTIC lines. Wealso report, for the first time, MSH6 mutations in threeoligodendroglioma samples and two treatment-na€�ve glio-ma cases, thus demonstrating that MSH6 mutations mayoccur in at least two subtypes of glioma, including GBM,and be present before radiotherapy and alkylator therapy.
Materials and MethodsPatient tumor samples and brain tumor–initiating cellculture
Fresh tumor samples were obtained from 26 adultpatients with GBM and anaplastic oligodendrogliomaduring their operative procedure, following informedconsent and approval by the University of Calgary (Cal-gary, AB, Canada) Ethics Review Board. Tumor tissue wasfrozen at �80�C for molecular analysis or used to cultureBTICs as previously described (11–14). In brief, BTIClines were initiated in defined culture serum-free medium(SFM). All cultures grew as nonadherent spheres exceptfor one line (BT117, which grew as a monolayer) andwere expanded and cryopreserved in 10% DMSO (Sigma-Aldrich) in SFM for use in later experiments. Authentica-tion and testing of all cell lines were performed perAmerican Association for Cancer Research recommenda-tions. Molecular characterization of all BTIC lines wasperformed as described below at initiation and beforeexperimental use. Thirteen tumor samples, from which 11paired BTIC lines were established, were obtained frompatients at recurrence following chemoradiotherapy(Table 1). The remaining samples were from treatment-na€�ve patients. For tumor 61, tumor tissue was availableboth at initial presentation and recurrence followingchemoradiotherapy.
MGMT promoter methylation assayDNA was extracted from tumor tissue and BTIC sphere/
monolayer cultures using DNeasy (Qiagen) accordingto the manufacturer’s instructions. Of note, 500 ng ofDNA was bisulfite converted using the Epitect Bisulfite Kit(Qiagen) according to the manufacturer’s instructions.Two microliters of the Epitect product was used for meth-ylation-specific PCR (MS-PCR) determination of MGMTpromotermethylation as previously described (3). Thermo-cycling conditions for the MGMT MS-PCR were 95�C for10 minutes initial denaturation, then 35 cycles of 95�Cfor 45-second denaturation, 45-second annealing, 72�C for45-second extension, followed by 72�C for 10-minute finalextension. The annealing temperatures were 61�C forMGMT-methylated and 58�C for MGMT-unmethylatedMS-PCR. PCR products were visualized by agarose gelelectrophoresis.
Translational RelevanceTemozolomide chemotherapy is an integral part of the
management of glioblastoma (GBM), but long-termtumor control cannot be achieved in most patients.Emerging evidence indicates that MSH6 and other mis-match repair proteins, in addition to MGMT, play animportant role in temozolomide resistance. We investi-gated endogenous MSH6 mutations in GBM and ana-plastic oligodendroglial tumor tissue and correspondingbrain tumor–initiating cell lines (BTIC). We documen-ted 11 previously unreported MSH6 mutations, includ-ing three mutations in oligodendrogliomas and twomutations in treatment-na€�ve gliomas. These previouslyundocumented findings expand the diversity of MSH6mutations in glioma and the implications of temozolo-mide treatment for MSH6-mutated patients. Inhibitionof MGMT with O6-benzylguanine and temozolomidetreatment of BTIC xenografts confirmed that endoge-nousMSH6mutations influenced temozolomide, inde-pendent of MGMT promoter methylation status. Ourresults suggest that endogenous MSH6 mutations maybe present before alkylator therapy, occur in at least twohistologic subtypes of glioma, and influence temozolo-mide sensitivity both in vitro and in vivo.
Novel MSH6 Mutations and Temozolomide Resistance in Glioma
www.aacrjournals.org Clin Cancer Res; 20(18) September 15, 2014 4895
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856
cDNA sequencingRNA was extracted from tumor tissue and BTIC cultures
with RNeasy (Qiagen) according to the manufacturer’sinstructions.Of note, 500ngof RNAwas reverse transcribedwith the SuperScript III First-Strand Synthesis System (Invi-trogen) using poly-T primers according to the manufac-turer’s instructions. Sequencing of EGFR, PTEN, and TP53cDNA was performed as previously described (13). Forsequencing of MSH6, 2 mL of cDNA was then used in two50-mL RT-PCR reactions (Platinum High Fidelity Taq; Invi-trogen) with primers designed in PrimerBlast (NCBI) toamplify overlapping 2100-bp fragments (fragment 1—for-ward: 50-AGC TCC GTC CGA CAG AAC GGT-30, reverse: 50-CCA CCT AGA GCA GAG AGG GCC A-30; fragment 2—forward: 50-GGG TGA TGT TAC CCC AGG TGC TTA-30,reverse: 50-CACCTT TGT CAGAAG TCAACTCAA AGC-30).Thermocycling conditions were 94�C for 5-minute initialdenaturation, then 35 cycles of 94�C for 30-second dena-turation, 62�C for 30-second annealing, 68�C for 3-minuteextension, followed by 68�C for 7-minute final extension.The RT-PCR products were purified by agarose gel electro-
phoresis and isolated with the QIAquick Gel Extraction Kit(Qiagen). A series of three primers were used to sequenceoverlapping regions of each fragment (fragment 1: primer 1-1: 50-TTG ATG ACA GCC CAA CAA GG-30, primer 1-2: 50-TTT CCT CAA TTC TTG TAC TCC TGG-30, primer 1-3: 50-TTC TTC CTC TTC TGGCTC TG-30; fragment 2: primer 2-1:50-GAT TTTCTT TCTGCTCTGAAGG-30, primer 2-2: 5-TATGTG TCG CCC AGT AAT TCT GTT G-30, primer 2-3: 50-TACAGT CAA ATC AGG AAA ACG ACC-30). For sequencing ofMSH2, 2-mL of cDNA was then used in two 50-mL RT-PCRreactions (Platinum High Fidelity Taq; Invitrogen) withprimers designed in PrimerBlast (NCBI) to amplify over-lapping fragments (fragment 1—forward: 50-GGC GGGAAA CAG CTT AGT GGG-3, reverse: 5-TAC TTG AAT GACGGA ACC GGT TAG-30; fragment 2—forward: 50-CGT GTCAAA TGG AGC ACC TGT TC-30, reverse: 50-CTT AAA ATATTT ATT TTA GTA CAT CAA ACA CC-30). RT-PCR productswere amplified as described above using annealing tem-peratures of 62�C for fragment 1 and 60�C for fragment 2. Aseries of primers were used to sequence overlapping regionsof each fragment (fragment 1: primer 1-1: 50-GGCGGGAAA
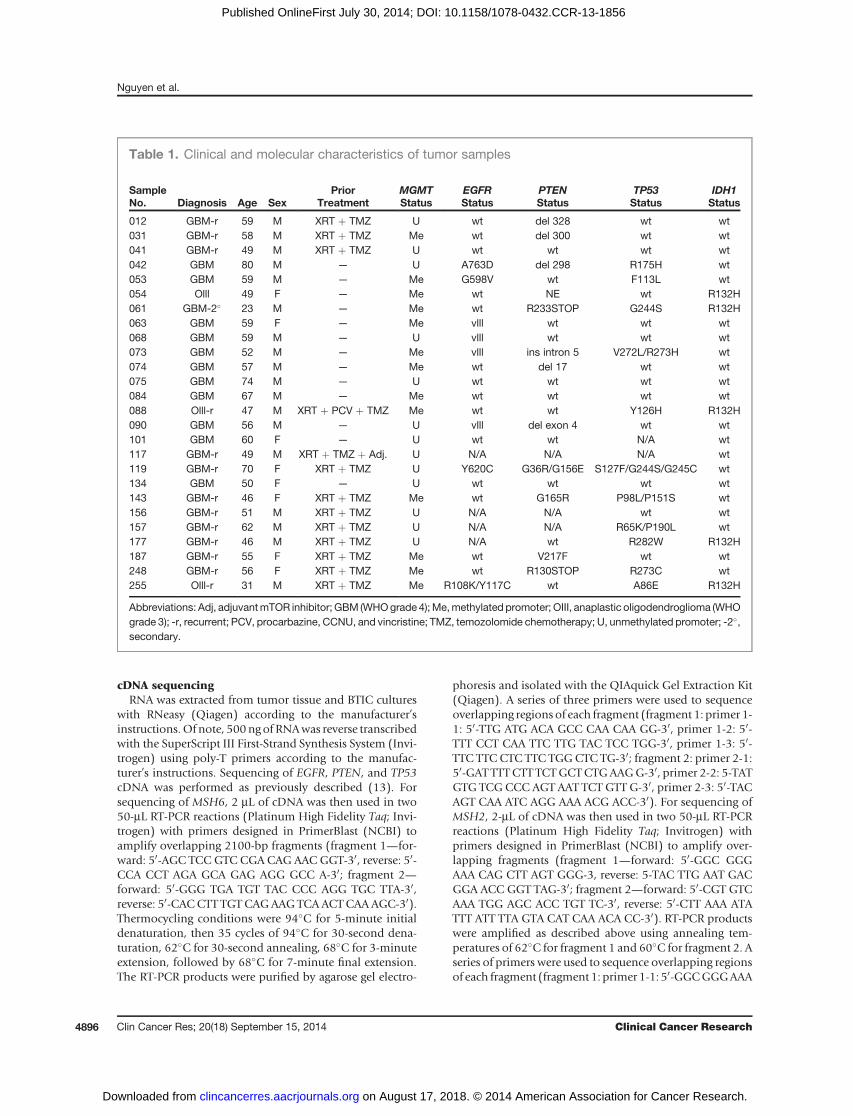
Table 1. Clinical and molecular characteristics of tumor samples
SampleNo. Diagnosis Age Sex
PriorTreatment
MGMTStatus
EGFRStatus
PTENStatus
TP53Status
IDH1Status
012 GBM-r 59 M XRT þ TMZ U wt del 328 wt wt031 GBM-r 58 M XRT þ TMZ Me wt del 300 wt wt041 GBM-r 49 M XRT þ TMZ U wt wt wt wt042 GBM 80 M — U A763D del 298 R175H wt053 GBM 59 M — Me G598V wt F113L wt054 Olll 49 F — Me wt NE wt R132H061 GBM-2� 23 M — Me wt R233STOP G244S R132H063 GBM 59 F — Me vlll wt wt wt068 GBM 59 M — U vlll wt wt wt073 GBM 52 M — Me vlll ins intron 5 V272L/R273H wt074 GBM 57 M — Me wt del 17 wt wt075 GBM 74 M — U wt wt wt wt084 GBM 67 M — Me wt wt wt wt088 Olll-r 47 M XRT þ PCV þ TMZ Me wt wt Y126H R132H090 GBM 56 M — U vlll del exon 4 wt wt101 GBM 60 F — U wt wt N/A wt117 GBM-r 49 M XRT þ TMZ þ Adj. U N/A N/A N/A wt119 GBM-r 70 F XRT þ TMZ U Y620C G36R/G156E S127F/G244S/G245C wt134 GBM 50 F — U wt wt wt wt143 GBM-r 46 F XRT þ TMZ Me wt G165R P98L/P151S wt156 GBM-r 51 M XRT þ TMZ U N/A N/A wt wt157 GBM-r 62 M XRT þ TMZ U N/A N/A R65K/P190L wt177 GBM-r 46 M XRT þ TMZ U N/A wt R282W R132H187 GBM-r 55 F XRT þ TMZ Me wt V217F wt wt248 GBM-r 56 F XRT þ TMZ Me wt R130STOP R273C wt255 Olll-r 31 M XRT þ TMZ Me R108K/Y117C wt A86E R132H
Abbreviations: Adj, adjuvantmTOR inhibitor; GBM (WHOgrade 4); Me,methylated promoter; OIII, anaplastic oligodendroglioma (WHOgrade 3); -r, recurrent; PCV, procarbazine, CCNU, and vincristine; TMZ, temozolomide chemotherapy; U, unmethylated promoter; -2�,secondary.
Nguyen et al.
Clin Cancer Res; 20(18) September 15, 2014 Clinical Cancer Research4896
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

CAG CTT AGT GGG-30, primer 1-2: 50-GCA CAT TGG ACATTC CTT CTT TTT CAG G-30, primer 1-3: 50-TAC TTG AATGAC GGA ACC GGT TAG-30; fragment 2: primer 2-1: 50-CGT GTC AAA TGG AGC ACC TGT TC-30, primer 2-2: 50-CTT AAA ATA TTT ATT TTA GTA CAT CAA ACA CC-30).Automated DNA sequencing was performed at the Univer-sity of Calgary Core DNA Services facility. All identifiedmutations were confirmed in at least two independentlyamplified RT-PCR products.
DNA sequencingGenomic DNA was extracted from patient blood, tumor
tissue, and BTIC cultures with DNeasy (Qiagen) accordingto themanufacturer’s instructions. Sequencing of IDH1wasperformed as previously described (14). For sequencing ofMSH6, 30-ng of DNA was used in two 50-mL PCR reactions(Platinum High Fidelity Taq; Invitrogen) with primersdesigned in PrimerBlast (NCBI) to amplify regions of exons4, 7, and 8 ofMSH6 containing mutations according to thecDNA sequence analysis (exon 4-1 forward: 50-ATC CCAAGC CCA CGT TAG TG-30, reverse: 50-TCA GTC TGT TCCACT CGT GC-30; exon 4-2 forward: 50-CCT CTG AGA ACTACA GTA AG-30, reverse: 50-CCA AAA CTG GGA GCC GGGTA-30; exon4-3 forward: 50-CTGTTCTCTTCAGGAAGGTC-30, reverse: 50-AGC CAT TGC TTT AGG AGCCG-30; exon 4-4 forward: 50-GAC CTC ATG GTT GTG CCT GA-30, reverse:50-GGT ATT CCA GGA GGC TCT GT-30; exon 7 forward: 50-CCCGGCCAATAATTGCATAGTC-30 reverse: 50-TGGTGAGTG CGT GCT CTA AA-30; exon 8 forward: 50-CCG ATGTTG CTT TTC TGT CCT-30, reverse: 50-TGG CTG GGG TCTTCA CAT TC-30). The PCR products were purified asdescribed above and sequenced bidirectionally using theforward and reverse primers for each respective fragment.
Western blottingBTICs were treated with O6-benzylguanine (BG; Sigma-
Aldrich) or DMSO (Sigma-Aldrich) for 72 hours and lysedin modified radioimmunoprecipitation assay buffer sup-plemented with Complete Mini Protease (Roche) and HaltPhosphatase (Thermo Scientific) inhibitor cocktails. Ofnote, 15 mg of protein was loaded on 10% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE)gels and transblotted to nitrocellulose membranes. Blotswere stained with mouse anti-MGMT (1:1,000; Santa CruzBiotechnology) and goat anti-actin (1:2,500; Santa CruzBiotechnology) antibodies diluted in 5% nonfat milk (Bio-Rad) in Tris-buffered saline with Tween 20. Horseradishperoxidase–conjugateddonkey anti-mouse IgGanddonkeyanti-goat IgG (Millipore) were used at 1:5,000. Bands werevisualized with the ECL Plus Western Blotting DetectionSystem and Hyperfilm (Amersham).
BTIC drug sensitivity assaysDissociated BTIC cultures were seeded at 2,500 to 5,000
cells per well in 96-well plates and treated with temozolo-mide (Merck), BG (Sigma-Aldrich), or vehicle (DMSO;Sigma-Aldrich). For MGMT depletion experiments, cellswere allowed to rest for 24 hours after plating, pretreated
with BG for 48 hours, and then treated with temozolomideat 72 hours. After 14 to 21 days, BTIC growth was assessedby performing the alamarBlue assay (Invitrogen) accordingto the manufacturer’s instructions. All culture experimentswere performed in triplicate with a minimum of 3 wells percondition.
Intracranial BTIC xenograftsBTIC spheres from BT073 (MGMT methylated/MSH6
wild-type), BT134 (MGMT unmethylated/MSH6 wild-type), and BT143 (MGMTmethylated/MSH6mutant) weredissociated to single-cell suspensions and 1� 105 cells werestereotactically implanted into the right striata of 6- to 8-week-old NOD/SCID mice as previously described (11–14). Seven days after BTIC implantation, mice were ran-domized to vehicle or temozolomide treatment cohorts(BT073: n ¼ 7 for vehicle and n ¼ 8 for temozolomide;BT134: n ¼ 7 for vehicle and n ¼ 7 for temozolomide;BT143: n ¼ 11 for vehicle and n ¼ 12 for temozolomide).Oral treatment with temozolomide (50 mg/kg; Merck) orvehicle (Ora-Plus; Galenova) was delivered daily 5 days aweek for 3 weeks. Mice were sacrificed upon significantweight loss or presentation of neurologic symptoms neces-sitating euthanasia as per University of Calgary animal careguidelines. Necropsy and cranial dissectionwere performedto confirm presence of tumor in all animals.
Statistical analysesResults are displayed as the mean � standard deviation.
Student t tests were performed for comparison of twoconditions and one-way ANOVAs followed by Tukey mul-tiple comparison tests were performed for comparison ofmultiple conditions. Fisher exact tests were used to testcorrelation between G�C to A�T transition frequency andMSH6 status and temozolomide exposure. Statistically sig-nificant differences in median survival were determined bythe log-rank test using GraphPad Prism.
ResultsMSH6mutationsarepresent inBTIC lines isolated fromboth MGMT-methylated and -unmethylated tumorsbefore and following chemoradiotherapy
To investigate MSH6 mutations in malignant glioma,we sequenced MSH6 cDNA in tumor samples and BTIClines cultured from a series of 26 patients with GBM oranaplastic oligodendroglioma (Table 1). Point mutations(Fig. 1 and Table 2) resulting in nonsynonymous aminoacid substitutions or protein truncationwere present innineof the samples. Most tumors contained only a singleMSH6mutation except for samples 143 and 248, which had threeand two mutations, respectively. MSH6 mutations wereheterozygous in all of the samples except for sample 88.A polymorphism involving a substitution of glutamate forglycine at codon 39, previously reported not to influencepatient survival (15), was detected in BT042, 054, and 088(data not shown). To exclude other uncommon poly-morphisms, sequencing of paired patient blood samplesfrom five tumors (119, 143, 157, 248, and 255) was
Novel MSH6 Mutations and Temozolomide Resistance in Glioma
www.aacrjournals.org Clin Cancer Res; 20(18) September 15, 2014 4897
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856
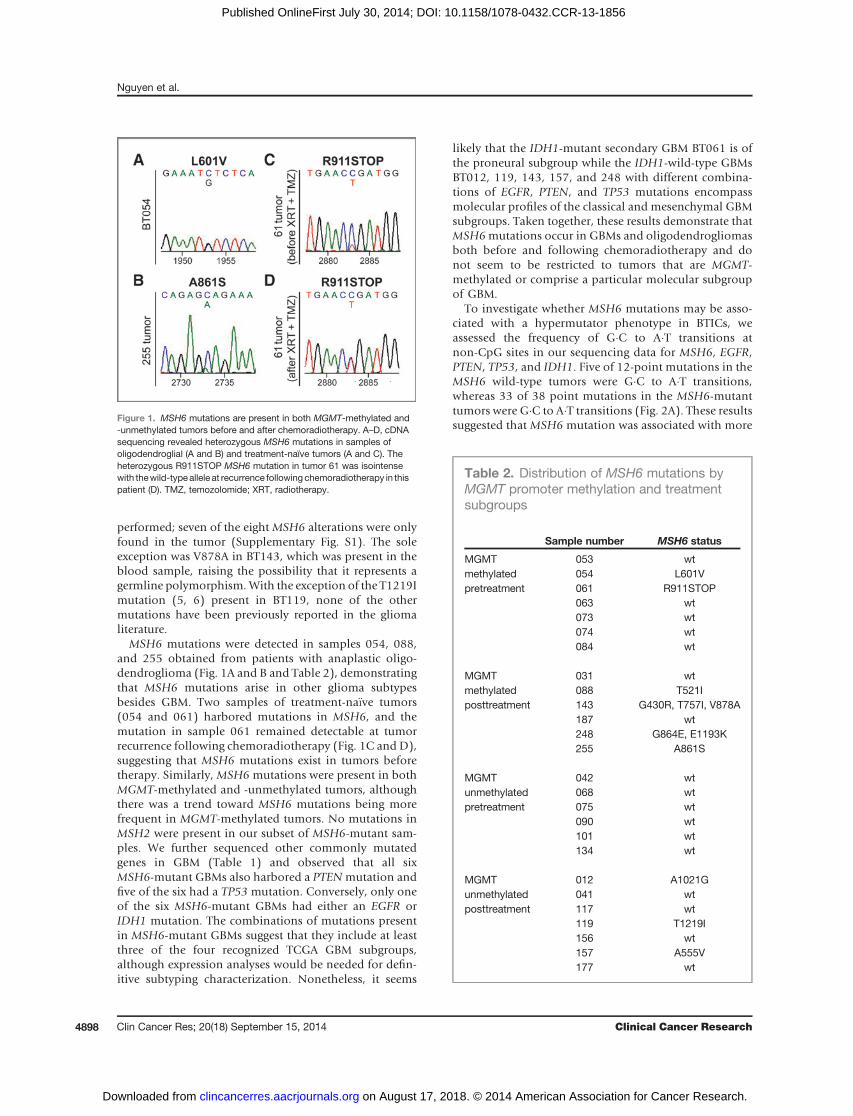
performed; seven of the eight MSH6 alterations were onlyfound in the tumor (Supplementary Fig. S1). The soleexception was V878A in BT143, which was present in theblood sample, raising the possibility that it represents agermline polymorphism. With the exception of the T1219Imutation (5, 6) present in BT119, none of the othermutations have been previously reported in the gliomaliterature.
MSH6 mutations were detected in samples 054, 088,and 255 obtained from patients with anaplastic oligo-dendroglioma (Fig. 1A and B and Table 2), demonstratingthat MSH6 mutations arise in other glioma subtypesbesides GBM. Two samples of treatment-na€�ve tumors(054 and 061) harbored mutations in MSH6, and themutation in sample 061 remained detectable at tumorrecurrence following chemoradiotherapy (Fig. 1C and D),suggesting that MSH6 mutations exist in tumors beforetherapy. Similarly, MSH6 mutations were present in bothMGMT-methylated and -unmethylated tumors, althoughthere was a trend toward MSH6 mutations being morefrequent in MGMT-methylated tumors. No mutations inMSH2 were present in our subset of MSH6-mutant sam-ples. We further sequenced other commonly mutatedgenes in GBM (Table 1) and observed that all sixMSH6-mutant GBMs also harbored a PTENmutation andfive of the six had a TP53 mutation. Conversely, only oneof the six MSH6-mutant GBMs had either an EGFR orIDH1 mutation. The combinations of mutations presentin MSH6-mutant GBMs suggest that they include at leastthree of the four recognized TCGA GBM subgroups,although expression analyses would be needed for defin-itive subtyping characterization. Nonetheless, it seems
likely that the IDH1-mutant secondary GBM BT061 is ofthe proneural subgroup while the IDH1-wild-type GBMsBT012, 119, 143, 157, and 248 with different combina-tions of EGFR, PTEN, and TP53 mutations encompassmolecular profiles of the classical and mesenchymal GBMsubgroups. Taken together, these results demonstrate thatMSH6mutations occur in GBMs and oligodendrogliomasboth before and following chemoradiotherapy and donot seem to be restricted to tumors that are MGMT-methylated or comprise a particular molecular subgroupof GBM.
To investigate whether MSH6 mutations may be asso-ciated with a hypermutator phenotype in BTICs, weassessed the frequency of G�C to A�T transitions atnon-CpG sites in our sequencing data for MSH6, EGFR,PTEN, TP53, and IDH1. Five of 12-point mutations in theMSH6 wild-type tumors were G�C to A�T transitions,whereas 33 of 38 point mutations in the MSH6-mutanttumors were G�C to A�T transitions (Fig. 2A). These resultssuggested that MSH6 mutation was associated with more
Table 2. Distribution of MSH6 mutations byMGMT promoter methylation and treatmentsubgroups
Sample number MSH6 status
MGMTmethylatedpretreatment
053 wt054 L601V061 R911STOP063 wt073 wt074 wt084 wt
MGMTmethylatedposttreatment
031 wt088 T521I143 G430R, T757I, V878A187 wt248 G864E, E1193K255 A861S
MGMTunmethylatedpretreatment
042 wt068 wt075 wt090 wt101 wt134 wt
MGMTunmethylatedposttreatment
012 A1021G041 wt117 wt119 T1219I156 wt157 A555V177 wt
Figure 1. MSH6 mutations are present in both MGMT-methylated and-unmethylated tumors before and after chemoradiotherapy. A–D, cDNAsequencing revealed heterozygous MSH6 mutations in samples ofoligodendroglial (A and B) and treatment-naïve tumors (A and C). Theheterozygous R911STOP MSH6 mutation in tumor 61 was isointensewith thewild-typeallele at recurrence followingchemoradiotherapy in thispatient (D). TMZ, temozolomide; XRT, radiotherapy.
Nguyen et al.
Clin Cancer Res; 20(18) September 15, 2014 Clinical Cancer Research4898
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856
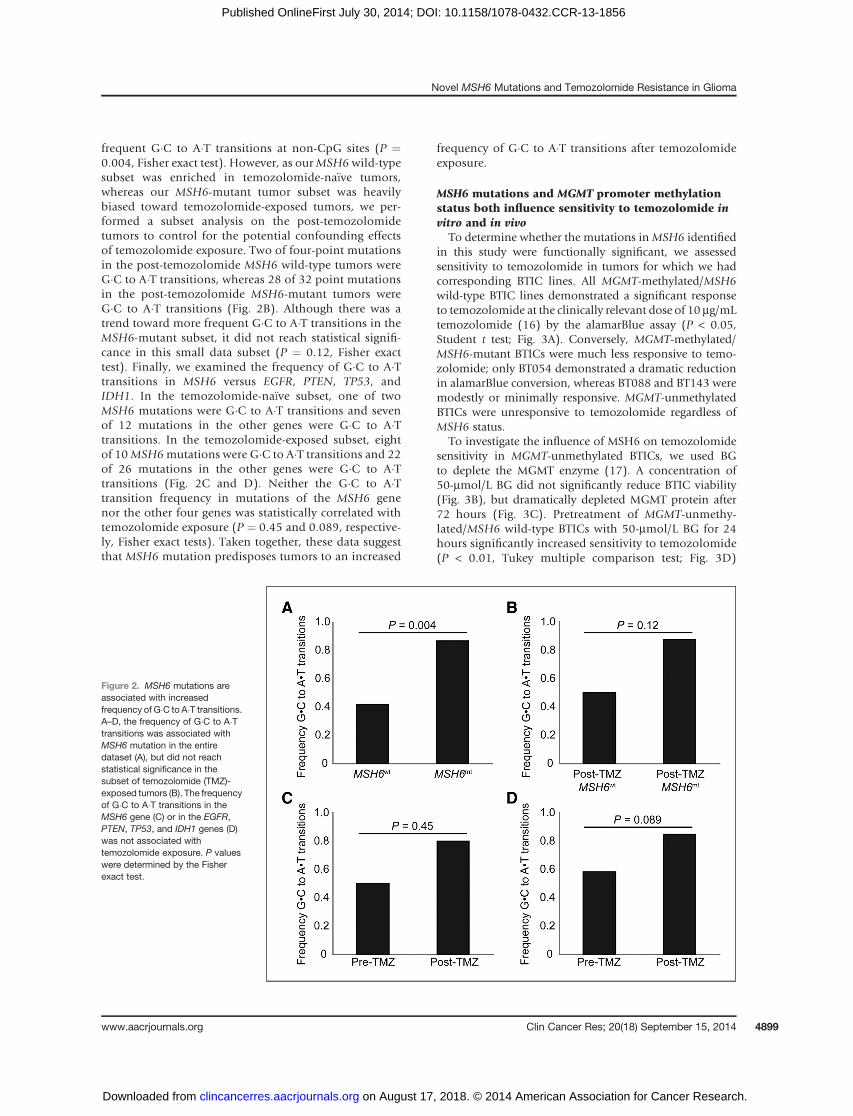
frequent G�C to A�T transitions at non-CpG sites (P ¼0.004, Fisher exact test). However, as ourMSH6 wild-typesubset was enriched in temozolomide-na€�ve tumors,whereas our MSH6-mutant tumor subset was heavilybiased toward temozolomide-exposed tumors, we per-formed a subset analysis on the post-temozolomidetumors to control for the potential confounding effectsof temozolomide exposure. Two of four-point mutationsin the post-temozolomide MSH6 wild-type tumors wereG�C to A�T transitions, whereas 28 of 32 point mutationsin the post-temozolomide MSH6-mutant tumors wereG�C to A�T transitions (Fig. 2B). Although there was atrend toward more frequent G�C to A�T transitions in theMSH6-mutant subset, it did not reach statistical signifi-cance in this small data subset (P ¼ 0.12, Fisher exacttest). Finally, we examined the frequency of G�C to A�Ttransitions in MSH6 versus EGFR, PTEN, TP53, andIDH1. In the temozolomide-na€�ve subset, one of twoMSH6 mutations were G�C to A�T transitions and sevenof 12 mutations in the other genes were G�C to A�Ttransitions. In the temozolomide-exposed subset, eightof 10MSH6mutations were G�C to A�T transitions and 22of 26 mutations in the other genes were G�C to A�Ttransitions (Fig. 2C and D). Neither the G�C to A�Ttransition frequency in mutations of the MSH6 genenor the other four genes was statistically correlated withtemozolomide exposure (P ¼ 0.45 and 0.089, respective-ly, Fisher exact tests). Taken together, these data suggestthat MSH6 mutation predisposes tumors to an increased
frequency of G�C to A�T transitions after temozolomideexposure.
MSH6 mutations and MGMT promoter methylationstatus both influence sensitivity to temozolomide invitro and in vivo
To determine whether the mutations inMSH6 identifiedin this study were functionally significant, we assessedsensitivity to temozolomide in tumors for which we hadcorresponding BTIC lines. All MGMT-methylated/MSH6wild-type BTIC lines demonstrated a significant responseto temozolomide at the clinically relevant dose of 10 mg/mLtemozolomide (16) by the alamarBlue assay (P < 0.05,Student t test; Fig. 3A). Conversely, MGMT-methylated/MSH6-mutant BTICs were much less responsive to temo-zolomide; only BT054 demonstrated a dramatic reductionin alamarBlue conversion, whereas BT088 and BT143 weremodestly or minimally responsive. MGMT-unmethylatedBTICs were unresponsive to temozolomide regardless ofMSH6 status.
To investigate the influence of MSH6 on temozolomidesensitivity in MGMT-unmethylated BTICs, we used BGto deplete the MGMT enzyme (17). A concentration of50-mmol/L BG did not significantly reduce BTIC viability(Fig. 3B), but dramatically depleted MGMT protein after72 hours (Fig. 3C). Pretreatment of MGMT-unmethy-lated/MSH6 wild-type BTICs with 50-mmol/L BG for 24hours significantly increased sensitivity to temozolomide(P < 0.01, Tukey multiple comparison test; Fig. 3D)
Figure 2. MSH6 mutations areassociated with increasedfrequency of G�C to A�T transitions.A–D, the frequency of G�C to A�Ttransitions was associated withMSH6 mutation in the entiredataset (A), but did not reachstatistical significance in thesubset of temozolomide (TMZ)-exposed tumors (B). The frequencyof G�C to A�T transitions in theMSH6 gene (C) or in the EGFR,PTEN, TP53, and IDH1 genes (D)was not associated withtemozolomide exposure. P valueswere determined by the Fisherexact test.
Novel MSH6 Mutations and Temozolomide Resistance in Glioma
www.aacrjournals.org Clin Cancer Res; 20(18) September 15, 2014 4899
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856
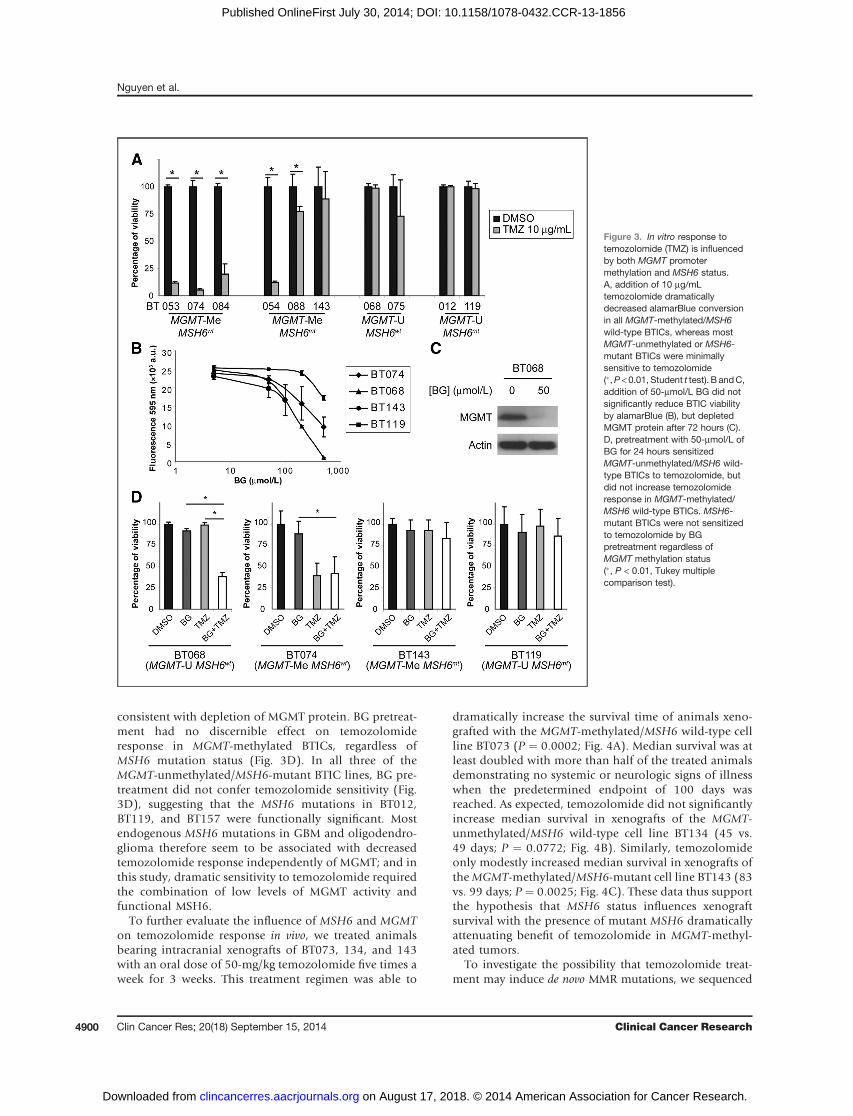
consistent with depletion of MGMT protein. BG pretreat-ment had no discernible effect on temozolomideresponse in MGMT-methylated BTICs, regardless ofMSH6 mutation status (Fig. 3D). In all three of theMGMT-unmethylated/MSH6-mutant BTIC lines, BG pre-treatment did not confer temozolomide sensitivity (Fig.3D), suggesting that the MSH6 mutations in BT012,BT119, and BT157 were functionally significant. Mostendogenous MSH6 mutations in GBM and oligodendro-glioma therefore seem to be associated with decreasedtemozolomide response independently of MGMT; and inthis study, dramatic sensitivity to temozolomide requiredthe combination of low levels of MGMT activity andfunctional MSH6.
To further evaluate the influence of MSH6 and MGMTon temozolomide response in vivo, we treated animalsbearing intracranial xenografts of BT073, 134, and 143with an oral dose of 50-mg/kg temozolomide five times aweek for 3 weeks. This treatment regimen was able to
dramatically increase the survival time of animals xeno-grafted with the MGMT-methylated/MSH6 wild-type cellline BT073 (P ¼ 0.0002; Fig. 4A). Median survival was atleast doubled with more than half of the treated animalsdemonstrating no systemic or neurologic signs of illnesswhen the predetermined endpoint of 100 days wasreached. As expected, temozolomide did not significantlyincrease median survival in xenografts of the MGMT-unmethylated/MSH6 wild-type cell line BT134 (45 vs.49 days; P ¼ 0.0772; Fig. 4B). Similarly, temozolomideonly modestly increased median survival in xenografts oftheMGMT-methylated/MSH6-mutant cell line BT143 (83vs. 99 days; P ¼ 0.0025; Fig. 4C). These data thus supportthe hypothesis that MSH6 status influences xenograftsurvival with the presence of mutant MSH6 dramaticallyattenuating benefit of temozolomide in MGMT-methyl-ated tumors.
To investigate the possibility that temozolomide treat-ment may induce de novo MMR mutations, we sequenced
Figure 3. In vitro response totemozolomide (TMZ) is influencedby both MGMT promotermethylation and MSH6 status.A, addition of 10 mg/mLtemozolomide dramaticallydecreased alamarBlue conversionin all MGMT-methylated/MSH6wild-type BTICs, whereas mostMGMT-unmethylated or MSH6-mutant BTICs were minimallysensitive to temozolomide(�,P <0.01, Student t test). B andC,addition of 50-mmol/L BG did notsignificantly reduce BTIC viabilityby alamarBlue (B), but depletedMGMT protein after 72 hours (C).D, pretreatment with 50-mmol/L ofBG for 24 hours sensitizedMGMT-unmethylated/MSH6 wild-type BTICs to temozolomide, butdid not increase temozolomideresponse in MGMT-methylated/MSH6 wild-type BTICs. MSH6-mutant BTICs were not sensitizedto temozolomide by BGpretreatment regardless ofMGMT methylation status(�, P < 0.01, Tukey multiplecomparison test).
Nguyen et al.
Clin Cancer Res; 20(18) September 15, 2014 Clinical Cancer Research4900
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

MSH6 and MSH2 in our xenograft tumors. De novo MSH6mutations were not identified in any of the xenografttumors. Interestingly, several different de novo MSH2muta-tions were present in the BT073 xenografts treated withtemozolomide, but absent in the xenografts treated withvehicle (data not shown). No MSH2 mutations were iden-tified in either the BT134 or 143 temozolomide-treatedxenografts. These data demonstrate that MSH2 mutationscan arise during the course of temozolomide treatment, butdid not seem to abrogate temozolomide response in theMGMT-methylated/MSH6 wild-type BT073 xenograftswithin the time frame of the experiment.
DiscussionTemozolomide has improved the prognosis for patients
with GBM (1). Although the addition of temozolomide toradiotherapy (XRT) has lengthened median survival onlymodestly, those with MGMT-methylated tumors seem tohave a greater degree of benefit from chemoradiotherapy.This enhanced benefit is reflected in the significantly greaterproportion of 3- and 5-year survivors after temozolomide/XRT, than XRT alone (18). Unfortunately, regardless ofMGMT methylation status, most patients eventually recurand these recurrences are often resistant to temozolomide.Recent evidence indicates that there are several potentialmechanisms that contribute to temozolomide resistance inadult and pediatric GBM, including activation of the baseexcision DNA repair pathway (19, 20) and phosphoinos-tide-3-kinase–mediated upregulation of the HOXA9/HOXA10 genes (21). Findings from the TCGA and othergroups implicate defects in the MMR pathway, frequentlythroughmutationofMSH6, as amediator of temozolomide
resistance in alkylator-exposed, MGMT-methylated GBMs(5–7). Here, we reaffirm the role of MSH6 mutations as amechanism of temozolomide resistance and also make thepreviously unreported observation of mutations in othersubgroups of patients, including those that have oligoden-drogliomas or GBMs that are treatment-na€�ve or MGMT-unmethylated. Although our dataset is relatively small,these novel findings potentially implicate preexistingnon-germline MSH6 mutations in early temozolomideresistance in some patients with oligodendroglioma andGBM.
The MSH6 mutations in cases 054, 088, and 255 are thefirst such alterations to be reported in oligodendrogliomas.The consequences of these alterations for response to temo-zolomide were evaluated in BTICs derived from 054 and088. In BT088, the T521I substitution seemed to result inobvious resistance to temozolomide compared with BTIClines with methylation of the MGMT promoter and wild-type MSH6. In contrast, the L601V substitution in BT054was not associated with resistance to temozolomide. It ispossible that this mutation is simply another functionallyneutral polymorphism similar to G39E (15); however,normal DNA from the patient was not available for com-parison to address this possibility. In addition, the observedfailure to confer resistance to temozolomidemay have beena result of the nature of a leucine to valine substitution. Bothamino acids are small, nonpolar, and neutral and hencemay not have dramatically affected MSH6 folding or func-tion. Nevertheless, the results with BT088 demonstrate thatMSH6 mutations can functionally influence response totemozolomide in oligodendroglioma. These findings sug-gest that MSH6 mutations may be a potential mechanism
Figure 4. In vivo response totemozolomide (TMZ) is influencedby both MGMT promotermethylation and MSH6 status.A–C, oral administration oftemozolomide dramaticallyincreased the survival of animalsbearing intracranial xenografts ofMGMT-methylated/MSH6 wild-type BT073 (A), but not MGMT-unmethylated/MSH6 wild-typeBT134 (B). Temozolomideadministration only modestlyincreased survival of animalsbearing xenografts of MGMT-methylated/MSH6-mutantBT143 (C; P values determinedby log-rank test).
Novel MSH6 Mutations and Temozolomide Resistance in Glioma
www.aacrjournals.org Clin Cancer Res; 20(18) September 15, 2014 4901
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

for temozolomide resistance inoligodendrogliomas, aswellas GBMs.
MMR defects have a well-established role in familialGBMs in patients with Turcot Syndrome type II (22) orconstitutional MMR-deficiency syndrome (23). However,in sporadic adult GBM, MMR mutations do not seem tobe an early, initiating event in tumor development. Pre-vious reports of MSH6 mutations in nonfamilial adultgliomas have been almost exclusively confined to post-treatment tumors (5, 7, 9, 10), with the exception of onemutation in the MMR gene MSH2 in a treatment-na€�veGBM sample in the TCGA dataset (5). Analysis of severalTCGA samples for which tissue was available both atinitial presentation and at recurrence following chemo-therapy seemed to demonstrate that MSH6 mutationswere detectable only after exposure to radio- and alky-lator therapy (7). However, it is unclear whether MSH6mutations may develop spontaneously and are thenselected for by alkylating agents or if they are in factinduced as a side effect of alkylator therapy. Our resultswith tumor 061 suggest that at least in some tumors it isthe former possibility. In tumor 061, we demonstrate aweak, but unequivocal, signal for the R911STOP muta-tion that was present before temozolomide exposure. Assuch, early in tumor evolution before exposure to alky-lator chemotherapy, MSH6 mutations may already bepresent, even if it is potentially only in a small fraction oftumor cells and below the level of detection by currentmethods. Our results thus provide some of the firstevidence that MSH6 mutations occur in treatment-na€�vetumors, and may help to explain the paradoxical failureof temozolomide and rapid recurrence in some MGMT-methylated gliomas.
Finally, in addition to the results reported by theTCGA, our data illustrate thatMSH6 mutations can occurin both MGMT-methylated and -unmethylated tumors.The potential role of MMR inactivation in MGMT-meth-ylated tumors is a seemingly intuitive mechanism fortumors lacking MGMT expression to ignore unrepairedO6-MeG lesions that otherwise would induce apoptosis.In tumors displaying the hypermutator phenotype (5) inwhich frequent G�C to A�T transitions arise at non-CpGsites, MSH6 deficiency would seem integral to both theaccumulation of these mutations and ongoing survival ofthe tumor cells. Conversely, the presence of MSH6 muta-tions in MGMT-unmethylated tumors seems controver-sial and functionally redundant given that these tumorsshould be able to readily repair O6-MeG lesions before anapoptotic response is initiated. However, two recentstudies with paired GBM specimens from patients atinitial presentation and at recurrence have demonstratedreduced expression of MSH6, MSH2, and PMS2 (24) orMLH1 (25) in the recurrent tumors compared with theircorresponding primaries. Although neither study furthersubdivided the results for MMR protein expression basedon MGMT methylation status, given that nearly 75% ofGBMs are unmethylated (5), it would seem likely that
alterations in MSH6 expression could occur with reason-able frequency in MGMT-unmethylated tumors. Theresults presented here thus build on these findings bydocumenting MSH6 mutations in three GBMs withMGMT promoters that clearly are unmethylated andexpress detectable MGMT protein on Western blotting(data not shown). Depletion of MGMT protein in thethree BTIC lines derived from these tumors failed toconfer temozolomide sensitivity. As such, MSH6 muta-tions likely are a mechanism for enhanced temozolomideresistance in some MGMT unmethylated tumors. Thismay be of particular clinical significance for dose-densetemozolomide or other treatment strategies aimed atincreasing response to alkylator chemotherapy in MGMT-unmethylated tumors.
Temozolomide is a cornerstone of the current standard ofcare for malignant glioma. However, the near universaldevelopment of resistance to alkylator therapies remainsa major obstacle to durable tumor control. Our findingssuggest that MMR deficiency may be more prevalent thanpreviously appreciated, including in treatment-na€�ve andMGMT-unmethylated GBMs and also in oligodendroglio-mas. These findings may thus help to explain seeminglyparadoxical resistance to temozolomide in some tumorsubgroups and reinforce the importance of developingadditional nonalkylator therapeutics to deal with MSH6-deficient glioma.
Disclosure of Potential Conflicts of InterestD.L. Senger and S.M. Robbins have ownership interest (including
patents) in Arch Biopartners Inc. No potential conflicts of interest weredisclosed by the other authors.
Authors' ContributionsConception and design: S.A. Nguyen, O.D.M. Stechishin, H.A. Luchman,S. WeissDevelopment of methodology: O.D.M. StechishinAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): O.D.M. Stechishin, D.L. Senger, S.M. RobbinsAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): S.A. Nguyen, O.D.M. Stechishin,H.A. Luchman, D.L. Senger, J.G. CairncrossWriting, review, and/or revision of the manuscript: S.A. Nguyen,O.D.M. Stechishin, H.A. Luchman, J.G. Cairncross, S. WeissStudy supervision: H.A. Luchman, J.G. Cairncross, S. WeissOther (conducted animal experiments): X.Q. Lun
AcknowledgmentsThe authors thank Rozina Hassam, Dorothea Livingstone, and Orsolya
Cseh for technical assistance. The patient samples were obtained through theCalgary Brain Tumour and Tissue Bank, generously supported by funds fromthe Clark H. Smith Family.
Grant SupportThisworkwas supported by grants to S.Weiss from the StemCellNetwork
and Alberta Cancer Foundation. S.A. Nguyen was the recipient of an AlbertaCancer Foundation scholarship.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received July 5, 2013; revised July 7, 2014; accepted July 14, 2014;published OnlineFirst July 30, 2014.
Nguyen et al.
Clin Cancer Res; 20(18) September 15, 2014 Clinical Cancer Research4902
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

References1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med
2008;359:492–507.2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn
MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomidefor glioblastoma. N Engl J Med 2005;352:987–96.
3. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M,et al. MGMT gene silencing and benefit from temozolomide in glio-blastoma. N Engl J Med 2005;352:997–1003.
4. Olson RA, Brastianos PK, Palma DA. Prognostic and predictivevalue of epigenetic silencing of MGMT in patients with high gradegliomas: a systematic review and meta-analysis. J Neurooncol2011;105:325–35.
5. TheCancerGenomeAtlas (TCGA)ResearchNetwork. Comprehensivegenomic characterization defines humanglioblastomagenes and corepathways. Nature 2008;455:1061–8.
6. York SJ, Modrich P. Mismatch repair-dependent iterative excision atirreparable O6-methylguanine lesions in human nuclear extracts. J BiolChem 2006;281:22674–83.
7. Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6mutations arise in glioblastomas during temozolomide therapy andmediate temozolomide resistance. Clin Cancer Res 2009;15:4622–9.
8. Hunter C, Smith R, Cahill DP, Stephens P, StevensC, Teague J, et al. Ahypermutation phenotype and somatic MSH6 mutations in recurrenthuman malignant gliomas after alkylator chemotherapy. Cancer Res2006;66:3987–91.
9. Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB,et al. Loss of themismatch repair proteinMSH6 in humanglioblastomais associated with tumor progression during temozolomide treatment.Clin Cancer Res 2007;13:2038–45.
10. Maxwell JA, Johnson SP, McLendon RE, Lister DW, Horne KS,Rasheed A, et al. Mismatch repair deficiency does not mediate clinicalresistance to temozolomide in malignant glioma. Clin Cancer Res2008;14:4859–68.
11. Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, et al.Proliferation of human glioblastoma stem cells occurs independentlyof exogenous mitogens. Stem Cells 2009;27:1722–33.
12. Kelly JJ,BloughMD,StechishinO,ChanJ,BeauchampD,PerizzoloM,et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10). NeuroOncol 2010;12:745–55.
13. Stechishin OD, Luchman HA, Ruan Y, Blough MD, Nguyen SA, KellyJJ, et al. On-target JAK2/STAT3 inhibition slows disease progressionin orthotopic xenografts of humanglioblastomabrain tumor stemcells.Neuro Oncol 2013;15:198–207.
14. Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C,Kelly JJ, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol 2012;14:184–91.
15. Pei C, Chen H, Jia X, Yan L, Zou Y, Jiang C, et al. A high frequency ofMSH6G268A polymorphism and survival association in glioblastoma.Int J Neurosci 2013;123:114–20.
16. Brock CS, Newlands ES, Wedge SR, Bower M, Evans H, Colquhoun I,et al. Phase I trial of temozolomide using an extended continuous oralschedule. Cancer Res 1998;58:4363–7.
17. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, RichJN, et al. Phase II trial of temozolomide pluso6-benzylguanine in adultswith recurrent, temozolomide-resistant malignant glioma. J Clin Oncol2009;27:1262–7.
18. Stupp R, Hegi ME,MasonWP, van den Bent MJ, TaphoornMJ, JanzerRC, et al. Effects of radiotherapy with concomitant and adjuvanttemozolomide versus radiotherapy alone on survival in glioblastomain a randomised phase III study: 5-year analysis of the EORTC-NCICtrial. Lancet Oncol 2009;10:459–66.
19. Tang JB, Svilar D, Trivedi RN, Wang X, Goellner EM, Moore B, et al. N-methylpurine DNA glycosylase and DNA polymerase {beta}modulateBER inhibitor potentiation of glioma cells to temozolomide. NeuroOncol 2011;13:471–86.
20. Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, MischelPS, et al. Alkylpurine-DNA-N-glycosylase confers resistance to temo-zolomide in xenograft models of glioblastoma multiforme and is asso-ciated with poor survival in patients. J Clin Invest 2012;122:253–66.
21. Gaspar N, Marshall L, Perryman L, Bax DA, Little SE, Viana-Pereira M,et al. MGMT-independent temozolomide resistance in pediatric glio-blastoma cells associated with a PI3-kinase-mediated HOX/stem cellgene signature. Cancer Res 2010;70:9243–52.
22. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM,et al. The molecular basis of Turcot's syndrome. N Engl J Med1995;332:839–47.
23. Ilencikova D, Sejnova D, Jindrova J, Babal P. High-grade brain tumorsin siblings with biallelic MSH6 mutations. Pediatr Blood Cancer2011;57:1067–70.
24. Felsberg J, ThonN, Eigenbrod S, Hentschel B, SabelMC,WestphalM,et al. Promoter methylation and expression of MGMT and the DNAmismatch repair genes MLH1, MSH2, MSH6 and PMS2 in pairedprimary and recurrent glioblastomas. Int J Cancer 2011;129:659–70.
25. Stark AM, Doukas A, Hugo HH, Mehdorn HM. The expression ofmismatch repair proteins MLH1, MSH2 and MSH6 correlates with theKi67 proliferation index and survival in patients with recurrent glio-blastoma. Neurol Res 2010;32:816–20.
www.aacrjournals.org Clin Cancer Res; 20(18) September 15, 2014 4903
Novel MSH6 Mutations and Temozolomide Resistance in Glioma
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856

2014;20:4894-4903. Published OnlineFirst July 30, 2014.Clin Cancer Res Stephanie A. Nguyen, Owen D.M. Stechishin, H. Artee Luchman, et al.
Promoter MethylationMGMTResistance Independently of Anaplastic Oligodendroglioma Contribute to Temozolomide
Mutations in Treatment-Naïve Glioblastoma andMSH6Novel
Updated version
10.1158/1078-0432.CCR-13-1856doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2014/07/31/1078-0432.CCR-13-1856.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/20/18/4894.full#ref-list-1
This article cites 25 articles, 8 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/20/18/4894.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/20/18/4894To request permission to re-use all or part of this article, use this link
on August 17, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 30, 2014; DOI: 10.1158/1078-0432.CCR-13-1856


















