
NONTEMPLATE-DEPENDENT POLYMERIZATION PROCESSES
50
Annu. Rev. Biochem. 2005. 74:433–80 doi: 10.1146/annurev.biochem.74.082803.133013 Copyright c 2005 by Annual Reviews. All rights reserved NONTEMPLATE-DEPENDENT P OLYMERIZATION PROCESSES: Polyhydroxyalkanoate Synthases as a Paradigm 1 JoAnne Stubbe, 1,2 Jiamin Tian, 1 Aimin He, 1 Anthony J. Sinskey, 2,3 Adam G. Lawrence, 2 and Pinghua Liu 1 Department of Chemistry, 1 Department of Biology, 2 and Department of Health Sciences and Technology, 3 Massachusetts Institute of Technology, Cambridge, Massachusetts 02139; email: [email protected], [email protected], [email protected], [email protected], [email protected], [email protected] Key Words starch, glycogen, polyphosphate, cyanophycin, rubber, PHA synthases, PHA depolymerases, phasin ■ Abstract This review focuses on nontemplate-dependent polymerases that use water-soluble substrates and convert them into water-insoluble polymers that form granules or inclusions within the cell. The initial part of the review summarizes briefly the current knowledge of polymer formation catalyzed by starch and glycogen syn- thases, polyphosphate kinase (a polymerase), cyanophycin synthetases, and rubber synthases. Specifically, our current understanding of their mechanisms of initiation, elongation (including granule formation), termination, remodeling, and polymer re- utilization will be presented. General underlying principles that govern these types of polymerization reactions will be enumerated as a paradigm for all nontemplate- dependent polymerizations. The bulk of the review then focuses on polyhydroxyalka- noate (PHA) synthases that generate polyoxoesters. These enzymes are of interest as they generate biodegradable polymers. Our current knowledge of PHA production and utilization in vitro and in vivo as well as the contribution of many proteins to these processes will be reviewed. CONTENTS INTRODUCTION ..................................................... 434 GENERAL OUTLINE .................................................. 435 1 Abbreviations used: ADP-Glc, ADP-glucose; CoA, coenzyme A; HB, 3(R)-hydro- xybutyrate; HB-CoA, 3(R)-hydroxybutyryl-CoA; IPP, isopentenyl pyrophosphate; MW, molecular weight; PHA, polyhydroxyalkanoate; PHB, polyhydroxybutyrate; PT- Fase, prenyl transferase; TEM, transmission electron microscopy; UDP-Glc, UDP-glucose; wt, wild type. 0066-4154/05/0707-0433$20.00 433 Annu. Rev. Biochem. 2005.74:433-480. Downloaded from arjournals.annualreviews.org by MASSACHUSETTS INST. OF TECHNOLOGY on 07/06/05. For personal use only.
Transcript of NONTEMPLATE-DEPENDENT POLYMERIZATION PROCESSES

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX10.1146/annurev.biochem.74.082803.133013
Annu. Rev. Biochem. 2005. 74:433–80doi: 10.1146/annurev.biochem.74.082803.133013
Copyright c© 2005 by Annual Reviews. All rights reserved
NONTEMPLATE-DEPENDENT POLYMERIZATION
PROCESSES: Polyhydroxyalkanoate Synthases as aParadigm1
JoAnne Stubbe,1,2 Jiamin Tian,1 Aimin He,1
Anthony J. Sinskey,2,3 Adam G. Lawrence,2
and Pinghua Liu1
Department of Chemistry,1 Department of Biology,2 and Department of Health Sciencesand Technology,3 Massachusetts Institute of Technology, Cambridge, Massachusetts02139; email: [email protected], [email protected], [email protected], [email protected],[email protected], [email protected]
Key Words starch, glycogen, polyphosphate, cyanophycin, rubber, PHAsynthases, PHA depolymerases, phasin
■ Abstract This review focuses on nontemplate-dependent polymerases that usewater-soluble substrates and convert them into water-insoluble polymers that formgranules or inclusions within the cell. The initial part of the review summarizes brieflythe current knowledge of polymer formation catalyzed by starch and glycogen syn-thases, polyphosphate kinase (a polymerase), cyanophycin synthetases, and rubbersynthases. Specifically, our current understanding of their mechanisms of initiation,elongation (including granule formation), termination, remodeling, and polymer re-utilization will be presented. General underlying principles that govern these typesof polymerization reactions will be enumerated as a paradigm for all nontemplate-dependent polymerizations. The bulk of the review then focuses on polyhydroxyalka-noate (PHA) synthases that generate polyoxoesters. These enzymes are of interest asthey generate biodegradable polymers. Our current knowledge of PHA production andutilization in vitro and in vivo as well as the contribution of many proteins to theseprocesses will be reviewed.
CONTENTS
INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434GENERAL OUTLINE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 435
1Abbreviations used: ADP-Glc, ADP-glucose; CoA, coenzyme A; HB, 3(R)-hydro-xybutyrate; HB-CoA, 3(R)-hydroxybutyryl-CoA; IPP, isopentenyl pyrophosphate;MW, molecular weight; PHA, polyhydroxyalkanoate; PHB, polyhydroxybutyrate; PT-Fase, prenyl transferase; TEM, transmission electron microscopy; UDP-Glc, UDP-glucose;wt, wild type.
0066-4154/05/0707-0433$20.00 433
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
434 STUBBE ET AL.
STARCH BIOSYNTHESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 437Biosynthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 440Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442
BACTERIAL GLYCOGEN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442Biosynthesis and Degradation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 443Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444
POLYPHOSPHATE BIOSYNTHESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444Biosynthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 445Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 445Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 445Utilization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 446
CYANOPHYCIN BIOSYNTHESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 446Pathway and Gene Organization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 447Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 447Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 447Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448Utilization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448
POLYISOPRENE BIOSYNTHESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448Candidates for Rubber Synthase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 450Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 451Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 452Regulation and Modern Methods of Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 452
PHA HOMEOSTASIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 452PHB Synthases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454Initiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456Elongation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 460Termination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 465Utilization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 467Regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 469
SUMMARY . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 475GENERAL CONCLUSIONS AND CHALLENGES . . . . . . . . . . . . . . . . . . . . . . . . . . 476
INTRODUCTION
What do a grease-laden French fry, an airplane tire, and a biodegradable shampoobottle all have in common? They all are composed of polymers made by naturethat play a central role in our everyday lives. Nature, using a few basic buildingblocks, has created biodegradable materials that can be generated from biorenew-able sources with a wide range of useful properties (1–3).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 435
Cellulose, starch, and glycogen all use glucose as a building block. Celluloseis the world’s most abundant organic substance. Starch is also ubiquitous and isthe most important source of energy for all living beings. Polymers using acetateas a building block (polyisoprenes, polyhydroxyalkanoates, and triacylglycerols)are also encountered in our everyday lives. Polypeptide polymers that are com-posed of two amino acids and not generated by ribosomes, are also found innature and have interesting properties. This review provides an overview of ourpresent understanding of the nontemplate-driven polymerization processes foundin nature. A summary of the systems being investigated is presented in Table 1.The specific focus will be on polymerases that use a soluble monomeric substrate(or substrates) that is transformed into an insoluble inclusion during the elonga-tion process. First, a brief overview of the common features of the synthases thatgenerate starch, bacterial glycogen, polyphosphate, L-arginyl-L-polyaspartate, andpolyisoprenes are described. We then examine in detail the β-hydroxyalkanoate(PHA) synthases, with a specific focus on the short-chain polyhydroxybutyrate(PHB) synthases, as a potential paradigm for all polymerases. We focus on themechanism of PHB formation, that is, initiation, elongation, and terminationprocesses in vitro and in vivo. We then examine the models for PHB granuleformation and utilization as well as the role of the proteins required for theseprocesses.
GENERAL OUTLINE
The general outline of each section is to present the biosynthetic pathway for poly-mer formation and the genes thus far identified in this process. We then describein detail the structural properties and mechanism of the synthase. The mecha-nisms of the initiation and priming processes are described in vitro and in vivo.Then the elongation process is addressed. Focus is placed on the phase transi-tion in which the growing soluble polymer becomes insoluble. The protein(s) thatcontrol this transition and the structures of the resulting inclusions (used inter-changeably with granules) are described, including the structures of the polymerswithin the inclusions. The proteins involved in remodeling of the polymers andthe effect of these proteins on the polymer properties are also presented. The na-ture of the termination process and the basis for the molecular weight range andpolydispersity of the polymers are discussed. In addition to the synthases, mostorganisms have depolymerases that are involved in polymer degradation and re-modeling. Regulation of synthesis and degradation are also discussed. Finally,potential contributions of new methods (genomics, metabolomics, mRNA profil-ing, and proteomics) to understand these complex processes are presented. In eachsystem, a reference to the most recent, comprehensive review is provided in theintroductory section as are additional references that have enlightened us since thisreview.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
436 STUBBE ET AL.
TA
BL
E1
Prop
ertie
sof
synt
hase
sin
volv
edin
nont
empl
ate-
depe
nden
tpol
ymer
izat
ions
Pol
ymer
MW
ofsy
ntha
sem
onom
er(k
Da)
Synt
hase
(nat
ive
stat
e)M
onom
erIn
itia
tion
Gra
nule
size
Pol
ymer
size
Ref
eren
ces
Star
ch60
to90
AD
P-G
lcM
alto
olig
osac
char
ide
oram
ylos
eV
aria
ble
(1–3
µm
to10
0µ
m)
Am
ylos
e:9
×10
4to
3×
106
Da
5,12
Am
ylop
ectin
:3
×10
8D
aa
Gly
coge
n(H
uman
mus
cle)
∼84
UD
P-G
lcG
lyco
geni
n(s
elf-
prim
ing)
Var
iabl
e>
1×
106
Da
15,2
0
Gly
coge
n(A
grob
acte
rium
tum
efac
iens
)
∼58
Dim
erA
DP-
Glc
Self
-pri
min
gN
ogr
anul
ede
tect
ed>
1×
106
Da
16,1
9
Poly
phos
phat
e∼8
0Te
tram
erA
TP
Olig
omer
icph
osph
ates
Sour
cede
pend
ent
∼7×
104
Da
27,3
1
Cya
noph
ycin
∼100
Dim
erA
sp&
Arg
Asp
-Arg
olig
omer
s0.
2to
0.5
µm
2.5
×10
4to
1×
105
Da
32,3
3,36
Rub
ber
(Hev
eabr
asil
iens
is)
∼33
Unk
now
nIP
PFP
Por
gera
nylg
eran
ylpy
roph
osph
ate
∼0.2
µm
,∼1
.0µ
m∼1
05D
aan
d∼1
06D
a41
PHB
∼64
(cla
ssI)
∼40
(cla
ssII
I)D
imer
(cla
ssI)
HB
-CoA
Self
-pri
min
g∼0
.5µ
m∼1
×10
6D
a13
Het
erot
etra
mer
(cla
ssII
I)
a Info
rmat
ion
sour
ce:h
ttp://
ww
w.ls
bu.a
c.uk
/wat
er/h
ysta
.htm
l(14
1).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 437
STARCH BIOSYNTHESIS
Starch is one of the major sources of calories in our diet, and over 600 commercialproducts are made by the starch extraction and processing industry. Starch struc-tures can be subdivided into those involved in long-term usage and those involvedin transitory usage. Long-term starch is the main carbon reserve and energy sourcein most plants. It is stored in underground plant organs, such as tubers or legumeseeds. Transitory starch in photosynthetic tissue, such as leaves, is synthesizedin the day and used at night and provides a buffer for sugar pools. A number ofexcellent reviews on the biosynthesis, structure, physical properties, and physio-logical roles of this polymer are available (4, 5). We cannot possibly do justice tothis huge body of work, but focus on the general principles and problems encoun-tered in studying starch synthases as related in general to all nontemplate-drivenpolymerizations.
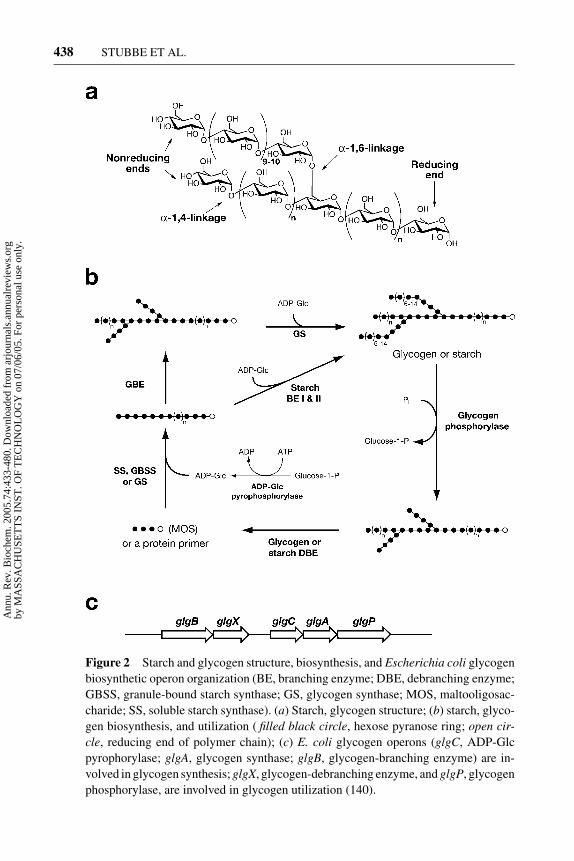
Starch is composed of glucose (GLC) polymers packaged in vivo as complex,semicrystalline, water-insoluble granules and is mainly found in plastids of theplant cells (Figure 1a). The granules are diverse in size and shape, and gran-ule morphology depends on the plant and the location within the plant. Starch iscomposed of two types of Glc polymers: amylose and amylopectin. Amylose ispredominantly α-(1,4)-linked Glc units with less than 1% α-(1,6) linkages, and itis 20% to 30% of the starch granule in starch-storing organs. Amylose is a linearpolymer that can form a left-handed helical coil. Amylopectin constitutes the re-mainder of the granule in which linear α-(1,4)-linked Glc units are joined togetherby 5% to 6% α-(1,6) linkages (Figure 2a). Starch is distinct from glycogen (see be-low) by the frequency of the α-(1,6) linkages. The branch linkages in amylopectinare clustered every 10–20 Glc units along the amylopectin structure and occur atregular intervals of about 90 A along the axis of the molecule. This branching al-lows highly ordered parallel arrays of double helical glucans to form at the root ofa unit cluster (4). Chains of about 45 units span two clusters, and chains of about 70units span three clusters. The double helices pack together in a regular manner toform crystalline lamellae that alternate with amorphous lamellae where the branch
Figure 1 Granule (bar size). (a) Starch from corn (10 µm) (139); (b) polyphosphatefrom Vibrio cholerae (0.5 µm) (31); (c) cyanophycin from Aphanocapsa 6308 (1 µm)(33); (d) rubber from Hevea brasiliensis (5 µm) (42); and (e) polyhydroxybutyrate(PHB) from Wautersia eutropha (1.0 µm) (13).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.
10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
438 STUBBE ET AL.
Figure 2 Starch and glycogen structure, biosynthesis, and Escherichia coli glycogenbiosynthetic operon organization (BE, branching enzyme; DBE, debranching enzyme;GBSS, granule-bound starch synthase; GS, glycogen synthase; MOS, maltooligosac-charide; SS, soluble starch synthase). (a) Starch, glycogen structure; (b) starch, glyco-gen biosynthesis, and utilization ( filled black circle, hexose pyranose ring; open cir-cle, reducing end of polymer chain); (c) E. coli glycogen operons (glgC, ADP-Glcpyrophorylase; glgA, glycogen synthase; glgB, glycogen-branching enzyme) are in-volved in glycogen synthesis; glgX, glycogen-debranching enzyme, and glgP, glycogenphosphorylase, are involved in glycogen utilization (140).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 439
points are located. Crystalline and amorphous lamellae together form growth rings.One growth ring is composed of tens of lamellae repeats. Recent studies have sug-gested that these structures can be remodeled by phosphorylation (6).
Biosynthesis
The starch synthases use ADP-glucose (ADP-Glc) as a substrate to form the α-(1,4)-glucans. ADP-Glc is synthesized by ADP-Glc pyrophosphorylase as shownin Figure 2b. There are two types of starch synthases: soluble starch synthaseand granule-bound starch synthase. The ADP-Glc can be added to either thenonreducing end of a Glc chain or the reducing end depending on the starchsynthase isozyme (5, 7). Expression analyses of plant genomes, accompaniedby knockout experiments, are revealing the complexity of the isoenzymes ofstarch synthases (8). The resulting α-(1,4)-glucans are then substrates for starch-branching enzymes that introduce α-(1,6) branches to yield the amylopectin poly-mers (4). Starch-debranching enzymes also are essential for starch biosynthe-sis. They can hydrolyze α-(1,6) branches either directly or indirectly. Directdebranching occurs in plants and bacteria, and the enzymes with different speci-ficity are called isoamylases or pullulanases. Indirect debranching occurs in yeastand animals (9).
Our chemical understanding of the mechanisms of the soluble starch synthaseand granule-bound starch synthase are still in a rudimentary stage. Both synthasesare glycosyl transferases. On the basis of sequence alignments, glycosyl trans-ferases are now divided into 50 families (10, 11). The amylopectin synthases frombacteria and plants are placed in family 5 and differ from the glycogen synthasesin mammals and fungi that are placed in family 3. Both the soluble starch syn-thase and granule-bound starch synthase catalyze their reactions with retention ofconfiguration. The evidence thus far, however, suggests that the enzymes use dif-ferent mechanisms with different roles for the conserved glutamates. The amylosesynthase is in general a granule-bound starch synthase, whereas the amylopectinsynthase is usually soluble (similar to glycogen synthase discussed below). Thesizes of the synthases range from 60 to 90 kDa, and no structural data on any ofthese proteins presently exist.
Initiation
Recent studies have provided insight into the different mechanisms of initiation ofthe soluble starch synthases and granule-bound starch synthases. In the case of thegranule-bound starch synthases, in vitro studies suggested two possible initiationmechanisms (12). The first is that maltooligosaccharides (2 to 7 residues) generatedby starch degradation enzymes can function as noncovalent primers. The secondis that branches of amylopectins can be cleaved to amylose that then serves as theprimer. Complementary studies in vivo, using a variety of mutants in Arabidopsisthaliana leaves in which maltooligosaccharide levels have been modulated, supportthe maltooligosaccharide-priming model (12). In the proposed mechanism, the
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
440 STUBBE ET AL.
starch synthase adds the monomer to the nonreducing end and does not involvecovalent catalysis. The Glu residues presumably function as general acid/basecatalysts (Figure 3a).
Elongation
Recent studies on starch granules from eight different plant sources provide con-vincing evidence for covalent catalysis through a conserved Glu. These exper-iments are presumably interrogating amylopectin biosynthesis (7). The authorscarried out pulse-chase experiments on intact granules. The results from theirexperiments have eliminated many favored models for initiation and elongation.Their results suggest that ADP-Glc is transferred to the protein to form a chemi-cally labile bond with the synthase. They provide further support that the additionof Glc from ADP-Glc to this linkage occurs from the reducing end. The mecha-nism they proposed, involving two covalent intermediates, is shown in Figure 3band has been called the two-site insertion mechanism. This mechanism is verysimilar to the one initially proposed for PHB synthases (13). This mechanism dif-fers from the established mechanism for glycogen biosynthesis in mammals andfungi, which uses a protein primer in which Glc is added from UDP-Glc throughthe nonreducing end (Figure 3c). Self-glycosylating proteins, with sequence simi-larities, have been identified in plant systems such as A. thaliana, but none of themcontains an appropriate N-terminal sequence for localization in the plastid wherestarch synthesis occurs. Whether they are protein primer equivalents in plants hasnot been established biochemically or genetically.
Subsequent to initiation and elongation, continued chain extension results information of granules that vary tremendously in size (1–3 µm to 100 µm) andshape, depending on the system (Figure 1a). The process by which these semicrys-talline structures are initiated remains a mystery. The granule formation is thoughtto involve self-assembly based on physical properties, rather than being directedby biology (4). This conclusion is based on the observation that most mutationsresulting in alterations in granule structure have been associated with the genesorganizing the carbohydrate chains, rather than scaffolding proteins. A very re-cent study in potato tubers implicates starch debranching enzymes, isoamylaseisozymes, in granule initiation (9). Antisense experiments to suppress specificisozymes resulted in greatly increased numbers of small granules. Accompanyingtransmission electron microscopy (TEM) studies revealed that these small gran-ules are contiguous to large granules. Controls revealed that isoamylases appear tosuppress initiation of glucan molecules that would crystallize to form new starchgranules (9). Despite the extensive knowledge of the enzymes involved in starchbiosynthesis, no one has been able to recapitulate formation of the amylose oramylopectin in vitro nor organize these polymers into a granule. Furthermore, nogroup has been able to generate starch granules in bacteria. In conclusion, a com-mon problem in nontemplate-dependent polymerization systems is our inability toclearly define function(s) of proteins due to unavailability of substrates experiencedby these proteins in vivo.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 441
Figure 3 Possible mechanisms involving covalent catalysis and/or general acid/base catal-ysis for starch and glycogen biosynthesis (open circle, reducing end of polymer chain; filledblack circle, pyranose ring; E, enzyme; P, protein; X, ADP or UDP) (7). (a) Elongation byadding a monomer to the nonreducing end of an oligosaccharide primer. (b) Elongation byadding a monomer to the reducing end. (c) Elongation by adding a monomer to the nonre-ducing end of an oligosaccharide attached to a protein primer.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
442 STUBBE ET AL.
Termination
The large number of enzymes involved in remodeling of the amylopectin chains,including phosphorylation at all stages of granule assembly (6), has made the ter-mination process difficult to examine, and thus almost no information is available.
BACTERIAL GLYCOGEN
Glycogen is a major reserve of carbohydrates in both prokaryotes and eukaryotes.However, glycogen in eukaryotes is made from UDP-Glc, whereas glycogen fromprokaryotes is made from ADP-Glc. Many bacterial species accumulate glycogenin either stationary phase or under nutrient-limited conditions. Photosynthetic bac-teria, however, also can produce glycogen in a transient form. In Escherichia coli,glycogen synthase is not essential for growth (14). The bacterial glycogen syn-thases are in the glycosyl transferase family 5, as is the amylopectin synthasediscussed above (11). Glycogen is similar in structure to amylopectin except thatthe α-(1,6) branch linkages occur more frequently at 10% to 12%, and as a conse-quence, the polymers have shorter chain lengths and twice the number of branchesFigure 2a (5). Bacterial glycogen, in contrast to starch, is homogeneous and watersoluble.
Biosynthesis and Degradation
Glycogen synthesis in bacteria depends on the actions of four enzymes: phos-phoglucomutase, ADP-Glc pyrophosphorylase (glgC ), glycogen synthase (glgA),and glycogen-branching enzyme (glgB). Glycogen degradation requires glycogenphosphorylase (glgP) and a debranching enzyme (glgX ) in addition to enzymesfor catabolizing maltooligosaccharides (Figure 2c).
Initiation
In animal and fungal cells, initiation is carried out by the self-glycosylating protein,glycogenin (Figure 3c) (15). UDP-Glc is the substrate for both glycogenin (thepriming protein) and glycogen synthase. The Km values for the substrate and themetal ion requirements for glycosyl transfer catalyzed by each of these proteinsare substantially different. The Km for UDP-Glc priming is µM, and the priming isMn2+ dependent; conversely, the Km for elongation is mM, and elongation is Mg2+
dependent. Glycogenin is glycosylated on tyrosine at the reducing end and extendsfrom its nonreducing end by 8 Glc units. The end Glc residue is then recognized byglycogen synthase. Glycogenin remains linked to the polymer through the entireelongation process.
Protein primer has not been identified in bacteria or plants through biochem-ical efforts or through genome searching with the glycogenin sequence. Stud-ies on Agrobacterium tumefaciens glycogen synthase, patterned after those on
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 443
glycogenin, suggested that this protein can self-prime (16). Studies with µM [14C]-ADP-Glc, followed by SDS-PAGE analysis of products, suggested that radiola-beled sugar is covalently attached to the glycogen synthase. The labeled materialcould be chased into a large molecular weight glycogen with cold ADP-Glc (mM).Thus, A. tumefaciens glycogen synthase is self-priming and appears to occur bycovalent glycosylation. Sequence similarities between A. tumefaciens glycogensynthase and other bacterial glycogen synthases suggest that this mechanism ofpriming may be general.
Elongation
As with starch synthases, bacterial glycogen synthase adds ADP-Glc (Km is mM)to the growing chain with retention of its configuration. Both the E. coli andA. tumefaciens glycogen synthases are close functional relatives to starch syn-thases. Two structures of glycosyl transferases have been solved, and they havebeen designated GT-A and GT-B. Members of the glycosyl transferase family 5are proposed to have a GT-B fold, and a threading model of the E. coli glycogensynthase has been reported (17). Sequence alignments reveal only a few conservedresidues, and their functions have not been mechanistically defined.
Site-directed mutagenesis studies have been carried out on conserved residues inthe E. coli glycogen synthase (D137A, R300A, K305A, H161A, and E377A). Thestudies with aspartate and glutamate mutants revealed complete loss of activity,whereas the other mutants have reduced activity (17). In the maize starch synthase,the equivalent of the conserved aspartate and glutamate both show no activity withD → N or E → Q mutations and 10% to 30% activity with the E → D or D → Emutations (18). The roles of these residues in initiation and elongation (covalentor acid/base catalysis vs structural) are still not clear, nor is it clear whether theADP-Glc adds to the nonreducing or the reducing end of the growing polymer assuggested by the recent studies on intact starch granules (7).
As with starch synthesis, bacteria (E. coli and cyanobacteria) have glycogen-branching enzymes that catalyze formation of the α-(1,6)-linkages. Recent geneticstudies, accompanied by analysis of product size and solubility, demonstrated thatcyanobacteria, in which the glycogen-branching enzyme gene has been deleted,synthesize amylose-like glycans (19). The lack of branching is predicted to causea decrease in the number of nonreducing ends that can be extended and, hence,reduction in the molecular weight of the polymer. Thus, an experiment to examinethe size of the polymer would provide insight into the mechanism of elongation.Consistent with the addition of Glc to the nonreducing end, the wt strain had amolecular weight of 6.6 × 107 Da, while the mutant strain had a molecular massof 4.7 to 5.6 × 103 Da.
The key to looking for different-sized polymers is understanding their solubilityproperties and how they change so that appropriate extractions and quantitativerecoveries of polymer are possible. Characterizing polymers produced by all ofthe polymerases discussed in this review is a challenging problem. It is interesting
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
444 STUBBE ET AL.
to speculate that the major difference between plants producing starch and bac-teria generating glycogen will be found, not in the soluble synthases, but in thesteps leading to polysaccharide aggregation into insoluble semicrystalline ma-terial. In neither cyanobacteria nor in E. coli have starch-like inclusions beendetected.
Termination
Investigators have proposed that the sizes of the glycogen polymers are stericallyself-limiting. The mass of rabbit muscle glycogen is approximately 107 Da. Mod-eling studies have predicted that glycogen could not grow past 12 branches becausepacking would limit access to the nonreducing ends required for growth (20).
POLYPHOSPHATE BIOSYNTHESIS
Inorganic polyphosphate is a linear polymer of inorganic phosphate linked byphosphoanhydride bonds (Figure 4a). A number of biological functions have beenproposed for this polymer. Polyphosphate can replace ATP in kinase reactions andmay be involved in regulation of levels of NTP and dNTP pools by serving as adonor for the NDPs or dNDPs (21). It can function as a reservoir of phosphates, achelator, and a buffer. The polymer has most often been associated with its role incoping with acute and prolonged environmental stress (depletion of amino acids,Pi, nitrogen, and changes in ionic strength). A number of excellent reviews haverecently been published (22–24).
Biosynthesis
The biosynthesis and degradation of polyphosphate involve polyphosphate ki-nase (PPK, the polymer synthetase) and exopolyphosphatase (PPX, polyphosphatedegradase). Both genes are found on the same operon (Figure 4b) (25). A numberof additional enzymes, such as endopolyphosphatase (26) and phosphotransferase,are also thought to be involved in degradation (22). The PPK1 from E. coli is thebest-characterized synthetase and converts ATP into polyphosphate (Figure 4a).
Figure 4 Polyphosphate biosynthesis. (a) E. coli polyphosphate kinase (PPK)and exophosphatase (PPX) catalyze polyphosphate synthesis and degradation,respectively. (b) E. coli operon (31).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 445
The reaction is reversible, and thus the polymer formation requires the couplingof ADP to another reaction (27). The PPK1 is a soluble homotetramer of subunitmolecular weight (MW) 80 kDa. In vivo the protein is membrane associated. Thereare 850 ± 120 molecules/cell (4.7 µM assuming the volume of an E. coli cell is3 × 10−16 L and the protein is soluble) when E. coli are grown in Luria-Bertanimedium (28). Recently, a second polyphosphate kinase (PPK2) has been foundin many bacteria, excluding E. coli (29). This enzyme can use GTP or ATP tomake polyphosphate and prefers Mn2+ instead of Mg2+ required for PPK1. PPK2is expressed in late-log phase of cell growth. Its function has been proposed tobe the synthesis of GTP from GDP, which is required for the massive synthesisof exopolysaccharides and alginate that are essential when the organism entersstationary phase (30).
Initiation
As with the other polymerases, efforts to find a small molecule or protein thatfunctions as an initiator in vivo have been unsuccessful. In vitro at low ATPconcentrations (µM) and with PPK1 at any concentration, the rate of polymerformation shows a lag phase that is suppressed by addition of (P)4 but not by (P)3
or (P)2 (27). Thus, (P)4 appears to function as a primer. These conditions are farremoved from the mM concentrations of ATP found inside the cell, and under mMATP, no lag phase is observed. At µM ATP concentrations, the involvement ofa phosphorylated enzyme as a primer has also been suggested (27). A phospho-rylated histidine in PPK1 has been detected in vitro using [γ -32P] ATP at 0◦C.Addition of cold ATP (mM) resulted in incorporation of 95% of [γ -32P] ATP intopolyphosphate, demonstrating the chemical competence of the phosphorylatedPPK1.
Elongation
At mM ATP, the elongation rate is processive, resulting in a polymer chain of∼750 residues in vitro. No intermediate polyphosphates were detected. Incubationof radiolabeled short-chain phosphate oligomers (2 to 400 residues in length)and addition of unlabeled ATP resulted in formation of an unlabeled 750 mer(27). Although the concentrations of polyphosphate in E. coli can change from100 µM to 50 mM under environmental stress, no granules of this material havebeen detected. Granules of polyphosphate have, however, been detected in thevacuole of yeast and can account for as much as 10% to 20% dry cell weight. InV. cholerae, granules are found in the cytosol (31) (Figure 1b).
Termination
The polymer chain length synthesized by PPK1 in vitro is very monodisperse.Nothing is known about the apparently exquisite control of the chain length.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
446 STUBBE ET AL.
Utilization
Control of the polymer length in vivo is predominantly regulated by the depoly-merase, PPX. E. coli PPX has also been extensively studied, and it is a dimer witha subunit MW of 58 kDa that is also membrane associated. The enzyme requiresMg2+, is stimulated by K+, and has a turnover number of 20 s−1. By removingthe orthophosphates from the polymer ends, the protein degrades the polyphos-phate made by PPK processively without any polyphosphates of intermediate chainlengths being detected. (25). The simultaneous presence of a synthase and a de-polymerase (PPK and PPX) is a theme that repeats itself in the biosynthesis ofstarch, glycogen, PHB, and cyanophycin. The regulatory mechanisms involved inpreventing futile cycling are not understood in any case.
CYANOPHYCIN BIOSYNTHESIS
Cyanophycin is a polyamide: L-arginyl-poly(L-aspartate). The amino acids argi-nine and aspartate are present in about a 1:1 ratio, and nearly all the β-carboxylgroups of the polyaspartate backbone are linked to the amino groups of arginineby an isopeptide bond (Figure 5a). This polypeptide, in contrast to proteins, isnot made on the ribosome. This polymer is most frequently found as insolubleinclusions in cyanobacteria, oxygenic photosynthetic prokaryotes that are capableof adapting to many environments (32). Survival of cyanobacteria under stressfulconditions, such as high light, or starvation for CO2, sulfur, or phosphorus, resultsin accumulation of cyanophycin granules in stationary phase (Figure 1c) (33).Under these conditions, the polyamide is thought to serve as a nitrogen reserve.
Figure 5 Cyanophycin biosynthesis. (a) Proposed mechanism for cyanophycin chain elon-gation catalyzed by cyanophycin synthase from Anabaena 29413 (n ≥ 3). (b) Synechocystissp. operon: cphB (cyanophycinase) and cphA (cyanophycin synthetase) (32, 36).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 447
However, in other cyanobacteria capable of fixing nitrogen, it has been suggestedthat cyanophycin serves as a dynamic reservoir of newly fixed nitrogen to bufferthe nitrogen-fixing cells (heterocysts) against environmental fluctuations (34, 35).In these systems, the polyamide is thought to play an integral role between nitrogenand carbon metabolism (32, 34).
Pathway and Gene Organization
The de novo synthesis of cyanophycin is catalyzed by a single protein, cyanophycinsynthetase. The reaction is shown in Figure 5a. Incubation of Mg2+•ATP withaspartate and arginine results in the formation of product. The loci of genes involvedin the synthesis and degradation of the polymer are found on an operon (Figure 5b)(32, 36). A theme that repeats itself in almost all of the systems discussed in thisreview. The synthetase has been purified and expressed from Anabaena variabilis(36) and Synechocystis sp. (32). The A. variabilis protein has a subunit MW of100 kDa and a native MW of 230 kDa (36). Sequence analysis and Clustal Walignments of all cyanophycin synthetases reveal that they have two ATP-bindingsites. The N-terminal domain (residues 127 to 424) is homologous to the ATP-grasp superfamily of enzymes. This family activates carboxylates for nucleophilicattack by phosphorylation with Mg2+•ATP. The C-terminal domain (residues 550to 800) is homologous to ATP enzymes involved in peptidoglycan biosynthesis(peptide ligases) (37). Although the structure is not available for any synthetase,structures of the ATP-grasp domain and the peptide ligase domain are available,and high sequence identity reveals that threading models could be made (37).Mechanistic studies have revealed that biosynthesis of the polymer proceeds by C-terminal elongation of the poly-aspartate backbone with the addition of aspartatefollowed by generation of the isopeptide bond with arginine. Each step requiresan ATP (Figure 5a) (32, 38). Covalent catalysis via the enzyme is not anticipatedgiven our understanding of the chemistry of the two superfamily members thatcompose the synthetase.
Initiation
In vitro cyanophycin synthesis requires a primer and either (α-Asp-Arg)3
or (α-Asp-Arg)2 can function in this capacity (32, 38). The mechanism of primingin vivo is unknown.
Elongation
Experiments have not yet addressed the issue of distributive versus processivepolymerization, nor has the issue of granule formation been investigated. Thegranules from Aphanaocapsa 6308 have been examined by electron microscopy(EM) using thin section and freeze fracture techniques (Figure 1c) (33). Theyare nonuniform in size and range from 0.2 to 0.5 µm. No membrane aroundthe granule is visible by either method. Both the synthetase and depolymerase
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
448 STUBBE ET AL.
(cyanophycinase) are soluble proteins, and there is no evidence for their associationwith the granules (33, 39).
Termination
The polymers generated in vivo are highly polydisperse with MWs ranging from25 to 100 kDa (32). In vitro polymerization or production of cyanophycin inE. coli results in smaller polymers of MW of 25 to 30 kDa (32).
Utilization
Cyanophycin is degraded by cyanophycinase, a 29.4-kDa protein (32). The cyano-phycinase hydrolyzes the polymer from its C terminus to release the dipeptide,Asp-Arg. This protein has been suggested to be a member of the serine hydrolasefamily, but the critical experiments to test this hypothesis have not been carriedout. Presumably there is an isoaspartyl peptidase that can hydrolyze the dipeptideto aspartate and arginine (32).
POLYISOPRENE BIOSYNTHESIS
Natural rubber is a raw material used in the manufacture of many products. Al-though more than 2500 plant species are known to produce rubber, Hevea brasilien-sis, the Brazilian rubber tree, is the only competitive source of commerciallyavailable natural rubber (40). Despite the increasing demand for natural rubber,the acreage for rubber trees has diminished in recent years, and thus there hasbeen interest in the development of additional sources of natural rubber. To guidethe generation of rubber-producing transgene plants, identification of the genesinvolved in its biosynthesis and regulation is essential.
Rubber is generated from the isopentenyl pyrophosphate (IPP) building block(Figure 6c). There are two biosynthetic pathways that can generate this monomer:the mevalonate pathway and the 1-deoxy-D-xylulose-5-phosphate/2-C-methyl-D-erythritol-4-phosphate pathway. The choice of pathway is dependent on the organ-ism (Figure 6a,b) (41). The polymer generated from IPP is a polyisoprene withthe double bonds having the cis configuration. The polymer chains, by unknownmechanisms, aggregate into rubber particles found in the latex vessels of rubbertrees (Figure 1d ) (42).
Candidates for Rubber Synthase
The identification and characterization of the genes and enzymes involved in rubberbiosynthesis have been slow relative to other homo-polymerization systems. Infact, most of the studies thus far reported begin with rubber particles. This startingpoint was based on early biochemical studies that demonstrated that fresh latexcould be separated by centrifugation into three phases (43). The bottom fraction
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 449
Figure 6 Rubber biosynthetic pathway (41). H. brasiliensis has two different pathwaysfor synthesis of dimethylallyl pyrophosphate (DMAPP) and IPP. (a) The mevalonate(MVA) pathway. (b) The 1-deoxy-D-xylulose-5-phosphate/2-C-methyl-D-erythritol-4-phos-phate (DXP/MEP) pathway. (c) Proposed rubber biosynthetic scheme.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
450 STUBBE ET AL.
(20% of the latex) contains membrane-bound organelles, called lutoids and Frey-Wyssling particles. The middle fraction is called the C-serum. The top fractionphase that contains the rubber particles was the focus of most investigators. Veryrecent biochemical and EM studies, however, have established that latex in thisfractionated form is unstable (43). These studies also suggest that the bottomfaction is required for initiation of polymer synthesis. This knowledge and thedevelopment of methods to store the fractions without destablization should greatlyfacilitate progress in identifying proteins involved in the early stages of polymerformation.
Distinct structurally and mechanistically from the trans-prenyl transferases(trans-PTFases) (44), information on the cis-prenyltransferases (cis-PTFases),making linkages similar to those bonds found in rubber, has only become avail-able very recently. Using the sequences from E. coli, Micrococcus luteus, andA. thaliana cis-PTFases, DNA primers from conserved sequences have been usedin conjunction with a polymerase chain reaction to pull out the correspondinggenes from RNA isolated from the latex of Hevea (45). The genes were taggedwith a (His)6 or thioredoxin tail, and the proteins were expressed in E. coli in allcases in inclusion bodies. Solubilization in denaturant and refolding gave a proteinof 33 kDa, subsequent to tag removal. This protein was inactive by itself, but whenincubated with the washed bottom fraction of the latex, large isoprene polymerswere convincingly observed for the first time. Thus, proteins and the membranesfound in this bottom fraction are essential for the initiation and, presumably, elon-gation catalyzed by the cis-PTFase. There have been many previous reports thatwashed rubber particles from the top fraction can add IPP to a growing rubberparticle (41). The turnover numbers using these preparations are low and variable,owing to missing factors. The discovery that the fresh bottom fraction of the latexis essential for initiation of polymerization (45) has thus set the stage to study thebiochemistry in vitro and in vivo of this interesting polymerase.
Initiation
The mechanism of polymer initiation in Hevea is unknown, although in vitroaddition of farnesyl pyrophosphate or geranylgeranyl pyrophosphate resulted inenhanced incorporation of [14C]-IPP in the presence of washed rubber particles.In goldenrod (Solidago altissima), the polymers are of sufficiently low molecularweight that NMR spectra of the isolated isoprenes allow a distinction betweenthe residues at its ends and the bulk monomer (41). Two trans isoprene unitsand a dimethylallyl group were detected, providing evidence in this system forsmall molecule priming in vivo. When a similar experiment was carried out onrubber isolated from Hevea leaves (not the rubber particles themselves), two transisoprene units were detected and there was no dimethylallyl unit. The absencecould imply that the dimethylallyl unit had been modified or that it simply was notpresent.
Structures of the M. luteus and E. coli undecaprenyl synthases (cis-PTFases)are available (46, 47) and thus homology modeling of the Hevea PTFase is
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 451
possible. The steady-state and presteady-state kinetic studies on undecaprenylsynthases have revealed a distributive mechanism for attaching the 11 units of IPPto generate product (48). Comparison of gene sequences of the polymerase andthe undecaprenyl pyrophosphate synthases reveals that the sizes of the plant andbacterial proteins are similar. Thus, if the mechanism of the elongation is proces-sive in the case of the polymerase, then components of the polymerase system areprobably missing.
Elongation
As noted above, elongation has been studied by most investigators using washedrubber particles. Isolation of these particles and characterization of the proteinsassociated with them by SDS-PAGE revealed two predominent proteins (41): one,MW 14.6 kDa, designated rubber elongation factor and the second, which has aMW of 24 kDa and is homologous to rubber elongation factor, has been desig-nated small rubber particle protein. Transcriptome analysis of rubber latex revealsthat these two proteins comprise up to 29% of the total expressed sequence tagsof the lactifier cells (49). Both of these proteins have been expressed, isolated,and antibodies generated. There have been no reports of evidence for an abundantprotein of MW 33 kDa, the putative cis-PTFase. Immunogold labeling studies andtransmission electron microscopy have revealed that small rubber particle proteinis located over the entire surface of the rubber particle (Figure 7a) (42). The lo-calization of small rubber particle protein had been elusive for many years. It wasonly by use of nonconventional fixation methods (a lot of trial and error) that asmall rubber particle protein was shown to be located on the granule surface. Ingeneral, isolation of each granule to preserve its constituents will have its ownidiosyncracies. This must be kept in mind in thinking about constitutents requiredfor granule initiation in all nontemplate-dependent polymerizations. With Hevea(Figure 1d ), TEM has revealed two subsets of spherical particles: one 0.2 µmand the other 1.0 µm in diameter. Rubber particles with their interior made of
Figure 7 Detection of purified granule-bound proteins using antibodies to surfaceproteins and gold-conjugated secondary antibodies. (a) Small rubber particle proteinon H. brasiliensis rubber particle surface (Bar = 200 nm) (42). (b) W. eutropha PHBgranules covered by phasin protein PhaP (bar = 1 µm) (62).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
452 STUBBE ET AL.
polyisoprenes and surface covered by small proteins (Figure 7a) are very reminis-cent of PHB granules covered by phasin proteins (Figure 7b).
Termination
Rubber appears to be a metabolic dead end because there have been no findings ofenzymes capable of breaking down the rubber in latex. In Hevea, the MW of thepolymer has a bimodal distribution with some polymers in the range of 106 Da andothers 105 Da. Both the Hevea polymers and the C55 to C120 oligomeric isoprenesappear to have well-controlled molecular weights (50, 51). In vitro studies revealthe size of the polymer is related to the relative concentrations of the putativeprimer (farnesyl pyrophosphate or geranylgeranyl pyrophosphate) and IPP (50).The higher the ratio of primer to substrate, the shorter the chains. Thus, it is likelythat the granule and its associated proteins in conjunction with the elongationprotein(s) will play a critical role in chain length control and will be different fromin vitro studies.
Regulation and Modern Methods of Analysis
Modern “omics” methods have allowed analysis of the transcriptome in latex andprovided information about metabolic activity in this specialized tissue (49). Theresults revealed that the latex is surprisingly unique and that only seven genefamilies accounted for >50% of the latex transcriptome. The cis-PTFase wasunexpectedly not present in the expressed sequence tag pool. Its absence resultedfrom their low levels of expression, as genes for four such cis-PTFases were clonedby screening the latex cDNA library. Finally, the second most abundant groupof transcripts was demonstrated to be associated with defense or stress genes,suggesting a function for these lactifier cells in the Hevea plant. The unexpectedpresence of the DXP/MEP pathway for IPP production suggests that this pathway(Figure 6b) (49), in addition to the more conventional MVA pathway (Figure 6a)for IPP production, might play a role in rubber biosynthesis.
PHA HOMEOSTASIS
PHAs are polyoxoesters generated from 3-hydroxyalkanoate coenzyme A esterswith loss of coenzyme A (CoA) concomitant with formation of each ester bond(Figure 8). The PHA polymers are deposited as insoluble inclusions or granuleswithin the cells (Figure 1e). These polyesters are generated in almost all
Figure 8 Polyhydroxyalkanoate (PHA) biosynthesis cat-alyzed by polyhydroxyalkanoate synthase, PhaC.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 453
bacteria under nutrient-limited growth conditions when a carbon source is read-ily available (2). The PHA production occurs in exponential, late exponential,or in stationary growth phases, depending on the organism. Accumulation ofPHA can reach as much as 85% of the dry cell weight (1). When the envi-ronment becomes more hospitable, the PHAs are degraded to the correspond-ing monomers, which are used as a source of energy for biosynthesis (supplyingNADH) and as biosynthetic building blocks. As with all the other polymers dis-cussed above, there are also conditions of growth in which PHB is generatedtransiently (1, 2).
PHAs have received much attention from the bioengineering community be-cause, depending on the size of alkyl side chain (R, Figure 8), the resulting polymershave properties that range from thermoplastics (R = H, methyl, ethyl) to elas-tomers (R = C3H7-C14H29). Furthermore, they are biodegradable (52, 53). Billionsof pounds of plastic waste are generated each year from the oil-based polyethy-lene and polypropylene plastics. Thus, there is a growing interest in generatingbiodegradable thermoplastic polymers such as the polyoxoesters from biorenew-able sources (54–56). Most effort is presently focused on producing these polymersin an economically competitive fashion. Understanding PHA homeostasis may beessential to the success of this endeavor. A number of recent reviews cover thebiochemistry and biology of the PHA synthesis and degradation as well as theprospects for making plastic factories (1, 53, 57–60).
In the past few years, genes involved in PHA homeostasis in many bacteriahave been identified. The organization of the genes in W. eutropha, and Allochro-matium vinosum are shown in Figure 9a, as these organisms are paradigms for theshort chain PHB synthases (designated, PhaC) that have been extensively studied
Figure 9 Three classes of polyhydroxyalkanoate (PHA) synthases and typical geneorganizations (74). (a) PHB biosynthetic gene clusters (phaC, synthase; phaA, β-ketothiolase; phaB, reductase; phaP, phasin protein; phaE, one component of the classIII synthase heterodimer). (b) Medium chain PHA synthase gene cluster (phaC1 andphaC2, synthases; phaZ, depolymerase; phaD, a protein with unknown function butrelated to a major granule-bound protein, PhaI).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
454 STUBBE ET AL.
Figure 10 Polyhydroxybutyrate (PHB) biosynthetic pathway (BktB, a β-ketothiolaseprimarily involved in β-ketovaleryl CoA synthesis).
(53). Additional genes involved in PHB biosynthesis are phaB and phaA, whichcode for acetoacetyl-CoA reductase and a thiolase, respectively (Figure 9a andFigure 10) (61). The phaABC genes are usually found in an operon and are highlyconserved (Figure 9) (57). In addition to proteins involved in polymer synthe-sis, phasin proteins that play an important role in granule formation have alsobeen identified (62–64). Recent genomic studies in W. eutropha suggest that it hasmultiple phasin proteins (65). In contrast to the other proteins involved in PHBhomeostasis, the phasins are not sequence conserved between organisms. Thusfar, two transcription factors, PhaR (63, 66–68) and PhaF (69), have been iden-tified. The regulation of transient PHB production in nutrient-rich conditions orPHB production under stress (nutrient-limited conditions) obviously requires theinvolvement of many transcription factors. The details of the regulatory mecha-nisms are an active area of investigation. When the limiting nutrient is providedand no carbon is available for growth, PHB is degraded as a source for carbonand energy. Three depolymerase genes phaZ1 (renamed phaZ1a) (70), phaZ2 (re-named phaZ1b) (71), and an oligomer hydrolase (renamed phaZ2) (72, 73) havebeen identified, and their functions were suggested by analysis of deletion strains.Several additional putative intracellular depolymerase genes from the genome ofW. eutropha have recently been identified according to sequence homology (65).
In addition to the class I and class III PHB synthases, there is another class ofsynthase that uses 3-hydroxyalkanoate derived from fatty acids as substrates (R =C3H7 to C14H29, Figure 8 and Figure 9b). The Pseudomonas oleovorans synthasehas served as the paradigm for this class of synthase. The synthase has only beenpurified from insoluble inclusions. Renaturation of the inclusions solublized withdenaturants results in an active synthase. However, the turnover number is verylow, only 1/103 of that of class I and class III synthases (81, 84). Consequently,this class of synthase will not be discussed further.
PHB Synthases
PHB is generated by the class I and class III synthases, which are classified by theirsubunit molecular weights (74). Both synthases use predominantly HB-CoA andcan also use 3-hydroxyvalerate-CoA as substrates. The class I enzymes, typifiedby the enzyme from W. eutropha, has a subunit MW of 64 kDa and is probably adimer during the elongation process (75–77). The class III enzymes, typified by theenzyme from A. vinosum, have two subunits each of MW 40 kDa, designated PhaC
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 455
(the synthase) and PhaE (78–80). PhaE has no sequence homology to any knownproteins. The active synthase is a tetramer of a 1:1 complex of PhaC:PhaE. All syn-thases have resisted purification from their host organism with one exception. Re-cently, a PHA synthase was purified from Thermus thermophilus, an organism withan optimum growth temperature at 70◦C (82). Its unusual subunit MW (55 kDa)and substrate specificity suggest that it may be a new class of synthase. Thus, allsynthases that have been examined in vitro in detail are from recombinant sources.The relationship between their properties in vitro with those in vivo is essentialto establish given that the location of these proteins, at least during elongationprocess, is on the surface of the granules. The turnover numbers of the class I andIII synthases are 40 U/mg (83) and 140U/mg (80), respectively, The synthase fromT. thermophilus has the highest specific activity thus far reported, 2050 U/mg (82).The recombinant class I synthase, while soluble, is ill behaved unless the nonionicdetergent Hecameg, well below its critical micelle concentration, is added to thebuffers used in the purification (75). In contrast, the class III PhaCPhaE synthase iswell behaved and soluble when expressed in E. coli. Both N-terminal His-taggedPhaC and PhaE were separately expressed in E. coli, and the individual subunitswere purified . PhaC had very low polymerization activity (0.1% the activity of thecoexpressed PhaCPhaE), while PhaE did not have detectable activity. Titration ofPhaC with the tagged PhaE eventually produced activity comparable to the coex-pressed proteins. The ratio of PhaC:PhaE, however, was 1:10 rather than 1:1; thusPhaE plays a role in vivo that cannot be recapitulated in vitro (80). PhaE may beplaying a role similar to Hecameg or a phasin protein.
Sequence alignments of all classes of synthases revealed a number of conservedresidues (Table 2) (13). In addition, Blast searches highlighted a 50-amino acidstretch of the class III synthase that was 42% sequence identical to bacterial li-pases, including the GXSXG “lipase” box (85). In the synthase, the serine that isknown to be involved in covalent catalysis in lipase is replaced with a cysteine.
TABLE 2 List of the conserved amino acidresidues in class I and III synthases
Class I (R. eutropha) Class III (A. vinosum)
S260 S90
C319 C149
G322 G152
D350 D177
W425 W248
D480 D302
G507 G330
H508 H331
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
456 STUBBE ET AL.
The similarity between lipases and synthases may be mechanistically informativefor a number of reasons. First, lipases catalyze triacylglycerol hydrolysis, and inorganic solvents, lipases can catalyze ester bond formation. Second, the turnoverof lipases is accelerated 100-fold by their binding to a micelle or membrane (86).Lipases serve as a paradigm for interfacial catalysis. PHB synthases, as shownbelow, are bound to the surface of PHB granules (79, 87) that appear to be coveredextensively with a monolayer of lipid. Hence, the synthases may also involve in-terfacial catalysis (88, 89). The requirement for Hecameg to solublize the class Isynthase suggests that it spends part of its time associated with a hydrophobiccellular component. Third, lipases are members of the α/β hydrolase superfamilyof proteins, and atomic resolution structures are available. A threading model forboth the class I and III synthases has been generated using sequence alignmentsand the lipase structures (85, 90). The three-dimensional threading models gen-erated for the synthases have provided the basis for mutagenesis studies. Fourth,the mechanism of lipases is well understood and involves covalent catalysis usingan active site serine that is activated for nucleophilic attack by a histidine (91).This information has facilitated formulation of possible mechanisms for PHBproduction by the synthases. Just as in the case of the starch synthases (Figure3) (7), many mechanisms are possible. Several working hypotheses that fit theavailable data and involve covalent catalysis with a cysteine and a histidine arepresented in Figure 11. Mechanism A involves an active site located at the inter-face of two protein monomers (76, 80, 92, 93). According to this proposal, twocysteines, one provided by each monomer, are involved in covalent catalysis. Ineach monomer, a histidine residue activates the cysteine residue for nucleophilicattack. Activation of the hydroxyl of HB-CoA for nucleophilic attack involves anaspartate that can function as a general base catalyst. In this model (Figure 11a),the growing polymer chain remains covalently attached during polymer forma-tion and switches from one monomer to the other on addition of the subsequenthydroxybutyrate unit. Thus, there is a single polymer chain per synthase dimer.This mechanism is actually very similar to one of the mechanisms postulatedfor starch synthase (Figure 3b). An alternative mechanism (Figure 11b) involvescovalent catalysis with a single cysteine (13). In this case, the second HB-CoAbinds noncovalently to the synthase, to generate a (HB)2-CoA, which then be-comes covalently attached to the cysteine with loss of CoA. The mechanism inFigure 11a is similar to that proposed for fatty acid biosynthesis. The mecha-nism in Figure 11b is based on our understanding of several polyketide synthases[chalcone (94) and surfactin synthase (95)]. Currently, we favor the mechanism inFigure 11b, although at present a distinction between these two mechanisms is notpossible.
Initiation
The mechanism of priming or initiation is presently not understood in vivo. As withthe other synthases discussed above, three different mechanisms of priming havebeen considered. One involved a protein primer [e.g., glycogenin for glycogen
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 457
Figure 11 Two proposed mechanisms for class I and class III synthases involving two (a) orone (b) covalent intermediate(s) (13). In the mechanisms, the active site cysteine is activatedfor nucleophilic attack on HB-CoA by a histidine. In both cases, ester bond formation requiresgeneral base catalysis by an aspartate.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
458 STUBBE ET AL.
biosynthesis (96)]. The second involved short oligomers [e.g., amylose synthase(12), polyphosphate synthetase (27), and cyanophycin synthetase (38)]. The thirdinvolved self-priming (A. tumefaciens glycogen synthase) (16).
To study initiation, an informative assay is essential. The assay for PHB syn-thase, like all of the synthases, is problematic. In the PHB synthase case, the re-action is monitored discontinuously by measuring CoA release with 5,5′-dithiobis(2-nitrobenzoic acid) (80). Alternatively, 3-[3H]-HB-CoA can be used to measureradiolabeled incorporation into polymer that is extracted into chloroform. In theformer assay, CoA can be released as the result of hydrolysis as well as polymer-ization, a problem especially when studying substrate analogs (83). In the secondassay, the length of the polymer changes as a function of time and governs itsextractability. The measurement of CoA release is simpler and is most frequentlyemployed.
Efforts to find a protein primer by adding crude extracts from a �phaC W. eu-tropha strain grown under PHB production conditions to recombinant W. eutrophaPhaC were unsuccessful (J. Tian, A. J. Sinskey, and J. Stubbe, unpublished results).A series of oligomers (HB)n-CoA (n = 2, 3) and a saturated trimer (sT-CoA) weresynthesized and examined as possible primers with both the class I and III enzymes(76, 80, 93). The structure of sT-CoA is
The sT-CoA, in which a radiolabel [3H in place of OH group] provides a wayto monitor reactions, behaves identically to the (HB)3-CoA. These compounds didfunction as primers with both classes of enzymes and provided the basis for themechanistic model in Figure 11a. The importance of a protein dimer in polymerelongation was proposed on the basis of the results from the following experiments.
Low concentrations of class I enzyme incubated with HB-CoA resulted in along lag phase prior to detection of CoA release. Incubation of the enzyme with(HB)n-CoA (n = 2, 3 and sT-CoA) followed by addition of HB-CoA showeda decrease in the lag phase and an increase in the rate of the CoA release (93).Incubation of [3H]-sT-CoA with synthase resulted in the covalent labeling of theprotein and conversion of the monomeric form of the protein (64 kDa) to thedimeric form (76, 93). [3H]-sT-CoA labeled the single conserved cysteine with astoichiometry of one [3H]-label per dimer of synthase. The [3H]-label could bechased into [3H]-PHB upon addition of HB-CoA, demonstrating chemical com-petence of this intermediate. This result also substantiated the mechanism that theincoming monomer adds to the acylated enzyme, that is, polymerization occursthrough hydroxyl attack on the activated carboxylate end. The stoichiometry ofthe labeling and the sT-CoA-mediated dimerization of synthase was the basis formechanism A (Figure 11) in which the growing polymer chain is at the interfaceof the dimer. A recent study in which a heterodimer composed of one wt subunitand one Cys319A mutant subunit of the synthase were used to examine the dimer-ization model concluded, probably incorrectly, that the dimer of the synthase isactive in initiation and elongation (77). However, a dimeric synthase with a single
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 459
active site is difficult to construct on the basis of lipase structures. Lipases areall monomers and have active sites that are deeply buried and connected to thesurface through a long narrow channel (97). For the model in Figure 11b to accountfor these results, only one of the two initially identical monomers can support apolymer chain. A structure of the synthase is required to resolve these issues.
The kinetics of CoA release catalyzed by the class III synthase is also multipha-sic: Initial rapid CoA release phase is followed by a slower phase. With the class IIIenzyme and the sT-CoA, the labeled enzyme was also isolatable, but the labelingwas not stoichiometric (0.2 equivalents), owing to chemical lability of the linkageto the conserved active site cysteine. However, measurement of CoA release fromincubation of sT-CoA with enzyme gave 0.5 label per PhaEPhaC, as with the class Isynthase. Furthermore, incubation of sT-CoA with synthase in the presence of asmall amount of HB-CoA (1 to 2 equivalents per equivalent of synthase) resulted intrapping the (HB)n trimer, tetramer, and pentamer covalently bound to the cysteine,suggesting that with this artificial primer initiation proceeds through a covalentintermediate and that elongation proceeds through the same covalent intermediate(80). Thus, oligomers of (HB)n-CoA can function as primers, but the relevance ofthese primers to the mechanism of initiation in vivo has not been established. Iden-tifying the primers in vivo should be possible by extraction of the PHB from thebacteria and examination of the end groups of the polymer using NMR methods.These studies are analogous to those carried out to determine the primers in poly-isoprene synthesis. To facilitate the end-group analysis, a W. eutropha strain wasconstructed in which the wt synthase was replaced by a mutant synthase disabled inelongation by an aspartate to alanine mutation. Culturing this strain under PHB pro-duction conditions may minimize the size of polymer and facilitate the end-groupidentification and consequently the identification of the small molecule primers.
The success of (HB)n-CoA as primers suggests that it is likely that self-primingcan occur with HB-CoA. Efforts to examine the priming process using the normalsubstrate [3H]-HB-CoA and class I synthase in a substrate to enzyme ratio (S:E)of 1:1 to 1:5 were unsuccessful. Most of the protein remained unmodified, anda small amount of the protein appeared covalently linked to a large molecularweight PHB polymer (75). These results suggested that in vitro the elongation rateis much greater than the initiation rate. No intermediate chain length (HB)n-linkedproteins were detected. An experiment with the class III synthase at low HB-CoAand synthase ratios gave similar results, which implies that in vitro the synthasesare not uniformly loaded with HB-CoA (98). In class III synthase, however, theelongation rate is more closely balanced to the initiation rate.
As noted above, with one exception, all the PHB synthases studied have beenfrom recombinant sources, and hence, the gold standard of the enzyme from thehost organisms is not available. Thus, in an effort to isolate the class I synthasefrom its host, using gene replacement methods in W. eutropha, the wt synthase genewas replaced with an N-terminal His-tagged version, preserving the upstream anddownstream regions of the gene. The same construct expressed in E. coli greatlyfacilitated isolation of the recombinant synthase (83). Despite expression of syn-thase in W. eutropha, detected by Western blotting, the His-tagged synthase was
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
460 STUBBE ET AL.
nonisolatable by Ni-affinity chromatography used to purify the same constructfrom E. coli. The kinetics of CoA release from the purified W. eutropha synthase(10% to 20% pure), however, showed no lag phase and exhibited kinetics similar tothose observed with the class III synthase. These results demonstrate that a proteinfactor, a small molecule primer, or protein localization within W. eutropha alteredits properties relative to those observed with the recombinant protein (98). Thistheme repeats itself in the synthase studies addressed in this review. The intracel-lular environment plays an important and still unclear role in PHB production.
Elongation
Mutagenesis studies on the synthases supported the importance of C319 (PhaCfrom W. eutropha) and C149 (PhaC from A. vinosum), identified with the sT-CoAstudies. H508 (PhaC from W. eutropha) and His 331 (PhaC from A. vinosum)were shown to be the general base catalyst involved in generating the thiolate ofthe active site cysteine (Table 2). Mutagenesis studies suggested that D480 (PhaCfrom W. eutropha) and D302 (PhaC from A. vinosum) played important roles asgeneral base catalysts in the elongation process (99). Studies with the class IIID302A synthase mutant slowed the rate of PHB formation 1000-fold relative tothat of the wt synthase. If this reduced rate is in fact associated with reducedelongation rates, as both of our mechanistic models imply (Figure 11a,b), self-priming of the synthase with HB-CoA might be detected because the elongationand initiation rates are nearly equivalent.
In fact, results of in vitro and in vivo studies with the D302A PhaC fromA. vinosum have given the first direct evidence for covalent labeling of the C149of PhaC by HB-CoA (99). Incubation of the D302A mutant with various substrateto enzyme ratios of [14C]-HB-CoA to PhaC (1:1 to 100:1) resulted in the detectionof acylated intermediates of different lengths by SDS-PAGE, autoradiography,and Western blotting. The PhaC alone migrated more rapidly than the (HB)n-PhaC. Protease digestion of the (HB)n-PhaC and analysis by HPLC of the reactionproducts from a reaction with HB-CoA:PhaCPhaE of 1:5 resulted in a peptidecontaining the C149 and oligomers, (HB)n (n = 3 to 10). Thus, this mutation(D302A) altered the rate of elongation relative to initiation, so that self-primingof PhaC occurred in a moderately uniform fashion. The role of D302 in chainelongation has also been tested in vivo by replacing the wt class I synthase inthe W. eutropha strain with the class III D302A-PhaCPhaE, maintaining the 5′-untranslated regulatory region of the class I synthase. The cells were grown andanalyzed by EM. Under conditions in which the wt strain generated 12 granules (onaverage 0.5 µm in diameter), this mutant strain generated only very small granules(Figure 12). These results provide strong support for the role of this aspartate as ageneral base catalyst and the role of self-priming in polymer initiation (99).
GRANULE FORMATION IN VITRO During the investigation of the elongation pro-cess in vitro, Gerngross & Martin (100) reported granule formation with the class Isynthase. Theoretical modeling of this process has also been reported (89, 101).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 461
Figure 12 TEM of W. eutropha with its synthase gene replaced with D302APhaCPhaE from A. vinosum. The mutant strain was cultivated under PHB produc-tion conditions for 24 h (magnification = 18933X).
Evidence will be provided below that these granules are not models for granuleformation in vivo and that the computer modeling studies based on these in vitrostudies are unlikely to be informative about the biology of the system.
GRANULE COMPOSITION Elongation is intimately linked to granule formation,and thus to study elongation one needs to understand the composition of the gran-ules, the kinetics of granule growth, and the structure of the PHB within thegranules. Granules were discovered by Lemoigne in 1926 and have been isolatedfrom cells by sucrose and glycerol gradient centrifugation (102, 103). These meth-ods are still used today for granule purification. As noted above, especially in thecase of the rubber synthase, the method of isolation is crucial, and this is an areawhere additional studies are essential (43). Proteins associated with the granuleshave only been partially identified. Steinbuchel’s lab (62) was the first to identifythe predominant granule-bound protein, PhaP (Figure 7b), which is 3% to 5% ofthe total cell protein under conditions in which the maximum PHB is produced(62, 63, 65). A very recent study, using genomic methods, has identified threeadditional PhaP homologs [P2 (20.2 kDa), P3 (19.6 kDa), P4 (20.2 kDa)], whichshare approximately 50% sequence similarity with the first PhaP (now designatedPhaP1). Under PHB production conditions (with fructose as the carbon source andwith limited nitrogen), the concentrations of these proteins are very low relative toPhaP1 (65). Knockout studies on phaP1 resulted in a 40% reduction in the amountof PHB produced under nutrient-limiting growth conditions, and furthermore, in-stead of 12 granules, most cells contained a single large granule (Figure 13b)(62, 64). Recent kinetic studies with the wt W. eutropha strain, cultured underPHB production and PHB utilization conditions in which PhaP concentrationswere determined by quantitative Western blotting using PhaP antibodies, demon-strate a strict correlation between the rate of PHB production and the rate of PhaPproduction (Figure 14) (104).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
462 STUBBE ET AL.
Figure 13 Transmission electron microscopy of W. eutropha mutant strains culti-vated under PHB production conditions for 72 h. (a) �phaR strain in which PhaP isoverexpressed (magnification = 21,000); (b) �phaP strain (magnification = 12,000).
Using TEM to measure granule and cell sizes and quantitative Western analysisto measure the amounts of PhaP, we have calculated that PhaP covers ∼30% ofthe granule surface when cells were grown in nutrient-rich medium (tryptic soybroth) (105). PhaP covers a similar amount of the granule surface when cellsare grown in nutrient-limited conditions in which PHB production is maximized(Figure 14) (104). A similar calculation, based on quantitative Western analyses,indicated that 1.6% and 1.3% of granule surface is covered by PhaC and PhaR,respectively, under nutrient-rich conditions (at 4 h, the time point with maximum
Figure 14 Relationship between PhaP accumulation (right axis) and PHB production(left axis) under nitrogen-limited conditions. The symbols used are filled circle, % celldry weight of PHB; and open diamond, amount of PhaP. The localization of PhaP onthe surface of granules has been established by immunogold-labeling experiments withPhaP antibodies (Figure 7b) (62).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 463
PHB production). Under PHB production conditions (at 24 h), PhaC and PhaReach covered ∼0.4% of the surface of granules. (104, 105). Thus, the surface ofthe granule is predominantly covered with PhaP and small amounts of PhaC, PhaR.EM studies of the granules from different organisms in the 1960s revealed a darkstaining coat on the exterior of the granule, and its thickness varied from 20 to200 A and was interpreted as being associated with a lipid monolayer (88). Thisinterpretation is supported by a 1.8-fold increase in lipid content (without changein lipid composition) in W. eutropha in the absence of PHB relative to W. eutrophawith maximum PHB produced (106). Thus, it is likely that PHB granules containboth PhaP and lipids on their surface.
The structure of PHB within the granule has also been investigated. AlthoughPHB isolated by most methods is in the crystalline form, solid-state NMR methodssuggest PHB is present in the amorphous form in vivo. This implies that the uniqueenvironment of the granule prevents polymer crystallization (107, 108). Insight intothe fine structure of the granules has started to be gained by using atomic forcemicroscopy on granules directly after disruption of the cell wall of W. eutropha(109). The investigators report a 40 A boundary layer on the granule, consistentwith a lipid monolayer. They also reported globular structures about 350 A indiameter with a central pore (150 A). The organized network of structures on thesurface was proposed to be associated with protein. Thus, we suggest that thesefactors might be associated with a not yet identified scaffold protein(s). Atomicforce microscopy studies have also been carried out on isolated granules treatedwith increasing concentrations of acetone. Under these conditions, fine structuresare apparent and are presumably associated with PHB. However, the 350 A/150 Afeatures are absent. The role of solvent in the production of these structures remainsto be studied (110). Imaging of granules without isolation is the desired approachgiven the potential lability of the interesting features.
BIOGENESIS OF THE GRANULE Two models for granule formation have been pro-posed (13). The first one is the micelle model, which suggests that the extendedPHB chains covalently bound to the synthase aggregate into a micelle. The secondmodel is the budding model,which suggests that the hydrophobic synthase bindsto the inner face of the plasma membrane, and the PHB granules bud from thismembrane, leading to granules with their surface covered by a lipid monolayer. Inboth cases, the PhaC and PhaP are on the exterior of the granule, which is consis-tent with experimental results. Recent kinetic EM studies suggested a third model(111). As revealed in Figure 15c, at early times when W. eutropha cells are placedin a PHB production medium, very small, discrete granules appear to be localizedto a mediation element located in the center of the bacterium. Over time, the gran-ules increase in size and appear to remain localized to this element (Figure 15d,e).By 24 h (Figure 15f ), this mediation element is no longer observed because ofeither masking by the large amounts of PHB or its degradation. The granule lo-calization and putative mediation element are also apparent under nutrient-richgrowth conditions. The basis for this localization is presently unknown. If it is not
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
464 STUBBE ET AL.
Figure 15 Kinetics of PHB granule formation in W. eutropha studied by TEM (mag-nification = 8,000 X). W. eutropha was first cultivated in a nutrient-rich medium (aand b) (105). (a) At 4 h, transient PHB production is maximum. (b) By 24 h, the sizeof the granules has decreased. This culture was then used to inoculate PHB productionmedium. The following time points after inoculation are presented: (c) 2.5 h, (d ) 5 h,(e) 9 h, ( f ) 24 h, (g) 73 h. Cells were then transferred to PHB utilization medium andexamined at 48 h (111).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 465
an artifact of TEM sample preparation, it will be central to understanding granuleformation and must be incorporated into any biogenesis model.
ROLE OF PhaP Given its properties, several roles have been postulated for PhaP.One function of PhaP is to control the number of granules. The results in Figure 13make this apparent. PhaP overexpression dramatically increases the number ofgranules (Figure 13a) whereas deletion of phaP results in the production of asingle large granule (Figure 13b) (62, 66). Under PHB production conditions, thegranules increase in size from 0 to 24 h (Figure 15), and from 5 to 20 h, the PhaPincrease correlates with the amount of PHB produced (Figure 14). The amountsof PHB and PhaP remain relatively constant from 24 h to 72 h (Figure 14). Theseobservations suggest a second function of PhaP: to control the surface area tovolume ratio of the granule. This control is essential for generation of a granulein which the PHB remains in an amorphous state and is consequently accessibleto degradation by the depolymerases in vivo. Third, the abundance of PhaP asso-ciated with granules has allowed the suggestion in Bacillus megaterium that thisprotein functions as a nitrogen storage source (112). A fourth role of PhaP is thatit provides protection to the host cell by contributing to coverage of the hydropho-bic surface of the polymer, preventing protein misfolding on the hydrophobicgranule (113).
Termination
One of the striking aspects of PHB polymers generated in vivo is their largemolecular weights and low polydispersity. These results are reminiscent of thoseobserved with polyisoprenes. Thus, the mechanism of the termination process andthe role of the proteins (PhaC, PhaP, and potentially PhaR) versus the role of theinherent physical properties of the polymer remain to be established in all thepolymers examined in this review. As noted in Figure 11, the growing polymer iscovalently attached to an active site cysteine. Thus, several questions can be posed.Does chain transfer occur by hydrolysis so that a single PhaC can be used more thanonce to make polymers within a granule? If this happens, what is the mechanismof chain transfer and is it governed by the conserved residues of the synthases(S and H in Table 2) whose functions have thus far remained a mystery. Second, isthere a protein involved in the termination process? In polyketide synthases, at theend of each biosynthetic pathway, there is a thioesterase that releases the naturalproduct. Could a similar thioesterase release PHB? The thioesterases are also α/β
hydrolase superfamily members and have structures and mechanisms similar tolipases. Third, depolymerases are present during the synthesis of the PHB (104).Could these depolymerases play a role in termination and perhaps reinitiation ofpolymer synthesis in a fashion similar to the postulated role for isoamylase instarch biosynthesis (9)?
Doi’s group (114, 115) proposed in 1992, on the basis of measurements availableat the time, that there were 18,000 molecules of PhaC per W. eutropha cell. [Given
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
466 STUBBE ET AL.
our recently calculated cell volume, this number of PhaC molecules would give aconcentration of 18 µM if PhaC is soluble and not membrane bound (111).] Theyargued, using this number and the rate of PHB production, that reinitiation oc-curs during granule formation under PHB production conditions. This conclusionwas confirmed by our recent more quantitative analysis, although our calculationssuggest that there are ∼250 PhaCs per cell (104, 105). This information alongwith knowledge of the PHB MW (∼106 Da) and PHB density (108, 116) alloweda more quantitative assessment of PhaC reinitiation events. There are 200–300 PHB molecules per PhaC molecule in an average cell (104). Under nutrient-rich conditions at 4 h (with maximum PHB production), this number is ∼60 PHBmolecules per PhaC molecule (105). Thus, it is now established that a chain trans-fer mechanism is occurring and that PhaC can be reused many times. The amountof PhaP has been determined using the same method, and the calculations indi-cated that there is ∼1 PhaP molecule per PHB molecule under both nutrient-richand nitrogen-limited conditions (104, 105). This unexpected result is very likelyto have important implications in a functional assignment for PhaP. Being able tomeasure the MW of the PHBs and to express a variety of PhaC mutants that differfrom the wt strain by a single mutation in PhaC under different growth conditionswill help elucidate the mechanism of chain transfer and the roles of PhaC and PhaPin this process.
Several additional experiments support the chain transfer mechanism and estab-lish that in vitro results differ from in vivo results. Studies with the phaAphaBphaCgenes on a plasmid behind an isopropyl-β-d-thiogalactopyranoside inducible pro-moter in E. coli demonstrated that the size of the PHB polymer was related to theamount of PhaC (117): the smaller the amount of PhaC, the larger the polymer.At low PhaC concentrations in E. coli, the MW of PHB reaches 3 × 106 Da,3 times that of PHB produced in W. eutropha. These results differ dramaticallyfrom studies in W. eutropha under nutrient-rich and PHB production conditions inwhich the concentrations of PhaC changed fourfold, but the size of the polymerremained unchanged [∼1 × 106 Da] (104, 105).
Insight into chain termination comes from in vivo experiments from Maddenet al. (118). They grew W. eutropha under PHB production conditions in the pres-ence of metabolites that can potentially function as chain transfer agents (glycerol,[2H]-propanol, ethylene glycol). After the resulting PHB polymers were isolated,the ends of the chains were examined by [2H] or 31P NMR methods. When W. eu-tropha was grown on glycerol, the polymer MW was substantially lower than PHBproduced when W. eutropha cells were grown on glucose. The end-group analysisrevealed glycerol, attached through its 1◦ and 2◦ alcohol, at the C terminus. The31P NMR method involved derivatization of the alcohol at one end and the car-boxylate at the other end of PHB polymer with 2-chloro-4,4,5,5,-tetramethyl-1,3,2dioxaphospholane. This derivatization removes the problems associated with thepreponderance of protons in the polymer, while retaining the sensitivity of protons(119). In one set of experiments, C termini of PHB, isolated from exponentiallygrowing W. eutropha cells, were detected without a corresponding modified alco-hol end. These results imply that there is a primer that remains to be identified
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 467
in vivo or, alternatively, that the derivatization method with the phosphorousreagent was not quantitative.
Finally, recent in vitro studies, using a hydroxybutyryl N-acetylcysteamine(CH3CHOHCH2COSCH2CH2NHCOCH3) as a substrate analog, have establishedthat chain termination occurs very frequently with this analog (A.G. Lawrence,J. Stubbe, and A.J. Sinskey, unpublished results). The MW of PHB is ∼75,000 Da,and it exhibits narrow polydispersity. 1H NMR studies revealed that C termini ofPHB contains N-acetylcysteamine and that there are no free carboxylates at theend. These studies indicate that chain termination and reinitiation occur in vitro.The mechanism of termination and reinitiation in vivo still remains a mystery, butit may make use of the conserved serine, histidine, and aspartate residues in allsynthases (Table 2).
Utilization
As in the case of soluble starch synthase, glycogen synthase, polyphosphate syn-thetases, and cyanophycin synthetases, the organisms that generate the polymerscan also degrade the polymers in time of need. Bacteria make both intracellularand extracellular PHA depolymerases. The extracellular enzymes have been ex-amined extensively because they are responsible for the biodegradability of thecrystalline PHB released from dead PHB-producing cells. Bacteria with these ex-tracellular depolymerases use the hydroxybutyrate generated as a building blockand the energy released for biosynthesis. An excellent review on the extracellulardepolymerase has recently appeared (58). Because the focus of this review is onPHB homeostasis, only the intracellular depolymerases will be discussed.
The first step in depolymerase identification requires a good enzymatic assay.The assays thus far reported for the intracellular depolymerase(s) are inadequatebecause they monitor HB, which is many steps removed from the first cleavageevents of polymer breakdown (70, 73). HB production is monitored using an HBdehydrogenase. The assays also typically contain an extracellular oligomer hydro-lase, an enzyme that converts (HB)n (n = 2 to 5) to the corresponding monomers,which can then be oxidized by the HB dehydrogenase. The substrates for depoly-merase assays cannot be natural granules because the intracellular depolymerasesare granule bound and copurify with granules. This copurification gives rise to highbackground activity. Therefore, artificial granules, made by solublizing crystallinePHB with detergents, are used (108). Alternatively, (HB)n (n = 2–5) are used assubstrate.
The identification of the intracellular depolymerases and their role in PHBhomeostasis are just beginning to be unraveled, and the story is complex. A numberof putative depolymerases have been identified and their functions analyzed bothin vitro and in vivo. Two depolymerases have been partially characterized in vitro:PhaZ1a (45 kDa) (70) and an oligomer hydrolase (PhaZ2, 78 kDa) (72, 73). Thespecific activity of both of these proteins is 0.1 µmol/min/mg. This number is verylow compared to in vivo rates of PHB degradation (see below). These in vitrostudies raise two concerns. The first is that the depolymerases are recombinant
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
468 STUBBE ET AL.
and isolated in soluble form. The biological studies thus far, however, suggestthat most of the depolymerases are granule bound. It is likely, as in the case oflipases, that interfacial catalysis will greatly increase the turnover numbers of theseenzymes. Second, the artificial granules used as substrates lack the proteins andlipid structure that may be essential for the depolymerases to function and couldthus also affect turnover rates.
The alternative substrates (HB)n (n = 2–5) used in these assays give very highrates for HB production: 50–350 µmol/min/mg. However, inside the cell, (HB)nshave not yet been identified, and therefore, the relevance of this assay to the biologyneeds to be established.
Recently, two additional genes homologous to PhaZ1a have been reported:PhaZ2 (renamed PhaZ1b) and PhaZ3 (renamed PhaZ1c) (71). The function forthese proteins has been assessed in isogenic strains in which each individual de-polymerase or multiple depolymerases have been deleted (71, 73). In addition, thecompleted genome sequences of Ralstonia solanacearum, Ralstonia metallidu-rans, and the partially completed sequence of W. eutropha now suggest that thereare five sequence homologous PhaZ1s (a, b, c, d, e) all with a MW of 45 kDa (65).Thus, at present, there are six candidate depolymerases whose roles remain to beelucidated. New ways of examining the phenotypes of deletion strains, isolatingof granules from deletion strains, and directly measuring H+ release from PHBgranule degradation as direct indicators of ester bond hydrolysis are crucial forsorting out their function.
Several recent studies have provided insights into the functions of some of theseproteins by comparison of the amount and the rate of PHB degradation in wt anddeletion strains grown in PHB utilization medium after being first grown in PHBproduction medium (71). Deletion of phaZ1a resulted in partial PHB degradation,whereas deletion of phaZ1b showed no difference in degradation relative to the wtstrain. The evidence suggesting that PhaZ1b plays a role in PHB degradation comesfrom studies using a double knockout (�phaZ1a�phaZ1b) strain, which exhibitedno degradation of PHB. Deletion of phaZ1c has, thus far, produced no detectablephenotype in vivo under PHB production or utilization conditions (71). Thus, inPHB utilization medium, both PhaZ1a and PhaZ1b act like depolymerases.
Under nutrient-rich conditions, the results for PhaZ1a and PhaZ1b differ. The�phaZ1a strain generates PHB at levels similar to that observed in the wt strain butretains higher amounts of PHB during late log and stationary phases (71). �phaZ1bstrain was indistinguishable from wt in its PHB levels (71). Recent studies by theSaito’s group (73), using �phaZ1a �phaZ2 strain, gave different results. Undernutrient-rich conditions, PHB production in a wt strain was maximum at 15%(dry cell weight) and increased to 20% in the mutant strain (73). The resultssuggested that synthases and depolymerases are active simultaneously. EM studiesunder nutrient-rich growth conditions substantiate the PHB degradation by cells.From 4 h (maximum PHB production) to 24 h, the size of the granules decreased(Figure 15a,b). These EM results agree with Saito’s conclusions that both thesynthases and depolymerases are active simultaneously in nutrient-rich medium.
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 469
Recently, the function of many of these proteins has been examined using tran-scription profiling (monitoring mRNA levels) and proteomics (monitoring proteinlevels) as a function of time under PHB production and PHB utilization conditions(104, 105). The results under PHB production conditions allow calculation of therate constant for PHB disappearance. This number measured in several experi-ments varied from 20 U to 40 U/mg, substantially faster than the turnover numbersmeasured in vitro (0.1 U/mg) (73). Thus, the present in vitro assays need to berefined as noted above. Western blotting with antibodies to PhaZ1a, PhaZ1b, andPhaZ2 have been used to interrogate the presence of these proteins under PHBproduction and utilization conditions. PhaZ1a and PhaZ2 are present during theentire time course for synthesis and utilization. However, PhaZ1b grows in aboutmidway during the PHB production period and then disappears rapidly within thefirst few hours in PHB utilization medium (104). In the �phaZ1a�phaZ1b strain,as noted above, the PHB levels are virtually identical to those in the wt strainat maximum PHB production (71). The former results suggest that PhaZ1b maybe involved in remodeling or termination of PHB, whereas the latter results sug-gest that PhaZ2 does not degrade PHB but probably degrades oligomers producedby PhaZ1a. Similar Western blotting experiments have been carried out undernutrient-rich conditions. Both PhaZ1a and PhaZ2 are present during productionand utilization. However, in contrast to the results in PHB production conditions,no PhaZ1b was detected in the nutrient-rich conditions (73, 105).
The presence of depolymerases and the synthase at the same time during PHBproduction and PHB utilization raises the issue of whether they are active simul-taneously, and if they are, why? Under nongrowth conditions, studies by Doi et al.(120) provided convincing evidence that the synthases and depolymerases wereworking simultaneously in W. eutropha. Conversely, studies by Haywood et al.suggested that PHB does not turnover in the steady state and that net degradationis kept to a minimum (121). In many of the systems described above, synthases anddepolymerases are present simutaneously. Major unsolved issues are assignment offunctions to these proteins and regulation of their activities to prevent futile cycling.
Regulation
The most recent review of regulation is that of Kessler & Witholt (122). As with theother synthases described in this review, it is clear that most studies have focusedon polymer production and utilization under nutrient-limited conditions. However,PHB production occurs transiently under nutrient-rich conditions as well. Theinterplay between transcriptional and metabolic regulation in PHB production hasbeen the focus of most studies.
TRANSIENT PHB PRODUCTION A few examples of transient PHB production arepresented. Azotobacter vinelandii is an obligate aerobe that fixes nitrogen. Thisorganism can also undergo differentiation to form desiccation-resistant cysts thatproduce both alginate (an exopolysaccharide) and PHB (123). In this organism,
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.
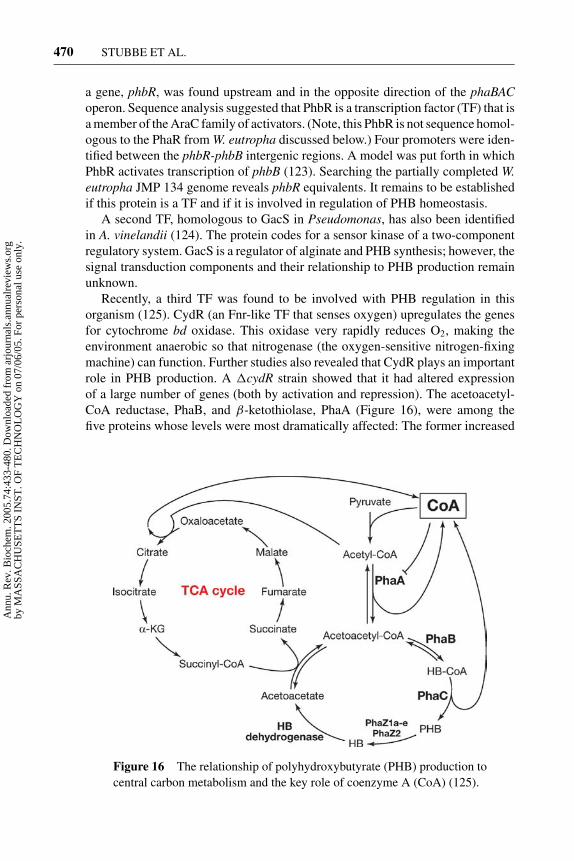
10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
470 STUBBE ET AL.
a gene, phbR, was found upstream and in the opposite direction of the phaBACoperon. Sequence analysis suggested that PhbR is a transcription factor (TF) that isa member of the AraC family of activators. (Note, this PhbR is not sequence homol-ogous to the PhaR from W. eutropha discussed below.) Four promoters were iden-tified between the phbR-phbB intergenic regions. A model was put forth in whichPhbR activates transcription of phbB (123). Searching the partially completed W.eutropha JMP 134 genome reveals phbR equivalents. It remains to be establishedif this protein is a TF and if it is involved in regulation of PHB homeostasis.
A second TF, homologous to GacS in Pseudomonas, has also been identifiedin A. vinelandii (124). The protein codes for a sensor kinase of a two-componentregulatory system. GacS is a regulator of alginate and PHB synthesis; however, thesignal transduction components and their relationship to PHB production remainunknown.
Recently, a third TF was found to be involved with PHB regulation in thisorganism (125). CydR (an Fnr-like TF that senses oxygen) upregulates the genesfor cytochrome bd oxidase. This oxidase very rapidly reduces O2, making theenvironment anaerobic so that nitrogenase (the oxygen-sensitive nitrogen-fixingmachine) can function. Further studies also revealed that CydR plays an importantrole in PHB production. A �cydR strain showed that it had altered expressionof a large number of genes (both by activation and repression). The acetoacetyl-CoA reductase, PhaB, and β-ketothiolase, PhaA (Figure 16), were among thefive proteins whose levels were most dramatically affected: The former increased
Figure 16 The relationship of polyhydroxybutyrate (PHB) production tocentral carbon metabolism and the key role of coenzyme A (CoA) (125).
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 471
fivefold and the latter fourfold. An integrated overview of the regulation is stillmissing.
Transient PHB production is also observed in the Acinetobacter spp., an organ-ism that is believed to be one of the major organisms responsible for enhancedbiological removal of phosphate from wastewater treated in alternating anaer-obic/aerobic activated sludge systems (126, 127). Under anaerobic conditions,breakdown of polyphosphate and synthesis of PHB occur, whereas during aero-bic conditions, breakdown of PHB and synthesis of polyphosphate ensue. Recentevidence suggests that phosphate starvation can positively effect the transcriptionof these phaAphaBphaCAC genes (128). In E. coli, PhoB activates transcriptionof the phosphate regulon by binding to an 18-bp consensus pho box sequencefound 10 bp upstream of the -10 region of promoters in the pho regulon. Twopho box consensus sequences have been found in the PHA biosynthetic genelocus (128).
Rhizobium etli uses PHB synthesis as an important component of their meta-bolism both in symbiosis and in free life. Studies with a transposon inserted intophaC (129, 130), in addition to being unable to make PHB, showed diminishedoxidative capacity, inability to grow on carbon sources (such as glucose or pyru-vate), and elevated levels of NADH/NADPH. By random Tn5 mutagenesis of the�phaC strains, they found that the interruption of aniA, a TF, restored the abilityof the cells to grow on both glucose and pyruvate. AniA is located adjacent to anddivergently transcribed from phaAphaB. Tn5 mutagenesis of aniA resulted in PHBproduction yield of only 40% relative to the wt strain and a many fold increase inexopolysaccharide biosynthesis. A proteomic analysis of this mutant relative to thewt strain showed failure to produce 759 proteins including PhaB (130, 131). AniAinfluences carbon flow and global protein expression. Genes encoding AniA-likeproteins have been found thus far only in the proteobacterial branch of eubacteria.A homologue designated PhaR has been found in W. eutropha (β subdivision) andin A. vinosum (γ subdivision).
STRESS-RELATED PHB PRODUCTION Regulation of PHB production under nitro-gen limiting conditions has been best studied to date. PHB production, however,also occurs under oxygen, phosphate, and Mg2+ limitations. Thus far, regulation atthe transcriptional level and the enzymatic level has been documented. However,the studies raise more questions than they answer.
TRANSCRIPTIONAL CONTROL The PhaR homologs from Paracoccus denitrificansand W. eutropha have been recently investigated both in vitro and in vivo (63,66–68, 132). The model of the mechanism by which PhaR is involved in PHBhomeostasis is shown in Figure 17. PhaR is proposed to function as a repressorof phaP transcription and a repressor of its own transcription under conditionsin which PHB does not accumulate. Once the accumulation of PHB is initiated,PhaR is proposed to dissociate from the promoter DNA and bind to oligomeric orpolymeric forms of (HB)n (68). PhaP transcription and translation is then initiated
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
472 STUBBE ET AL.
Figure 17 Regulation of PhaP expression by PhaR. PhaR is a putative repressor ofphaP transcription and derepression occurs only in the presence of active PhaC (13).
as is the transcription and translation of PhaR. Under PHB production conditions,the granules generated are covered with PhaP (30%) and small amounts of PhaR(0.4%) (104). Polymer degradation results in release of PhaR that can then represstranscription of phaP. The role of PhaR has been investigated in vitro and in vivoin P. denitrificans and W. eutropha.
In P. denitrificans, PhaR is a 22-kDa protein that has been shown to bind tothe upstream regions of both phaP and phaR by DNAse I footprinting in vitro(68). The consensus binding sequences, located at two sites, have been shownto be TAAAATTTTTCTGCACCGCAGCAAGAAAAC located immediately up-stream of -35 and TGCAATGCTGCGGTGCAGAAAGTA located between -10and -35 from the phaP transcriptional starting site. These two sites are -60 to -115bps upstream from the phaP translational start site. The PhaR-binding site on thephaR promoter (TCATGCTGCAAATGCACTGCCGG) is located immediatelydownstream of the -10 site from the transcriptional start and -10 to -30 bps fromthe translational start. The conserved nucleotides are shown in bold font. On thephaP promoter, the palindromic sequences are separated by three nucleotides,whereas on phaR promoter the palindromic sequence is separated by four nu-cleotides. It remains to be determined whether this difference is responsible forthe decreased affinity of PhaR for the phaR promoter relative to the phaP promoter.In vitro gel mobility shift assays were used to demonstrate the binding of PhaRto the DNA sequences defined above, and to monitor the effects of (HB)n on themobility shift (68). Gel shifts with PhaR were observed, although no Kds were re-ported. The concentration of PhaR used to observe the shift was 350 nM. (HB)30,crystalline PHB, and relatively high concentrations of artificial PHB granules(prepared using detergents) all resulted in the disruption of the observed mobilityshift. Addition of natural granules had no effect. In addition, a number of metabo-lites were also investigated for their ability to disrupt the DNA-protein complexformation. CoA, acetyl-CoA, HB-CoA, NAD+, NADP+, NADH, NADPH, acetyl
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 473
phosphate, PEP, citrate, and polyphosphate were unable to alter the observed mobil-ity shift. Thus, the mechanism of derepression is unusual and is suggested to involveoligomeric HBs.
Similar in vitro studies have recently been reported on PhaR isolated from W.eutropha (63). DNAse I footprinting studies suggested that PhaR binds upstream ofphaP at two sites and upstream of phaR at one site. Mobility shifts of DNA with theputative DNA-binding sites were also carried out using 5 µM PhaR. Although thestarting DNA disappeared, bands with slower migrating properties (DNA-proteincomplexes) were not detected. To test the role of PHB in derepression of PhaR,artificial granules were prepared and incubated with PhaR, and the granules werethen isolated. SDS-PAGE of the proteins from the isolated granule detected PhaR.More recent studies have been able to measure a Kd of 138 nM for PhaR binding tothe phaP promoter, using gel shift methods under physiological conditions (P. Liu,A.G. Lawrence, A.J. Sinskey, J. Stubbe, unpublished results). DNase I footprintingexperiments and sequence analysis revealed at least two binding sites for PhaR up-stream of phaP. The first binding site (AATTGTTGCGGCGCACCAAATAAG)is located immediately downstream of the -10 site from the phaP transcriptionalstart. The second site (GTTTGTGCATTGCACAAAATCCAC) is located imme-diately upstream to -35 site of the phaP transcriptional start. Both PhaR-bindingsequences within the phaP promoter are palindromic. No PhaR binding to a DNAfragment generated based on the sequence upstream of the phaR gene was de-tected. A Kd of 138 nM is relatively high for a TF binding to DNA and raises anumber of issues. First, what is the concentration of PhaR inside the cell and is itsufficiently high to function as a repressor protein? Second, is PhaR posttransla-tionally modified, and could this modification affect the affinity for DNA? Third,are there additional protein factors that can act in conjunction with PhaR? Theseissues remain unresolved.
The strongest evidence that PhaR is a TF that regulates phaP transcription isbased on in vivo studies with �phaR, �phaR�phaC, �phaC, and wt strains ofW. eutropha (66). As noted above, the wt strain accumulates PHB in a fashionstrictly coordinated with PhaP accumulation (Figure 14). In contrast, the phaRdeletion strain accumulated PhaP at levels three times higher than wt strain underPHB production conditions at 24 h. One interpretation of these results is that PhaRis a repressor of PhaP expression and that PhaR specifically prevents accumulationof PhaP in cells that are not producing PHB. Transfer of W. eutropha phaR, phaP,and phaCAB genes into E. coli was shown to be sufficient to reconstitute thisPhaR/PhaP regulatory system (66).
In W. eutropha, studies with �phaR and �phaR�phaP strains showed 25%and 50% reductions in PHB accumulation (% cell dry weight) relative to the wtstrain (66). Thus, it appears that PhaR has a PhaP-independent pathway as wellas a PhaP-dependent pathway to affect PHB production. The sequence similaritybetween PhaR and AniA and the proposed function of AniA suggest that PhaRmay also be a global regulator that responds to either carbon flux or to nitrogenlimitation, or both. Further studies are required. The in vitro and in vivo studies
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
474 STUBBE ET AL.
suggest that PhaR is in fact a TF. However, many studies are required to elucidateits detailed role under different growth conditions and in the signal transductionnetwork in PHB homeostasis regulation.
A different type of TF, PhaF, has been identified in bacteria containing theclass II synthases that make PHAs (R = 3 to 14, Figure 8) (69, 134). The Cterminus of PhaF is homologous to the H1 protein family of TFs [a histone-like family with amino acid repeats (AAKP)] typified by AlgP. PhaF has beenpartially characterized in P. oleovorans (69). PhaF and phaI are genes downstreamfrom the operon containing phaC1phaZphaC2 and phaD (coding for a synthase,depolymerase, a second synthase, and a phasin) involved in PHA production (134).The N terminus of PhaF is homologous to PhaI, and both of these proteins havebeen localized to the surface of PHA granules. The N terminus is thus proposedto be a PHA-binding domain. Analysis of �phaF strains resulted in increasedtranscription of the phaC1 and phaI genes. Thus, the model is that PhaF is anegative regulator. The binding site for PhaF has thus far not been identified, noris there any experimental evidence that PhaF binds to DNA. The carbon sourceused for growth of P. oleovorans also regulates levels of the phaC1 transcript.In the presence of octanoate, PhaC1 is expressed, whereas in the presence ofglucose or citrate, no PhaC1 transcript is observed. The phaF transcript is presentunder all of these growth conditions. The model is that when growth occurs onglucose or citrate, no PHA is produced, and thus PhaF functions as a repressor.This regulation is distinct from that described for W. eutropha. In both cases,understanding regulation at the transcription level is in its infancy.
ENZYMATIC CONTROL W. eutropha has been the organism of choice for investi-gation of PHB homeostasis. Regulation of carbon flux into PHB production occursat multiple levels, and understanding this regulation is essential for understandingthe physiological functions of PHB and for production of new plastics by bioengi-neering. The role of acetyl-CoA and its relationship to central metabolism andPHB homeostasis is shown in Figure 16. In addition to transcriptional control,metabolite control of enzymatic activities has also been studied.
Regulation of PHB synthesis at the enzymatic level is well documented. FreeCoA plays a central role in polymer synthesis. Studies in vitro demonstrate thatCoA inhibits the thiolase (PhaA) with complex kinetics (135). Some investiga-tors have reported that CoA inhibits the synthase (PhaC) in vitro. In our hands,inhibition was not observed. Polymer synthesis in vivo has also been shown to bestimulated by changes in the NAD(P)H/NAD+ ratios (136). Consistent with thismodel is the inhibition of citrate synthase by NADH/NADPH (137). Increasedlevels of NADH would decrease the levels of CoA and alleviate CoA inhibition ofPhaA, thus increasing the flux of acetyl-CoA toward PHB (Figure 16). Additionalsupport for this model comes from experiments with an W. eutropha strain con-taining an isocitrate dehydrogenase mutation. This mutation results in low TCAcycle activity and, consequently, an increased rate of PHB production (122). Ex-periments in W. eutropha have been carried out in which the levels of enzymatic
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 475
activity of PhaA and PhaB have been altered using a plasmid construct. In strainsin which these activities have been increased, the rate, but not the amount, of PHBproduction is also increased (138). Altering the levels of PhaC had no effect on therates of PHB production. Thus, at the enzyme level, the redox status of the cell andthe concentrations of CoA play important regulatory roles. With current analyticaltools available, it would be of interest to measure the kinetics and the concen-trations of metabolites [CoA, NAD(P)H, acetoacetyl-CoA, HB-CoA] during thePHB production phase.
Our understanding of regulation even in the best-studied system, W. eutropha,is still rudimentary. The mechanism controlling the rate of substrate delivery andthe rate of PhaP production can both influence the rate and amounts of PHBproduced. The extent to which metabolites control enzymatic activities and theimportance of this level of control relative to transcriptional control remain to beelucidated. The complexity of the regulation at the transcriptional level is apparentand probably involves integration of two-component signaling systems and a vari-ety of repressors and activators. Also, the mechanism to minimize futile cycling ofPHB production/utilization, owing to the simultaneous presence of synthase anddepolymerases, is still a mystery.
SUMMARY
PHA formation is one of the simplest homopolymerization reactions. The synthasemechanism involves covalent catalysis with an essential cysteine. The proteincan be primed with (HB)2 or 3-CoA in vitro. (HB)2 or 3 can be generated by PHBdepolymerases. However, no specific enzyme that can generate the CoA ester ofthese species has yet been identified, which suggests that this priming mechanismis unlikely in vivo. Thus, an in vivo initiation mechanism is likely to involve self-priming with HB-CoA. Identification of an aspartate that plays an essential role inactivating the hydroxyl group for ester bond formation and the ability to replace thewt synthase with any mutant in W. eutropha offer a way to interrogate the primingprocess in vivo. The mechanism of granule formation requires a phasin proteinwhose rate of production is correlated with rate of PHB production. The ratio ofPhaPs to PHB polymer chains is approximately 1:1. The surface of the granuleappears to be covered with a lipid monolayer and granule-associated protein. PhaPcovers as much as 30% of the granule surface. PhaC, PhaZ1a, PhaZ1b, and PhaRwere found to be associated with the granule as well. However, they contributelittle to the surface coverage. There are many fewer PhaCs than PHB molecules percell, and therefore a mechanism for PHB chain reinitiation is required. Recent EMstudies monitoring the kinetics of PHB production in PHB production conditionsprovided the first insight into a mechanism for granule formation. The EM studiesrevealed a time-dependent increase in granule size at the early phases of granuleformation, and the small granules appear to be associated with some mediationelement within the cell. The structure is also apparent when PHB is producedtransiently under nutrient-rich conditions. Identification of the mediation element
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
476 STUBBE ET AL.
may provide insight into this process in vivo. The availability of deletion strainsof many of the genes involved in PHB homeostasis and the ability to monitorPHB production by various microscopies suggest that many of the secrets of PHBproduction and utilization will soon be unveiled.
GENERAL CONCLUSIONS AND CHALLENGES
1. Nature has used simple building blocks (acetate, amino acids, sugars, andphosphates) as components of storage polymers in times of stress. Thesepolymers are also generated transiently.
2. Generation of polymers as insoluble inclusions from soluble substrates avoidsproblems with changes in osmolarity within the cell but generates challengesfor their production and reutilization.
3. All synthases (polymerases) are very simple proteins and carry out dehydra-tion reactions. Mechanisms of polymerization (initiation, elongation, andtermination), however, have resisted elucidation, owing to difficulties ofassaying a changing product with time and of the recapitulation of phasetransitions in vitro. Studies in vitro often do not reflect activities in vivo anddemonstrate that location of the polymerase inside the cell and, perhaps,additional factors are key to allowing these proteins to make polymers ofvery high molecular weight.
4. No structures of any of these polymerases are available.
5. Isolation of granules without disruption of their components has proven chal-lenging. Understanding their composition is essential to understand theirbiogenesis. Whether granules self-assemble on the basis of the physicalproperties of the polymers or whether their formation is carefully orches-trated with scaffold proteins and/or kinetic control of substrate and proteinproduction is not yet understood.
6. The mechanism of the control of the MW and polydispersity of the polymersmade in vivo remains a challenging question.
7. Despite our understanding of primary metabolism, it is amazing how little weunderstand about metabolic and transcriptional regulation of homopolymer-ization processes in bacteria. Polymerases and depolymerases are presentsimultaneously during polymer production, and the regulatory mechanismsto prevent futile cycling are not understood. Perhaps functions of these pro-teins have been misassigned given the difficulty of designing informativeassays.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM49171 to JoAnne Stubbe and AnthonyJ. Sinskey, and by NIH grant 5T32GM08334 to Jiamin Tian. This project has been
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 477
a collaboration between the Stubbe and Sinskey groups, and we thank all of ourcoworkers who have worked on this project over the years.
The Annual Review of Biochemistry is online athttp://biochem.annualreviews.org
LITERATURE CITED
1. Madison LL, Huisman GW. 1999. Micro-biol. Mol. Biol. Rev. 63:21–53
2. Dawes EA, Senior PJ. 1973. Adv. Micro-bial Physiol. 10:135–267
3. Steinbuchel A. 2003. Curr. Opin. Micro-biol. 6:261–70
4. Smith AM. 2001. Biomacromolecules 2:335–41
5. Ball SG, Morell MK. 2003. Annu. Rev.Plant Physiol. Plant Mol. Biol. 54:207–33
6. Blennow A, Nielsen TH, Baunsgaard L,Mikkelsen R, Engelsen SB. 2002. TrendsPlant Sci. 7:445–50
7. Mukerjea R, Yu L, Robyt JF. 2002. Car-bohydr. Res. 337:1015–22
8. Hirose T, Terao T. 2004. Planta 220:9–169. Bustos R, Fahy B, Hylton CM, Seale R,
Nebane NM, et al. 2004. Proc. Natl. Acad.Sci. USA 101:2215–20
10. Cid E, Geremia RA, Guinovart JJ, FerrerJC. 2002. FEBS Lett. 528:5–11
11. Coutinho PM, Deleury E, Davies GJ, Hen-rissat B. 2003. J. Mol. Biol. 328:307–17
12. Zeeman SC, Smith SM, Smith AM. 2002.Plant Physiol. 128:1069–76
13. Stubbe J, Tian J. 2003. Nat. Prod. Rep.20:445–57
14. Preiss J, Romeo T. 1989. Adv. Microbial.Physiol. 30:183–238
15. Alonso MD, Lomako J, Lomako WM,Whelan WJ. 1995. FASEB J. 9:1126–37
16. Ugalde JE, Parodi AJ, Ugalde RA. 2003.Proc. Natl. Acad. Sci. USA 100:10659–63
17. Yep A, Ballicora MA, Preiss J. 2004.Biochem. Biophys. Res. Commun. 316:960–96
18. Nichols DJ, Keeling PL, Spalding M,
Guan H. 2000. Biochemistry 39:7820–25
19. Yoo SH, Spalding MH, Jane JL. 2002.Carbohydr. Res. 337:2195–203
20. Goldsmith E, Sprang S, Fletterick R.1982. J. Mol. Biol. 156:411–27
21. Kuroda A, Kornberg A. 1997. Proc. Natl.Acad. Sci. USA 94:439–42
22. Kornberg A, Rao NN, Ault-Riche D.1999. Annu. Rev. Biochem. 68:89–125
23. Kulaev I, Kulakovskaya T. 2000. Annu.Rev. Microbiol. 54:709–34
24. Shiba T, Tsutsumi K, Ishige K, NoguchiT. 2000. Biochemistry 65:315–23
25. Akiyama M, Crooke E, Kornberg A. 1993.J. Biol. Chem. 268:633–39
26. Sethuraman A, Rao NN, Kornberg A.2001. Proc. Natl. Acad. Sci. USA 98:8542–47
27. Ahn K, Kornberg A. 1990. J. Biol. Chem.265:11734–39
28. Akiyama M, Crooke E, Kornberg A. 1992.J. Biol. Chem. 267:22556–61
29. Zhang H, Ishige K, Kornberg A. 2002.Proc. Natl. Acad. Sci. USA 99:16678–83
30. Ishige K, Zhang H, Kornberg A. 2002.Proc. Natl. Acad. Sci. USA 99:16684–88
31. Ogawa N, Tzeng CM, Fraley CD, Korn-berg A. 2000. J. Bacteriol. 182:6687–93
32. Oppermann-Sanio FB, Steinbuchel A.2002. Naturwissenschaften 89:11–22
33. Allen MM, Weathers PJ. 1980. J. Bacte-riol. 141:959–62
34. Li H, Sherman DM, Bao S, Sherman LA.2001. Arch. Microbiol. 176:9–18
35. Picossi S, Valladares A, Flores E, HerreroA. 2004. J. Biol. Chem. 279:11582–92
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
478 STUBBE ET AL.
36. Ziegler K, Diener A, Herpin C, Richter R,Deutzmann R, Lockau W. 1998. Eur. J.Biochem. 254:154–59
37. Dementin S, Bouhss A, Auger G, ParquetC, Mengin-Lecreulx D, et al. 2001. Eur.J. Biochem. 268:5800–7
38. Berg H, Ziegler K, Piotukh K, Baier K,Lockau W, Volkmer-Engert R. 2000. Eur.J. Biochem. 267:5561–70
39. Simon RD. 1971. Proc. Natl. Acad. Sci.USA 68:265–67
40. Cornish K. 2001. Nat. Prod. Rep. 18:182–89
41. Ohya N, Koyama T. 2001. In Biopoly-mers: Vol. 2: Polyisoprenoids, ed. T Ko-yama, A Steibuchel, pp. 73–109. Wein-heim: Wiley-VCH
42. Singh AP, Wi SG, Chung GC, Kim YS,Kang H. 2003. J. Exp. Bot. 54:985–92
43. Wititsuwaannakul D, RattanapittayapornA, Koyama T, Wititsuwaannakul R. 2004.Macromol. Biosci. 4:314–23
44. Ogura K, Koyama T. 1998. Chem. Rev.98:1263–76
45. Asawatreratanakul K, Zhang YW, Witit-suwannakul D, Wititsuwannakul R, Taka-hashi S, et al. 2003. Eur. J. Biochem.270:4671–80
46. Fujihashi M, Zhang YW, Higuchi Y, LiXY, Koyama T, Miki K. 2001. Proc. Natl.Acad. Sci. USA 98:4337–42
47. Ko TP, Chen YK, Robinson H, Tsai PC,Gao YG, et al. 2001. J. Biol. Chem. 276:47474–82
48. Pan JJ, Chiou ST, Liang PH. 2000. Bio-chemistry 39:10936–42
49. Ko JH, Chow KS, Han KH. 2003. PlantMol. Biol. 53:479–92
50. Cornish K, Castillon J, Scott DJ. 2000.Biomacromolecules 1:632–41
51. Oh SK, Han KH, Ryu SB, Kang H. 2000.J. Biol. Chem. 275:18482–88
52. Byrom D. 1987. Trends Biotechnol. 5:246–50
53. Steinbuchel A, Hein S. 2001. Adv.Biochem. Eng. Biotechnol. 71:81–123
54. Page WJ. 1995. Can. J. Microbiol.41(Suppl. 1):1–3
55. Gerngross TU. 1999. Nat. Biotechnol. 17:541–44
56. Gross RA, Kalra B. 2002. Science 297:803–7
57. Rehm BH. 2003. Biochem. J. 376:15–33
58. Jendrossek D, Handrick R. 2002. Annu.Rev. Microbiol. 56:403–32
59. Aldor IS, Keasling JD. 2003. Curr. Opin.Biotechnol. 14:475–83
60. Steinbuchel A, Lutke-Eversloh T. 2003.Biomed. Eng. J. 16:81–89
61. Peoples OP, Sinskey AJ. 1989. J. Biol.Chem. 264:15293–97
62. Wieczorek R, Pries A, Steinbuchel A,Mayer F. 1995. J. Bacteriol. 177:2425–35
63. Potter M, Madkour MH, Mayer F,Steinbuchel A. 2002. Microbiology 148:2413–26
64. York GM, Stubbe J, Sinskey AJ. 2001. J.Bacteriol. 183:2394–97
65. Potter M, Muller H, Reinecke F, Wiec-zorek R, Fricke F, et al. 2004. Microbiol-ogy 150:2301–11
66. York GM, Stubbe J, Sinskey AJ. 2002. J.Bacteriol. 184:59–66
67. Maehara A, Doi Y, Nishiyama T, TakagiY, Ueda S, et al. 2001. FEMS Microbiol.Lett. 200:9–15
68. Maehara A, Taguchi S, Nishiyama T, Ya-mane T, Doi Y. 2002. J. Bacteriol. 184:3992–4002
69. Prieto MA, Buhler B, Jung K, Witholt B,Kessler B. 1999. J. Bacteriol. 181:858–68
70. Saegusa H, Shiraki M, Kanai C, Saito T.2001. J. Bacteriol. 183:94–100
71. York GM, Lupberger J, Tian J, LawrenceAG, Stubbe J, Sinskey AJ. 2003. J. Bac-teriol. 185:3788–94
72. Saegusa H, Shiraki M, Saito T. 2002. J.Biosci. Bioeng. 94:106–12
73. Kobayashi T, Shiraki M, Abe T,Sugiyama A, Saito T. 2003. J. Bacteriol.185:3485–90
74. Rehm BH, Steinbuchel A. 1999. Int. J.Biol. Macromol. 25:3–19
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
NONTEMPLATE-DRIVEN POLYMERIZATION 479
75. Gerngross TU, Snell KD, Peoples OP,Sinskey AJ, Cushai E, et al. 1994. Bio-chemistry 33:9311–20
76. Jia Y, Yuan W, Wodzinska J, Park C,Sinskey AJ, Stubbe J. 2001. Biochemistry40:1011–19
77. Zhang S, Kolvek S, Lenz RW, Good-win S. 2003. Biomacromolecules 4:504–9
78. Liebergesell M, Steinbuchel A. 1992. Eur.J. Biochem. 209:135–50
79. Liebergesell M, Sonomoto K, MadkourM, Mayer F, Steinbuchel A. 1994. Eur.J. Biochem. 226:71–80
80. Muh U, Sinskey AJ, Kirby DP, Lane WS,Stubbe J. 1999. Biochemistry 38:826–37
81. Qi Q, Steinbuchel A, Rehm BH. 2000.Appl. Microbiol. Biotechnol. 54:37–43
82. Pantazaki AA, Tambaka MG, Langlois V,Guerin P, Kyriakidis DA. 2003. Mol. Cell.Biochem. 254:173–83
83. Yuan W, Jia Y, Tian J, Snell KD, MuhU, et al. 2001. Arch. Biochem. Biophys.394:87–98
84. Rehm BH, Qi Q, Beermann BB, HinzHJ, Steinbuchel A. 2001. Biochem. J.358:263–68
85. Jia Y, Kappock TJ, Frick T, Sinskey AJ,Stubbe J. 2000. Biochemistry 39:3927–36
86. Berg OG, Cajal Y, Butterfoss GL, GreyRL, Alsina MA, et al. 1998. Biochemistry37:6615–27
87. Gerngross TU, Reilly P, Stubbe J, SinskeyAJ, Peoples OP. 1993. J. Bacteriol.175:5289–93
88. Lundgren DG, Pfister RM, Merrick JM.1964. J. Gen. Microbiol. 34:441–46
89. Steinbuchel A, Aerts K, Babel W, FollnerC, Liebergesell M, et al. 1995. Can. J. Mi-crobiol. 41(Suppl. 1):94–105
90. Rehm BH, Antonio RV, SpiekermannP, Amara AA, Steinbuchel A. 2002.Biochim. Biophys. Acta 1594:178–90
91. Karlsson M, Contreras JA, Hellman U,Tornqvist H, Holm C. 1997. J. Biol. Chem.272:27218–23
92. Griebel R, Smith Z, Merrick JM. 1968.Biochemistry 7:3676–81
93. Wodzinska J, Snell KD, Rhomberg A,Sinskey AJ, Biemann K, Stubbe J. 1996.J. Am. Chem. Soc. 118:6319–20
94. Austin MB, Noel JP. 2003. Nat. Prod.Rep. 20:79–110
95. Bruner SD, Weber T, Kohli RM,Schwarzer D, Marahiel MA, et al. 2002.Structure 10: 301–10
96. Bruner SD, Weber T, Kohli RM,Schwarzer D, Marahiel MA, et al. 2002.Structure 10:301–10
97. Lang D, Hofmann B, Haalck L, HechtHJ, Spener F, et al. 1996. J. Mol. Biol.259:704–17
98. Tian J, Sinskey AJ, Stubbe J. 2005. Bio-chemistry. In press
99. Tian J, Sinskey AJ, Stubbe J. 2005. Bio-chemistry 44:1495–503
100. Gerngross TU, Martin DP. 1995. Proc.Natl. Acad. Sci. USA 92:6279–83
101. Jurasek L, Marchessault RH. 2004. Appl.Microbiol. Biotechnol. 64:611–17
102. Lemoigne M. 1925. Ann. Inst. Pasteur 39:144
103. Lemoigne M. 1926. Bull. Soc. Chim. Biol.8:770
104. He A, Lawrence AG, Liu P, Tian J,Sinskey AJ, Stubbe J. 2005. J. Bacteriol.In press
105. Tian J, He A, Lawrence AG, Liu P,Sinskey AJ, Stubbe J. 2005. J. Bacteriol.In press
106. Thiele OW, Dreysel J, Hermann D. 1972.Eur. J. Biochem. 29:224–36
107. Shaw GL, Melby MK, Horowitz DM,Keeler J, Sanders JKM. 1994. Int. J. Biol.Macromol. 16:59–63
108. Horowitz DM, Sanders JKM. 1994. J. Am.Chem. Soc. 116:2695–702
109. Dennis D, Liebig C, Holley T, ThomasKS, Khosla A, et al. 2003. FEMS Micro-biol. Lett. 226:113–19
110. Sudesh K, Gan Z, Matsumot KO, Doi Y.2002. Ultramicroscopy 91:157–64
111. Tian J, Sinskey AJ, Stubbe J. 2005. J. Bac-teriol. In press
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

10 May 2005 10:57 AR AR261-BI74-16.tex XMLPublishSM(2004/02/24) P1: JRX
480 STUBBE ET AL.
112. McCool GJ, Cannon MC. 1999. J. Bacte-riol. 181:585–92
113. Bohmert K, Balbo I, Steinbuchel A, Tis-chendorf G, Willmitzer L. 2002. PlantPhysiol. 128:1282–90
114. Kawaguchi Y, Doi Y. 1992. Macro-molecules 25:2324–29
115. Doi Y, Kawaguchi Y, Koyama N, Naka-mura S, Hiramitsu M, et al. 1992. FEMSMicrobiol. Rev. 103:103–8
116. Mas J, Pedros-Alio C, Guerrero R. 1985.J. Bacteriol. 164:749–56
117. Sim SJ, Snell KD, Hogan SA, Stubbe J,Rha C, Sinskey AJ. 1997. Nat. Biotechnol.15:63–67
118. Madden LA, Anderson AJ, Shah DT, As-rar J. 1999. Int. J. Biol. Macromol. 25:43–53
119. Spyros A, Argyropoulos DS, Marches-sault RH. 1997. Macromolecules 30:327–29
120. Doi Y, Segawa A, Kawaguchi Y, KuniokaM. 1990. FEMS Microbiol. Lett. 55:165–69
121. Haywood GW, Anderson AJ, Dawes EA.1989. FEMS Microbiol. Lett. 57:1–6
122. Kessler B, Witholt B. 2001. J. Biotechnol.86:97–104
123. Peralta-Gil M, Segura D, Guzman J,Servin-Gonzalez L, Espin G. 2002. J.Bacteriol. 184:5672–7
124. Castaneda M, Guzman J, Moreno S, Es-pin G. 2000. J. Bacteriol. 182:2624–28
125. Wu G, Moir AJ, Sawers G, Hill S,Poole RK. 2001. FEMS Microbiol. Lett.194:215–20
126. Kortstee GJ, Appeldoorn KJ, Bonting CF,van Niel EW, van Veen HW. 1994. FEMSMicrobiol. Rev. 15:137–53
127. Seviour RJ, Mino T, Onuki M. 2003.FEMS Microbiol. Rev. 27:99–127
128. Schembri MA, Bayly RC, Davies JK.1995. J. Bacteriol. 177:4501–7
129. Dunn MF, Araiza G, Encarnacion S, delCarmen Vargas M, Mora J. 2002. J. Bac-teriol. 184:2296–99
130. Encarnacion S, del Carmen Vargas M,Dunn MF, Davalos A, Mendoza G, et al.2002. J. Bacteriol. 184:2287–95
131. Encarnacion S, Guzman Y, Dunn MF,Hernandez M, del Carmen Vargas M,Mora J. 2003. Proteomics 3:1077–85
132. Maehara A, Ueda S, Nakano H, YamaneT. 1999. J. Bacteriol. 181:2914–21
133. Deleted in proof134. Klinke S, de Roo G, Witholt B, Kessler B.
2000. Appl. Environ. Microbiol. 66:3705–10
135. Oeding V, Schlegel HG. 1973. Biochem.J. 134:239–48
136. Lee IY, Kim MY, Chang JN, Young HP.1995. FEMS Microbiol. Lett. 131:35–39
137. Henderson RA, Jones CW. 1997. Arch.Microbiol. 168:486–92
138. Lee Y-H, Park J-S, Huh T-L. 1997.Biotechnol. Lett. 19:771–74
139. Rendleman JA Jr. 2000. Biotechnol. Appl.Biochem. 31(Part 3):171–78
140. Preiss J, Romeo T. 1994. Prog. NucleicAcid Res. Mol. Biol. 47:299–329
141. London South Bank Univ. 2004. Wa-ter structure and behavior. http://www.lsbu.ac.uk/water/hysta.htmlA
nnu.
Rev
. Bio
chem
. 200
5.74
:433
-480
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
MA
SSA
CH
USE
TT
S IN
ST. O
F T
EC
HN
OL
OG
Y o
n 07
/06/
05. F
or p
erso
nal u
se o
nly.

P1: JRX
April 29, 2005 5:26 Annual Reviews AR261-FM
Annual Review of BiochemistryVolume 74, 2005
CONTENTS
FROM PROTEIN SYNTHESIS TO GENETIC INSERTION,Paul Zamecnik 1
THE BIOCHEMISTRY OF PARKINSON’S DISEASE,Mark R. Cookson 29
APPLICATIONS OF DNA MICROARRAYS IN BIOLOGY,Roland B. Stoughton 53
ZONA PELLUCIDA DOMAIN PROTEINS, Luca Jovine, Costel C. Darie,Eveline S. Litscher, and Paul M. Wassarman 83
PROLINE HYDROXYLATION AND GENE EXPRESSION,William G. Kaelin Jr. 115
STRUCTURAL INSIGHTS INTO TRANSLATIONAL FIDELITY,James M. Ogle and V. Ramakrishnan 129
ORIGINS OF THE GENETIC CODE: THE ESCAPED TRIPLET THEORY,Michael Yarus, J. Gregory Caporaso, and Rob Knight 179
AN ABUNDANCE OF RNA REGULATORS, Gisela Storz, Shoshy Altuvia,and Karen M. Wassarman 199
MEMBRANE-ASSOCIATED GUANYLATE KINASES REGULATE ADHESIONAND PLASTICITY AT CELL JUNCTIONS, Lars Funke, Srikanth Dakoji,and David S. Bredt 219
STRUCTURE, FUNCTION, AND FORMATION OF BIOLOGICALIRON-SULFUR CLUSTERS, Deborah C. Johnson, Dennis R. Dean,Archer D. Smith, and Michael K. Johnson 247
CELLULAR DNA REPLICASES: COMPONENTS AND DYNAMICS AT THEREPLICATION FORK, Aaron Johnson and Mike O’Donnell 283
EUKARYOTIC TRANSLESION SYNTHESIS DNA POLYMERASES:SPECIFICITY OF STRUCTURE AND FUNCTION, Satya Prakash,Robert E. Johnson, and Louise Prakash 317
NOD-LRR PROTEINS: ROLE IN HOST-MICROBIAL INTERACTIONS ANDINFLAMMATORY DISEASE, Naohiro Inohara, Mathias Chamaillard,Christine McDonald, and Gabriel Nunez 355
v
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.

P1: JRX
April 29, 2005 5:26 Annual Reviews AR261-FM
vi CONTENTS
REGULATION OF PROTEIN FUNCTION BY GLYCOSAMINOGLYCANS—ASEXEMPLIFIED BY CHEMOKINES, T.M. Handel, Z. Johnson, S.E. Crown,E.K. Lau, M. Sweeney, and A.E. Proudfoot 385
STRUCTURE AND FUNCTION OF FATTY ACID AMIDE HYDROLASE,Michele K. McKinney and Benjamin F. Cravatt 411
NONTEMPLATE-DEPENDENT POLYMERIZATION PROCESSES:POLYHYDROXYALKANOATE SYNTHASES AS A PARADIGM,JoAnne Stubbe, Jiamin Tian, Aimin He, Anthony J. Sinskey,Adam G. Lawrence, and Pinghua Liu 433
EUKARYOTIC CYTOSINE METHYLTRANSFERASES, Mary Grace Golland Timothy H. Bestor 481
MONITORING ENERGY BALANCE: METABOLITES OF FATTY ACIDSYNTHESIS AS HYPOTHALAMIC SENSORS, Paul Dowell, Zhiyuan Hu,and M. Daniel Lane 515
STRUCTURE AND PHYSIOLOGIC FUNCTION OF THE LOW-DENSITYLIPOPROTEIN RECEPTOR, Hyesung Jeon and Stephen C. Blacklow 535
COPPER-ZINC SUPEROXIDE DISMUTASE AND AMYOTROPHIC LATERALSCLEROSIS, Joan Selverstone Valentine, Peter A. Doucette,and Soshanna Zittin Potter 563
THE STRUCTURE AND FUNCTION OF SMC AND KLEISIN COMPLEXES,Kim Nasmyth and Christian H. Haering 595
ANTIBIOTICS TARGETING RIBOSOMES: RESISTANCE, SELECTIVITY,SYNERGISM, AND CELLULAR REGULATION, Ada Yonath 649
DNA MISMATCH REPAIR, Thomas A. Kunkel and Dorothy A. Erie 681
GENE THERAPY: TWENTY-FIRST CENTURY MEDICINE, Inder M. Vermaand Matthew D. Weitzman 711
THE MAMMALIAN UNFOLDED PROTEIN RESPONSE, Martin Schroderand Randal J. Kaufman 739
THE STRUCTURAL BIOLOGY OF TYPE II FATTY ACIDBIOSYNTHESIS, Stephen W. White, Jie Zheng, Yong-Mei Zhang,and Charles O. Rock 791
STRUCTURAL STUDIES BY ELECTRON TOMOGRAPHY: FROM CELLSTO MOLECULES, Vladan Lucic, Friedrich Forster,and Wolfgang Baumeister 833
PROTEIN FAMILIES AND THEIR EVOLUTION—A STRUCTURALPERSPECTIVE, Christine A. Orengo and Janet M. Thornton 867
Ann
u. R
ev. B
ioch
em. 2
005.
74:4
33-4
80. D
ownl
oade
d fr
om a
rjou
rnal
s.an
nual
revi
ews.
org
by M
ASS
AC
HU
SET
TS
INST
. OF
TE
CH
NO
LO
GY
on
07/0
6/05
. For
per
sona
l use
onl
y.


















