





Classification and analysis of the GCP inspection findings of GCP ...

NON-INTERVENTIONAL STUDIES?What are they?How do they differ from Clinical Trials?
Stuart McCully, PhD 16th January 2011

DISCLAIMER
Although this material contains information of a legal nature, ithas been developed for informational purposes only and doesnot constitute legal advice or opinions as to the currentoperative laws, regulations, or guidelines of any jurisdiction. Inaddition, because new standards are issued on a continuingbasis, this training material is not an exhaustive source of allcurrent applicable laws, regulations, and guidelines relating toclinical research. While reasonable efforts have been made toassure the accuracy and completeness of the informationprovided, researchers and other individuals should check withlocal authorities and/or research ethics committees beforestarting clinical research activities

Topics Covered
1. Introduction: A Snapshot of Key Events a. The Nazi Medical Experimentsb. The Nuremberg Code and the Declaration of
Helsinkic. The Tuskegee Syphilis (Observational) Studyd. Ethical Principles in Biomedical Research
2. Clinical Research: Clinical Trials vs NIS a. Research or Audit/Service Evaluation?b. Research: Interventional or Non-Interventional?c. What is a Clinical Trial?d. The Regulatory Framework: Clinical Trialse. The Regulatory Framework: NISf. What is a Non-Interventional Study?g. Country-Specific Regulation of NIS: UKh. How and Why are they Different?i. “GCP-lite”j. Ethical Principles in NISk. The Principle of Observation

• Nazi Medical Experiments• Nuremberg Code• Declaration of Helsinki• The Tuskegee Syphilis Study• Ethical Principles
Introduction: A Snapshot of Key Events

The Nazi Medical Experiments
• Experiments on twins• Freezing experiments• Malaria experiments• Mustard gas experiments• Sulfonamide experiments• Sea water experiments• Sterilization experiments• Experiments with poison• Incendiary bomb
experiments• High altitude experiments

The Nuremberg Code (1947) & The Declaration of Helsinki (1964)
• Voluntary consent is absolutely essential
• Studies should be based on good science
• The benefit should outweigh the risks
• Participants should be at liberty to withdraw
• The experiment should be conducted by scientifically qualified persons
• Research protocols should be reviewed by an independent committee

The Nuremberg Code (1947)
The voluntary consent of the human subject is absolutely essential.
This means that the person involved should have legal capacity to give consent; shouldbe so situated as to be able to exercise free power of choice, without the interventionof any element of force, fraud, deceit, duress, over-reaching, or other ulterior form ofconstraint or coercion; and should have sufficient knowledge and comprehension ofthe elements of the subject matter involved, as to enable him to make anunderstanding and enlightened decision. This latter element requires that, before theacceptance of an affirmative decision by the experimental subject, there should bemade known to him the nature, duration, and purpose of the experiment; the methodand means by which it is to be conducted; all inconveniences and hazards reasonablyto be expected; and the effects upon his health or person, which may possibly comefrom his participation in the experiment.
The duty and responsibility for ascertaining the quality of the consent rests upon eachindividual who initiates, directs or engages in the experiment. It is a personal duty andresponsibility which may not be delegated to another with impunity.
Source: The Nuremberg Code

The Declaration of Helsinki (1964)
• Research with humans should be based on laboratory and animal experimentation
• Research protocols should be reviewed by an independent committee
• Informed Consent is necessary
• Research should be conducted by medically/ scientifically qualified individuals
• Risks should not exceed benefits
The Interests of the Patient should prevail over those of science and society

The Tuskegee Syphilis (Observational) Study
• In 1932 the US Government misled 623 African-Americans into participating into a study of untreated syphilis
• The government induced these men to participate in a study in which the government represented that the participants were being treated for whatever their ailments were– They were never told what their ailment was– They never gave their consent to be involved in a study– Nor did they realise they were part of a study until the
story broke in July 1972– Treatment was knowingly withheld for 40 years
Fred D Gray – Attorney, 8th April 1997

The Tuskegee Syphilis (Observational) Study
“Men who were poor and African American, withoutresources and with few alternatives believed they hadfound hope when they were offered free medical care bythe United States Public Health Service. They werebetrayed.
For 40 years, hundreds of men were betrayed, along withtheir wives and children, along with a community in MaconCounty, Alabama, the City of Tuskegee, the fine universitythere, and the larger African American community. TheUnited States government did something that was wrong –deeply, profoundly, morally wrong. It was an outrage to ourcommitment to integrity and equality for all of our citizens.”
President Bill Clinton, 16th May 1997

The Tuskegee Syphilis (Observational) Study
“Medical professionals willingly and intentionally lethuman beings suffer from a treatable, and then later acurable illness. These researchers knew that mercuryand arsenic compounds could treat the disease, but theTuskegee men did not receive the medicine. Later theresearchers knew that penicillin could cure the disease,but again, the Tuskegee men did not get the medicine.They didn’t get treated until the 40 year study wasdiscovered and stopped amid public outcry in 1972. Itwas a disgraceful episode for American Scientists.”
Vice President Al Gore, 16th May 1997

The Tuskegee Syphilis (Observational) Study
This case reaffirmed the principle that prior informed consentshould be obtained from individuals before they are allowed toparticipate in human experimentation

Ethical Principles in Biomedical Research
• Protect the rights, safety and well-being of human subjects
• Independent ethical review
• Informed consent
• Balance the benefits, harms and risks of study participation
• Safeguard confidentiality
• Ensure scientific credibility

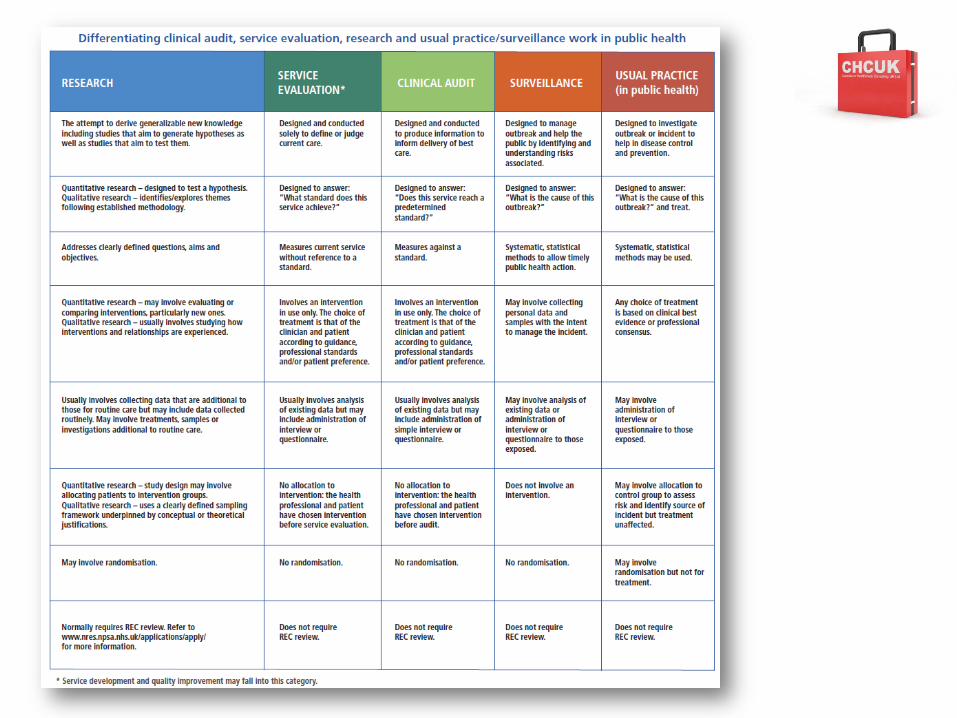
Research or Audit/Service Evaluation?
• NRES reviews research proposals to protect the rights and safety of research participants and enables ethical research which is of potential benefit to science and society
• Patients expect HCPs to undertake audit and service evaluation as part of quality assurance. These involve minimal additional risk, burden or intrusion for participants and are regulated outside of NRES.
• Research may involve greater risk, burden or intrusion for participants than standard clinical practice.– Research requires review by a REC– Research Governance Framework for Health and Social Care
Source: NRES Leaflet – Defining Research

Research or Audit/Service Evaluation?
Research or Audit? Audit • Outside scope of NRES
Research
Interventional or Non-Interventional?
• Requires REC Review• Requires R&D Approval
Interventional(i.e., Clinical Trial)
• Requires MHRA Approval
Non-Interventional(e.g., Observational study)
• Does Not Require MHRA Approval

Research: Interventional or Non-Interventional?
Research or Audit? Audit • Outside scope of NRES
Research
Interventional or Non-Interventional?
• Requires REC Review• Requires R&D Approval
Interventional(i.e., Clinical Trial)
• Requires MHRA Approval
Non-Interventional(e.g., Observational study)
• Does Not Require MHRA Approval

What is a Clinical Trial?
“Clinical trial” means any investigation in human subjects, other than a non-interventional trial, intended:
a) To discover or verify the clinical, pharmacological or other pharmacodynamic effects of one or more medicinal products,
b) To identify any adverse reactions to one or more such products, or
c) To study absorption, distribution, metabolism and excretion of one or more such products,
With the object of ascertaining the safety or efficacy of these products
(As per Regulation 2(1) of SI 2004/1031 (as amended))

From Human Abuse to Regulated Use
Tuskegee Syphilis Study
1932-1972
1938 - 1945WWII
Nuremberg Code
1947
1957 -1961
Thalidomide
Declaration of Helsinki
1964
Directive 65/65/EC
1965
1996
ICH E6
Clinical Trials
Directives
2001 & 2005
Cause(Abuse)
Effect(Regulations)

Good Clinical Practice: ICH E6
1. Glossary
2. The Principles of GCP
3. Ethics Committees
4. Investigator
5. Sponsor
6. Clinical Trial Protocol & Amendments
7. Investigator’s Brochure
8. Essential Documents for the Conduct of a Clinical Trial

Clinical Trials & GCP2001/20/EC2005/28/EC
Safety ReportingENTR/CT 3
Competent Authority SubmissionsENTR/CT 1
IEC SubmissionsENTR/CT 2
EU Detailed Guidance
Document Retention2001/83/EC (as amended)[Annex 1, Part I, Section 5.2.c]
GMP2003/94/EC[Annex 13]
2001/83/EC (as amended)
Data Privacy95/46/EC
Paediatric StudiesEthical Considerations…
1901/2006/EC1902/2006/EC
EU Supporting Regulations & Directives
EU Implementing Directives
UK Law
The Medicines for Human Use (Clinical Trials) Regulations
2004SI 2004/1031
AmendmentsSI 2004/3224, SI 2005/2754, SI 2005/2759, SI 2006/1928, SI 2006/2984, SI 2008/941, SI 2009/1164, SI 2010/1882IMP vs Non-IMP
Interventional or Non-Interventional?Decision Tree
GCP GuidelinesICH E6
Ethics of Human ResearchDeclaration of Helsinki (1996)
Data Protection Act 1998TMF Content & Archiving
Copyright © Dr Stuart McCully, 2009-2010All Rights Reserved
ICH Efficacy (E) Series
The Medicines Act 1968
Regulation of Clinical Trials: UK
Overview of Clinical Trial Regs (UK) – 31st August 2010

Clinical Trials Directive2001/20/EC
The Medicines for Human Use (Clinical Trials) Regulations
2004SI 2004/1031
ICH GCP GuidelinesICH E6
Ethics of Human ResearchDeclaration of Helsinki (1996)
Copyright © Dr Stuart McCully, 2009All Rights Reserved
Clinical Research: How is it Regulated?
Overview of Clinical Trial Regs (UK) – 7th September 2009
EU Implementing Directives
Global GCP Guidelines
Bioethical Principles
UK Law
GCP Directive2005/28/EC
The Medicines for Human Use (Clinical Trials) Amendments
Regulations 2006SI 2006/1928
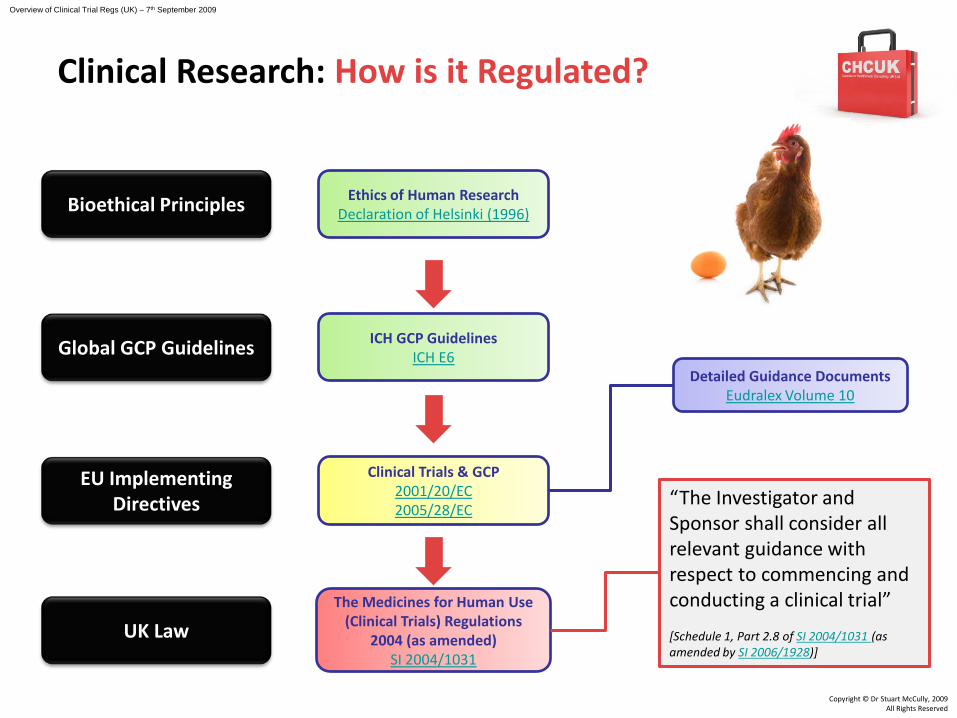
Clinical Trials & GCP2001/20/EC2005/28/EC
The Medicines for Human Use (Clinical Trials) Regulations
2004 (as amended)SI 2004/1031
ICH GCP GuidelinesICH E6
Ethics of Human ResearchDeclaration of Helsinki (1996)
Copyright © Dr Stuart McCully, 2009All Rights Reserved
Clinical Research: How is it Regulated?
Overview of Clinical Trial Regs (UK) – 7th September 2009
“The Investigator and Sponsor shall consider all relevant guidance with respect to commencing and conducting a clinical trial”
[Schedule 1, Part 2.8 of SI 2004/1031 (as amended by SI 2006/1928)]
Detailed Guidance DocumentsEudralex Volume 10
EU Implementing Directives
Global GCP Guidelines
Bioethical Principles
UK Law

Country-Specific Regulation of NIS: UK
Article 1.1 of the Clinical Trials Directive (2001/20/EC)which is implemented into UK law by SI 2004/1031states that “this Directive does not apply to non-interventional trials”.

How and Why are they Different?
“The purpose for excluding these [NIS] trials from the scope ofthe Directive 2001/20/EC is that these trials are typically of alower risk than interventional clinical trials.
Moreover, this restriction shall ensure that medical activitieswhich are normal clinical practice and as such part of the generalmedical surveillance of a patient are excluded from the scope ofthe Directive 2001/20/EC.”
(Source: Eudralex Volume 10 - Questions and Answers, Version 7.0, September 2010)

What is a Non-interventional Study?
“non-interventional trial” means a study of one or more medicinal products which have a marketing authorisation, where the following conditions are met:
a) The products are prescribed in the usual manner in accordance with the terms of that authorisation
b) The assignment of any patient involved in the study to a particular therapeutic strategy is not decided in advance by a protocol but falls within current practice,
c) The decision to prescribe a particular medicinal product is clearly separated from the decision to include the patient in the study
(As per Regulation 2(1) of SI 2004/1031 (as amended))

What is a Non-interventional Study?
“non-interventional trial” means a study of one or more medicinal products which have a marketing authorisation, where the following conditions are met:
d) No diagnostic or monitoring procedures are applied to the patients included in the study, other than those which are ordinarily applied in the course of the particular therapeutic strategy in question, and
e) Epidemiological methods are to be used for the analysis of the data arising from the study
(As per Regulation 2(1) of SI 2004/1031 (as amended))

What is a Non-interventional Study?
Source: ICH E2E – Pharmacovigilance Planning (2004)

The Regulatory Framework: NIS
The regulatory requirements for non-interventional studies differ from clinical trials.
As a minimum:
• Highest standards of professional conduct and confidentiality must be maintained – Source: Eudralex Volume 9A, Part I, Section 7.7
With regards to conduct:
• In observational studies, the investigator “observes and evaluates results of ongoing medical care without 'controlling' the therapy beyond normal medical practice.”– Source: ICH E2E, Section 3.2.1

Country-Specific Regulation of NIS: UK
Applicable Legislation: Non-interventional studies (NIS) are controlled by acombination of legislation and codes of practice in the UK.
SI 2004/1031 (as amended) • Legal definition of “NIS”
• MHRA Approval Not Required
Research Governance
Framework for Health and Social
Care
• REC Approval Required• R&D Approval Required
ABPI COP(Industry Code) • Essential Criteria for Industry-
Sponsored NIS
Declaration of Helsinki/ ICH E6 NRES
• REC Approval• Protocol• Informed Consent• Annual Progress Reports• Substantial Amendments• SAE Reporting• End of Trial Notification• Final Study Report
Fundamental Ethical Principles:• Protect rights, safety and well-
being of patients• Independent Ethical Review• Informed Consent• Protocol• Scientific Credibility
SI 1994/3144 (as amended) • Safety Reporting Requirements
Eudralex Volume 9A
(IPSE GPP/ ICH E2E)
• Ethical Considerations• Data Confidentiality• Professional Conduct• Safety Reporting in NIS• Protocol• Final Report

Medicinal ProductsEC/726/2004 (as amended)2001/83/EC (as amended)
International Supporting Guidance
Document Retention2001/83/EC (as amended)[Annex 1, Part I, Section 5.2.c]
Data Privacy95/46/EC
EU Supporting Regulations & Directives
European Directives
ICH GuidelinesICH E2EICH E6
Ethics of Human ResearchDeclaration of Helsinki (1996)
Copyright © Dr Stuart McCully, 2009 - 2010All Rights Reserved
Regulation of Non-Interventional Studies: UK
Overview of NIS Regs (EU) – 8th Oct 2010
The Medicines for Human Use (Clinical Trials) Regulations
2004 (as amended)SI 2004/1031
Data Protection Act 1998
UK Law & Guidance
MHRA Website
ABPI Code of Practice (2008)
NRES SOPs
NHS Research Governance Framework(s)
The Medicines for Human Use (Marketing Authorisations etc) Regulations 1994 (as
amended)SI 1994/3144
IPSE Guidelines• Good Pharmacoepidemiology Practices• Data Privacy• GRACE Principles (Endorsed by IPSE)
General Considerations for Post-Authorisation Studies and Post-
Authorisation safety StudiesEudralex Volume 9a [Chapter I.7]
• Safety Reporting• Protocol and Report Guidance• Competent Authority Notification• Ethical Considerations• Promotion of Medicinal Products• Participation of Healthcare Professionals • Procedures for Complaints
Non-Interventional Study or Clinical Trial?Eudralex Volume 10 - Decision Tree
EFPIA Code of PracticeOctober 2007
CIOMS International Ethical Guidelines for Epidemiological Studies 2009

Industry Guidance: ABPI Code of Practice (2008)

ABPI Code of Practice (2008): Non-Interventional Studies of Marketed Products
According to Clause 13 of the ABPI Code of Practice (2008), non-interventional studies (NIS) that areprospective in nature and involve the collection of patient data must comply with all of the following criteria:
1. The study is conducted for a scientific purpose2. There is a written protocol3. There are written contracts between the Study Sponsor and Healthcare Professionals and/or Institutions4. Any remuneration provided must be reasonable and reflect fair market value5. The study protocol should be submitted for review in those countries where the Ethics Committees are
prepared to review the document6. Data protection legislation must be complied with7. The Company’s Scientific Service must approve the protocol and must supervise the conduct of the study8. The study results must be analysed and summaries be made available within a reasonable period of time
to the Company’s Scientific Service and the Healthcare Professionals who participated in the study9. If the study shows results that are important for the assessment of benefit-risk profile of the medicinal
product, the summary report should be immediately forwarded to the relevant Competent Authority10. Sales Representatives may only be involved in an administrative capacity and such involvement must be
under the supervision of the Company’s Scientific Service
Also: • Companies are encouraged to publicly disclose the summary details and results of non-interventional
studies in a manner consistent with the parallel obligations for clinical trials• Companies are encouraged to apply the same requirements (to the extent applicable) to all other types of
studies including epidemiological studies, registries and other studies that are retrospective in nature
Copyright © Dr Stuart McCully, 2009 - 2010All Rights Reserved
Overview of NIS Regs (EU) – 29th June 2010

How and Why are they Different?
Clinical Trials
Post-Authorisation Studies
Non-Interventional Studies
Higher Risk
Lower Risk
Marketing Authorisation
Pre-
Aut
hori
satio
nPo
st-A
utho
risat
ion
Post-Approval Commitment (PAC)
Sponsor Driven
Post-Approval Commitment (PAC)
Sponsor Driven
Risk Management Plan (RMP)
Post-Approval Safety Study (PASS)
Post-Approval Study (PAS)

How and Why are they Different?
Clinical Trial• Ethical principles (DoH/GCP)
– Ethical review• Novel/ “unknown” drug• Higher risk to subject• Interventional• Clinical practice driven by
protocol• Harmonised GCP Framework and
regulations to ensure safety of subject and scientific credibility
• Strictly regulated due to higher risk– Routine monitoring, audits and
inspections
Non-Interventional Study • Ethical principles (DoH/GCP)
– Ethical review• Known/ Authorised drug• Lower risk• Non-interventional• Routine clinical practice – not
driven by protocol• No harmonised legal framework
(currently) due to lower risk to subject however greater focus on data protection and scientific credibility
• Less strictly regulated– Routine audits

Good Clinical Practice: ICH E6
1. Glossary
2. The Principles of GCP
3. Ethics Committees
4. Investigator
5. Sponsor
6. Clinical Trial Protocol & Amendments
7. Investigator’s Brochure
8. Essential Documents for the Conduct of a Clinical Trial

“GCP-lite”
• No IMP• SmPC rather than IB• Simplified Protocol• Simplified Informed Consent• Ethics Review• No Competent Authority
Approval (e.g., MHRA or IMB)• Simplified TMF/ISF• Simplified CRF• Reduced Monitoring

Ethical Principles in Non-Interventional Studies
For non-interventional post-authorisation safety studies, the Marketing Authorisation Holders andInvestigators should follow relevant national legislation in those Member States where this exists, inaddition to the guidance given here.
The highest possible standards of professional conduct and confidentiality must always be maintainedand legislation on data protection followed (see Directive 95/46/EC). The Patient’s right toconfidentiality is paramount. The Patient’s personal identifiers should be replaced by a code in the studydocuments, and only authorised persons should have access to identifiable personal details if dataverification procedures demand inspection of such details. Responsibility for the retrieval of informationfrom personal medical records lies with the Healthcare Professional(s) responsible for the patient’s care.Such information from medical records should be provided to the Marketing Holder, who is thereafterresponsible for the handling of such information.
It is recommended that non-interventional post-authorisation safety studies are referred to an EthicsCommittee. Studies conducted entirely using records not containing any personal identifiers e.g.anonymised records) may not require an ethical review of individual study protocols. Nationalguidelines in this respect should be followed where they exist.
According to European data protection legislation, explicit consent is required when the study plans tocollect data containing personal identifiers, though some exceptions are envisaged.
(Source: Eudralex Volume 9A, Part I, Section 7.7)

The Principle of Observation
Observational Study:• Non-experimental study*• Epidemiological study
that does not involve intervention*
• Observe ‘real-life’ scenarios– E.g., a record of outcomes
in routine clinical practice• Supplement existing data
on authorised drugs or existing conditions
* CIOMS International Ethical Guidelines for Epidemiological Studies, 2009 (ISBN 92-9036-081-X)


















