
NMR eHand 13C) and IR Studies of Dioxouranium(VI...
3
Indian Journal of Chemistry Vol. 23A, July 1984, pp. 602-604 NMR e Hand 13C) and IR Studies of Dioxouranium(VI) Complexes with a Bidentate Pyridyl-Pyrazole Ligand s C NA YAK, V CHAKRA VORTTY & K C DASH- Department of Chemistry, Utkal University, Vani Vihar , Bhubaneswar 751004 Receired 18 January 1984; rerised and accepted 21 Februarv 1984 The bidentate ligand, 3,5-dimethyl-I(2' -pyridyl)pyrazole (DMPP), reacts with U0 2 (NO J h and U0 2 (NCSj, in ethanol to form yellow, crystalline, diamagnetic and non-electrolytic complexes [UO,(DMPP)(N0 3 )2J and [U0 2 (DMPP),(NCShJ. The IR spectra indicate coordination of the thiocyanato group through the N-end and of the nitrato group through the O-atoms in a bidentate manner. The IR bands also suggest the presence of linear U0 2 2 + moiety. The I Hand 13C NMR spectra adequately confirm the above assignments. The mass spectra of the complexes show the molecular ion peak and the fragmentation pattern of the ligand, but not of the original complexes. As a part of the continuing investigations from our laboratory on biologically active Iigands I -3. we report here the preparation and characterisation of dioxouranium(VI) complexes with another biologi- cally-active ligand, 3,S-dimethyl-I(2' -pyridyl)pyrazole (DMPP). UOiN0 3 h was a BDH (England) reagent. U0 2 (NCSh was prepared by the metathesis reaction of UOz{N0 3 h and KCNS in ethanolic medium". The ligand (DMPP) was synthesised by the condensation of acetylacetone with 2-hydrazinopyridine. The ligand, obtained as a light yellow liquid (b.p. 74''C. O.4mm of Hg), was characterised by IR. NMR and mass spectra 5. The ligand (DMPP, I) and the metal salt (uranyl nitrate or thiocyanate) were mixed in appropriate stoichiometric ratio (I: I or 2: I) and the mixture was refluxed for 3 hr. The solvent was then concentrated and the resulting solution slowly cooled to obtain yellow or orange-yellow crystalline materials. These were collected on a frit, washed with ethanol and then with ether and finally dried in vacuo. These complexes are easily soluble in common organic solvents. [UOiN0 3 h(DMPp)] This compound is obtained as bright-yellow crystalline plates (m.p. nOT) [Calc. for UOsCloH IINs (M.W. S67): C, 21.16; H, \.94; N, 12.34. Found C, 21.13; H, 2.04; N, 12.41%]. The compound is a non-electrolyte in MeOH at 21"C, AM 602 =28.2mhoscm 2 mol-I.A I: I electrolyte in MeOH has a AM value" of '" 120-140 mhos em? mol -I. [U0 2 (NCS)z{DMPP)2] This compound is orange-yellow crystalline (m.p.166°q. [Calc. for U02C22HnNsS2 (M.W. 732): C, 36.06; H, 3.00; N, IS.30. Found C, 36.18;H, 3.03; N, IS. 12%]. The compound is a non-electrolyte in MeOH at 21°C, AM=74.81mhoscm 2 mol- l . Somewhat higher AM value is obtained due to partial solvolysis. IR spectra (4000-200cm -I) were recorded in nujol on a Beckmann-12 double-beam spectrophotometer. Mass spectra were recorded on a Varian MAT SM-l BH spectrometer operating at an electron energy of 70eV. The IH NMRspectra were measured at 60MHz with a Varian EM 360 equipment using TMS as an external standard. The 13C NMR spectra were recorded on a JEOL FX-60 spectrometer operating in the F.T. mode at IS.08 MHz. The probe temperature was 40 ± I DC with broad band decoupling. The shifts are taken relative to internal CD 3 0D resonating at 47.041 ppm down field to TMS7. The two complexes synthesised viz., [UOiN0 3 h(DMPP)] and [UOz{NCSh(DMPPh] are shown to be eight-coordinated on the basis of analytical data, electrical conductance and vibrational as well as resonance spectroscopy. The presence of a linear UO~ + group is confirmed on the basis of IR bands at 910-920,810 and 2S0-260cm -I correspond- ing to the vas(U-O), vs(U-O) and <5(O-U-O) modes, respectively of the linear UO~ + group'':". The v(CN) band of UOiNCSh(DMPPh is split into components observed at 20S6 and 2022 em -I; the v(CS) band is observed at 820cm -I and the J(NCS) is observed at 481 cm -I, all suggesting coordination of the - NCS group through the hard -N end 1 0. UOiNO 3h(DMPP) exhibits IR bands due to the N0 3 group at IS20, 1479, IOt9, 918 and 820cm- 1 indicating its bidentate coordination behaviour 11. The molar conductivity values in MeOH indicate that the nitrato complex is a non-electrolyte, whereas the thiocyanato complex has a value somewhat higher than that expected for a non-electrolyte, although substantially less than that for a I: I electrolyte". This
Transcript of NMR eHand 13C) and IR Studies of Dioxouranium(VI...

Indian Journal of ChemistryVol. 23A, July 1984, pp. 602-604
NMR eHand 13C) and IR Studies ofDioxouranium(VI) Complexes with a
Bidentate Pyridyl-Pyrazole Ligand
s C NA YAK, V CHAKRA VORTTY & K C DASH-
Department of Chemistry,Utkal University, Vani Vihar , Bhubaneswar 751004
Receired 18 January 1984;rerised and accepted 21 Februarv 1984
The bidentate ligand, 3,5-dimethyl-I(2' -pyridyl)pyrazole (DMPP),reacts with U02(NOJh and U02(NCSj, in ethanol to form yellow,crystalline, diamagnetic and non-electrolytic complexes[UO,(DMPP)(N03)2J and [U02(DMPP),(NCShJ. The IR spectraindicate coordination of the thiocyanato group through the N-endand of the nitrato group through the O-atoms in a bidentate manner.The IR bands also suggest the presence of linear U022 + moiety. TheI Hand 13C NMR spectra adequately confirm the aboveassignments. The mass spectra of the complexes show the molecularion peak and the fragmentation pattern of the ligand, but not of theoriginal complexes.
As a part of the continuing investigations from ourlaboratory on biologically active Iigands I -3. we reporthere the preparation and characterisation ofdioxouranium(VI) complexes with another biologi-cally-active ligand, 3,S-dimethyl-I(2' -pyridyl)pyrazole(DMPP).
UOiN03h was a BDH (England) reagent.U02(NCSh was prepared by the metathesis reactionof UOz{N03h and KCNS in ethanolic medium". Theligand (DMPP) was synthesised by the condensation ofacetylacetone with 2-hydrazinopyridine. The ligand,obtained as a light yellow liquid (b.p. 74''C. O.4mm ofHg), was characterised by IR. NMR and massspectra 5.
The ligand (DMPP, I) and the metal salt (uranylnitrate or thiocyanate) were mixed in appropriatestoichiometric ratio (I: I or 2: I) and the mixture wasrefluxed for 3 hr. The solvent was then concentratedand the resulting solution slowly cooled to obtainyellow or orange-yellow crystalline materials. Thesewere collected on a frit, washed with ethanol and thenwith ether and finally dried in vacuo. These complexesare easily soluble in common organic solvents.
[UOiN03h(DMPp)]This compound is obtained as bright-yellow
crystalline plates (m.p. nOT) [Calc. forUOsCloH IINs (M.W. S67): C, 21.16; H, \.94; N,12.34. Found C, 21.13; H, 2.04; N, 12.41%]. Thecompound is a non-electrolyte in MeOH at 21"C, AM
602
=28.2mhoscm2mol-I.A I: I electrolyte in MeOHhas a AM value" of '" 120-140 mhos em? mol -I.
[U02(NCS)z{DMPP)2]This compound is orange-yellow crystalline
(m.p.166°q. [Calc. for U02C22HnNsS2 (M.W. 732):C, 36.06; H, 3.00; N, IS.30. Found C, 36.18;H, 3.03; N,IS. 12%]. The compound is a non-electrolyte in MeOHat 21°C, AM=74.81mhoscm2mol-l. Somewhathigher AM value is obtained due to partial solvolysis.
IR spectra (4000-200cm -I) were recorded in nujolon a Beckmann-12 double-beam spectrophotometer.Mass spectra were recorded on a Varian MAT SM-lBH spectrometer operating at an electron energy of70eV. The IH NMRspectra were measured at 60MHzwith a Varian EM 360 equipment using TMS as anexternal standard. The 13C NMR spectra wererecorded on a JEOL FX-60 spectrometer operating inthe F.T. mode at IS.08 MHz. The probe temperaturewas 40 ± IDC with broad band decoupling. The shiftsare taken relative to internal CD30D resonating at47.041 ppm down field to TMS7.
The two complexes synthesised viz.,[UOiN03h(DMPP)] and [UOz{NCSh(DMPPh]are shown to be eight-coordinated on the basis ofanalytical data, electrical conductance and vibrationalas well as resonance spectroscopy. The presence of alinear UO~ + group is confirmed on the basis of IRbands at 910-920,810 and 2S0-260cm -I correspond-ing to the vas(U-O), vs(U-O) and <5(O-U-O)modes, respectively of the linear UO~ + group'':". Thev(CN) band of UOiNCSh(DMPPh is split intocomponents observed at 20S6 and 2022 em -I; thev(CS) band is observed at 820cm -I and the J(NCS) isobserved at 481 cm -I, all suggesting coordination ofthe - NCS group through the hard - N end 10.
UOiNO 3h(DMPP) exhibits IR bands due to the N03group at IS20, 1479, IOt9, 918 and 820cm-1
indicating its bidentate coordination behaviour 11.The molar conductivity values in MeOH indicate
that the nitrato complex is a non-electrolyte, whereasthe thiocyanato complex has a value somewhat higherthan that expected for a non-electrolyte, althoughsubstantially less than that for a I: I electrolyte". This

situation perhaps arises due to the partial solvolysis ofthe thiocyanato complex in MeOH. (In the solid statethere is no evidence for the presence of ionic CNS fromthe IR spectra).
The 1H NMR spectra show three sharp singlets inthe intensity ratio of 3: 3: I at <51.8, 2.0 and 5.26 ppmcorresponding to the protons of 3-methyl and 5-methylgroups, and the proton at position 4 of the pyrazolering!", respectively. The somewhat downfield shift ofthe 5-methyl signal is perhaps caused by thediamagnetic anisotropic effect of the pyridine nucleus.The characteristic signals in the ligand and thecomplexes at <56.26, 6.92, 7.32 and 7.65 ppm, eachcorresponding to a single proton, are assigned to theprotons at positions 3', 5',4' and 6', respectively, of thepyridine ring'? (Table1).
The proton decoupled 15.08 MHz 13CNMR datafor the ligand and the complexes are identical and poseno problem for assignments (Table 2). The 13C NMRspectrum of [UOiNCSh(DMPPhJ is shown in Fig. I.The 13C shifts have been assigned on the basis of dataobtained using the shift reagents Eutfod), andPr(fodhl3 -18. The protons undergo a somewhatgreater down field shift on complexation as comparedto 13C. As expected, the 13C signals for the carbons at2',3 and 5 positions are relatively weak and the NCScarbons are not observed.
NOTES
9
10
6
8
Fig. 1·_· 13C NMR spectrum of U02(NCS)2(DMPPjz In CDJOD
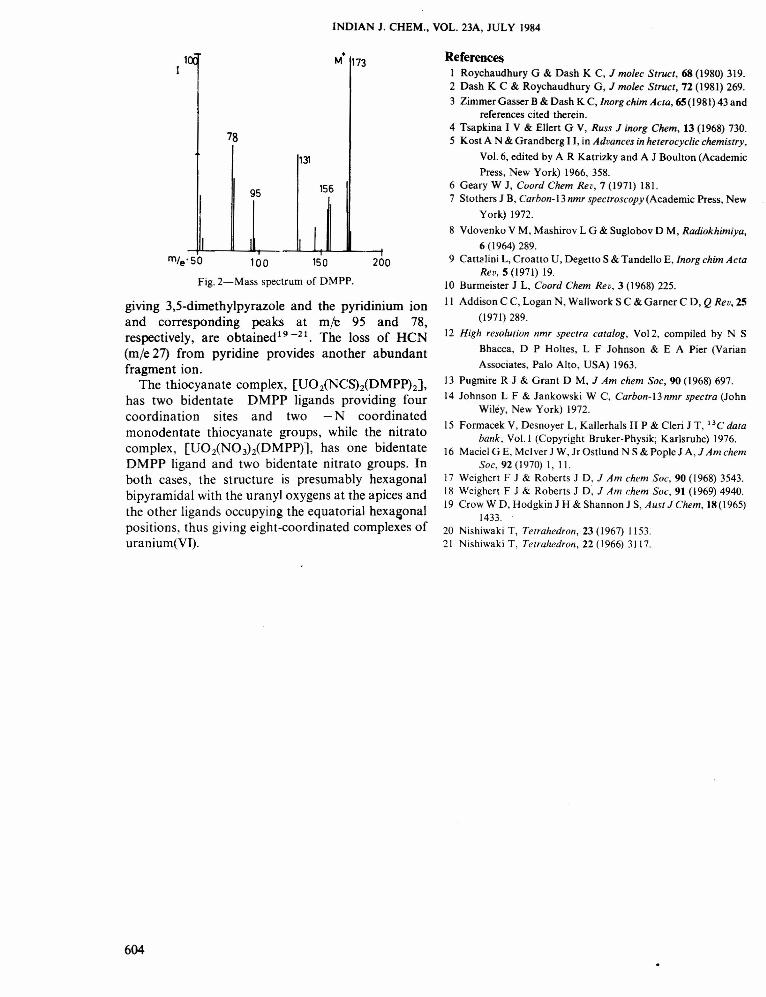
The mass spectra of the complexes do not show themolecular ion peak or any peak assignable to an ionwith the metal ion. In all cases, the molecular ion peaksdue to the ligand and its fragmentation products wereobtained. The molecular ion M + (m/e 173) constitutesthe base peak and an abundant (M - I)+ at m.e 172 is aprominent feature. Subsequent loss of a - CH3 group(m/e 15) and then of a further - CH 3 group alongwitha HCN group (the protonated form of acetonitrile)(m/e 42) give fragments at m/e 158 and 131, respec-tively. The DMPP ligand (Fig. 2) ultimately breaks up
Table I--PMR Data for DMPP and Its Complexes in CD 30D (ppm)':"DMPP,b' DMPP U02(N03jzDMPP U02(NCSliDMPPjz Assignment
1.99 2.03 2.09 2.19 S-Me1.63 1.73 1.85 1.93 .i-Me5.26 5.49 5.69 5.83 4-H
'-I The H-3', H-5', H-4', and H-6' signals are obtain~d at 6.26, 6.926.7.22 and 7.65 ppm, respectively for the neat ligand and around thesame regions for the complexes.
,b'Spectrum of pure neat ligand.
Table2-13CNMR Data for DMPP and Its Complexes in CD30D(ppm)(alSignal DMPP UOiNO,liDMPPl U02(NCSl2(DMPPjz Assignment
No.1. 151.97 152.10 151.97 C-c2. 149.24 149.31 149.24 C-33. 146.77 146.84 146.77 C-64. 140.92 141.05 140.92 Co)5. 137.93 137.93 137.93 C-46. 120.85 120.85 120.85 C-57. 115.78 115.84 115.78 C-J8. 107.52 107.59 107.52 C-49. 11.64 11.76 11.69 3- and
10. 11.24 11.24 11.24 S-methylcarbon
{a} All values in ppm relative to internal CD30D resonating at47.041 ppm from TMS.
--------_.-_._-----
603
INDIAN J. CHEM., VOL. 23A, JULY 1984
+M 173HXr
78131
15695
II
100 150 200
Fig. 2-Mass spectrum of DMPP.
giving 3,5-dimethylpyrazole and the pyridinium ionand corresponding peaks at mle 95 and 78,respectively, are obtained 19-21. The loss of HCN(m/e 27) from pyridine provides another abundantfragment ion.
The thiocyanate complex, [UOz{NCShCDMPPh],has two bidentate DMPP ligands providing fourcoordination sites and two - N coordinatedmonodentate thiocyanate groups, while the nitratocomplex, [UOz{N03)2(DMPP)], has one bidentateDMPP ligand and two bidentate nitrato groups. Inboth cases, the structure is presumably hexagonalbipyramidal with the uranyl oxygens at the apices andthe other ligands occupying the equatorial hexagonalpositions, thus giving eight-coordinated complexes ofuranium(VI).
604
References1 Roychaudhury G & Dash K C, J molec Struet, 68 (1980) 319.2 Dash K C & Roychaudhury G, J molee Struct, 72 (1981) 269.3 Zimmer Gasser B & Dash KC,lnorgchim Acta, 65(1981)43 and
references cited therein.4 Tsapkina I V & Ellert G V, Russ J inorg Chem, 13 (1968) 730.5 Kost A N & Grandberg r I, in Advances in heterocyclic chemistry,
Vol. 6, edited by A R Katrizky and A J Boulton (Academic
Press, New York) 1966, 358.6 Geary W J, Coord Chem Rev, 7 (1971) 181.7 Stothers J B, Carbon-13 nmr spectroscopy (Academic Press, New
York) 1972.
8 Vdovenko V M, Mashirov L G & Suglobov 0 M, Radiokhimiya,6 (1964) 289.
9 Cattalini L, Croatto U, Degetto S & Tandello E, Inorg chim ActaRev,S (1971) 19.
10 Burmeister J L, Coord Chem Rev, 3 (1968) 225.
11 Addison C C, Logan N, Wallwork S C & Garner C 0, Q Rev, 25(1971) 289.
12 High resolution nmr spectra catalog, Vol2, compiled by N S
Bhacca, D P Holtes, L F Johnson & E A Pier (Varian
Associates, Palo Alto, USA) 1963.
I3 Pugmire R J & Grant D M, JAm chem Sac, 90 (1968) 697.
14 Johnson L F & Jankowski W C, Carbon-I3nmr spectra (JohnWiley, New York) 1972.
15 Formacek V, Desnoyer L, Kallerhals H P & Cieri J T, DC databank, Vol. I (Copyright Bruker-Physik; Karlsruhe) 1976.
16 Maciel G E, McIver J W,JrOstlund N S & PopleJ A,J AmchemSoc, 92 (1970) I, II.
17 Weigher! F J & Roberts J 0, J Am chem Soc, 90 (1968) 3543.18 Weighert F J & Roberts J 0, JAm chem Sac, 91 (1969) 4940.19 Crow W D, Hodgkin J H & Shannon J S, Aust J Chem, 18 (1965)
1433.20 Nishiwaki T, Tetrahedron, 23 (1967) 1153.21 Nishiwaki T, Tetrahedron, 22 (1966) 3117.


















