
New genetic therapies for inherited neuromuscular diseases
31
New genetic therapies for inherited neuromuscular diseases Francesco Muntoni Dubowitz Neuromuscular Centre UCL Institute of Child Health & Great Ormond Street Hospital London, UK
Transcript of New genetic therapies for inherited neuromuscular diseases

New genetic therapies for inherited neuromuscular diseases
Francesco MuntoniDubowitz Neuromuscular Centre
UCL Institute of Child Health& Great Ormond Street Hospital
London, UK

Francesco Muntoni: disclosuresDuchenne trials• CI of AON clinical trials with AVI / Sarepta. • PI of Prosensa / Biomarin sponsored AON trials• PI of PTC phase II and III sponsored trials. • PI of Pfizer phase II clinical trial (myostatin inhibition)• CI of Summit Phase I and II trials. • CI of Esperare Phase I trial
Spinal Muscular Atrophy trials
• PI of Trophos SMA III trial• PI of Ionis/ Biogen AON Phase III study
• Other• Avexis, Biogen, Sarepta, Pfizer, PTC, Roche, Summit, Wave ad-hoc SAB

Dramatic impact of genetic therapies for Duchenne muscular dystrophy (DMD)and Spinal Muscular Atrophy (SMA)
Dealing with mutant RNA. Splice-switching antisense oligonucleotides for:
- exon skipping for DMD deletions- exon inclusion in SMA
Replacing genes with gene therapy
- AAV gene therapy for DMD- AAV gene therapy for SMA type I

Topics discussed
• Why antisense oligomers for DMD and SMA?
• Emerging data from AAV gene therapy
• Implications for clinical practice

• Most common muscular dystrophy of childhood
• X-linked, 1 in 3,500 male births• 26,000 new cases each year, ~ 100 in UK• De novo mutations hamper genetic
efforts to reduce incidence• Economic cost: at least $ 120,000 per
year
The two most common neuromuscular diseases in children
DMD SMA
• Most common lethal genetic disorder of childhood
• Autosomal recessive, 3 different levels of severity
• Cumulative incidence: 1 in 6,500 -1,10.000 births
• Heterozygous carrier frequency: 1:37 – 1: 60 in Caucasian populations

• Onset in first years of life, however mean age of diagnosis 4.5 years
• Progressive weakness affecting legs> arms• Inability to run, progressive difficulties
raising from the floor, going upstairs• Grossly elevated creatine kinase (CK)• Loss of ambulation 9.513.5 years (steroids)
Main clinical features
DMD SMA 1• Onset by 6 months of age• Diagnostic delay of ~2.5 months• Sitting posture never acquired• Severe respiratory muscle weakness• Feeding difficulties• Mean age of survival 9 months
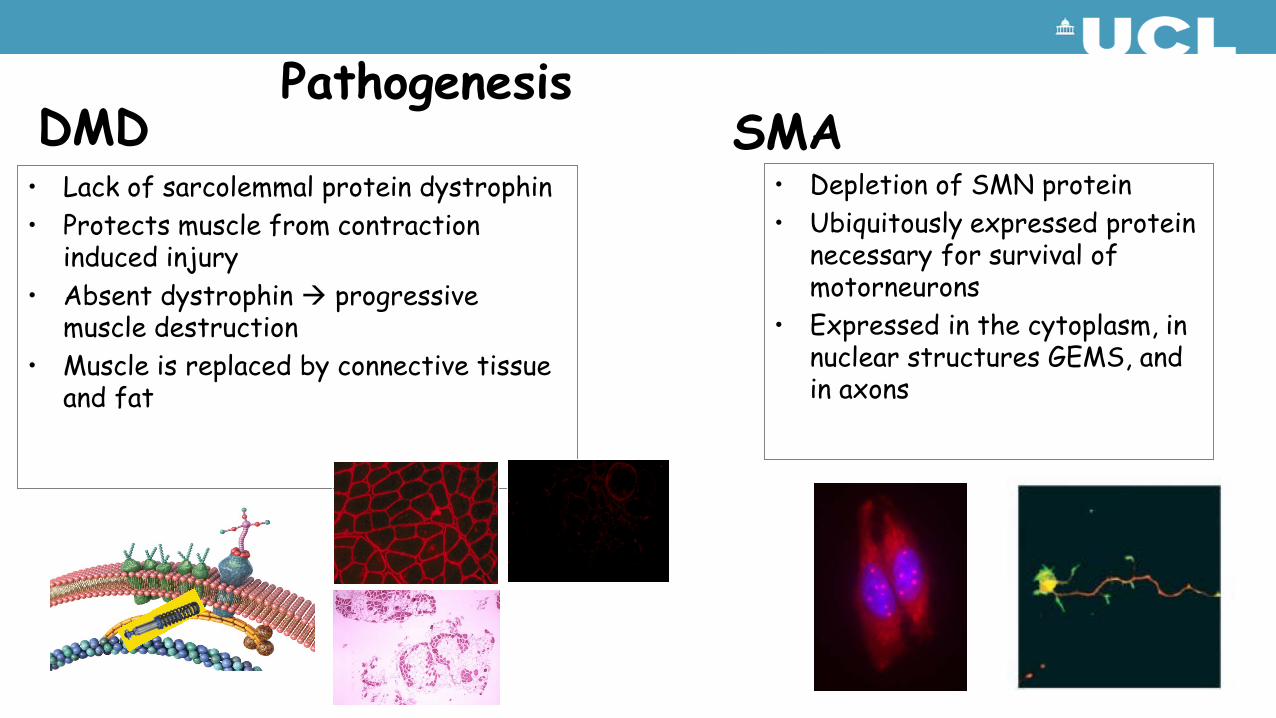
• Lack of sarcolemmal protein dystrophin• Protects muscle from contraction
induced injury• Absent dystrophin progressive
muscle destruction• Muscle is replaced by connective tissue
and fat
PathogenesisDMD SMA
• Depletion of SMN protein• Ubiquitously expressed protein
necessary for survival of motorneurons
• Expressed in the cytoplasm, in nuclear structures GEMS, and in axons

DMD genetics• 65% out-of-frame DMD deletions• 10% out-of-frame duplications• 10-15%% nonsense mutations• Remaining: small mutations
Becker MD genetics
In framedeletions

Antisense oligonucleotides to
induce exon skipping 52
Antisense oligonucleotides: to modify splicing of the DMD gene
Skipping exon 51: 15% of all DMD boys carrying deletions
Ann Neurol. 2013;74:637-47.
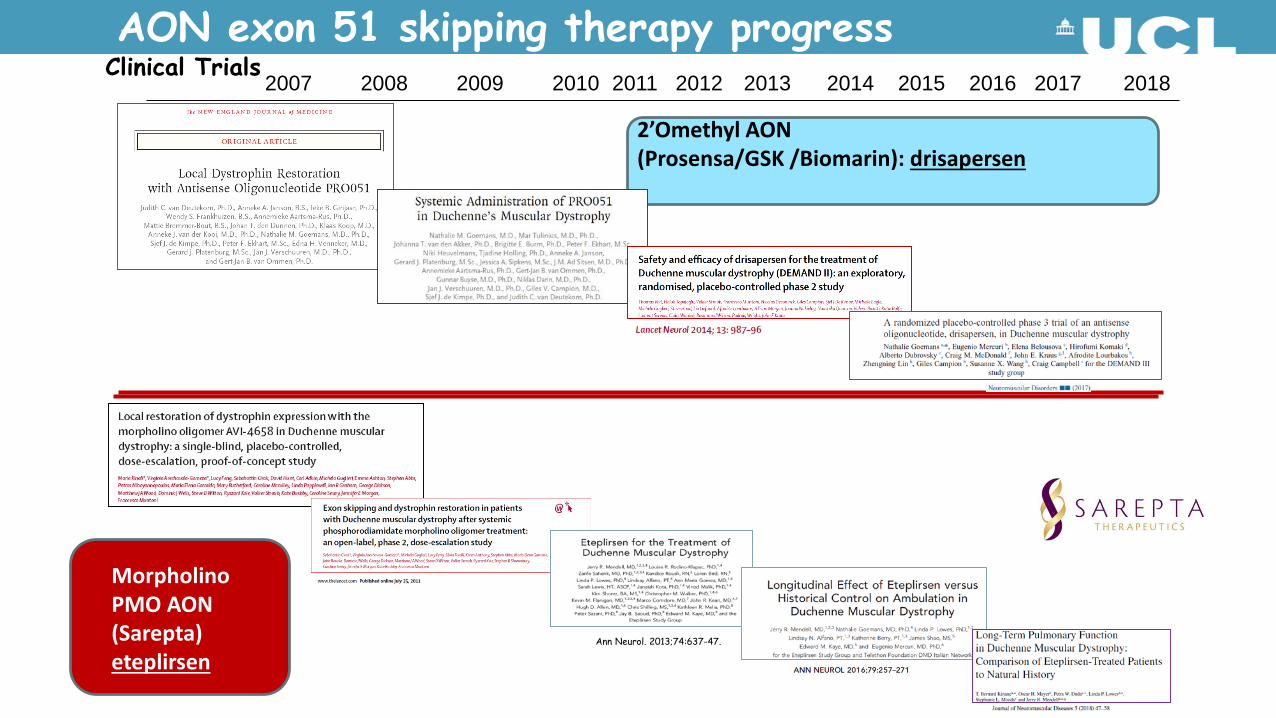
2’Omethyl AON(Prosensa/GSK /Biomarin): drisapersen
Morpholino PMO AON(Sarepta)eteplirsen
AON exon 51 skipping therapy progress2007 2008 2009 2010 2011 2012 2013 2014 2015 2016 2017 2018
Clinical Trials
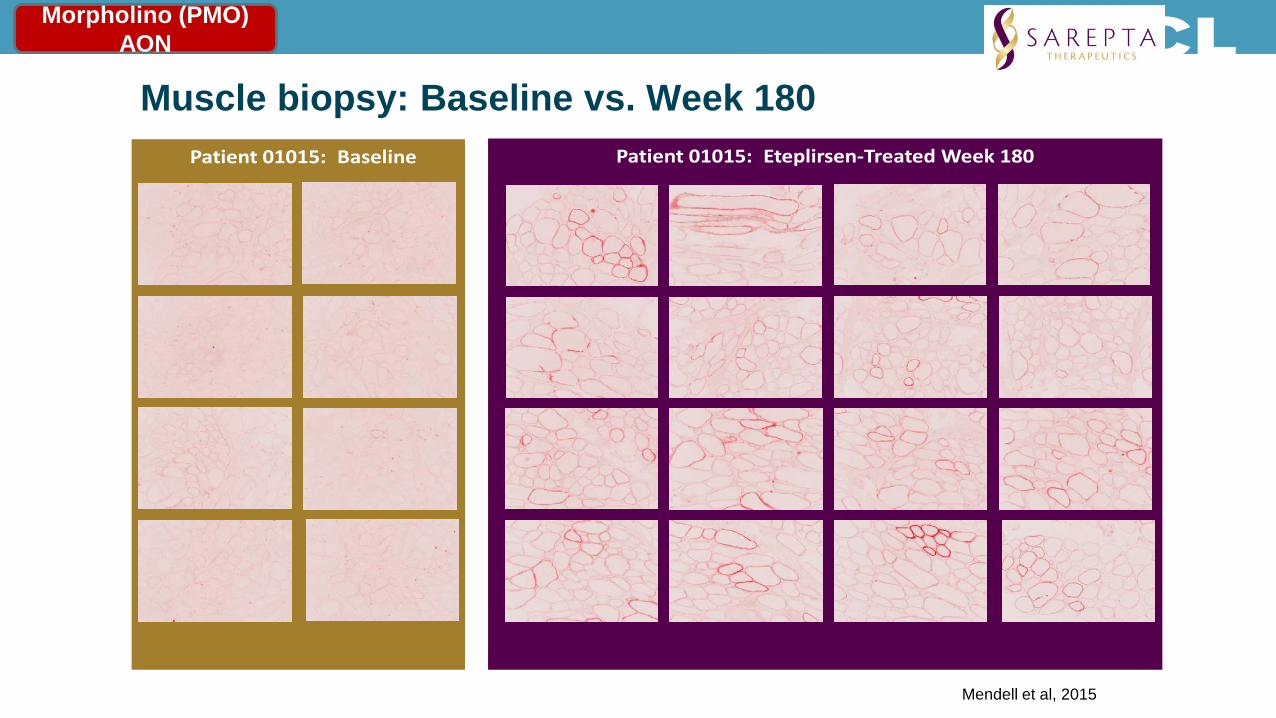
Muscle biopsy: Baseline vs. Week 180 Patient 01015: Eteplirsen-Treated Week 180Patient 01015: Baseline
Mendell et al, 2015
Morpholino (PMO) AON

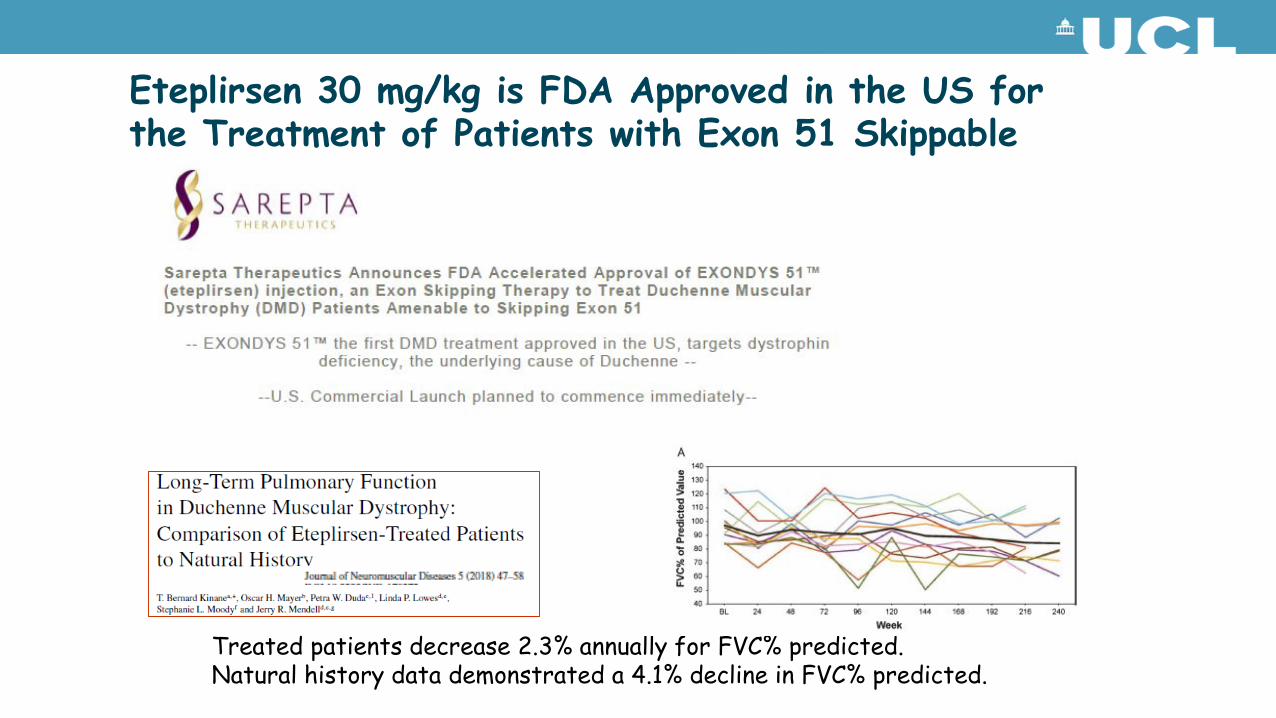
Eteplirsen 30 mg/kg is FDA Approved in the US for the Treatment of Patients with Exon 51 Skippable DMD
Treated patients decrease 2.3% annually for FVC% predicted.Natural history data demonstrated a 4.1% decline in FVC% predicted.

AON for DMD: rapidly evolving field:
1. Targeting additional exons to extend this approach to patients with other deletions
2. Next generation AONs

Targeting other DMD exons: SKIP-NMD Consortium
• Advocacy groups, research, healthcare providers and industry
• Francesco Muntoni project lead (UCL). Individual site leads are as follows:
• Institut de Myologie (Laurent Servais)• University of Newcastle upon Tyne (Volker Straub)• Università Cattolica del Sacro Cuore (Eugenio Mercuri)• Royal Holloway & Bedford New College (George Dickson)- in
vitro sequence selection & steering
Consultants for Research in Imaging & Spectroscopy

16
Increase in Sarcolemma localization of newly expressed dystrophin
BASELINE BIOPSY
ON-TREATMENTBIOPSY: Wk 48
Note: Indirect immunofluorescence staining of tissue cryosections was performed using MANDYS106 and anti-laminin 2 alpha antibodies

Targeting exons other than 51
Mor
phol
ino
AON
Nex
t gen
erat
ion
AO
N
AON: where is the field going
DMD
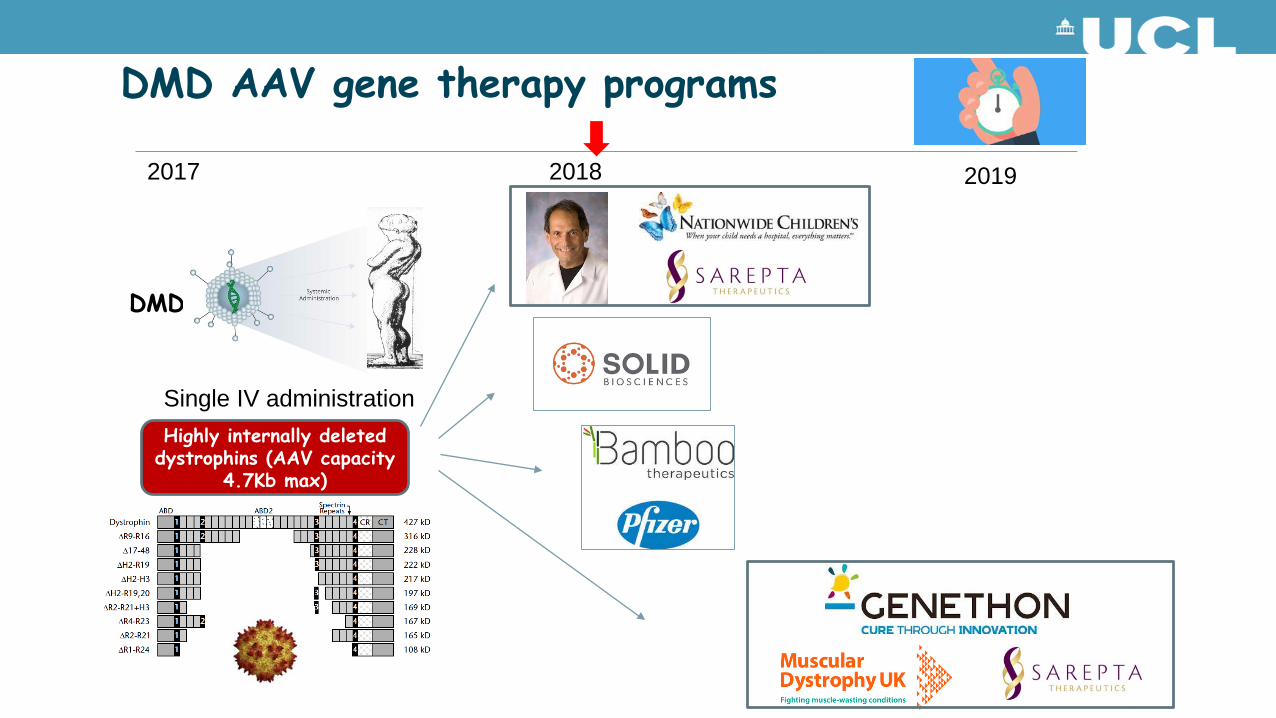
DMD AAV gene therapy programs
2017 2018 2019
Highly internally deleted dystrophins (AAV capacity
4.7Kb max)
Single IV administration


Full-length
Chromosome 5
SMN1
SMN2
SMN-FL SMND7 SMN-FL
SMN1SMN2
Full-length38kD
Truncated & less stable
~10% ~90% ~100%
Gene
mRNA
Protein
SPINAL MUSCULAR ATROPHY
AONs to induce exon 7 inclusion
SMN2 Gene
1 2 2 3 4 5 6 8 SMN2 mRNA
1 2a 2b 3 4 5 6 7 8
C to T
Functional Protein
7
IONIS-SMNRx

Intratechal administration of the antisense oligonucleotide spinraza
• AONs do not cross the blood brain barrier• Intratechal administration required• After a loading phase (4 IT injection in 2 months),
the long half life of the AON enables infrequent dosing

Motor performance
Infants on nusinersen achieved motor milestones unexpected for individuals with Type I SMA
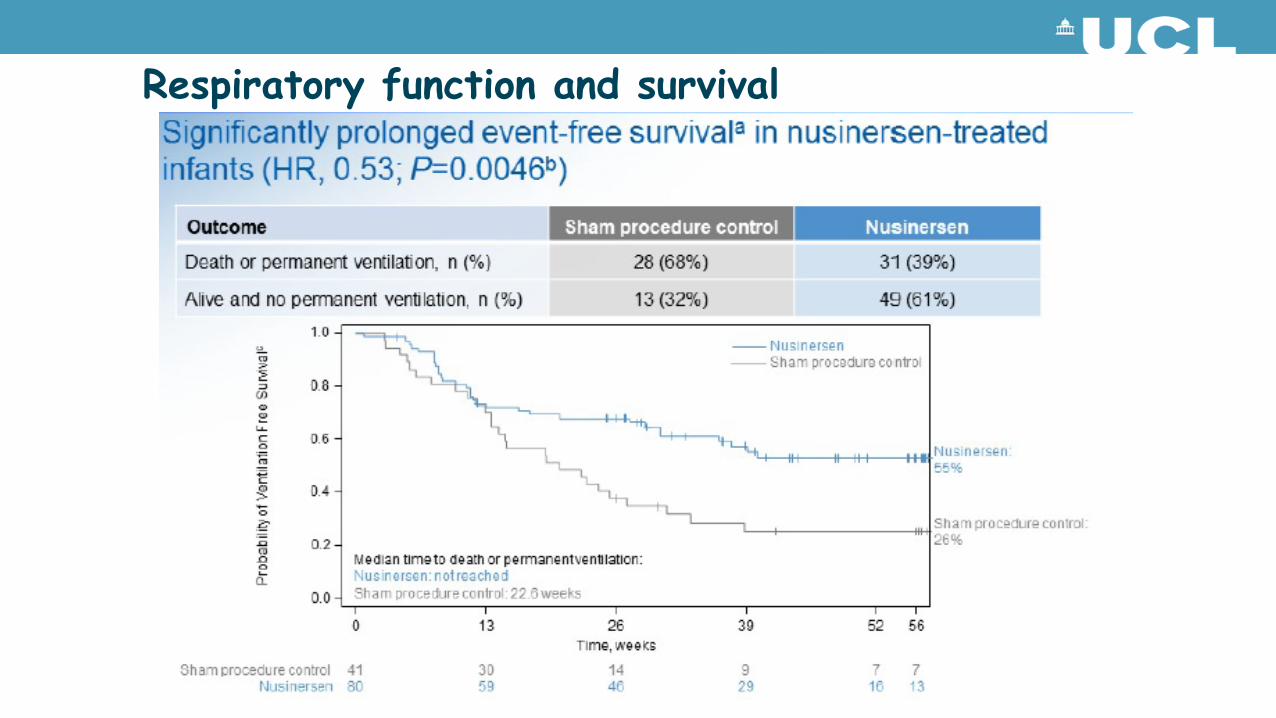
Respiratory function and survival



AAV9 for SMA gene therapy
Jerry Mendell (Columbus, Ohio) is pursuing AAV gene therapy in infants with SMA I
Single IV injection
Two doses used:A. Cohort 1: 6.7x1013 vg/kgB. Cohort 2: 2.0x1014vg/kg
Inclusion• 9 months/6 months of age¹ or younger at
infusion• Bi-allelic SMN1 gene deletions or point
mutations• 2 copies of SMN2• Onset of disease at birth to 6 months of
age

COHORT 2 (n=12)Baseline Age (months): 3.1 [median], 3.4 [mean]Current Age (months): 20.2 [median], 20.7 [mean]Mean CHOP INTEND Increase: 24.7 points
Dashed lines for individual patients denote missed or partial CHOP-INTEND assessments
Early intervention and dose appear to affect response
0
10
20
30
40
50
60
0 3 6 9 12 15 18 21 24 27 30Age (months) 1 month = 30 days
60
50
40
2/12
10/12
11/12Cohort 2 (proposed therapeutic dose)
Rapid Response in Cohort 2• CHOP INTEND Increase at Month 1: 9.8 [mean]
• CHOP INTEND Increase at Month 3: 15.4 [mean]
Max score is 64
AVXS-101: CHOP-INTEND Motor Function Scores

Cohort 2 Achieved Motor Milestones Not Seen in the Natural History of SMA Type 1

Concluding remarks (1)• Outstanding progress in the therapeutic approaches for DMD and
SMA in the last few years
• Second generation AON therapies now in clinic trials, and AAV gene therapy gaining rapid traction
• The size of therapeutic response in SMA is unprecedented, but also linked to the interval between symptom onset and initiation of treatment
• Reducing the diagnostic delay for these conditions now has very tangible implications
• Consider newborn screening

Concluding remarks (2)• While success and tolerability of these approaches encourages their
continuing development, both their long term safety, and cost/ benefit will require careful analysis
• The cost of some of these intervention is considerable, and this might preclude their access to our patients
• NHSE and NICE do not currently have a swift system to allow the assessment of emerging therapies for these devastating childhood conditions
• While cautious and measured counselling to families is imperative, there are reasons to change from pessimism to optimism for these patients, although there is a lot more work to do

AcknowledgmentClinical and research team atGOSH/ ICH
Matthew Wood, OxfordGeorge Dickson, RHUL


















