
Neuropathology of vascular cognitive impairment · Neuropathology of vascular cognitive impairment...
61
Neuropathology of vascular cognitive impairment Prof. Isidro Ferrer, Institut Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital Universitari de Bellvitge, Universitat de Barcelona, CIBERNED, Hospitalet de LLobregat; Spain
Transcript of Neuropathology of vascular cognitive impairment · Neuropathology of vascular cognitive impairment...

Neuropathology of vascular cognitive impairment
Prof. Isidro Ferrer, Institut Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital Universitaride Bellvitge, Universitat de Barcelona, CIBERNED, Hospitalet de LLobregat; Spain

Démence vasculaire. Forme diffuse sénile

Démence vasculaire. Forme diffuse sénile (A). Forme thalamique (B). Forme postérieure (C). Démencesénile mixte (D)
A
B C
D

Cerebral infarction: T2, FLAIR
Carothid thrombosis

Acute infarct of the territory of the middle cerebral artery. Kluver-Barrera

Acute stage: neuron shrinkage Subacute stage: reactive astrocytes, macrophages
Cellular responses following ischemia and infarction. Necrosis is the mechanism of cell death in the infarct core

Cell death at the penumbra of the area of infarction is accompanied by Bax translocation to the mitochondria, cytochrome C release from the mitochondria to the cytosol, caspase 3 activation and cells with apoptotic morphology
Cell death at the penumbra has molecular and morphological features of caspase-mediated apoptosis
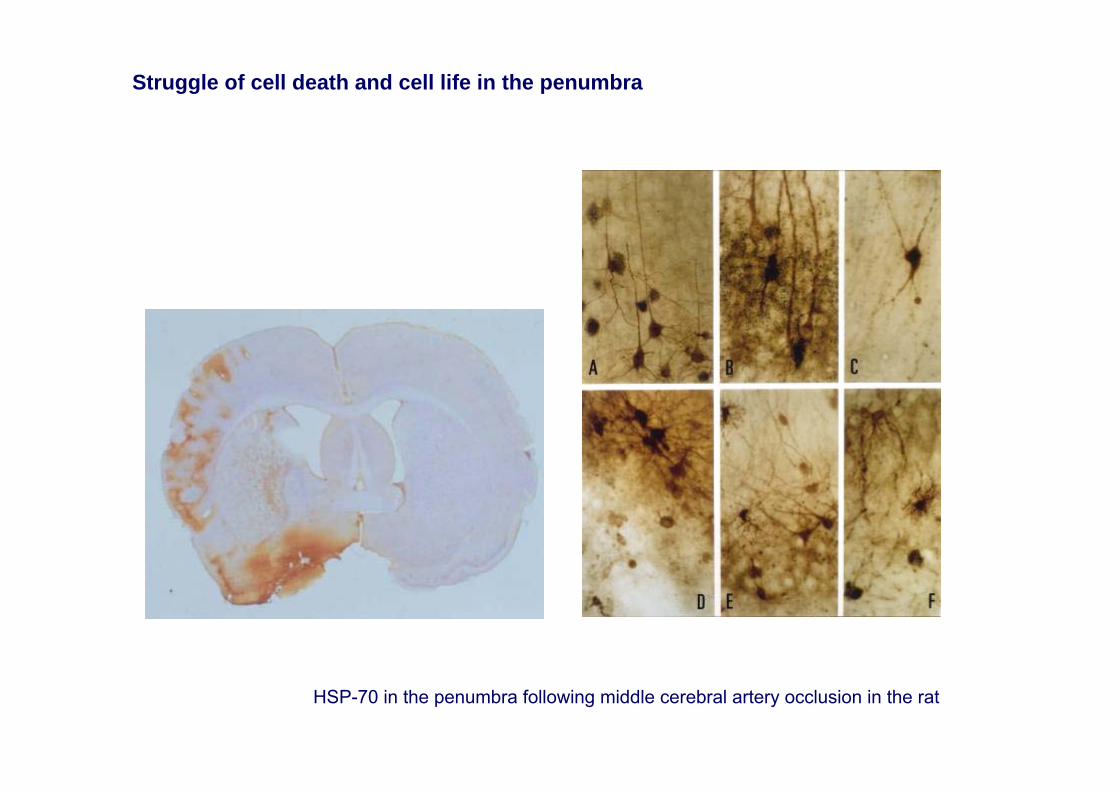
HSP-70 in the penumbra following middle cerebral artery occlusion in the rat
Struggle of cell death and cell life in the penumbra

Cerebral edema. Involvement of astrocytes

Cerebral edema. Involvement of the oligodendroglia and myelin

Necrotizing edema
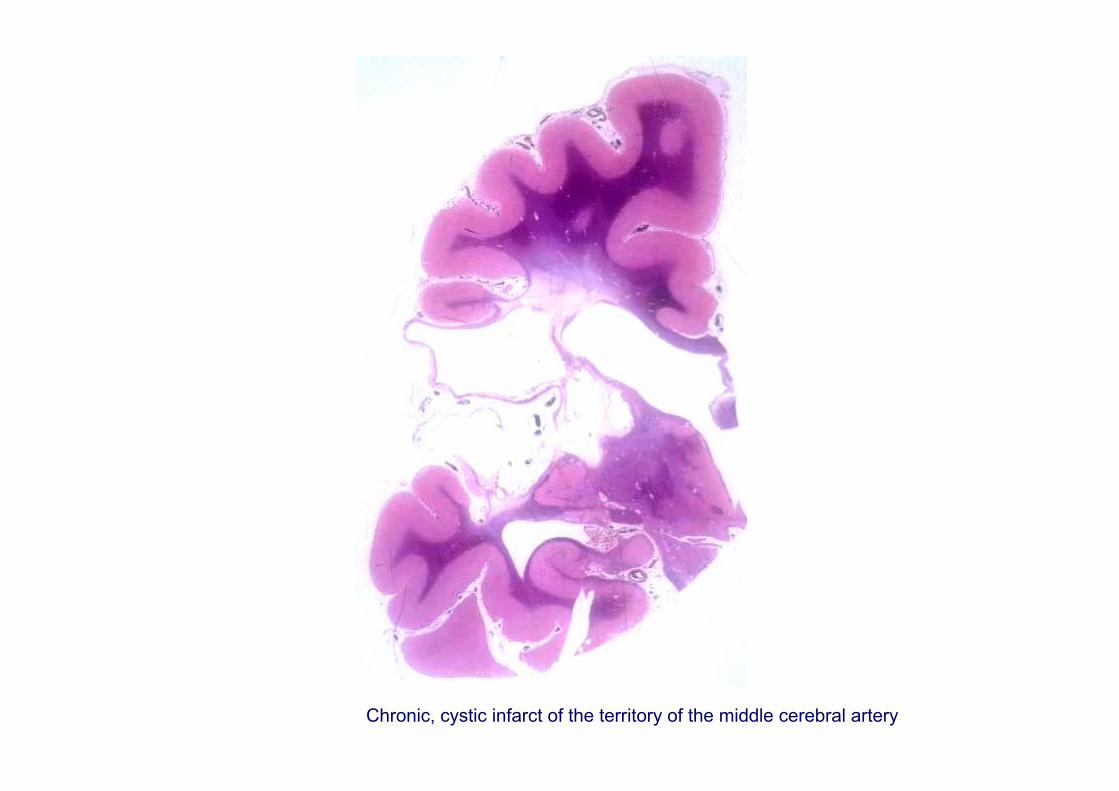
Chronic, cystic infarct of the territory of the middle cerebral artery
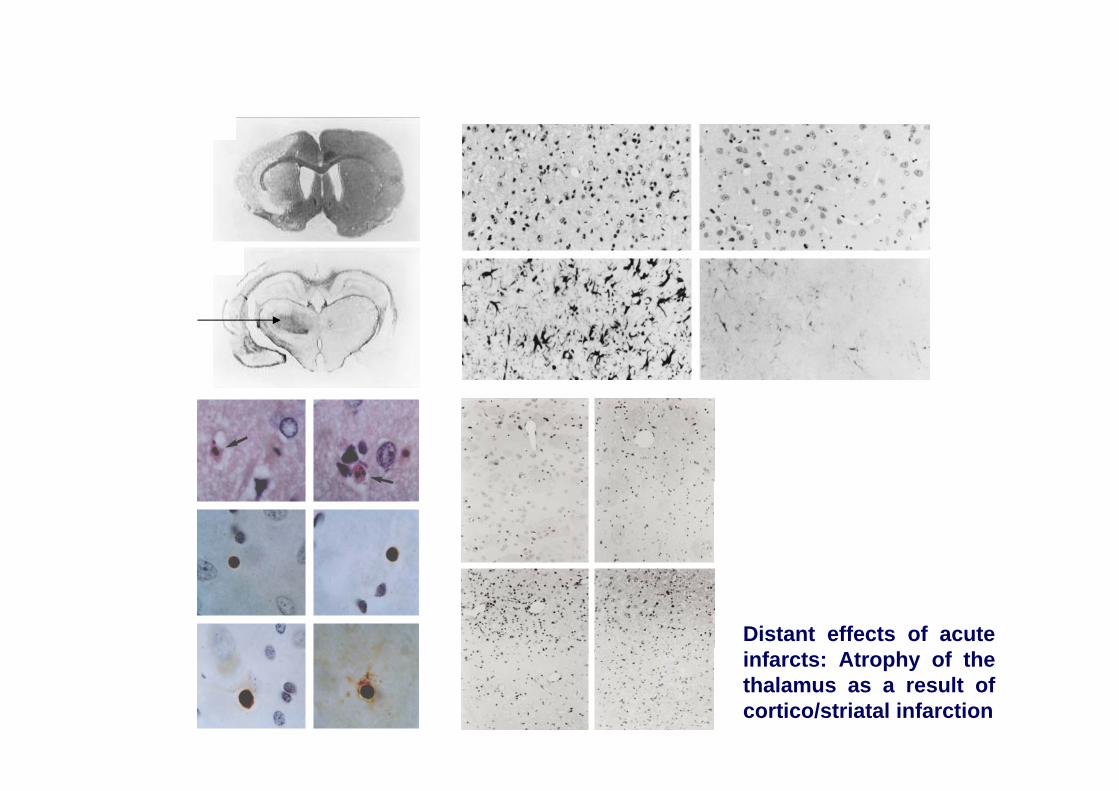
Distant effects of acute infarcts: Atrophy of the thalamus as a result of cortico/striatal infarction

Dementia post-ischemia
Causes: global ischemia resulting from cardiac arrest, or severe hypotension, including accidents during anesthesia
Condicioning factors: transient or permanent ischemia, duration of the ischemia, degree of ischemia, body temperature, glucose levels
Distribution of lesions: a. pattern of watershed areas of the large brain blood vesselsb. Generalized involvement involving the hippocampus (Sommer sector), cerebral
cortex, striatum, thalamus and Purkinje cells of the cerebellumc. Mixed patter d. Selective involvement of the cerebral white matter

Dementia post-ischaemia following cardiac arrest. Involvement of the cerebral cortex, basal ganglia and cerebellum

Dementia post-ischemia following cardiac arrest with a pattern of major involvement of watershed areas of the territories of the cerebral arteries
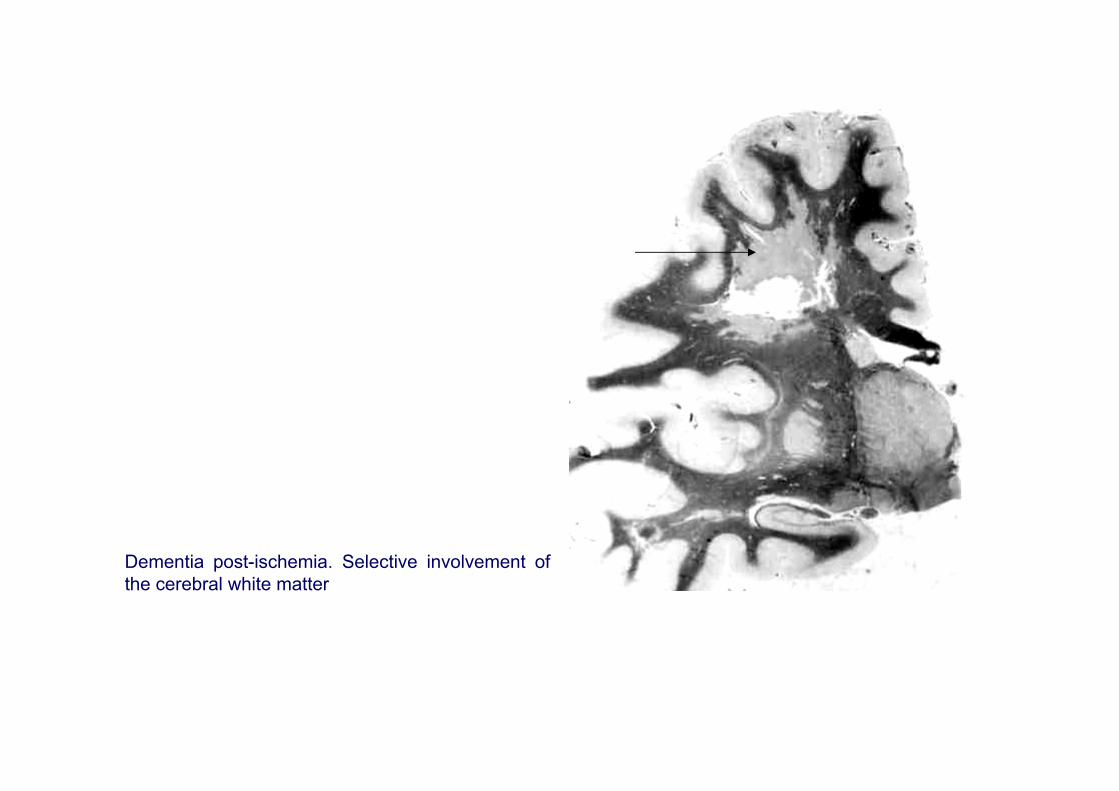
Dementia post-ischemia. Selective involvement of the cerebral white matter

Post-anoxic brain. Cerebral edema, nerve cell death, spongiosis
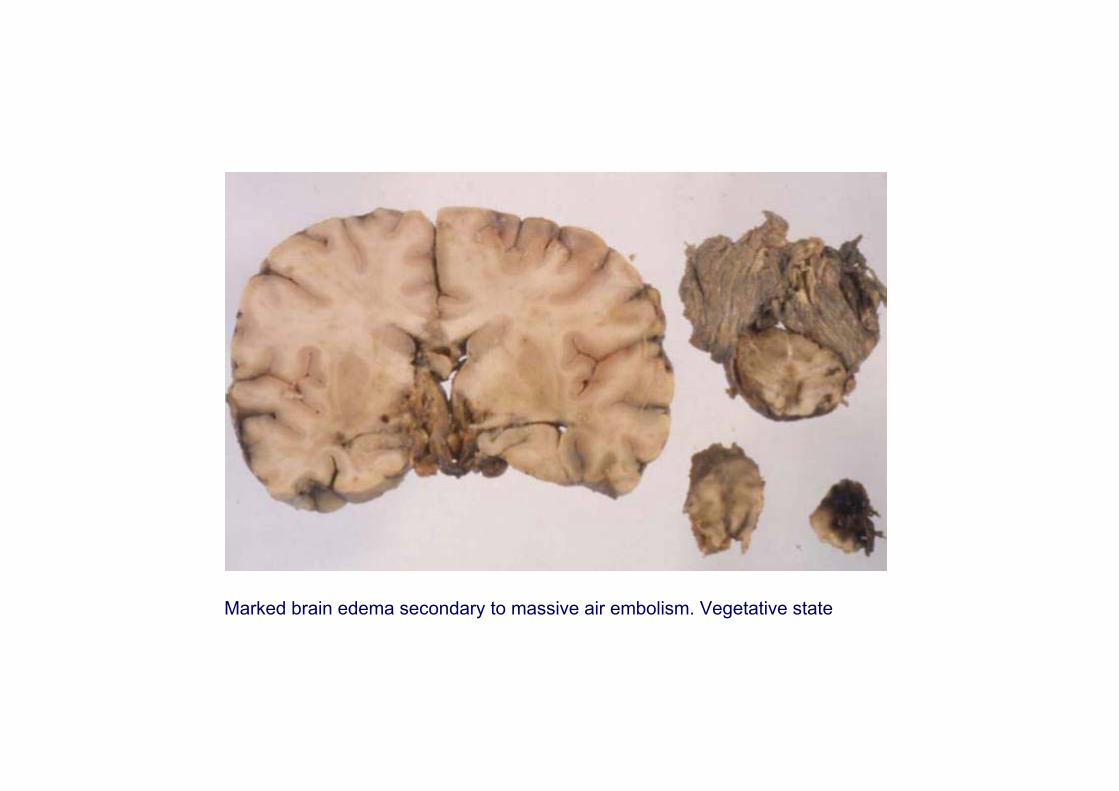
Marked brain edema secondary to massive air embolism. Vegetative state

Diseases of the blood vessels that may cause cognitive impairment anddementia of vascular origin
1. Atherosclerosis2. Hypertensive angiopathy3. Small blood vessel disease4. Inflammatory diseases of the blood vessels5. Infectious diseases of the blood vessels6. Sneddon syndrome7. Amyloid angiopathies8. CADASIL9. CARASIL (Maeda syndrome)10. Other diseases of the blood vessels
•Hereditary endotheliopathy with retinopathy, nephropathy and stroke (HERNS)•Cerebroretinal vasculopathy (CRV)•Hereditary vascular retinopathy (HVR) (linked to 3p21.1-p21.3)•Hereditary infantile hemiparesia with arteriolar retinopathy and leukoencefalopathy (HIHRATL) (notlinked to 3p21)•Fibromuscular dysplasia•Moya-moya disease: • sporadic (90%)
• familiar (linked to 3p24.2-p26, 6, 17)11.Vascular malformations12. Neoplastic disesaes13. Mitochondrial encephalopathies: MELAS

Multi-infarct encephalopathy
Lacunes
Binswanger disease (syndrome)
Mesial sclerosis of circulatory origin
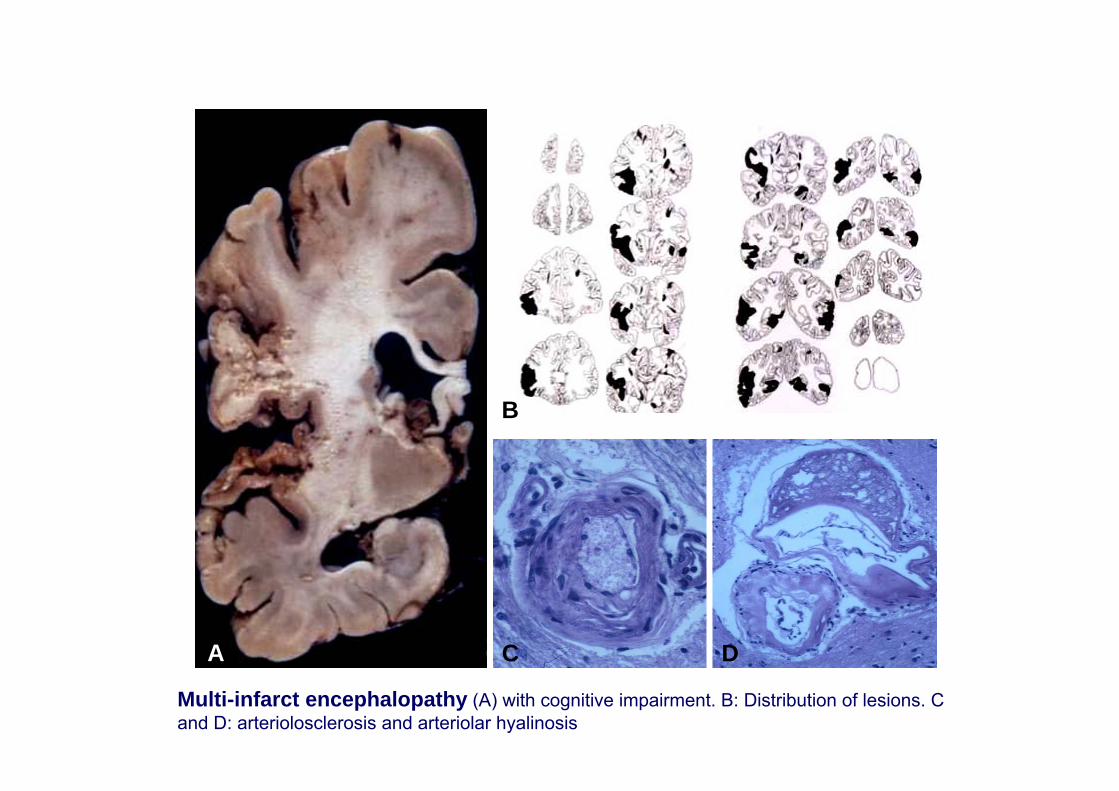
A D
B
C
Multi-infarct encephalopathy (A) with cognitive impairment. B: Distribution of lesions. C and D: arteriolosclerosis and arteriolar hyalinosis
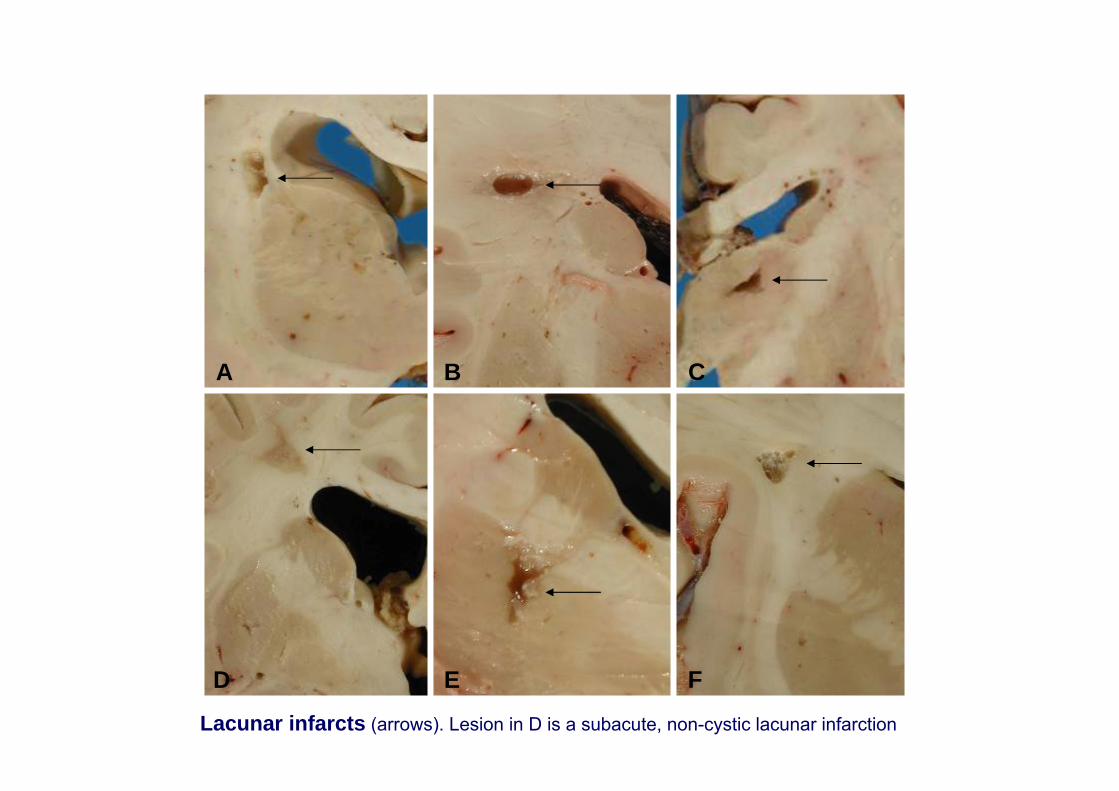
D E F
CBA
Lacunar infarcts (arrows). Lesion in D is a subacute, non-cystic lacunar infarction

Perforant blood vessels

A. Status cribosus; B. Lacuna; C and D: Perivascular dilatations

0
20
40
60
80
100
120Porcentaje
30-40 40-50 50-60 60-70 70-80 80-90
Edad (años)
Palidez sust blanca Hialinosis Arteriosclerosis
Estado criboso Lagunas PS y DNFhyalinosis
lacunesatherosclerosis senile plaques and neurofibrillary tanglesstatus cribosus
age (years)
white matter involv

Bilateral temporal infarction involving the auditory area.
Multiple bilateral lacunes in the cerebral white matter, striatum and thalamus.
Progressive cognitive impairment

++Infarcts in the territory of thepostero-inferior cerebellar artery(PICA)
++Pontine lacuna
++/+++++Leukoencephalopathy
+/+++/-Infarcts in the territory of theposterior cerebral artery
+++ bilateral++/+++ unilateralInfarcts in the territory of theSylvian artery
++/+++ +/-Thalamic infarcts
++/++++/-Infarcts in the corpus callosum
++/++++/-Infarcts in the hippocampus
++/+++++Striatal infarction
++++Lacuna in the centrum semiovale
With dementiaWithout dementiaType of lesion
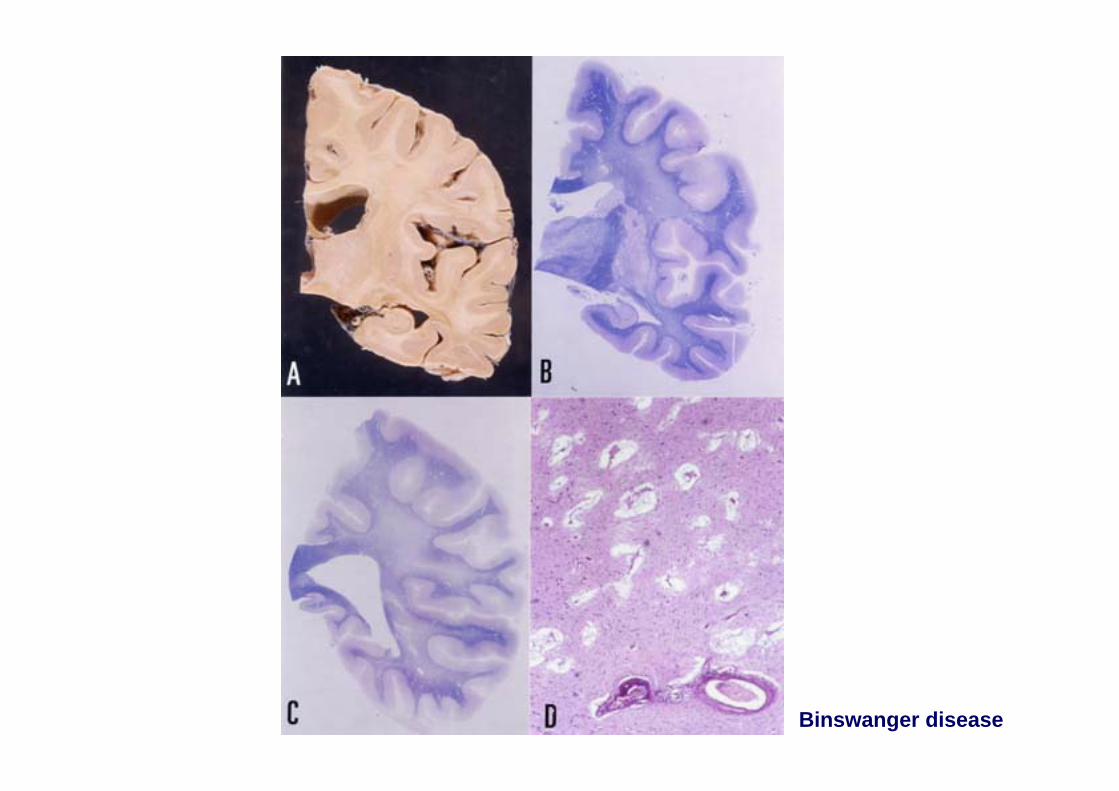
Binswanger disease

A B
Hippocampal sclerosis

Cerebral amyloid angiopathies with dementia
Sporadic forms:
Alzheimer’s disease (AD)Sporadic cerebral amyloid angiopathy (SCAA)
Familial forms:
Familial Alzheimer’s disease linked with mutations in AβPP, PS1, PS2Hereditary cerebral hemorrhage with amyloidosis Dutch type (HCHWA-D): AβPP
mutation (codón 693)Hereditary cerebral hemorrhage with amyloidosis, Icelandic type (HCHWA-I):
mutaciones in Cyst CFamilial amyloid polyneuropathy/meningo-vascular amyloidosis (FAP/MVA) associated
with mutations in the transthyretin (TTR) genePrion diseases with cerebral amyloid angiopathy associated with PRNP mutationsFamilial amyloidosis Finnish type (FAF) associated with mutations in the gene of
gelsolinAmyloid angiopathies associetd with mutations in the gene BRI (chromosome 13)
• Familial British dementia (FBD)• Familial Danish dementia (FDD)

A
D
CB
FE G
Amyloid angiopathy. A-C: Severe involvement of the cerebral white matter. D: βA1-17; E: βA1-24; F: βA1-40; G: βA1-42

Familial cerebral amyloidosis; familial Alzheimer’s disease
Cause: Mutaciones en AβPP (codons 692-694), PS1, PS2
Clínical manifestations:
A692G in AβPP (Flamish mutation): cerebral hemorrhages and dementia
E693K in AβPP (Italian mutation): cerebral hemorrhages and dementia
E693G in AβPP (Artic mutation): dementia
D694N in AβPP (lowa mutation): dementia and leukoencephalopathy
N141I in PS2 (Volga-German mutation): dementia
Mutations beyond codon 200 in PS1: dementia
Δ9 y ΔI83/ΔM84 en PS1: dementia, spastic paraparesis
Patology: Alzheimer’s disease and amyloid angiopathy with βA4 and cystatin C deposition. Δ9 y ΔI83/ΔM84 en PS1: cotton wool amyloid plaques
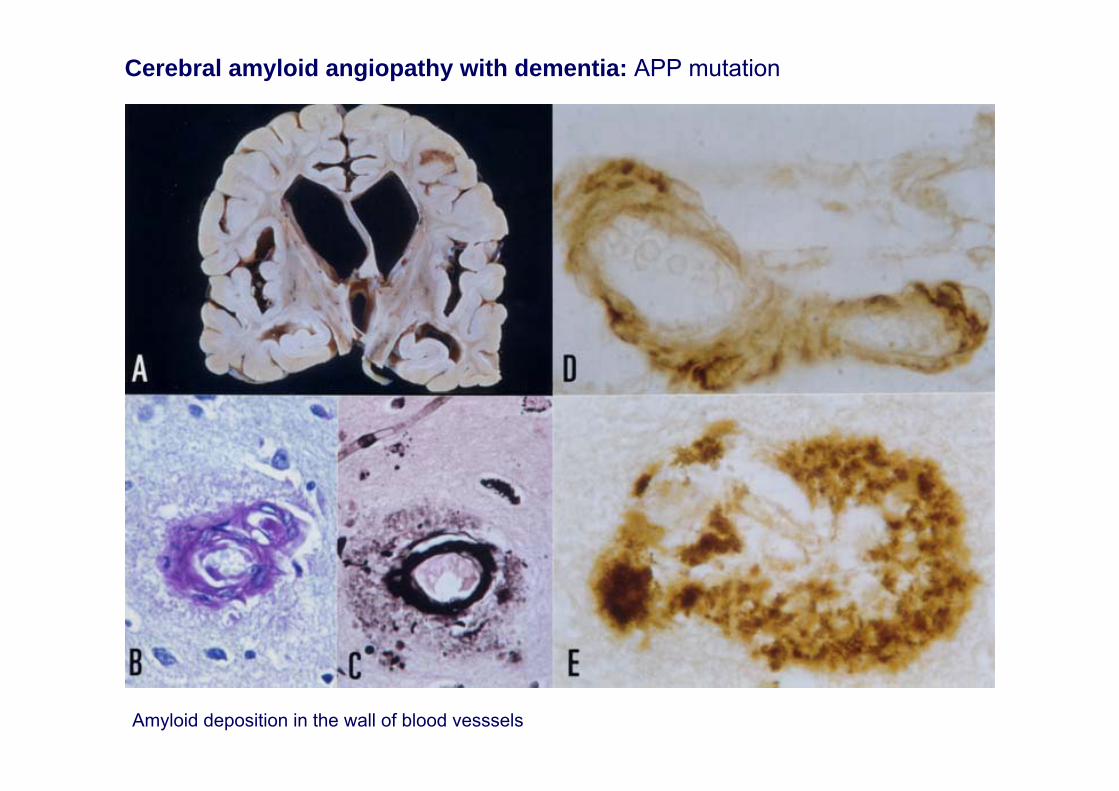
Amyloid deposition in the wall of blood vesssels
Cerebral amyloid angiopathy with dementia: APP mutation

Hereditary cerebral hemorrhage with amyloidosis Duch type (HCHWA-D)
Cause: Mutation codon 693 (glutamine for glutamic acid in AβPP
Clínical features: Autosomic dominant inheritance. Fatal unique hemorrhage orseveral small cerebral hemorrhages leading to cognitive impairment and dementia
Pathology: Deposits of βA4 amyloid wild type and mutant β-amyloid (and cystatinC) in meningeal and cerebral blood vessesl, and β-amyloid plaques with ubiquitinbut rarely accompanied by hyper-phosphorylated tau deposition
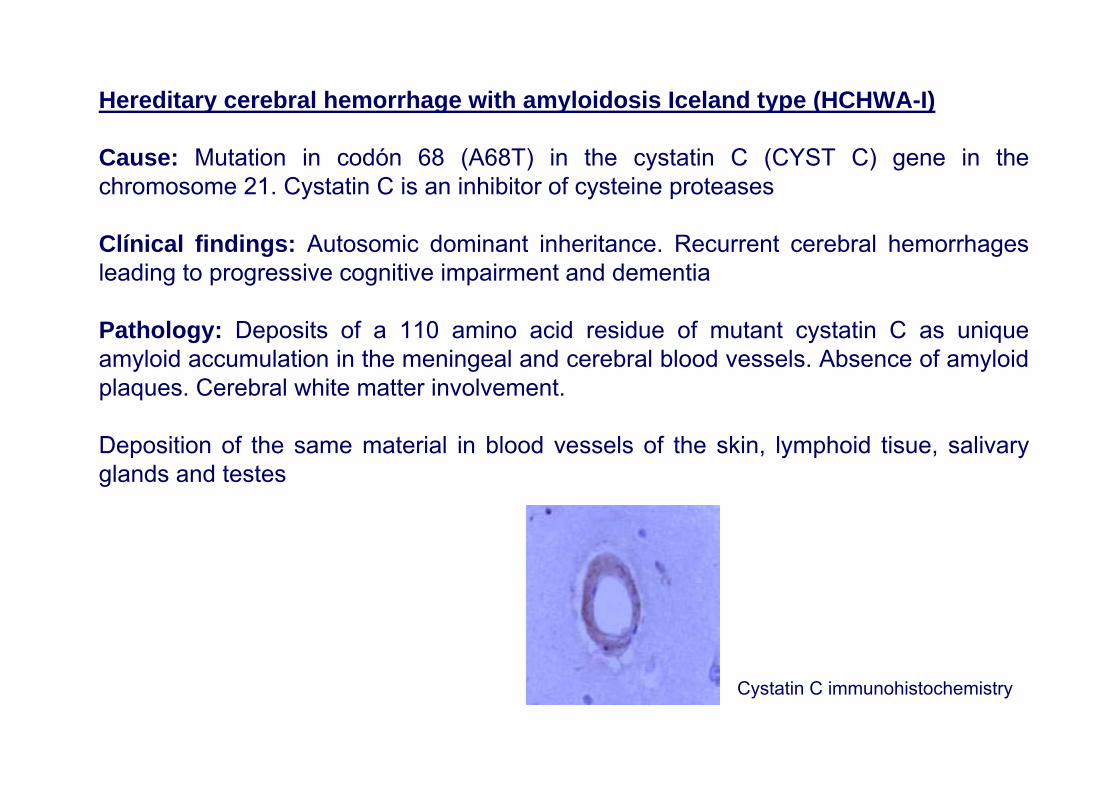
Hereditary cerebral hemorrhage with amyloidosis Iceland type (HCHWA-I)
Cause: Mutation in codón 68 (A68T) in the cystatin C (CYST C) gene in thechromosome 21. Cystatin C is an inhibitor of cysteine proteases
Clínical findings: Autosomic dominant inheritance. Recurrent cerebral hemorrhagesleading to progressive cognitive impairment and dementia
Pathology: Deposits of a 110 amino acid residue of mutant cystatin C as uniqueamyloid accumulation in the meningeal and cerebral blood vessels. Absence of amyloidplaques. Cerebral white matter involvement.
Deposition of the same material in blood vessels of the skin, lymphoid tisue, salivaryglands and testes
Cystatin C immunohistochemistry

Familial amyloidosis Finnish type
Cause: Mutations (G654A or G654T) in the gelsolin (Gel) gene in chromosome 9. Gelsolin is an actin binding protein
Clínical manifestations: Autosomal dominnat inheritance. Peripheral neuropathy, facial paresia, bulbar paralysis, ataxia, cognitive deterioration, corneal dystrophy andcutaneous lesions
Pathology: Deposition of fragment 173-243 and 173-225 of mutant gelsolin as uniqueaccumulation of amyloid in meningeal and cerebral blood vessels, extracellulardeposits in the duramatter, spinal nerve roots, sensorial ganglia. Demyelination of theposterior spinal fascicles and of the cerebral white matter
Deposition of the same material in the basal membranes of blood vessels in everyorgan
Antequera D, Vargas T, Ugalde C, Spuch C, Molina JA, Ferrer I, Bermejo-Pareja F, Carro ECytoplasmic gelsolin increases mitochondrial activity and reduces Aβ burden in a mouse model of Alzheimer’s diseaseNeurobiol Dis 36: 42-50 (2009)
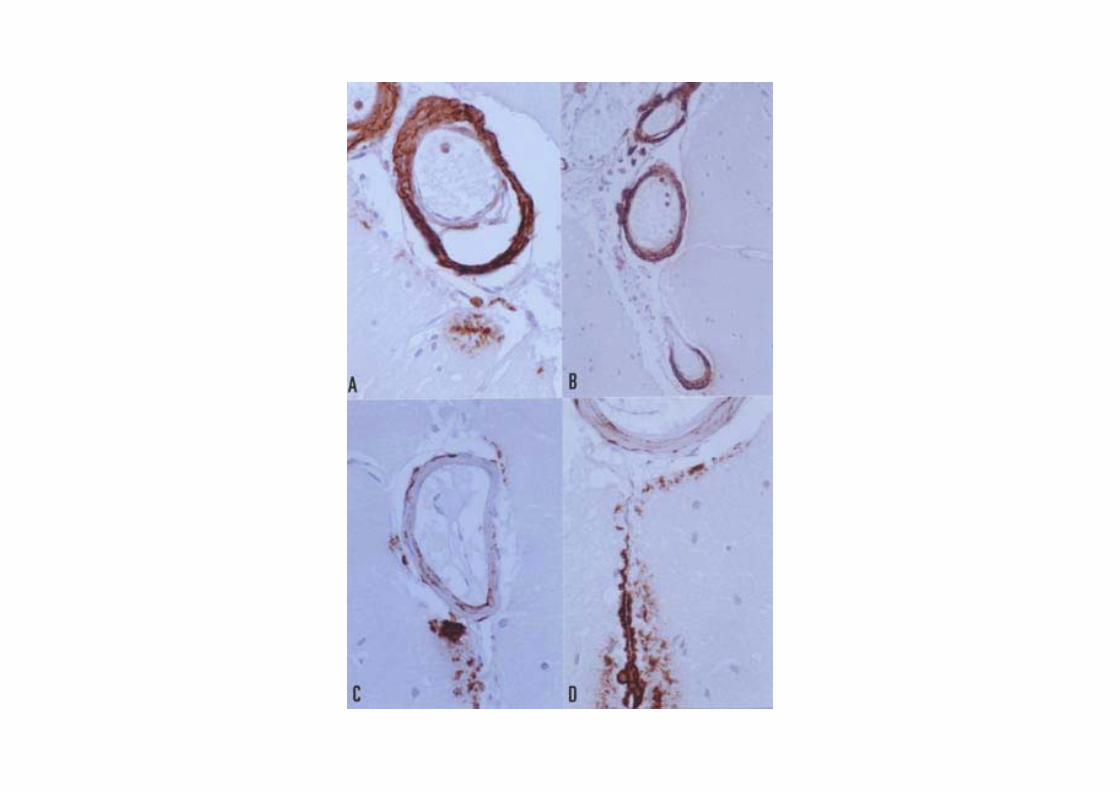

British familiar dementia
Cause: Substitution of a base of the codon stop of the BRI gene in the chromosome 3 (BRI2Stop267Arg). Furine effect upon the mutant protein triggers fibrillogenesis of the 34 amino acid subunit (ABri).Clinical symptoms: Dominant autosomic inheritance. Dementia, progressive spastic paraparesia and ataxia; cerebral hemorrhage rare.Pathology: Deposition of ABri with a characteristic carboxy-terminal. Amyloidangiopathy and perivascular plaques. Degenerative changes in the brain with abundant neurofibrillary tangles. Involvement of the cerebral white matter.
Danish familiar dementia
Cause: Duplication of ten nucleotides between codons 265 and 266 of the BRI gene (BRI2795-796Ins-TTTAATTTGT). Furine effect upon the mutant protein gives rise to a peptide of 4kDa (ADan) with specific carboxy-terminal.Clinical symptoms: Dominant at autosomic inheritance. Cataracts and ocular hemorrhages; followed by deafness, ataxia and dementia about twenty years later. Pathology: Deposition of amyloid ADan and, particularly pre-ADan (negative for tioflavin), and smaller amounts of βA4 in the blood vessel walls. Amyloid plaques in the cerebrum and cerebellum. Neurofibrillary tangles similar to those found in AD. Retinal degeneration due to amyloid angiopathy.

Inflammatory diseases of the blood vessels
Primary angiitis of the CNSGiant cell arteritisTakayasu arteritisKawasaki diseaseBuerger diseaseSneddon syndrome
Systemic vasculitis and autoimmune diseases
•Systemic lupus erythematosus•Panarteritis nodosa•Vasculitis associated with anti-neutrophil cytoplasm antibodies (ANCA)•Sjögren syndrome•Behçet syndrome•Rheumatoid arthritis
Infectious diseases: bacteria, M. tuberculosis, T. pallidum, fungi,amoebiasis, Plasmodium falciparum

Increased thickness and fibrosis of the skin blood vessels in Sneddon syndrome
Sneddon syndrome

A
B C
Vasculitis of the carotid artery secondaryto bacterial meningitis

A B
Tuberculous meningitis, vasculitis and cerebral infarct of the middle cerebral artery

A B C
Vasculitis and septic necrosis by Aspergillus

Primay angiitis
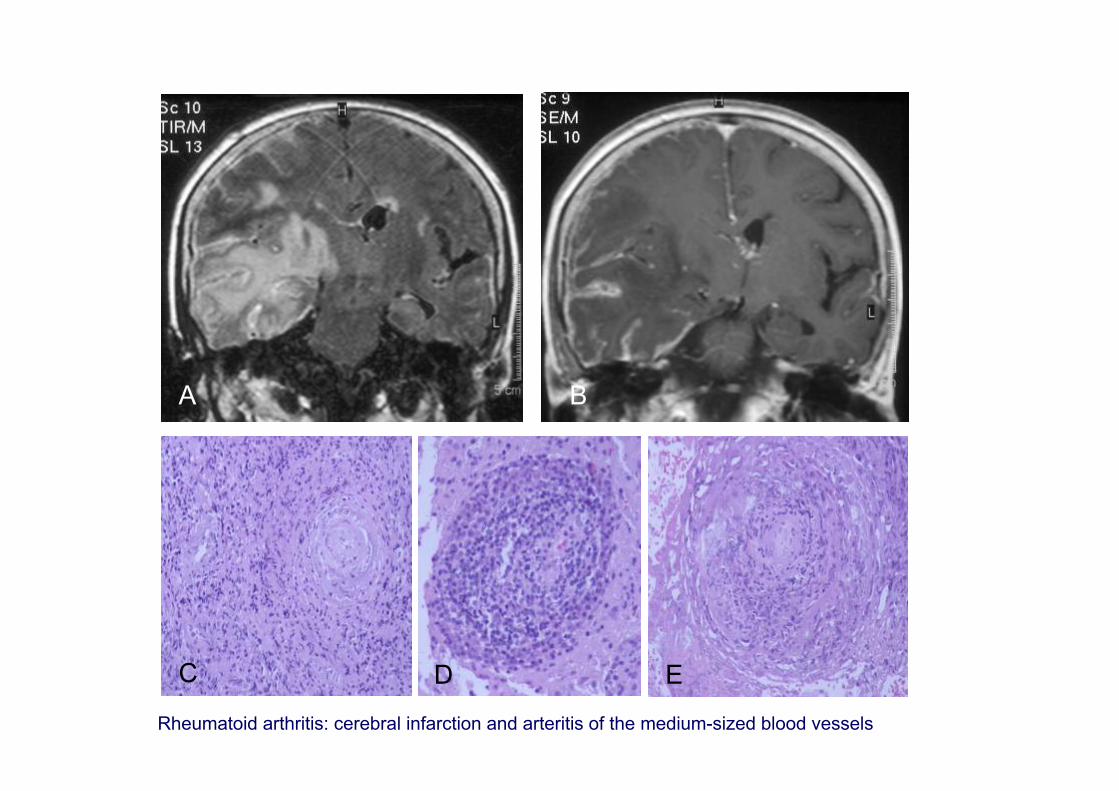
A B
C D E
Rheumatoid arthritis: cerebral infarction and arteritis of the medium-sized blood vessels

Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
Age at onset: between 25 and 70 years, peak around 40-50 years, high individual variations even in the same family. Sex and males equally affected
Clinical symptoms: migraine with aura, transient ischemic attacks or stroke; cognitive impairment; psychiatric symptoms; others: epilepsy
Genetics: mutations in Notch3 at chromosome 19p13.1-13.2 (more than 100 mutations)
Imaging: Hyperintensities in the white matter in T2 weighed and fluid attenuation inversion recovery (FLAIR) sequence MRI and variable number of small infarcts in white matter and basal ganglia
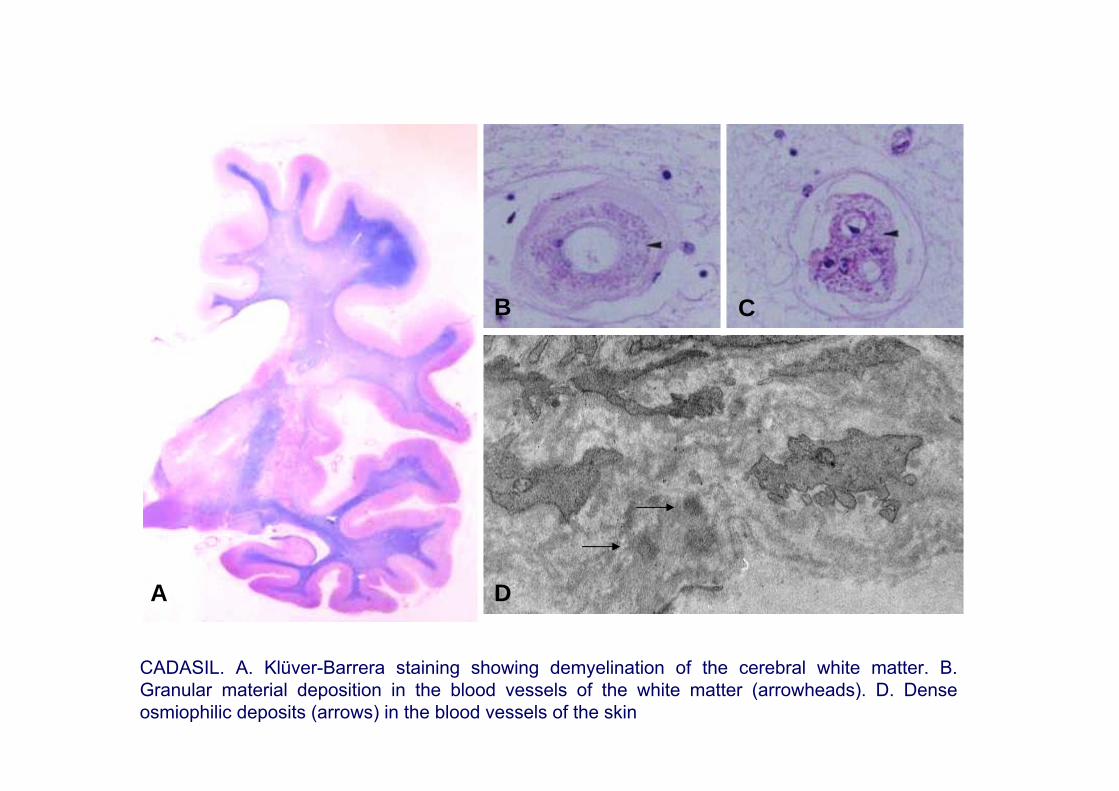
Pathology: Thickening of small blood vessels, degeneration of the smooth muscle cells (reduction of α-actin expression), accumulation of granular material in the blood vessels wall, and accumulation of N3ECD (extracellular domain of Notch)
Biopsy: Deposits of granular osmiophilic material as seen by electron microscopy in the walls of the arterioles in the skin, muscle and nerves; immunoreactivity for N3ECD
D
C
A
B
CADASIL. A. Klüver-Barrera staining showing demyelination of the cerebral white matter. B. Granular material deposition in the blood vessels of the white matter (arrowheads). D. Dense osmiophilic deposits (arrows) in the blood vessels of the skin

CARASIL: Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
Severe atrophy of the white matter with enlargement of the lateral ventricles (A), demyelination as revealed with myelin basic protein immunohistochemistry (B), thickening of the blood vessel walls with no abnormal granular deposition or amyloid deposits (C).
A B C

Neoplastic diseases as a cause of cognitive impairment of vascular origin
Intravascular lymphoma B or T (malignant hemangioendotheliomatosis)
Lymphomatoid granulomatosis
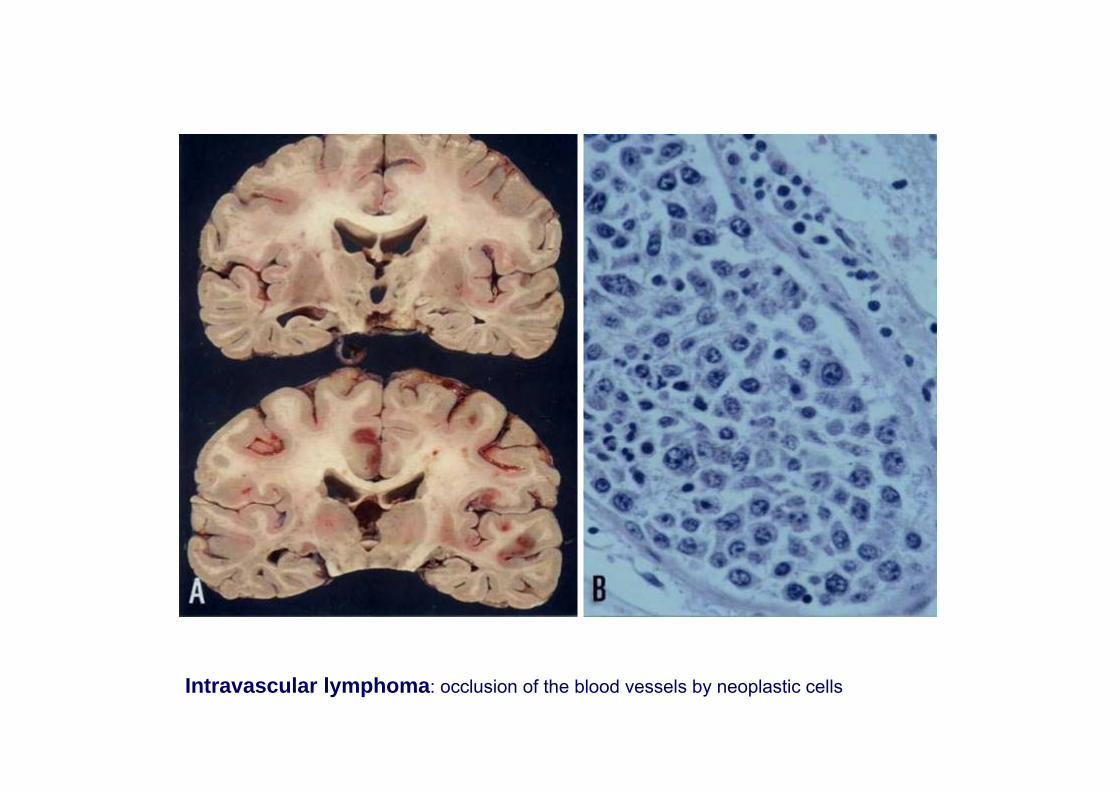
Intravascular lymphoma: occlusion of the blood vessels by neoplastic cells

Cerebral autosomic dominant arteriopathy with subcortical infarcts andleukoencephalopathy No-Notch: linked to chromosome 7
CARASIL (Maeda syndrome): Cerebral autosomic recessive arteriopathy withsubcortical infarcts and leukoencefalopathy
Other cerebro-vascular diseases
•Hereditary endotheliopathy with retinopathy, nephropathy and stroke(HERNS)
•Cerebroretinal vasculopathy (CRV)
•Hereditary vascular retinopathy (HVR) (linked to 3p21.1-p21.3)
•Hereditary infantile hemiparesia with arteriolar retinopathy andleukoencefalopathy (HIHRATL) (not linked to 3p21)
•Fibromuscular dysplasia
•Moya-moya disease: • sporadic (90%)• familiar (linked to 3p24.2-p26, 6, 17)

Vascular encephalopathies in children
•Post-anoxic encephalopathy with involvement of the white matter; periventricular leukoencephalomalacia
•Predominant involvement of the basal ganglia
•Predominant involvement of the cerebral cortex: laminar necrosisof the cerebral mantle
•Thalamic sclerosis
•Mesial sclerosis of perinatal origin
•Multicystic encephalomalacia
•Other types: cortico-pontine
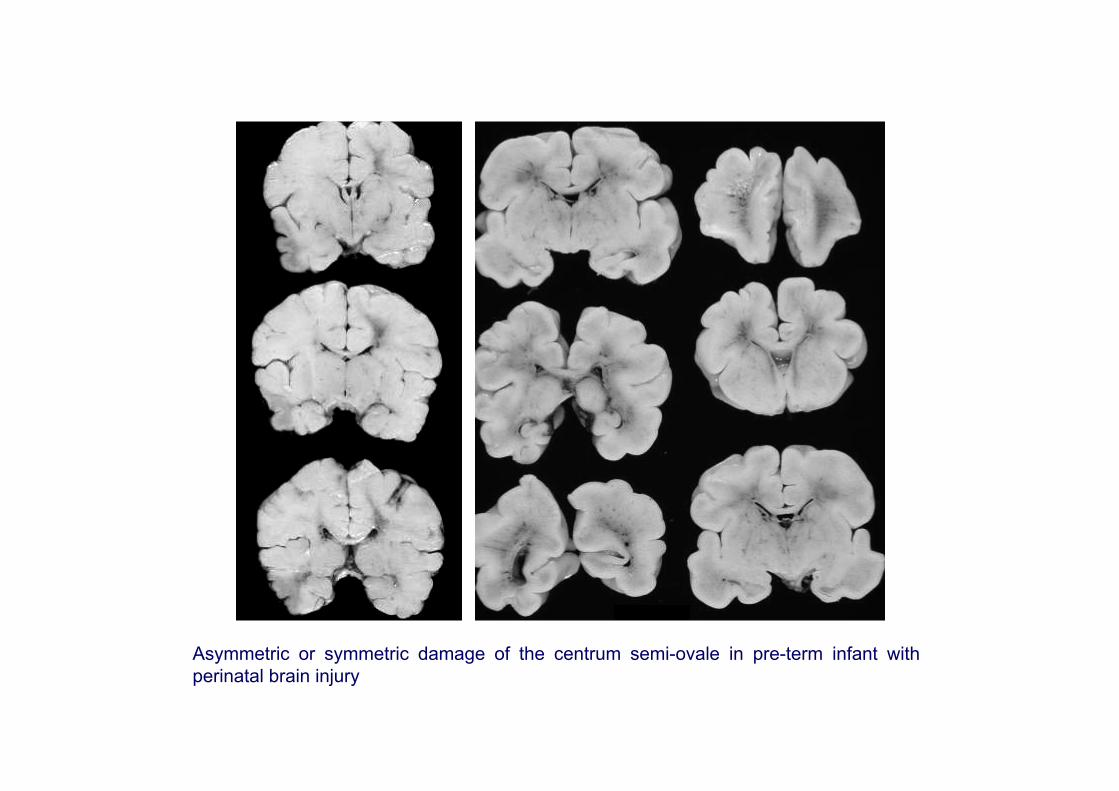
Asymmetric or symmetric damage of the centrum semi-ovale in pre-term infant with perinatal brain injury
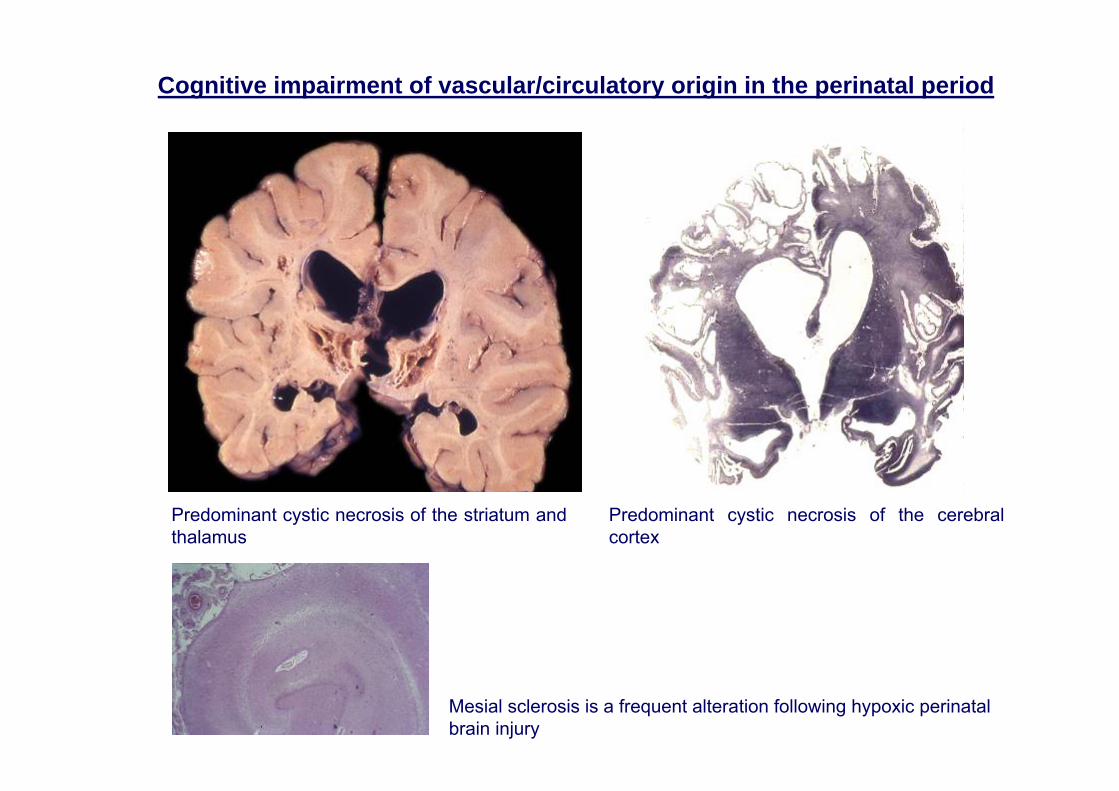
Predominant cystic necrosis of the striatum and thalamus
Predominant cystic necrosis of the cerebral cortex
Mesial sclerosis is a frequent alteration following hypoxic perinatalbrain injury
Cognitive impairment of vascular/circulatory origin in the perinatal period

Perinatal ischemia in the rat brain causes necrosis and apoptosis

Perinatal hypoxic encephalopathy
A: coronal section, Klüver-Barrera staining; B: cerebral cortex; C: putamen; D: hippocampus
A
C DB

Laminar necrosis of the cortical mantle (A and B), with severe spongiosis and neuron loss in best preserved regions (C)
CA
B
Laminar necrosis of the cerebral cortex due to perinatal hypoxia, reminiscent of Alper’s syndrome

Neuron loss, massive astrocytosis and micro-calcification in the thalamus
Perinatal encephalopathy with predominant thalamic involvement
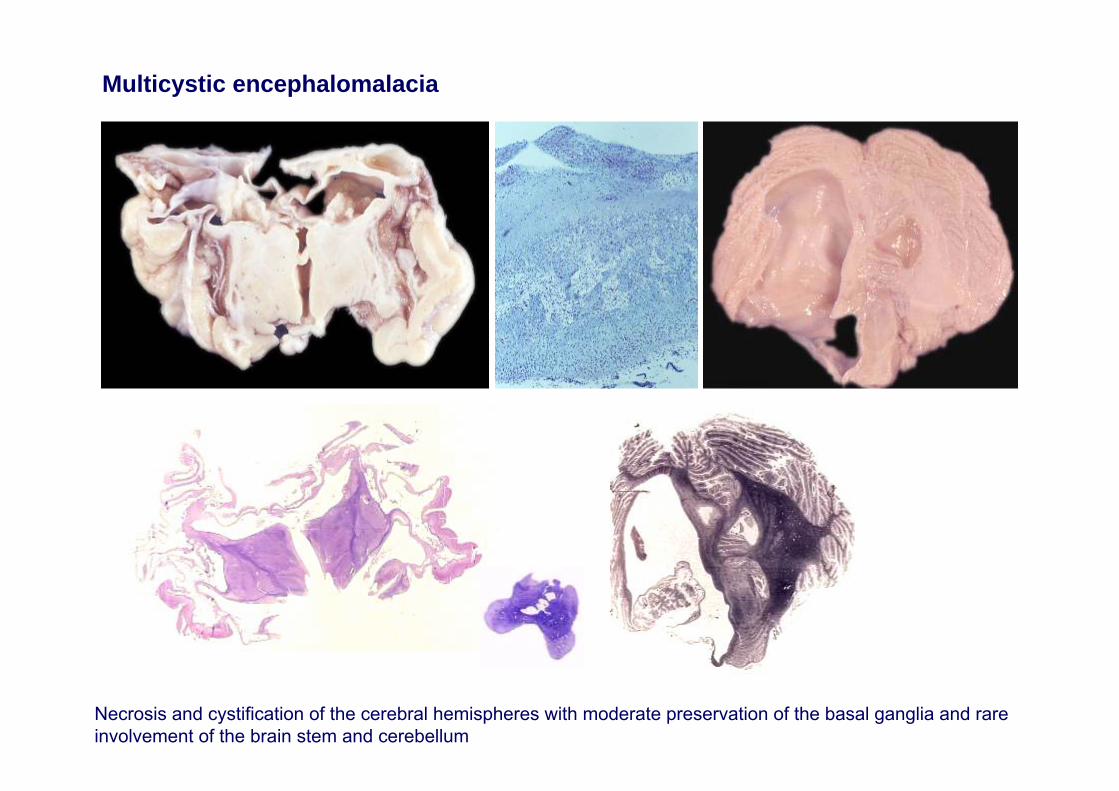
Multicystic encephalomalacia
Necrosis and cystification of the cerebral hemispheres with moderate preservation of the basal ganglia and rare involvement of the brain stem and cerebellum



















