
Epilepsy recent classification and definitions, dr. amit vatkar, pedaitric neurologist
Upload
dr-amit-vatkarCategory
view
287download
3
Dr. Amit VatkarMBBS, DCH, DNB Pediatrics
Fellow in Pediatric Neurology, MumbaiTrained in Neurophysiology & Epilepsy, USA
Contact No. : +91-8767844488Email: [email protected]
An approach to a child with Neuro-
Degenerative disorders

WHAT ARE THEY?
A group of inherited metabolic disorders.
Rare and untreatable - Mostly
Familial - Diagnosis is important for genetic
counselling.
Classification is difficult.
Difficult to remember all.

THE FIRST STEP
Differentiate fixed lesions from progressive
ones.
Earlier - Slowing in the rate of acquisition of
New Skills.
Later - Actual loss of acquired skills.
Rule out other disease processes like Infection,
Neoplasm, Hydrocephalus etc.,

• Regression: when child loses previously acquired
skills and milestones
• A progressive deterioration of neurological functions
with loss of speech,vision,hearing or locomotion ,often
associated with seizure, feeding and intellectual
impairment

• Neuroregressive /neurodegenerative disorders are a
group of heterogeneous diseases which results from
specific genetic, biochemical defect, chronic viral
infection, toxic substances
• Involves both the gray matter and white matter

• Dementia, used for neurodevelopmental regression in
children, is associated with loss of memory, ability to
think, understand and recognize along with
personality changes or distressing behaviour

Gray matter & White matter
Striations seen in white mater

White matter
Contains mostly myelinated axons
Appears pinkish white to the naked eye (myelin is
composed largely of lipid tissue veined with capillaries)
A 20 year-old male has a 176,000 km of myelinated axons
in his brain while that of a female is 149,000 km
connect various grey matter areas (the locations of nerve
cell bodies) of the brain to each other, and carry nerve
impulses between neuron

Gray matter
Major component of the CNS having a grey –brown
color(due to capillary blood vessels & neurinal cell bodies)
Consists of
neuronal cell bodies( in contrast to white matter)
neuropil (dendrites and both unmyelinated axons and
myelinated axons)
glial cells (astroglia and oligodendrocytes) & capillaries.

• The grey matter includes regions of the brain involved
in
– muscle control,
– sensory perception such as seeing and hearing,
memory, emotions, and speech.

Gray matter Disease White matter Disease
Processing center Represents networking
between these centers
Primarily involve neurons±
histologic evidence of
abnormal metabolic products--
> neuronal death and
secondary axon degeneration
Myelin is disrupted either
destruction of normal myelin
or biochemically abnormal
myelin production

Differentiatingfeatures
White matterdisorders
Gray matterdisorders
Age of onset Usually late(childhood) Usually early(infancy)
Head size May have megaenchepaly
Usually microcepaly
Seizures Late , rare Early, severe
Cognitive functions Initially normal Progressive dementia
Peripheral neuropathy
Early demyelination Late, axonal loss
Spasticity Early, severe Later, progressive
Reflexes Absent(neuropathy) or exaggerated(long tracts)
Normal or exaggerated

Differentiatingfeatures
White matterdisorders
Gray matterdisorders
Cerebellar signs Early,prominent late
Fundal examination May show optic atrophy
Retinal degeneration
EEG Diffuse delta slowing Epileptic form discharges
EMG Slowed nerve conduction velocity
Usually normal
Evoked potentials(VEP, ABR)
Prolonged or absent Usually normal
ERG Normal Abnormal
EEG=electroencephalogram, EMG= electomyography , VEP=visualevoked potential, ABR= auditory brain stem response,ERG= electroretinogram
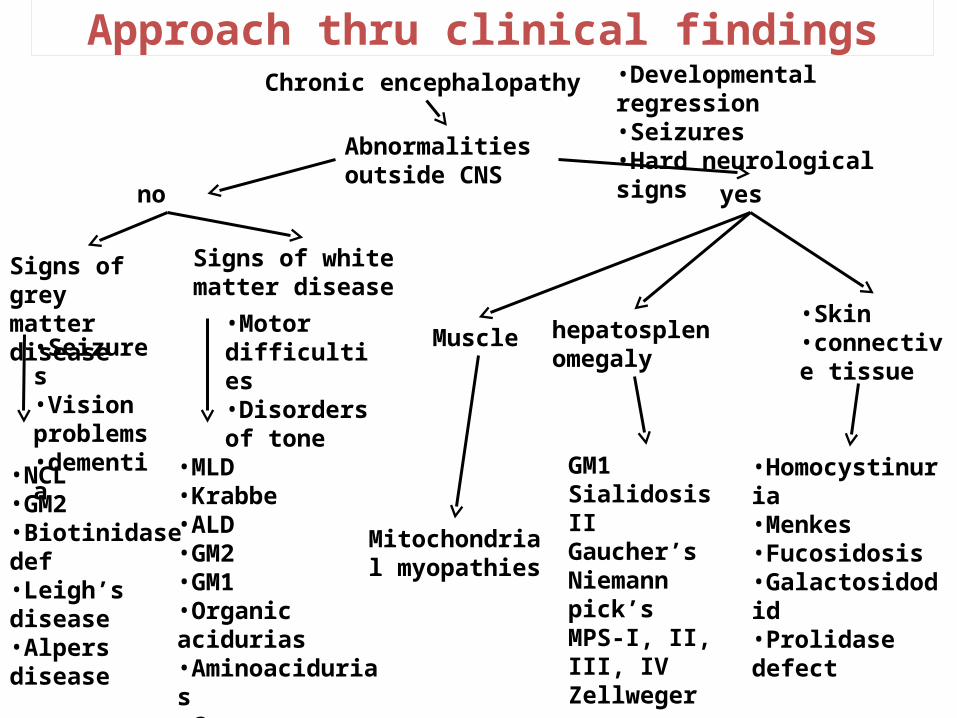
Approach thru clinical findings
Abnormalities outside CNS
Chronic encephalopathy
•Seizures•Vision problems•dementia
yes
•Developmental regression•Seizures•Hard neurological signs
no
Signs of white matter disease
•Homocystinuria•Menkes•Fucosidosis•Galactosidodid•Prolidase defect
Mitochondrial myopathies
Muscle
GM1Sialidosis IIGaucher’sNiemann pick’sMPS-I, II, III, IVZellweger
hepatosplenomegaly
•Skin•connective tissue
Signs of grey matter disease
•NCL•GM2•Biotinidase def•Leigh’s disease•Alpers disease
•MLD•Krabbe•ALD•GM2•GM1•Organic acidurias•Aminoacidurias•Canavan
•Motor difficulties•Disorders of tone

Classification
• Gray matter: fits, decrease HMF
– EEG: early abnormality
– MRI Brain: cortical atrophy
• White matter: blindness ,Gait disturbances ,Motor signs-Spasticity ,optic atrophy
,ataxia , pappiledema
– EEG: late abnormality
– MRI Brain: Demyelination
– Nerve conductance + Evoke potentials• Basal Ganglia :Dystonia,Involantary movements• Spinocerebellar degeneration: Ataxia

Classification of neurodegenerative brain disease
Inherited Acquired
Focal manifestations
Both
White matter
Gray matter Metabolic
Infections

Acquired causes
• Infections
SSPE
Progressive Rubella
Syndrome
Chronic HIV
• Metabolic
Chronic lead poisoning
Hypothyroidism
Vit B12 & E deficiency
Drugs (anticonvulsant)

Inherited causes
• Gray matter involvement: seizure, dementia, visual loss, intellectual
impairment. Spike & sharp waves in EEG
• A. Gray matter involvement with visceromegaly
– GM1 Gangliosides-Infantile , generalized , juvenile
– Sandholf disease (GM2)
– Niemann pick Disease( Sphingolipid storage disease)
– Sialidosis
– MPS
– Gaucher disease( Sphingolipid storage disease)

SCHEMATIC FOR INHERITED METABOLIC DISORDERS OF INFANCY
Dysmorphic Features
Urine Berry Spot Test
Negative Positive
Mucopoly saccharidoses Oligosaccharidoses
Mental Retardation Urinary Sialic Acid Excretion
No Yes Increased Normal
SCHIE
Morquio
Maroteux -Lamy
Hurler
Hurler Schie
Hunter
San Filippo
B-Glucuronidase
Mucosulfatidosis
Mucolipidoses I
Mucolipidoses II
Mucolipidoses III
GM1 Gangliosidosis
Fucosidosis
Mannosidosis
Aspartyl-Glucosaminuria
Mucolipidosis (IV)

• B. Gray matter diseases involvement without
visceromegaly
– Tay Sach disease (GM2)
– Rett Syndrome
– Neuronal curoid lipofuscinosis
– Menke’s kinky hair disease

White matter involvement
• Spasticity , optic atrophy, ataxia ,peripheral neuropathy .Seizure
, dementia are the late manifestations. Slow waves in EEG
• A. Leukodystrophies
– Metachromatic leukodystrophy
– Krabbe disease
– Adrenoleukodystrophy
– Alexander disease , Cannavan disease,P.Merzbacker disease

• B. Acquired causes/ Demyelinating
– Multiple sclerosis
– Schilder’s disease
– Devic disease

• Basal Ganglia
– Wilson's disease
– Dystonia muscular Deformans
– Huntington’s Disease
• Spinocerebellar
– Friedrich’s Ataxia
– Ataxia Telangiectasia

Krabbe disease(Globoid Cell Leukodystrophy)
• AR
• Galactocerebroside-B-galactosidase
• Myelin loss & presence of globoid bodies in white matter
• Symptom appear by 6 months
– Irritability Vomiting
– Bouts of hyperpyrexia Alteration in body tone
– Convulsions UMN signs with absent jerks

Krabbe disease
• Investigations
– Leukocyte & Skin fibroblast enzyme level
– CSF: Elevated protein
– NCV: Markedly delayed
– Prenatal diagnosis: by assays in chorionic villi or
amniotic fluid cell culture

Metachromatic leukodystrophy
• AR
• Arylsulfatase A deficiency
• Onset in late infancy
– Hypotonia
– Absent deep tendon reflexes
– Optic atrophy
– Decorticate posture

Juvenile MLD
• Onset at 5-10 yr
– Deterioration in school performance
– Alteration in personality
– Incontinence
– Incoordination,Dysarthria
– Spasticity
– Generalized tonic clonic seizures

Gaucher disease
• Commonest
• Galactocerebrocidase deficiency
• Infantile form (neuropathic)
– Nuchal rigidity & opisthotonus
• Juvenile form (non-neuropathic)
• Diagnosis
– Bone marrow examination
– WBCs or fibroblast cultures & enzyme level

Niemann pick disease
• Accumulation of sphingomyelin
• Onset 1st yr
• Mental retardation,Hepatosplenomegaly,Cherry red
spot
• Diagnosis
– Foam cells in bone marrow
– WBCs or fibroblast cultures & enzyme level

Neuronal ceroid lipofuscinosis
• AR
• Storage of auto fluorescent hydrophobic material in
lysosomes of neurons & other tissue
• 3 subtypes
– Infantile
– Late infantile
– Juvenile

Neuronal ceroid lipofuscinosis
• S/S
– Myoclonic epilepsy
– Visual symptoms
– Cerebellar ataxia
– Dementia
– Pigmentary abnormalities in retina
• Max .age 10 yr

Neuronal ceroid lipofuscinosis
• Investigation
• Skin biopsy:
– Ultra structural abnormalities
• Cortical biopsy:
– Distended neurons
– Staining for ceroid & lipofuscinosis

Leucodystrophy
• Inherited white matter disease
• Defect in myelin synthesis
• involve the brain, spinal cord and peripheral nervesinvolve the brain, spinal cord and peripheral nerves. .
• Classic dysmyelinating disorders are
Adrenoleukodystrophy, Metachromatic Leucodystrophy,
Krabbe's disease and neuroaxonal dystrophy

Adrenoleucodystrophy
• Life-threatening disorder
• Occurring in males affecting the white matter &
adrenal gland.
• Very long chain fatty acids accumulate in the cells &
tissues causing myelin sheath damage as well as
dysfunction of the adrenal gland.
• X-linked form is the commonest

• Neonatal ALD is not X-lined, is associated
with liver and adrenal dysfunction and is a
severe disorder
• Diagnosis is made by estimation of very
long-chain fatty acids in plasma

Metachromatic leukodystrophy
• Deficiency of Aryl sulphatase A or Sphingolipid activator
protein (Saponin-B)= accumulation of lipid sulphatide in
the myelin sheath, brain and peripheral nerve.
• Cognitive function is minimally affected initially followed
by motor difficulties and dysarthria.
• Diagnosis is by estimating Aryl sulphatase A in leukocytes
or fibroblasts. CSF protein concentration is grossly
elevated

Peroxysomal disorders
• Children with peroxysomal disorders - Zellweger
syndrome :dysmorphic features &severe psychomotor
retardation, sensory neuronal deafness, peripheral
neuropathy & hepatocellular degeneration.
• Diagnosed by demonstrating the elevated very long
chain fatty acids, pipecolic acid and bile and
derivatives.

Mitochondrial disorders
• A defect in the oxidative phosphorylation in
the inner mitochondrial membrane
involving the respiratory chain which is
encoded by two genomes, the mt DNA and
nuclear DNA

Rett syndrome
• Onset in 1st yr
• Females affected only
– Regression of language & motor milestones
– Repetitive hand wringing movements
– Loss of purposeful & spontaneous movements
– Gen. tonic clonic seizures
– Max. age 2-3 yr

Creatin deficiency syndromes
• Patients with these disorders develop delay /
regression /mental retardation along with severe
defects in expression and repetitive speech.
• Symptoms are due to severe depletion of
creatine/phosphocreatine in the brain.

Sialidosis
• AR
• Lysosomal Neuraminidase deficiency
• Accumulation of sialic acid-oligosaccharide complex
• Type 1: late onset
• Type 2
– Infantile
– Juvenile

Sialidosis
• Intractable Myoclonic seizures
• Cherry red spots
• Somatic involvement
– Coarse facial features
– Corneal clouding
– Dystonia multiplex

Mucopolysaccharidosis
• Absence of variety of lysosomal hydrolases
• Degradation of MPs defective
• Abnormally large amount excreted in urine

Mucopolysaccharidosis
• Coarse facial features , Dwarfism , Kyphoscoliosis
• Hepatosplenomegaly , Cardiovascular abnormalities
• Neurologic abnormalities
– Type I Hurler syndrome
– Type II Hunter syndrome
– Type III Sanfilippo
– Type VII


Friedreich ataxia
• AR• Early teenage
– Ataxia– Dysarthria– Pes cavus– Decreased proprioception, vibration– Absent reflexes with upgoing plantars– Nystagmus– Hypertrophic cardiomyopathy

Lesch Nyhan syndrome
• X-linked recessive
• Deficiency of hypoxanthine guanine
phosphoribosyl transferase
• Formation of excess of uric acid
• Normal until late in 1st yr

Lesch Nyhan syndrome
• S/S
– Psychomotor retardation
– Chorioethetosis
– Spasticity
– Severe self mutilation
– Gouty arthritis
– Renal calculi


Wilson’s disease
• AR
• Degenerative disease of basal ganglia
• Inborn error of copper metabolism
– Academic deterioration
– Behavioral changes , Dysarthria ,Dysphasia
– Drooling, Dystonia
• K F rings

Subacute Sclerosing Panencephalitis
• Progressive slow viral infection of CNS by measles virus
– Personality changes
– Aggressive behavior
– Myoclonic seizures
• Investigation
– Anti measles Ab in CSF
– EEG

An approach to a child with regression of
milestones

History
History of present illness:Onset/Age of onsetFits ,Clumsiness or difficulty in gaitDeterioration of HMFAtaxia or imbalanceHeadache,Blindness,Vomiting, deafnessChange in personality and behaviourDeteriorance in school performanceIncreased startle response or hyperacusis

• Birth history:
– Term/preterm
– Postnatal complications
• Meningitis
• Head trauma
• kernicterus

• Developmental history:– Detailed development history- decide whether there is
delayed development milestones or regression of milestones
• Family history:H/o of consanguinity
Family history of neurological disorder
Early or unexplained death
Nature of the neurological manifestations should be clarified

• Classically , the loss of previously acquired
milestones(regression) marks the onset of
most Neurodegenerative brain disease with
subsequent progressive neurological
deterioration

Clinical examination
General physical examination
Dysmorphism: Zellweger syndrome, Neonatal adrenal
leukodystrophy, coarse facial features(MPS)
OFC –microcepaly (gray matter disease)
Megaenchepaly – certain white mater
disorder(Cannavan & Alexander)
Jaundice
Enlarged tongue

Skin & hair ( Hartnup Diseases-pellagra like skin rash, Menkes
disease-kinky hair)
Examination of the spine- for associated complications
(scoliosis)
Contractures of joints
Systemic examination:
Hepatosplenomegaly
Chest deformity
Cardiomyopathy

Neurological manifestation
Age group
Early infantile Late infantile Juvenile
Psychomotor Regression/ Dementia Deteriorating scholastic performance
GM 1 gangliosidosis GM 2 gangliosidosis Menke's diseaseNiemann-Pick disease type A I-cell disease
Niemann-Pick disease type C Neuronal ceroid lipofuscinosis Rett syndrome
Sanfilippo disease Late onset MLDJuvenile Krabbe disease Juvenile GM 1 and GM 2 gangliosidosis Niemann-Pick disease type C Gaucher type III
Extrapyramidal symptoms Choreoathetosis Dystonia
Lesch-Nyhan syndrome Pelizaeus-Merzbacher disease Glutaric aciduria type I
Ataxia telangiectasia Wilson disease Hallervorden-Spatz disease Huntington disease Neuronal ceroid lipofuscinosis
Speech disturbances
Rett Syndrome ALD
Psychosis
GM 2 gangliosidosis Wilson disease Sanfilippo disease Niemann-Pick disease type CNeuronal ceroid lipofuscinosis Hallervorden-Spatz Huntington chorea

Neurological examination
• Higher mental function, signs of raised ICP• Speech, memory• Cranial nerves• Gait• Motor system:
– Tone-hypo/ hypertonia,Deep tendon reflexes– Motor spasticity
• Sensory loss /neuropathy• Abnormal /involuntary movements

Eye examination
• Optic atrophy(white matter- due to demyelination)
• Retinal degeneration(gray matter)- as the retinal
receptors are neuronal cells): Cherry red spot,
retinitis pigmentosa
• Cataracts
• Telengiectasias
• K.F ring

DECIDE
• REGRESSION AND NOT DELAYREGRESSION AND NOT DELAY
• AGE ABOVE 2 YEARS OR LESS THAN 2 YEARSAGE ABOVE 2 YEARS OR LESS THAN 2 YEARS
• VISCEROMEGALYVISCEROMEGALY
• NEUROPATHYNEUROPATHY
• GRAY OR WHITE MATTER DISEASEGRAY OR WHITE MATTER DISEASE

Investigations- to identify the underlying diagnosis & examining the associated complications
• Complete Blood picture-pancytopenia, vacuolated
lymphocytes,acanthocytes
• ABGs-metabolic acidosis(organic acidopathies, urea cycle
defects, mitochondrial encephalopathies)
• S/E (Anion gap), for adrenal
insufficiency(adrenoleukodystrophy)
• Ammonia level,LFTs,RFTs

• Special tests:
– Lactate & pyruvate levels, Lysosomal enzyme level
– WBCs, Fibroblast enzyme level
– Wilson’s disease-serum ceruloplasmin level, serum copper
– Amino acids
– Urinary organic acids
– Uric acid level

• Urine
– Reducing substances, Organic acids,24 hr (MPS)
• Imaging
– Skull & Vertebrae, Long bones
– CT/MRI
• Biopsy
– Skin, Bone marrow, nerve, brain

• Diagnosis
– Important for genetic counseling
• Outcome
– Invariably fatal

Neuro degenerative Disorders- summary
30/12/10 Dr.Hariaharan.V, RMMC 67
Disorder Biochemical defect incidence
Lysosomal storage disorders-Sphingolipidoses
GM1 gangliosidosis-Infantile b- Galactosidase 1:1,00,000
-Juvenile b- Galactosidase
GM2 Gangliosidoses-Tay-Sach’s Hexosaminidase A 1 in 3,00,000
-Sandhoff’s Hexosaminidase A & B 1:3,00,000
-Krabbe’s Galactocerebrosidase 1 in 1,00,000
Juvenile GM2 gangliosidosis hexosaminidase
Adult GM2 gangliosidosis hexosaminidase
Metachromatic Leukodystrophy 1:40,000
Late Infantile Arylsulfatase A
Juvenile Arylsulfatase A
Neuronal ceroid Lipofuscinosis 1:50,000
Infantile Palmitoyl Protein Thioesterase Tripeptidyl Peptidase 1Late infantileJuvenile
AdrenoLeukodystrophy VLCFA oxidation(auto dom) 1:40,000
Sialadosis Neuraminidase 1:40,00,000

Symptom complex Disorder Investigation Tissue
Coarse facies, ostosis, hepatosplenomegaly, hernias, corneal clouding
Late infancy, childhood • MPS, Fucosidosis, Mannosidosis, Mucolipidosis,
• MPS spot • U
• MPS electrophoresis • U
• GAG quantification • U
• Oligosaccharides • U
• Enzyme activity • L,F,CVS, AF
Extrapyramidal symptoms, hepatitis, portal hypertension, KF ring
• Gaucher disease • Niemann Pick disease • Wilson Disease
• Bone marrow examination
• Enzyme activity • L,F, CVS, AF
• Copper • U, S, Liver
• Ceruloplasmin
• Liver biopsy
Cognitive regression, ataxia, seizures, visual impairment
• Adrenoleukodystrophy • Neuronal ceroid lipofuscinosis • Mitochondrial disorders • Subacute sclerosing pan-encephalitis
• Lactate • S
• Biopsy •Skin, rectal, conjunctival
• Nerve conduction
• MRI
• Lactate •S, CSF
• MRI, MRS
• Immunoglobulins • S, CSF
Supporting investigations Electroencephalogram, electroretinogram, electromyography, nerve conduction studies, Magnetic resonance imaging, magnetic Resonance spectroscopy, DNA study

Management
• Directed towards the treatment of the underlying disorder,
other associated features and complications• Supportive :The treatable complications :
• feeding difficulties, Gastoresophageal reflux• spasticity, drooling• skeletal deformities, and recurrent chest infections• epilepsy, sleep disorder, behavioral symptoms
• A multidisciplinary approach(pediatrics, neurology, genetics, orthopedics, physiotherapy, and occupational therapy.

Specific treatment
Neurodegenerativedisorders
Specific treatment modality
Krabbe leukodystrophyKrabbe leukodystrophy Bone marrow transplantation
Metachromatic Metachromatic leukodystrophyleukodystrophy
Bone marrow transplantation
AdrenoleukodystrophyAdrenoleukodystrophy Glyceryl trioleate and trierucate,steroids for adrenal insufficiency, diet low in VLCFA, bone marrow transplantation
MucopolysaccharidosisMucopolysaccharidosis Bone marrow transplantation,recombinant human -L-iduronidaseα
Menkes kinky hair Menkes kinky hair syndromesyndrome
Copper sulfate

Neurodegenerative
disorders
Specific treatment modality
Mitochondrial encephalopathiesMitochondrial encephalopathies Nicotinamide, riboflavin,
dichloroacetate, L-carnitine, CoQ10
Wilson diseaseWilson disease D- penicillamine, trietine, zinc acetate,
liver transplantation
Refsum diseaseRefsum disease Reduction of phytanic acid intake
Lesch-Nyhan diseaseLesch-Nyhan disease Allopurinol
Fabry’s DiseaseFabry’s Disease Recombinant human α galactosidase
A

Counseling the families and educating the public about these
potentially preventable disorders is very important

Dr. Amit VatkarPediatric Neurologist, Navi Mumbai
MBBS, DNB
Email: [email protected] No.: +91-8767844488
Visit us at: http://pediatricneurology.in/
THANK YOU !


















