
Mutational Analysis Demonstrates Different Functional Roles for the ...
9
THE JOURNAL OF Ilromor~~~ CHEMISTRI Vol. 269, No. 47, Issue of November 25, pp. 29537-29545, 1994 Printed in U.S.A. Mutational Analysis Demonstrates Different Functional Roles for the Two A~P-~in~ing Sites in ClpAp Protease from E ~ c ~ ~ ~ c ~ i coli* (Received for publication,June 28,1994, and inrevised form, September 16, 1994) Satyendra K. Singh and Michael R. MauriziS From the Laboratory of Cell Biology, NCI, National Institutes of Health, Bethesda, Marylalzd 20892 ClpA, the regulatory subunit of Clp protease from Escherichia coli, has two ATP-binding sites in non- homologous regions of the protein, referred to as do- main I and domain 11. We have mutated the invariant lysine in the ATP-binding sites of domain I and domain II and studied the enzymatic properties of the purified mutant ClpA proteins. Thedomain I mutant, ClpA- KZZOQ, was unable to form a hexamer in the presence of nucleotide,but the comparable domain I1 mutant, ClpA- K501Q, associated into a hexamer in the presence ofATP, indicating that nucleotide binding to domain I favors a conformation required to stabilize the quaternary struc- ture of ClpA ClpA-K!220Q was defective in ATPase activ- ity and in the ability to activate protein and peptide degradation by ClpP, but thedefects could be partially overcome by formation of hybrid hexamers with wild- type ClpA. Another domain I mutant, ClpA-K%ZOR, readily formed hexamers in the presence of ATP and retained 260% of the wild-typeATPase activity and abil- ity to activate ClpP. These results indicate that hexamer formation is a prerequisite for expression of enzymatic activity. Domain I1 mutants ClpA-K501Q and ClpA- K501R had very low ATPase activity (40% of wild-type) and a severe defect in activation of protein degradation, which requires ATP hydrolysis. Domain I1 mutants were able to activate ClpP to degrade a peptide whose degra- dation required nucleotide binding but not hydrolysis. Nucleotide binding to domain I1 of ClpA is important to form a productive complex with ClpP, and domain I1 appears to be primarily responsible for an energy-re- quiring stepin the catalytic cycle unique to the degra- dation of large proteins. ATP-dependent proteases are responsible for the major part of protein degradation that occurs in the cytoplasm of bacteria and in the cytosol of eukaryotic cells (13). Two families of ATP-dependent proteases have been identified in Escherichia coli and other bacteria. Lon protease is an oligomer of identical subunits and has a single ATPase active site and aproteolytic active site within the same polypeptide chain 13-51. In contrast, the Clp protease family is composed of two types of subunits: the proteolytic active site resides in ClpP, an oligomer of 212 identical subunits, and the ATPase activity resides in either ClpA or ClpX, which interacts with ClpP to form a functional protease with unique substrate specificity (5-7). In eukaryotic cells, it appears that the proteolytic core, called the proteasome (8,9), is associated with a number of different regulatory pro- * The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C.Section1734solelyto indicate this fact. Xnstitute, Bldg. 37, Rm. 1B07,37 Convent Dr. MSC 4255, Bethesda, MD $To whom correspondence should be addressedNationalCancer 20892-4255. Tel.: 301-496-7961; Fa: 301-402-0~50. teins, at least some of which are ATPases (10, 111, to form the 26 S ATP-dependent protease (11-14). The topologyofATP- dependent proteases from prokaryotes and eukaryotes may be very similar despite differences in sequences and exact subunit compositions (15). Degradation of many proteins by Lon protease requires ATP hydrolysis (16-18), but degradation of peptides and some pro- teins is activated by ATPbinding alone (18,191. ATP plays two roles in activating Lon protease: as an allosteric effector to open the proteolytic active site, and as a substrate whose hydrolysis is required to drive some unfavorable step, possibly involving unfolding of protein substrates, in the reaction (3-5). Because the ATPase and proteolytic activities of Clp protease reside on different subunits, it has been possible to delineate the dual roles ofATP in activating protein degradation. ClpP by itself has a limited activity against very short peptides (20,211, but protein and polypeptide degradation by ClpP requires in- teraction with either ClpA or ClpX. We have shown that bind- ing of ATP or a non-hydrolyzable analog of ATP produces a conformationalchange that stabilizes a hexameric form of ClpA and allows the protein to interact with ClpP (5). The binding of hexameric ClpA to ClpP opens the active site of ClpP and in- creases its catalytic efficiency, allowing degradation of interme- diate length peptides but not large proteins (20).Activation of degradation of large proteins shows a strict requirement for ATP hydrolysis (22-24). The presence of an allosteric site on ClpA for binding protein and peptide substrates is clearly evi- dent from the ability of proteins and peptides to affect the ATPase activity of ClpA in the absence of ClpP (20-24). The protein-stimulated ATPase activity of ClpA, together with the sequence similarity between ClpA and ClpB, a protein that may act in vivo as amolecular chaperone, suggested that ClpA may function as a molecular chaperone in presenting sub- strates to ClpP. Recently,ClpAhas been shown to bind to the P1 phage replication protein RepA and to display the same ATP- dependent chaperone activity as DnaJ and DnaK in converting RepA from an inactive to anactive conformation in vitro (25). The sequence of ClpA has revealed the presence of two ATP- binding site consensus sequences in two separate regions that have little homology to each other, but are well conserved in the ClpA homologs of other organisms (26). The separation of two ATP-binding site consensus sequences in distinct regions sug- gests the possibility that ClpA is composed of two structural domains, each with an ATP-binding site as its primary func- tional feature. To understand the functions of two distinct ATP- binding domains in ClpA, random mutations at the invariant lysine residue in each domains were made by oligonucleotide- directed mutagenesis. In this paper we describe the effects of these mutations on the structure and ATPase activity of ClpA and on the ability of ClpA to activate ClpP to degrade different substrates. 29537
Transcript of Mutational Analysis Demonstrates Different Functional Roles for the ...

T H E JOURNAL OF I l r o m o r ~ ~ ~ CHEMISTRI Vol. 269, No. 47, Issue of November 25, pp. 29537-29545, 1994 Printed in U.S.A.
Mutational Analysis Demonstrates Different Functional Roles for the Two A ~ P - ~ i n ~ i n g Sites in ClpAp Protease from E ~ c ~ ~ ~ c ~ i u coli*
(Received for publication, June 28,1994, and in revised form, September 16, 1994)
Satyendra K. Singh and Michael R. MauriziS From the Laboratory of Cell Biology, NCI, National Institutes of Health, Bethesda, Marylalzd 20892
ClpA, the regulatory subunit of Clp protease from Escherichia coli, has two ATP-binding sites in non- homologous regions of the protein, referred to as do- main I and domain 11. We have mutated the invariant lysine in the ATP-binding sites of domain I and domain II and studied the enzymatic properties of the purified mutant ClpA proteins. The domain I mutant, ClpA- KZZOQ, was unable to form a hexamer in the presence of nucleotide, but the comparable domain I1 mutant, ClpA- K501Q, associated into a hexamer in the presence ofATP, indicating that nucleotide binding to domain I favors a conformation required to stabilize the quaternary struc- ture of ClpA ClpA-K!220Q was defective in ATPase activ- ity and in the ability to activate protein and peptide degradation by ClpP, but the defects could be partially overcome by formation of hybrid hexamers with wild- type ClpA. Another domain I mutant, ClpA-K%ZOR, readily formed hexamers in the presence of ATP and retained 260% of the wild-type ATPase activity and abil- ity to activate ClpP. These results indicate that hexamer formation is a prerequisite for expression of enzymatic activity. Domain I1 mutants ClpA-K501Q and ClpA- K501R had very low ATPase activity ( 4 0 % of wild-type) and a severe defect in activation of protein degradation, which requires ATP hydrolysis. Domain I1 mutants were able to activate ClpP to degrade a peptide whose degra- dation required nucleotide binding but not hydrolysis. Nucleotide binding to domain I1 of ClpA is important to form a productive complex with ClpP, and domain I1 appears to be primarily responsible for an energy-re- quiring step in the catalytic cycle unique to the degra- dation of large proteins.
ATP-dependent proteases are responsible for the major part of protein degradation that occurs in the cytoplasm of bacteria and in the cytosol of eukaryotic cells (13). Two families of ATP-dependent proteases have been identified in Escherichia coli and other bacteria. Lon protease is an oligomer of identical subunits and has a single ATPase active site and a proteolytic active site within the same polypeptide chain 13-51. In contrast, the Clp protease family is composed of two types of subunits: the proteolytic active site resides in ClpP, an oligomer of 212 identical subunits, and the ATPase activity resides in either ClpA or ClpX, which interacts with ClpP to form a functional protease with unique substrate specificity (5-7). In eukaryotic cells, it appears that the proteolytic core, called the proteasome (8,9), is associated with a number of different regulatory pro-
* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Xnstitute, Bldg. 37, Rm. 1B07,37 Convent Dr. MSC 4255, Bethesda, MD $To whom correspondence should be addressed National Cancer
20892-4255. Tel.: 301-496-7961; Fa: 301-402-0~50.
teins, at least some of which are ATPases (10, 111, to form the 26 S ATP-dependent protease (11-14). The topology of ATP- dependent proteases from prokaryotes and eukaryotes may be very similar despite differences in sequences and exact subunit compositions (15).
Degradation of many proteins by Lon protease requires ATP hydrolysis (16-18), but degradation of peptides and some pro- teins is activated by ATP binding alone (18,191. ATP plays two roles in activating Lon protease: as an allosteric effector to open the proteolytic active site, and as a substrate whose hydrolysis is required to drive some unfavorable step, possibly involving unfolding of protein substrates, in the reaction (3-5).
Because the ATPase and proteolytic activities of Clp protease reside on different subunits, it has been possible to delineate the dual roles ofATP in activating protein degradation. ClpP by itself has a limited activity against very short peptides (20,211, but protein and polypeptide degradation by ClpP requires in- teraction with either ClpA or ClpX. We have shown that bind- ing of ATP or a non-hydrolyzable analog of ATP produces a conformational change that stabilizes a hexameric form of ClpA and allows the protein to interact with ClpP (5). The binding of hexameric ClpA to ClpP opens the active site of ClpP and in- creases its catalytic efficiency, allowing degradation of interme- diate length peptides but not large proteins (20). Activation of degradation of large proteins shows a strict requirement for ATP hydrolysis (22-24). The presence of an allosteric site on ClpA for binding protein and peptide substrates is clearly evi- dent from the ability of proteins and peptides to affect the ATPase activity of ClpA in the absence of ClpP (20-24). The protein-stimulated ATPase activity of ClpA, together with the sequence similarity between ClpA and ClpB, a protein that may act in vivo as a molecular chaperone, suggested that ClpA may function as a molecular chaperone in presenting sub- strates to ClpP. Recently, ClpAhas been shown to bind to the P1 phage replication protein RepA and to display the same ATP- dependent chaperone activity as DnaJ and DnaK in converting RepA from an inactive to an active conformation in vitro (25).
The sequence of ClpA has revealed the presence of two ATP- binding site consensus sequences in two separate regions that have little homology to each other, but are well conserved in the ClpA homologs of other organisms (26). The separation of two ATP-binding site consensus sequences in distinct regions sug- gests the possibility that ClpA is composed of two structural domains, each with an ATP-binding site as its primary func- tional feature. To understand the functions of two distinct ATP- binding domains in ClpA, random mutations at the invariant lysine residue in each domains were made by oligonucleotide- directed mutagenesis. In this paper we describe the effects of these mutations on the structure and ATPase activity of ClpA and on the ability of ClpA to activate ClpP to degrade different substrates.
29537

29538 Mutational Analysis of the 7bo ATPase Sites in ClpA
EXPERIMENTAL. PROCEDURES Materials-ATP and ATPyS' were obtained from Sigma, MonoQ and
Monos were from Pharmacia Biotech Inc., and agarose, phenol, and SDS were obtained from Life Technologies, Inc. All other chemicals used in the present study were reagent grade. Oligonucleotides used for mutagenesis (K220f, d(5'-GGGGGAATCGGTGTCGGT(A/C/G)(A/C/G/ T)AACCGCGATTGCGG-3'), K220b, d(5'-CCGCAATCGCGGTT(T/C/G/ A)(T/C/G)ACCGACACCAGATTCCCCC-3'), and K501b, d(5"ATGC- CCAAAGCTTI'CGAAAGCTGTACCGTCACGTCTGTT(A/C/GI") (TIC/ G)CCCGACCCCGGTAGG-3')) were synthesized on an Applied Biosys- tems model 380A DNA Synthesizer and purified using Sep-Pack car- tridges as described by the manufacturer. ClpA sequencing primers used for polymerase chain reactions (PCR) and for dideoxy sequencing of double-stranded DNA templates were described in an earlier study (27). Polymerase chain reactions were performed using the GeneAmp Kit (Perkin Elmer) and DNA sequencing was done with Sequenase (United States Biochemical Corp., Cleveland, OH).
Bacterial Strains and Plasmids-E. coli DH5a F' (28) was used for subcloning and mutagenesis procedures. SG22030 (c1pP::cat AclpA clpB:: kan Agal) was used for expression and purification of mutant ClpA proteins. The in vivo expression of phenotypes for clpA mutants was studied in the isogenic strains SG25501 (clpA') and SG25518 (cZpA) containing an rbsA::lacZ translational fusion; the fusion protein has been shown to be a substrate for Clp protease in vivo.2 Plasmid pWPC3.1, a derivative of pWPC3 (27) that carries a 3.0-kilobase BamHI-PstI fragment containing the clpA gene, was used as a template for PCR mutagenesis. pSK16 is a recombinant derivative of pWPC3.1 obtained by moving an NdeI-PstI fragment containing the clpA gene into pUC18. E. coli SG strains, derivatives of E. coli MC4100, were kindly provided by S. Gottesman.
Standard Procedures-Previously described methods were used for assay of ATP-dependent proteolytic activity (23), ATPase assays (241, ClpP propeptide degradation assays (20), protein assays (231, SDS-gel electrophoresis (291, Western blotting (241, and preparation of PHI- casein (30). Small scale cultures of transformants were grown at 37 "C in Luria broth with 100 pg/ml ampicillin. p-Galactosidase activity was assayed as described by Miller (31).
Oligonucleotide-directed Mutagenesis-The lysine residue at position 220 in the ATP-binding domain 1 of ClpA was randomly mutated by overlap extension, using PCR (32). Briefly, a set of two complementary primers, K220f and K220b, and two external primers (SFF-2 and SFB-6) (27) annealing upstream and downstream of the domain I ATP- binding consensus sequence in pWPC3.1 were used. Two independent PCR reactions were carried out, one for each pair of primers (mis- matched and external). Amplification of DNA fragments was performed using 0.2-0.25 n~ of the primers. Low melting agarose (1% Sea Plaque or 3% Nusieve, FMC-BioProducts) was used to purify the amplified fragments. Bands were cut out, DNA extracted, and mixed in a fusion PCR reaction (32). The amplified fragments were purified on 1% normal agarose gel (Geneclean 11, Bio IOI), and cloned back in pWPC3.1 using MluIIDraIII restriction sites. Transformants were screened for the pres- ence of the MluIIDraIII restriction fragment and then for ClpA produc- tion by Western blotting. Sequencing of the plasmids identified eight different mutants (K220Q, K220R, K220P, K220T, K220L, K2201, K220V, and K220H) in which bases encoding lysine 220 were changed to CAA, AGA, CCA, ACA, CTA, ATA, GTA, and CAT, respectively. Only the mutants K220Q and K220R (Fig. 1) were selected for further study. The 323-base pair MluI-EcoRV fragments from plasmids with the K220Q and K220R mutations were inserted in place of the corresponding frag- ment in the clpA gene of pSK16, and the entire fragment was sequenced to ensure that no other mutations were present.
Lysine 501 in the second ATP domain of ClpA was mutagenized using PCR as described above. Plasmid pWPC3.1 was used as a template for amplification with SFF-4 (27) and K501b primers. The amplified frag- ment was purified on 1% low melting agarose gel and cloned in pSK16 using DraIILIHindIII restriction sites. Mutant plasmids were identified and screened as above and sequenced using clpA internal sequencing primers. A total of seven different mutations were obtained, K501Q, K501R, K501P, K501T, K501L, K5011, and K501A, in which the lysine codon AA4 was changed to CAA, AGA, CCA, ACA, CTA, ATA, and GCA, respectively. The K501Q and K501R (Fig. 1) mutants were selected for further study, and the entire cloned PCR fragment was sequenced to ensure that only the desired mutation was present.
' The abbreviations used are: ATPyS, adenosine-5'-0-(3-thiotriphos- phate); PCR, polymerase chain reaction; DIP, diisopropylphosphoryl.
M. R. Maurizi and S. Gottesman, unpublished results.
GESGVGKT GPTGVGKT Q R
Q R
FIG. 1. Schematic drawing of the primary structure of ClpA and location of site-directed mutations. The regions of ClpA were defined by amino acid sequence similarities to highly conserved pro- teins from eukaryotes and prokaryotes (26). Part A of the Walker-type ATP-binding consensus sequence (35) for the two most highly conserved regions are shown and the amino acid replacements for the invariant lysines in each consensus are indicated. The single-letter code for amino acids is used. E. coli ClpA and ClpB (26) have the following percent identical and similar amino acids in each region: NH, terminus, 23% identical and 22% similar; domain I, 54% identical and 20% similar; domain II,53% identical and 22% similar; COOH terminus, 29% iden- tical and 26% similar.
To construct the double mutant (ClpA-K220Q/K501Q), in which both ATP-binding domains carry lysine to glutamine substitutions, the MluI- EcoRV fragment from the plasmid encoding ClpA-K220Q was moved into the plasmid encoding ClpA-K501Q. The construct was confirmed by sequencing through the two mutated sites.
Expression and Purification of Mutant ClpA Proteins-Plasmids en- coding the domain I or I1 ATP-binding site mutants were transformed into E. coli SG22030, and the cultures were screened for ClpA expres- sion by Western blotting. For purification of the mutant ClpA proteins, transformants in SG22030 were grown at 37 "C in super broth contain- ing 100 pglml ampicillin in 10-liter batches, and cell paste was stored a t -70 "C until used. The mutant ClpA proteins were purified essentially as described for wild-type ClpA in our earlier publications (20, 33). Purified mutant ClpA proteins were estimated to be 295% pure as analyzed by SDS-polyacrylamide gel electrophoresis and Western blotting (23).
Gel Filtration Chromatography on Superose 6-Purified wild-type and mutant ClpA proteins were separately run on a Superose-6 gel filtration column (1 x 30 cm) equilibrated with buffer B containing either 0.25 or 0.3 M KCI, in the absence or the presence of 25 mM MgCI, and 2 mM ATP. ClpA proteins (200 pg of each) in buffer B containing 0.3 M KC1 were passed through the column at a flow rate of either 0.3 or 0.4 mumin, and fractions were collected a t I-min intervals. The ClpA pro- teins were monitored at 280 nm, and estimates of protein in the frac- tions were made with the Bradford reagent. The fractions containing ClpA were confirmed by SDS-polyacrylamide gel electrophoresis and Western blotting procedures with anti-ClpA serum.
RESULTS
Mutations a t the Invariant Lysine Residue in Both Domain Z and IZ Affect ClpA-dependent Proteolysis in Vivo-Activity of the mutant ClpAs was tested in vivo by transforming clpA mutant hosts with plasmids encoding the mutant proteins and assaying the level of an RbsA-LacZ fusion protein that is a substrate for ClpAP protease.' The inverse relation between ClpA activity and p-galactosidase activity of the fusion would not be expected to be a simple linear function, but this assay does allow substantial changes in ClpA activity in vivo to be detected. Table I shows that introducing wild-type ClpA or the mutant ClpA-K220R on a multicopy plasmid into ClpA- cells restored degradation of RbsA-Lac2 as indicated by the de- creased yield of p-galactosidase activity, but another domain I mutant, ClpA-K220Qt had no detectable activity in vivo. EX- pressing the domain 11 mutants from a multicopy plasmid led to a slight drop in RbsA-Lac2 activity, suggesting that domain I1 mutants might express very low activity in vivo. This activity was not evident at high cell densities and may depend on the physiological state of the cell (data not shown). Mutants with both ATP sites mutated to either arginine (K220RK501R) or glutamine (K220Q/K501Q) were completely inactive. Thus, in domain I, the seventy of the defect caused by mutating the invariant lysine depends on the particular amino acid substi- tuted in its place, whereas domain I1 appears more stringent in

I I . .. 3 . 7 . '' ' l luo ATPase Sites in ClpA
A.
(84 kDa)
dye front -+
Mutatzonal mzalyszs or tne
TABLE I In vivo degradation of an RbsA-LucZ fusion protein
A ClpA- E. coli strain SG25518 which carries an rbsA2::lacZ fusion was transformed with plasmids encoding wild-type or mutant ClpA proteins. Cells were grown in tryptone broth a t 32 "C, permeabilized, and assayed for P-galactosidase activity. Average specific activities in Miller units (31) are shown for cells a t mid-exponential phase (OD,, = 0.6) in two separate cultures. For most cultures, specific activity in- creased in parallel about 2-fold during growth.
ClpA on plasmid a-Galactosidase
Miller units None (pUC18) 205 t 10 Wild-type 25 f 5
K220Q K220R
K501Q K501R
K220Q/K501Q K220FVK501R
185 f 10 35 f 10
165 2 10 175 f 10
210 f 10 200 f 10
requiring a lysine at that position. This result suggests a dif- ference between the two domains in the requirement for ATP binding and/or hydrolysis to promote proteolysis. Transforma- tion of wild-type cells with plasmids encoding mutant ClpA proteins did not interfere with the degradation of the RbsA- LacZ by wild-type ClpA (data not shown), suggesting that either mixed hexamers are not stably formed in vivo or that the mixed hexamers are active.
Purification and Assay of Mutant ClpA Proteins-Mutant ClpA proteins with arginine or glutamine substituted for lysine in domain I (ClpA-K220R or ClpA-K220Q) or domain I1 (ClpA- K501R or ClpA-K501R) were purified by the same procedure used for wild-type ClpA. The double mutant ClpA-K22OQ/ K501Q was also isolated. Mutant ClpA proteins were purified to more than 95% homogeneity, and the identity of the major 84-kDa protein in each preparation was confirmed by Western blotting with anti-ClpA antibody (Fig. 2). The mutant ClpA proteins were assayed for basal and substrate activated ATPase activity and for the ability to activate ClpP to degrade protein substrates, which requires ATP hydrolysis, or to de- grade a propeptide substrate, which requires nucleotide bind- ing but not ATP hydrolysis.
Table I1 reports a summary of the activities of the purified mutant ClpA proteins compared with the wild-type ClpA. In either domain, glutamine served less well than arginine in place of the invariant lysine. In domain 11, both mutations produced severe defects in ATPase and protein degradation; however, in domain I, the arginine mutant was -70% as active as wild-type ClpA whereas the glutamine mutant was practi- cally devoid of activity. The difference in the effects of identical substitutions in the two domains could reflect specific binding properties of the sites or could indicate that the two domains are responsible for different nucleotide-dependent reactions. Because ATP binding promotes self-association of ClpA and formation of the active complex with ClpP (5, 23, 34), i t was important to determine if any of the mutants were affected in assembly of ClpAl? before interpreting the results of the enzymatic assays.
Hexamer Formation by Wild-type and Mutant ClpA Pro- teins-ClpA-K22OQ and ClpA-K501Q were tested for the abil- ity to form a stable hexamer upon addition of ATP by gel fil- tration on Superose 6 in the absence and presence of ATP (Fig. 3). The elution position of ClpA-K220Q was not altered in the presence of ATP, indicating that this mutant did not form a stable hexamer under these conditions. In addition, light scat- tering and electron microscopic analysis indicated that ClpA-
29539
U.
FIG. 2. SDS-gel electrophoresis of mutant ClpA proteins. Mutant ClpA proteins were purified from cells in which they were
A , proteins stained with Coomassie Blue. From left to right, the lanes overexpressed from the endogenous ClpA promoter on plasmid pSK16.
contained 1 pg of wild-type ClpA, ClpA-K22OV, ClpA-K220R, ClpA- K220Q, ClpA-K501R, ClpA-K50lQ, and ClpA-K220Q/K501Q. B, West- ern blot of purified mutant ClpA proteins using anti-ClpA antiserum. The order was the same as above, with 0.1 pg of protein in each lane.
TABLE I1 Activities of purified wild-type and mutant ClpA proteins
cedures" in the presence of 4-6 mM ATP which was near saturating for Assays were conducted as described under "Experimental Pro-
all mutant proteins except ClpA-K220Q. ATPase was assayed in the absence of ClpP (basal) and in the presence of 5 pg of ClpP. In casein degradation assays, assays with limiting ClpA (0.2 pg of wild-type or 1 pg of mutant ClpA) had 5-7 pg of ClpP; assays with excess ClpA con- tained 5-10 pg of ClpA proteins and 0.1 pg of ClpP. Propeptide degra- dation assays contained -125 pg of FAPHMAISTV, 0.1 pg of ClpP, and 5-10 pg of ClpA proteins in 250 p1 of assay buffer.
ATP Casein hydrolysis degradation Propeptide
Basal +clpp Limited Excess excess ClpA degradation,
ClpA ClpA
9
Wild-type 100 90 100 100 100
Domain I mutants K220R 60 99 32 68 100 K220Q 0 0 0 0 0
Domain I1 mutants K501R 9 8 6 8 K501Q 9 8 2 4 290
290
Double mutant K220Q/K501Q 1 1 1 2 1
K220Q had no detectable ability to interact with ClpP (data not shown). Since binding but not hydrolysis of nucleotide is re- quired for formation of ClpA hexamers or the ClpAP complex, these data indicate either that the glutamine substitution greatly impairs binding of ATP or that binding of ATP to the mutant site does not produce the conformational change re- quired to stabilize the hexamer. Data presented below suggest that binding affinity is affected by the glutamine mutation. ClpA-KBOlQ behaved the same as wild-type ClpA in forming a hexamer in the presence of ATP, indicating that ATP binding to the domain I1 site is not required to promote hexamer forma- tion. Mutants with arginine in either domain I or I1 did form hexamers in the presence of ATP (Fig. 3), and light scattering measurements indicated that these mutant proteins formed complexes with ClpP in the presence of ATP (data not shown).

29540
500K 150K 68K
0.2
0.1
0
0.1
Tho ATPase Sites in ClpA
ATPs (mM)
0 10 15 20 Elution volume (mi)
FIG. 3. Gel filtration of wild-type and mutant ClpA proteins. Purified ClpA proteins (100-200 pg) were loaded onto a Superose 6 gel filtration column equilibrated in buffer B with 0.3 M KC1 and 1 mM dithiothreitol with (panel A) or without (panel B ) 2 RIM ATP and 25 rn MgCl,. The column was run at 0.3 mumin, and the absorbance of the effluent was monitored at 280 nm. Wild-type (. . . .1; K220Q (-1; K220R (- I - . 4; K501Q (- - - -1; K501R (- - -1. The profiles for K220R and K501R, which were identical to that of wild-type ClpA, are not shown in panel B.
Thus, the substitution of arginine for lysine does not prevent binding of ATP to the mutated domain I site. The assembly defect of ClpA-K220Q explains the complete absence of activity in most assays.
ATP-de~enden~ Protein ~egrada~~on-case in degradation was assayed under conditions in which the mutant ClpA pro- teins were either limiting or in excess. Activities with limiting ClpA are sensitive to defects in any of the steps required for ClpA activation of ClpP, such as nucleotide-dependent self- assembly of ClpA into hexamers and association of ClpA hexa- mers with ClpP, conformational effects of ClpA on CIpP, or chaperone-like activity of ClpA on protein substrates. Defects in assembly might be overcome by using excess ClpA to drive the ClpA into a complex with ClpP, whereas defects in one of the latter activities would not. The domain I mutant ClpA- K220R had low activity when present in limiting amounts but retained 5040% of the wild-type activity when assayed with the mutant ClpA in excess (Table 11). ClpA-K220Q, whether limiting or in excess, was completely devoid of activity, as ex- pected for a mutant that is drastically defective in hexamer formation. These results confirm that the domain I ATP site is important for promoting assembly of the active ClpA-ClpP com- plex and indicate that the K220R mutation produces only a mild defect in nucleotide-promoted assembly with ClpR The proteolytic activities of domain I1 mutants ClpA-K501Q and ClpA-KSOlR were low whether the mutant ClpA was limiting or in excess (Table 11), indicating that the defect in the domain I1 ATP site affects a second nucleotide-promoted step needed for protein degradation.
Basal ATPase Activity-Basal ATPase, measured in the ab- sence of ClpP and protein substrate, paralleled the loss of ac- tivity in promoting casein degradation. ClpA-K220Q was com- pletely inactive, and ClpA-KSOlQ had only 8-9% of the wild- type basal ATPase activity (Table 11). ClpA-K220Qnc501Q, with glutamine in place of lysine in both domains, had ~ 2 % wild-type ATPase activity. Since it is likely that the glutamine mutation in domain I1 completely abolishes ATP hydrolysis at that site, the low activity seen with ClpA-KSOlQ is probably expressed by the non-mutated domain I site. The residual ATPase activity expressed by domain I in ClpA-K501R was nearly the same as that seen with ClpA-K501Q (<lo% of wild-
1
X
0.5 >
0 0 1 2 3 4 0 0.05 0.1 0.15
FIG. 4. Propeptide cleavage by mutant ClpAproteins and ClpP in the presence of varying amounts ofATPyS. Propeptide (FAPH- U W V ) degradation was measured in standard assay mixtures with excess (5-15 pg) ClpA and limiting (0.1 pg) ClpP. Degradation was measured by the appearance of specific cleavage products as described (20). A, activity a t different ATPyS concentrations. B, expanded view of paneE A at low ATPyS concentrations. Wild-type (0); K220Q (VI; K220R (0); K501R (0); K501Q (A); K220QK501Q (Dl.
type), which also indicates that either the glutamine or argi- nine mutation eliminates ATP hydrolysis at the mutated site. In contrast, domain I mutant K220R, which can form hex- amers, expressed high ATPase activity. ClpA-K220R retained a significant (260% wild-type) basal ATPase activity (Table II), suggesting that more than 50% of the basal ATPase activity is expressed by domain I1 in wild-type ClpA.
ATPase Activity of the CLpAP Complex-Complex formation between ClpA and ClpP requires nucleotide binding but not hydrolysis, and there is no stimulation ofATPase activity when ClpP is added to ClpA (22, 33). Addition of ClpP to ATPase assays with wild-type ClpA caused a slight decrease in activity (Table II), and ClpP also slightly inhibited ATPase activity of domain I1 mutants. In contrast, ATPase activity of the domain I mutant ClpA-K220R was significantly increased when as- sayed in the presence of ClpP. A similar increase was seen for another domain I mutant, ClpA-K22OV (data not shown), but no effect of adding ClpP was seen with ClpA-K220Q, presum- ably because of the severe assembly defect of this mutant. The assays were carried out under conditions in which ATP was saturating and activity was proportional to mutant ClpA con- centrations, indicating that the increased ATPase activity rep- resented an increase in the VmaX for the mutant ClpA-ClpP complex. One explanation for this effect is that the ClpA-K220R hexamer does not have a completely native structure capable of expressing maximal ATPase activity, and interaction with ClpP promotes a conformational change in the mutant hexamer toward the wild-type structure.
Activation of Propeptide (FAPHMALVPV) Cleavage-ClpA activates ClpP to degrade a synthetic decapeptide which spans the processing site of pro-ClpP in vivo (20). Cleavage of the peptide is activated to the same extent by wild-type ClpA in the presence of ATP or a non-hydrolyzable analog of ATP (201, in contrast to degradation of proteins such as casein, which shows a strict requirement for ATP hydrolysis. In the presence of excess mutant protein and ATP, the domain I mutant ClpA- K220R activated propeptide degradation as well as wild-type ClpA (Table I1 and Fig. 4). ClpA-K220Q, however, was inactive under the standard conditions for propeptide cleavage, al- though activity was seen when a high concentration of ATP analog was used (Fig. 4). These results are consistent with the requirement for nucleotide binding to domain I for formation of the ClpA-ClpP complex and activation of propeptide cleavage. The substitution of glutamine for lysine not only lowers the affinity for nucleotide binding considerably but also affects the response to different nucleotides because ATP concentrations as high as 6 mM failed to activate ClpA-K220Q (Table 11). Domain I1 mutants were as active as wild-type ClpA in pro-

!ll.uo ATPase Sites in ClpA 29541
TABLE I11 ATP concentration dependence of ClpA activities 6,$
0.1 pg of ClpP, 4-10 ~.lg of ClpA proteins, and varying amounts of ATP Casein degradation was measured in standard assay mixtures with
(0.05-8 mH). ATPase was assayed in the absence of ClpP with 0.4 pg of wild-type ClpA or 1-5 pg of mutant ClpA proteins with varying concen- trations of [-p3'P1ATP. The So,, for ATP in each assay was determined from the midpoint of plots of initial velocity uersus ATP.
ATP hydrolysis, basal
Casein degradation, excess ClpA
Wild-type
Domain I mutants K220R K220Q
m M
0.25 0.25
1.1 NDa
0.30 ND"
Domain I1 mutants K501R 0.13 0.60 K 5 O l Q 0.17 0.50
a No activity detected at the highest concentration of ATP used.
moting propeptide degradation in the presence ofATP (Table 11) but required higher concentrations of nucleotide for maximum activity (Fig. 4). The high propeptide degrading activity of the domain I1 mutants indicates that these proteins can interact productively with ClpP and suggests, therefore, that their low activity in casein degradation reflects a defect in a step requir- ing ATP-hydrolysis involved in the binding or positioning of large proteins for degradation.
ATP Concentrat~n Dependence-Table I11 summarizes the half-maximal on cent rations of ATP required to activate the various activities of the mutant ClpA proteins. Although a com- plete analysis of the affinities of nucleotides must await direct binding studies, the changes in So,, for ATP with the different mutants probably indicate decreased binding affinities for ATP at the mutant sites. The data are also most consistent with a model in which binding of ATP occurs but hydrolysis of ATP is negligible at the mutant sites. The So,5 in the ATPase assay should reflect the nucleotide requirement for self-association of ClpA or for binding at the catalytic site, whichever is higher. Since proteolysis can be assayed when ClpA is not fully as- sembled by using an excess of ClpA over ClpP, the for ATP should reflect binding at the catalytic site. Domain I mutants showed a high S,, for ATP compared to wild-type ClpA when ATPase activity was assayed, but the ATP requirement for either casein or propeptide degradation, assayed with excess ClpA, was similar to that of wild-type ClpA (Table 111). These results are consistent with a model in which binding of ATP at domain I affects self-association of ClpA, which is necessary for ATPase activity. Mutations in domain I shift the nucleotide requirement for assembly and hence So,5 for ATPase to higher concentrations. When the assembly defect of domain I mutants is by-passed by using excess mutant protein, ATP apparently binds with the same affinity to the normal domain I1 in the assembled complex. These data confirm that binding and pos- sibly hydrolysis at the domain I1 site is necessary to activate proteolysis. Domain I1 mutants had a So,5 for ATPase activity similar to that of wild-type ClpA (Table 111), as expected if ATPase activity and self-assembly were dependent on the non- mutated domain I site. If ATP hydrolysis does not occur at the mutant domain I1 site, the ATPase activity of the domain I site must be sufflcient to satisfy the energy-requiring step in the slow proteolysis seen with domain I1 mutants. Nevertheless, the domain I1 mutants required higher ATP concentrations to activate proteolysis, which suggests that nucleotide must be bound to the domain 11 site to activate proteolysis even to the low extent (<lo%) seen with these mutants.
0 2 4 6 8 1 0 0 2 4 6 0 1 0
Ratio (MutanWild-type)
type Clpk A, excess ClpA. Competition between wild-type and Fm. 5. Addition of domain I1 ClpA mutants to assays of wild-
mutant CIpA for binding to limiting ClpP was tested by assaying casein degradation in a standard reaction mixture with 0.1 pg of ClpP and 0.5 pg of wild-type ClpAin the presence of increasing amounts (0.5-5.5 pg) of ClpA-K501Q. B, limiting CIpA. Casein degradation (U) waa measured in 50 pl of reaction mixtures with standard assay solu- tions except that a-casein was 100 pg/ml. Assays contained 7 pg of CIpP, 0.2 pg of wild-type ClpA, and increasing amounts (0.2-2.2 pg) of CIpA- K501Q. AWase activity (0) was determined in standard reaction mix- tures with 0.2 pg of wild-type ClpAAnd increasing amounts (0.2-2.2 pg) of ClpA-K501Q.
Hybrid Hemmers of Wild-type and a Domain II Mutant Have Only Slightly Decreased Activation of ClpP for ATP-&pendent Proteolysis-To study subunit interactions within the hexamer of ClpA and their importance in activating ClpP, ClpA was assayed for ATPase activity and for ATP-dependent casein deg- radation in the presence of varying amounts of CIpA-KSOlQ. Casein degradation was assayed either under limiting ClpP conditions or in the presence of excess ClpP. When wild-type ClpA was in slight excess over ClpP, increasing amounts of mutant ClpA proteins inhibited casein degradation (Fig. 5A). Excess ClpA-K501Q resulted in a residual activity that was comparable to that of the mutant alone, indicating that mutant hexamers can compete effectively with wild-type ClpAfor ClpP. The partial inhibition seen when mutant ClpA was present at the same concentration as the wild-type ClpA could reflect com- petition between wild-type and mutant hexamers for binding to limiting amounts of ClpP or the formation of hybrid hexamers between mutant and wild-type ClpA dimers in which the mutant phenotype is dominant. To test the latter possibility, proteolysis assays were per-
formed with limiting amounts of wild-type ClpA and increasing amounts of mutant ClpA but with ClpP in excess of the total ClpApresent (Fig. 5B). Under these conditions, since ClpP was present to combine with all wild-type and mutant hexamers, changes in activity observed upon addition of mutant ClpA proteins must be due to interactions between mutant and wild- type ClpA proteins. Addition of excess ClpA-K501Q caused a reduction of -40% of the activity in both protein degradation and ATPase activities. These data suggest that hybrid hexa- mers are formed between wild-type and mutant subunits. Since considerable activity (60-70%) was retained even when domain I1 mutant subunits predominated, ClpA subunits or pairs of subunits may constitute the functional units within hexamers.
Evidence for Cooperative Interactions between Domain I Re- gions in ClpA Hexamers and Cooperative Effects on ClpA-ClpP Interactions-Premixing of the domain I mutant ClpA-K220Q with wild-type ClpA resulted in a complete inhibition of basal ATPase activity, which is measured in the absence of ClpP (Fig. 6). Thus, ClpA-K220Q, which cannot form hexamers itself, can interact with wild-type ClpA and alter the conformation of the
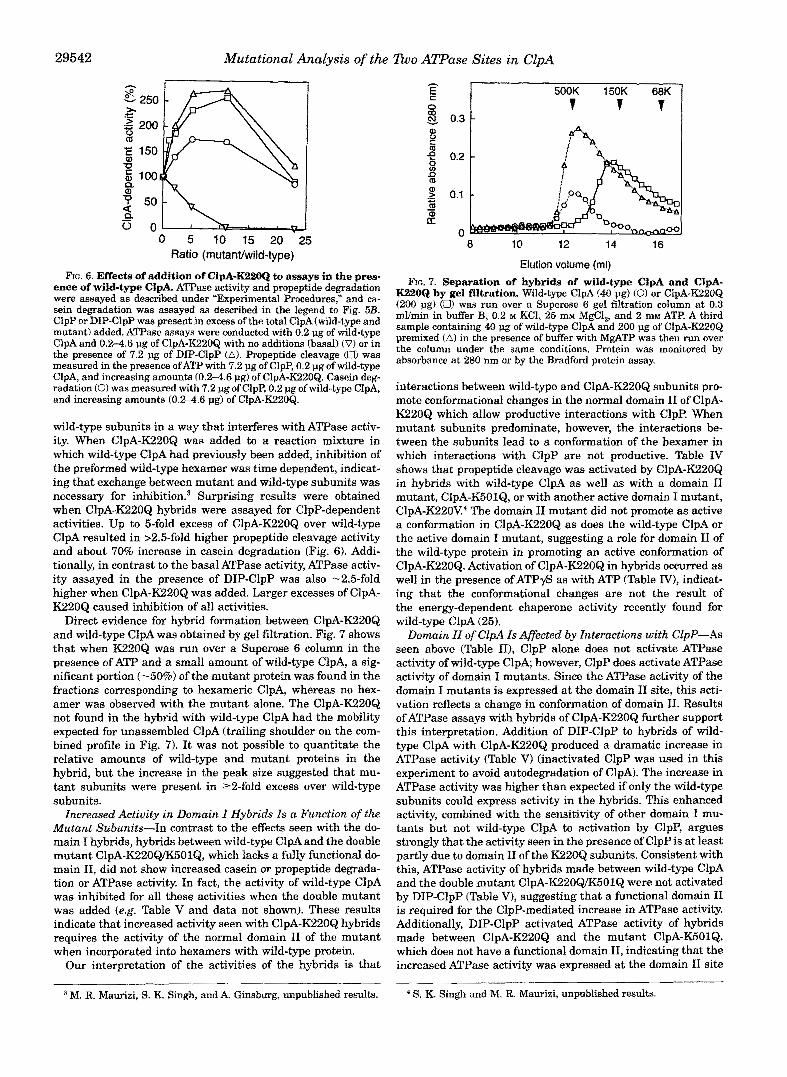
29542 Mutational Analysis of the Tho ATPase Sites in ClpA
-0 5 10 f5 20 -25 Ratio (mutan~wild-type)
FIG. 6. Effects of addition of ClpA-K22OQ to assays in the pres- ence of wild-me ClpG ATPase activity and propeptide degradation were assayed as described under ~ E x ~ r i m e n t ~ Procedures,” and ca- sein degradation was assayed as described in the legend to Fig. 5B. ClpP or DIP-ClpP was present in excess of the total ClpA(wi1d-type and mutant) added. ATPase assays were conducted with 0.2 pg of wild-type ClpA and 0.24.6 pg of ClpA-K220Q with no additions (basal) (VI or in the presence of 7.2 pg of DIP-ClpP (A). Propeptide cleavage (a) was measured in the presence ofATP with 7.2 pg of ClpP, 0.2 pg of wild-type ClpA, and increasing amounts (0.2-4.6 pg) of ClpA-K220Q. Casein deg- radation (0) was measured with 7.2 pg of ClpP, 0.2 pg of wild-type ClpA, and increasing amounts (0.2-4.6 pg) of ClpA-K22OQ.
wild-type subunits in a way that interferes with ATPase activ- ity. When ClpA-K220Q was added to a reaction mixture in which wild-type ClpA had previously been added, inhibition of the preformed wild-type hexamer was time dependent, indicat- ing that exchange between mutant and wild-type subunits was necessary for inhibition? Surprising results were obtained when ClpA-K220Q hybrids were assayed for ClpP-dependent activities. Up to &fold excess of ClpA-KZ20Q over wild-type ClpA resulted in 12.5-fold higher propeptide cleavage activity and about 70% increase in casein degradation (Fig. 6). Addi- tionally, in contrast to the basal ATPase activity, ATPase activ- ity assayed in the presence of DIP-ClpP was also -2.5-fold higher when ClpA-K22OQ was added. Larger excesses of ClpA- K220Q caused inhibition of all activities.
Direct evidence for hybrid formation between ClpA-KZZOQ and wild-type ClpA was obtained by gel filtration. Fig. 7 shows that when K220Q was run over a Superose 6 column in the presence of ATP and a small amount of wild-type ClpA, a sig- nificant portion (-50%) of the mutant protein was found in the fractions corresponding to hexameric ClpA, whereas no hex- amer was observed with the mutant alone. The ClpA-K220Q not found in the hybrid with wild-type ClpA had the mobility expected for unassembled ClpA (trailing shoulder on the com- bined profile in Fig. 7). It was not possible to quantitate the relative amounts of wild-type and mutant proteins in the hybrid, but the increase in the peak size suggested that mu- tant subunits were present in 22-fold excess over wild-type subunits.
Increased Activity in ~ a ~ a i n I Hybrids Is a Function of the ~ u t a n t Subunits-In contrast to the effects seen with the do- main I hybrids, hybrids between wild-type ClpA and the double mutant ClpA-K220QiK501Q, which lacks a fully functional do- main 11, did not show increased casein or propeptide degrada- tion or ATPase activity, In fact, the activity of wild-type ClpA was inhibited for all these activities when the double mutant was added (e.g. Table V and data not shown). These results indicate that increased activity seen with ClpA-KZ20Q hybrids requires the activity of the normal domain I1 of the mutant when incorporated into hexamers with wild-type protein.
Our interpretation of the activities of the hybrids is that
M. R. Maurizi, S. K. Singh, and A. Ginsburg, unpublished results.
8 10 12 14 16
Elution volume (ml)
K22OQ by gel filtration. Mfild-type ClpA (40 pg) (Of or ClpA-K220Q FIG. 7. Separation of hybrida of wild-type ClpA and ClpA-
(200 pg) (U) was run over a Superose 6 gel filtration column at 0.3 mumin in buffer B, 0.2 M KCI, 25 mM MgCl,, and 2 mM Am. A third sample containing 40 pg of wild-type ClpA and 200 pg of ClpA-K220Q premixed (A) in the presence of buffer with MgATP was then run over the column under the same conditions. Protein was monitored by absorbance at 280 nm or by the Bradford protein assay.
interactions between wild-type and ClpA-K220Q subunits pro- mote conformational changes in the normal domain I1 of ClpA- K220Q which allow productive interactions with ClpP. When mutant subunits predominate, however, the interactions be- tween the subunits lead to a conformation of the hexamer in which interactions with ClpP are not productive. Table Tv shows that propeptide cleavage was activated by ClpA-K220Q in hybrids with wild-type ClpA as well as with a domain I1 mutant, ClpA-K501Q, or with another active domain I mutant, C1pA-K220V4 The domain I1 mutant did not promote as active a conformation in ClpA-K220Q as does the wild-type ClpA or the active domain I mutant, suggesting a role for domain I1 of the wild-type protein in promoting an active conformation of ClpA-K220Q. Activation of ClpA-K220Q in hybrids occurred as well in the presence of ATPyS as with ATP (Table IVj, indicat- ing that the conformational changes are not the result of the energy-dependent chaperone activity recently found for wild-type ClpA (25). ~ o ~ ~ i n If of ClpA Is Affected by I n t e ~ ~ c t ~ o n s with ClpP-AS
seen above (Table II), ClpP alone does not activate ATPase activity of wild-type ClpA; however, ClpP does activate ATPase activity of domain I mutants. Since the ATPase activity of the domain I mutants is expressed at the domain I1 site, this acti- vation reflects a change in conformation of domain 11. Results of ATPase assays with hybrids of ClpA-K220Q further support this interpretation. Addition of DIP-ClpP to hybrids of wild- type ClpA with ClpA-K22OQ produced a dramatic increase in ATPase activity (Table V) (inactivated ClpP was used in this experiment to avoid autodegradation of ClpAj. The increase in ATPase activity was higher than expected if only the wild-type subunits could express activity in the hybrids. This enhanced activity, combined with the sensitivity of other domain I mu- tants but not wild-type ClpA to activation by ClpP, argues strongly that the activity seen in the presence of ClpP is at least partly due to domain I1 of the K220Q subunits. Consistent with this, ATPase activity of hybrids made between wild-type ClpA and the double mutant ClpA-K220Q/K501Q were not activated by DIP-ClpP (Table V), suggesting that a functional domain 11 is required for the ClpP-mediated increase in ATPase activity. Additionally, DIP-C1pP activated ATPase activity of hybrids made between ClpA-K220Q and the mutant ClpA-K501Q, which does not have a functional domain 11, indicating that the increased ATPase activity was expressed at the domain 11 site
* S. K. Singh and M. R. Maurizi, unpublished results.

~ u t ~ t i o ~ a l AnaZysis of the
TABLE IV Activation of propeptide cleavage by ClpA-K220Q hybrids
Propeptide degradation was measured in 125 pl of standard assay buffer with 62.5 pg of FAPHMALVPV and 7 pg of ClpP. &says had fixed amounts of wild-type ClpA (0.2 pg), ClpA-K501Q (0.2 pg?, or ClpA- K220V (0.5 pg? and increasing amounts of domain 1 mutant ClpA- K220Q and either 4 mM ATP or 1 mM ATPyS as indicated. _ _ _ _ _ _ ~
Ratio of ClpAX22OQ to aetive CIpA
Active ClpA added
Wild-type CIDA ClpA-K501Q ClpA-K22OV .. - ~~
% deavagelrninlpg ClpA -
A. Assays with ATP 0 2.0 0.50 1.7 1 3.4 0.69 ND" 2 3.7 1.2 ND 5 4.6 2.2 4.0
11 5.1 2.2 ND B. Assays with ATPyS
0 2.3 ND 1.2 5 4.7 ND 3.1
a ND, not done.
TABLE V Effects of ClpP interaction on the ATPase actiuity of ClpAproteins
and hybrids made with wild-type and mutant ClpA proteins
standard conditions with 0.2 pg of wild-type ClpA or with 2.5-5 pg of ATPase activity was measured in a reaction volume of 50 pl under
ClpA-K220Q, ClpA-K501Q, or the double mutant ClpA-K220Q/K501Q. Hybrids were formed by adding a 5-fold excess of ClpA-K220Q or ClpA- K220Q/K5~1Q to wild-type ClpA or a 2-fold molar excess of ClpA- K220Q to CIpA-K5OlQ. In paraIlel assays, ClpP that had been in- activated with diisopropylfluoro phosphate (DIP-ClpP) was added in 22-fold molar excess over the total ClpA.
ATPase activity
-DIP-ClpP +DIP-ClpP
B
___ .-
No addition Wild-type 100 a5 K220Q 0 0 K501Q 10 8 K220QK501Q 0.2 0.2
CIpA-K22OQ hybrid with Wild-type 42 167 CIpA-K5OlQ 0.7 7
ClpA-K220Q/K501Q hybrid with Wild-type 69 47
of ClpA-K220Q. Thus, interaction of ClpP with hybrids not only overcomes the inhibitory effects of interactions between mu- tant and wild-type subunits but induces an active conformation of domain I1 in the mutant subunits themselves.
DISCUSSION Enzymes with Walker-type nucleotide binding motifs (35)
carry out widely diverse functions (36-38). Substitution of the invariant lysine defined in part A of the consensus, G(XJGK (T/S), where Xis any amino acid, usually has adverse affects on the binding or hydrolysis of ATP, but the seventy of the defect on the enzymatic function coupled to ATP binding or hydrolysis can be quite variable and is dependent on the particular amino acid replacement (39-42). In some cases, such as the PriA pro- tein, mutation of the invariant lysine does not prevent nucleo- tide-dependent macromolecular assembly reactions but blocks enzymatic activities that depend on nucleotide hydrolysis (43).
Included in the family of proteins with Walker-type motifs are many proteins that have two ATP-binding sites encoded in a single polypeptide chain. For some proteins, such as the transmembrane conductance regulator (CFTR) or the multi- drug transporter, these ATPase sites arose by duplication and fusion of a single site so that the two halves of the molecule are
7200 ATPase Sites in ClpA 29543
very similar to each other. In other proteins, the ATP-binding sites are located in non-homologous regions of the protein, which appear to have arisen by fusion of two different ATPases. In such proteins as members of the Clp family (27) and the DNA repair enzyme UvrA (371, each domain can be expected to contribute a unique function to the overall activity carried out by the protein. For example, mutagenesis has indicated that ATP hydrolysis at the C O O H - ~ ~ i n a l nucleotide site of UvrA is required to release nucleoprotein complexes from undamaged DNA (37).
In this paper we have shown that both ATPase sites of ClpA are needed for activation of protein degradation by ClpP and have begun to elucidate the individual roles for the ATPase domains. Studies on the ClpAIp, homolog from yeast, ~ S P 1 0 4 , also indicate that both sites are needed for activity in vivo (44). It has been proposed that Clp ATPases have a protein unfolding or disaggregating activity (26, 45, 46). For ClpB-like proteins, the chaperone activity may be the sole physiological activity because no evidence that ClpB directly activates protein deg- radation has as yet been found. ClpA, on the other hand, func- tions in vivo and in vitro to activate protein degradation by ClpP (22-24). In addition, recent data demonstrate that ClpA can act aIone as a molecular chaperone or protein unfolding enzyme (25), and it appears likely that this chaperone activity is an essential part of the activity of ClpA in promoting protein degradation. The studies reported in this paper have focused on the roles of the ATP-binding site in assembly of the active proteol~ic complex and on the requirement for nucleotide hydrolysis for protein degradation.
Mutants altered in the ATPase sites were assayed for ClpA hexamer f o ~ a t i o n , ATPase activity, activation of propeptide cleavage (which does not require ATP hydrolysis), and activa- tion of protein degradation (which does), The failure of the domain I mutant ClpA-KZZOQ to form a hexamer in the pres- ence of high concentrations of ATP could reflect inability either to bind nucleotide or to undergo a conformational change upon nucleotide binding. We favor the latter interpretation because the lysine to glutamine substitution in the other domain re- duced nucleotide affinity but did not completely eliminate bind- ing. In either case, this mutant points to the essential role of domain I nucleotide binding in stabilizing the ClpA hexamer and forming the active complex with ClpP. Another domain I mutant (ClpA-K220R) was slightly impaired in enzymatic ac- tivity when mutant ClpA was limiting. Less direct evidence indicates that domain I1 also contributes to the stability of the ClpA hexamers. For example, domain I1 mutants disso- ciated somewhat during gel filtration and show a marked non- linearity of ATPase activity versus mutant enzyme concentra- tion (data not shown). In yeast, HSP104 mutants with amino acid substitutions in domain 11 are defective in forming hex- amers (471, but it may be that for ClpA domain I makes a relatively greater contribution to the hexamer stability.
Since the ATPase activity of the mutated site in domain I mutants should at least be severely impaired, the ability of these mutants and hybrids made between them and wild-type ClpA to activate proteolysis indicates that ATP hydrolysis at the domain I ATPase site is not essential, although it may be needed for maximal rates of proteolysis. The requirement for domain I may also vary with the protein substrate, depending on the recognition elements or on the degree of unfolding re- quired to initiate degradation. Possibly, domain I is responsible for only 8-9% of the total basal ATPase activity in wild-type ClpA. The ATPase activities of domain I mutants suggest that 265% of the ATP hydrolysis occurs at the domain IT site in the wild-type enzyme, a result consistent with the crucial role for ATP hydrolysis at domain I1 in promoting protein degradation.

29544 ~ u t a t ~ o ~ a l Analysis of the
The activity observed may have been lower than the expected 90% because the mutation in domain I affected the conforma- tion of the ClpA hexamer. ATPase activity of the domain I mutants was also stimulated in the presence of ClpP and pro- tein substrates, which correlates with the ability of those mu- tants to activate protein degradation.
Properties of domain 11 mutants which lose >90% of the ATPase activity and >90% of the ability to activate protein degradation by ClpP also indicate that the domain I1 ATPase site plays a major role in the activation of protein degradation. The defect in proteolysis is directly linked to reduced ATPase activity because domain I1 mutants can activate degradation of substrates, such as the propeptide, for which binding but not hydrolysis of ATP is required. In addition, the increase in the So,, for ATP in the protein degradation assay for domain 11 when CIpA is in excess suggests that ATP hydrolysis at the domain 11 site is needed for proteolysis. The activity of domain I mutants must therefore be a function of the normal domain I1 sites in those proteins. When ClpA-K220R (Table 111) and an- other domain I mutant, K220V (data not shown) were assayed with excess mutant ClpA, the So,& for ATP was the same as that for wild-type ClpA, reflecting the activating effect of ATP hy- drolysis at the normal domain I1 site in these mutants. Propep- tide degradation in the presence ofATP analogs also shows that saturation of the defective domain 11 nucleotide site is needed for maximal activation of ClpP and requires higher concentra- tions of ATPyS to reach saturation.
Enzymatic properties of hybrid hexamers indicate the impor- tance of subunit interactions in the activity of ClpA and ClpAP protease. The domain I mutant ClpA-K220Q cannot form a hexamer by itself, but interaction with wild-type CIpA induces a conformation that allows ClpA-K220Q to form a hybrid hex- amer. Inhibition of basal ATPase activity of wild-type ClpA in domain I mutant hybrids confirms that the active form of ClpA is a multimer, most likely a hexamer, and indicates that do- main I interactions play a role in promoting an active confor- mation of ClpA. For hybrids formed between domain 11 mu- tants and wild-type ClpA, conformational effects of interactions between mutant and wild-type subunits slightly interferes with the activity of wild-type subunits. Since wild-type ClpA sub- units in mixed hexamers with domain I1 mutants have ATPase activity and are active in promoting proteolysis by ClpP, these enzymatic activities may be carried out by individual subunits or pairs of subunits within wild-type ClpA hexamers.
The conformational effects of ClpP interactions are shown dramatically by the restoration ofATPase activity to both wild- type and domain I mutant subunits within the hybrids when ClpP or DIP-ClpP was added (Table V). In other domain I mutants, such as ClpA-K220R, the normal domain I1 also ap- pears to be in a less active conformation, which is altered by interaction with ClpP (Table 11). In hybrids formed between domain I1 mutants and wild-type ClpA, interaction with ClpP does not restore activity to the mutant subunits because of the defective ATPase site in domain 11. The simplest i n t e~ re t a t ion of these results is that the activating effect of ClpP is mediated by a direct interaction between ClpP and domain I1 of the mutant, which converts it to a conformation more closely re- sembling that of wild-type ClpA. This model is supported by the shift in the nucleotide concentration dependence of ClpP acti- vation for the domain 11 mutants (Table 111 and Fig. 4). Alter- natively, ClpP could interact with domain I of ClpA, and do- main 11 could be activated as a consequence of changes in interactions between the two domains.
Sedimentation velocity ultracentrifugation3 and electron mi- croscopic studies of ClpAP' indicate that when ClpP is in excess
M. Kessel and M. R. Maurizi, unpublished results.
ltuo ATPase Sites in ClpA
the complex has one ClpA hexamer bound to ClpP, and when ClpA is in excess there are two hexamers of ClpA bound to ClpP. Activity measurements, however, indicate that there is no dif- ference in the specific activity of ClpP for casein degradation in the 1:l and the 2:l complex, which suggests that the second hexamer of wild-type ClpA may be redundant for casein deg- radation. Table If shows that m u ~ t i o n s i n both domains ad- versely affect the activity of the 1:l complex (limiting ClpA). For domain I but not domain I1 mutants, however, activity was higher in the presence of excess mutant CIpA, that is, the 2:l complex appears more active than the 1:1 complex, suggesting that the second mutant hexamer contributes to the activation
ClpP can be activated in vivo and in vitro by either ClpA or ClpX (6, 7). Since ClpX is about half the size of ClpA and has only a single ATPase site, the crucial activities needed for ATP- dependent proteolysis could in theory be carried out by either domain of ClpA. ClpX bears greater sequence similarity with the COOH-terminal domain of ClpA, and results of these stud- ies suggest that the COOH-terminal domain of ClpA has most of the ATPase activity and plays the primary role in the ATP- consuming steps in promoting proteolysis. What is the role of the other domain in ClpA? Domain I could function primarily to stabilize the complex of ClpA and ClpP. Since binding of ATP is sufficient to promote assembly and the intrinsic ATPase activ- ity of domain I is low, the energy costs of maintaining the complex could be quite moderate and the resulting benefits in eficiency and regulation perhaps considerable. I t seems more likely, however, that the domain I has a more important role. One possibility is that domain I has the chaperone or unfolding fhnction recently described for ClpA (25). This activity enables ClpA to interact with a variety of partially unfolded proteins and to initially screen them for folding competence. Proteins that are slightly misfolded and can quickly resume a native conformation would be released, but proteins that are more grossly misfolded would interact more extensively with the protein-binding site in domain I and be passed to domain 11. Domain I1 would then act to further unfold or to translocate the protein substrate into the active site of ClpP, much as ClpX does. Domain I of ClpA would thus provide a scanning or proof- reading function that allowed ClpA to select from a wide spec- trum of potential substrates. The prediction would be that ClpX would have a much more restricted substrate range, and, in fact, ClpX in vitro does not promote the degradation of such heterologous proteins as a-casein (7). Further studies are un- derway to define the substrate range and the protein recogni- tion functions of ClpA.
of Clpp.
ments on the manuscript, Mark W. Thompson for helpful discussions Acknowledgments-We thank Susan Gottesman for advice and com-
and for providing the propeptide used in these studies, and William P. Clark for providing plasmid pWPC3.1.
REFERENCES 1. Goldberg, A. L., and John, A. C. S. (1976) ARRU. Rev. Biochem. 45,747-803 2. Hershko, A,, and Ciechanover, A. (1992) Annu. Reo. Biochem. 61,7614307
4. Goldberg, A. L. (1992) Eur: J . Biochem. 203,9-23 3. Gottesman, S., and Maurizi, M. R. (1992) Microbiol. Rev. 56,592421
5. Maurizi, M. R. (1992) Erperientia 48, 178-201 6. Gottesman, S., Clark, W. P., Crecy-Lagard, V. D., and Maurizi, M. R. (1993) J.
7. Wojtkowiak, D., Georgopoulos, C., and Zylicz, M. (1993) J. Biol. Chem. 268,
8. Rivett, A. J. (1989) Arch. Biochem. Biophys. 268,143 9. Orlowski, M. (1990) Biochemistry 29,10289-10297
Biol. Chem. 268,22618-22626
22609-22617
10. Dubiel, W., Ferrell, It, Pratt, G., and Rechsteiner, M. (19921 J. BioZ. Chem.
11. Eytan, E., Ganoth, D.,Armon, T., and Hershko, A. (1989) Proc. Natl. Acad. Sci.
12. Driscoll, J., and Goldberg, A. L. (1989) Proc. Natl. Acad. Sci. U. S. A. 86,
13. Ganoth, D., Leshinsky, E., Eytan, E., and Hershko, A. (1988) J. Biol. Chem.
267,22699-22702
U. S. A. 86,7751-7755
787-791
263, 12412-12419

Mutational Analysis of the ? b o ATPase Sites in ClpA 29545 14. Hoffman, L., Pratt, G., and Rechsteiner, M. (1992) J. Biol. Chem. 267,22362-
15. Rechsteiner, M., Hoffman, L., and Dubiel, W. (1993) J. Biol. Chem. 268,6065-
16. Goldberg, A. L., and Waxman, L. (1985) J. Biol. Chem. 260, 12029-12034 17. Waxman, L., Fagan, J. M., Tanaka, K., and Goldberg, A. L. (1985) J. Biol.
18. Waxman, L., and Goldberg, A. L. (1986) Science 232, 500-503 19. Waxman, L., and Goldberg, A. L. (1982) Proc. Natl. Acad. Sci. U. S. A. 79,
20. Thompson, M. W., and Maurizi, M. R. (1994) J. Biol. Chem. 269,18201-18208 21. Woo, K. M., Chung, W. J., Ha, D. B., Goldberg, A. L., and Chung, C. H. (1989)
22. Hwang, B. J., Woo, K. M., Goldberg, A. L., and Chung, C. H. (1988) J. Biol. J . Biol. Chem. 264,2088-2091
23. Katayama-Fujimura, Y., Gottesman, S., and Maurizi, M. R. (1987) J. Biol. Chem. 263,8727-8734
24. Katayama, Y., Gottesman, S., Pumphrey, J., Rudikoff, S., Clark, W. P., and Chem. 262,44774485
25. Wickner, S., Gottesman, S., Skowyra, D., Hoskins, J., McKenney, K., and Maurizi, M. R. (1988) J. Biol. Chem. 263, 15226-15236
26. Gottesman, S., Squires, C., Pichersky, E., Carrington, M., Hobbs, M., Mattick, Maurizi, M. R. (1994) Proc. Natl. Acad. Sci. U. S. A , , in press
J. S., Dalrymple, B., Kuramitsu, H., Shiroza, T., Foster, T., Clark, W. P., Ross, B., Squires, C. L., and Maurizi, M. R. (1990) Proc. Natl. Acad. Sci.
27. Gottesman, S., Clark, W. P., and Maurizi, M. R. (1990) J. Biol. Chem. 265, U. S. A. 87, 3513-3517
28. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: a 7886-7893
Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
22368
6068
Chem. 260, 11994-12000
4883-4887
29. Maurizi, M. R., Trisler, P., and Gottesman, S. (1985) J. Bacteriol. 164, 1124-
30. Maurizi, M. R. (1987) J. Biol. Chem. 262,2696-2703 31. Miller, J. H. (1992) A Short Course in Bacterial Genetics: a Laboratory Manual
and Handbook for Escherichia coli and Related Bacteria, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
32. Ho, S. H., Hunt, H. D., Horton, R. M., Pullen, J. K., and Pease, L. R. (1989) Gene (Amst. 77,51-59
33. Maurizi, M. R., Thompson, M. W., Singh, S. K, and Kim, S.-H. (1994) Methods
1135
34. Maurizi, M. R. (1991) Biochem. SOC. Dans. 19,719-723 35. Walker, J. E., Saraste, M., Runswick, M. J., and Gay, N. J. (1982) EMBO J. 1,
Enzymol. 244, 314331.
945-951 36. Kaur , P., and Rosen, B. P. (1992) J. Biol. Chem. 267, 19272-19277 37. Thiagalingam, S., and Grossman, L. (1991) J. Biol. Chem. 266, 11395-11403 38. Klose, M., Schimz, K L., van der Wolk, J., Driessen,A. J., and Freudl, R. (1993)
39. Haber, L. T., and Walker, G. C. (1991) EMBO J. 10, 2707-2715 40. Sung, P., Higgins, D., Prakash, L., and Prakash, S. (1988) EMBO J. 7, 3263-
J. Biol. Chem. 268,4504-4510
3269 41. Seelig, A,, Kloetzel, P.-M., Kuehn, L., and Dahlmann, B. (1991) Biochem. J.
42. Hsieh, S., and Julin, D. A. (1992) Nucleic Acids Res. 20,5647-5653 43. Zavitz, K. H., and Marians, K. J. (1992) J. Biol. Chem. 267,6933-6940 44. Parsell, D. A., Sanchez, Y., Stitzel, J. D., and Lindquist, S. (1991) Nature 353,
45. Squires, C., and Squires, C. L. (1992) J. Bacteriol. 174, 1081-1085
47. Parsell, D. A., Kowal, A. S., and Lindquist, S. (1994) J. Biol. Chem. 269, 46. Parsell, D. A., Kowal, A. S., and Lindquist, S. (1994) Nature, in press
280,225-232
270-273
4480-4487


















