
Symmetry reduction for molecular dynamics simulation of an ...

See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/268263168
Molecular Symmetry and Dynamics 31.1 DYNAMICS AND SPECTRA OF
MOLECULAR ROTORS
Article · January 2006
DOI: 10.1007/978-0-387-26308-3_32
CITATION
1
READS
43
1 author:
Some of the authors of this publication are also working on these related projects:
PHYSICS View project
William George Harter
University of Arkansas
122 PUBLICATIONS 2,429 CITATIONS
SEE PROFILE
All content following this page was uploaded by William George Harter on 07 February 2015.
The user has requested enhancement of the downloaded file.

31
Molecular Symmetry and DynamicsWilliam G. Harter
Department of Physics, University of Arkansas, Fayetteville, Arkansas 72701
31.1 DYNAMICS AND SPECTRA OF MOLECULAR ROTORS 37831.1.1 Rigid Rotors 37931.1.2 Molecular States Inside and Out 37931.1.3 Rigid Asymmetric Rotor Eigensolutions and Dynamics 380
31.2 ROTATIONAL ENERGY SURFACES ANDSEMICLASSICAL ROTATIONAL DYNAMICS 381
31.3 SYMMETRY OF MOLECULAR ROTORS 38431.3.1 Asymmetric Rotor Symmetry Analysis 384
31.4 TETRAHEDRAL-OCTAHEDRAL ROTATIONALDYNAMICS AND SPECTRA 38531.4.1 Semirigid Octahedral Rotors and Centrifugal Tensor
Hamiltonians 38531.4.2 Octahedral and Tetrahedral Rotational Energy Surfaces 38631.4.3 Octahedral and Tetrahedral Rotational Fine Structure 38731.4.4 Octahedral Superfine Structure 387
31.5 HIGH RESOLUTION ROVIBRATIONAL STRUCTURE 38931.5.1 Tetrahedral Nuclear Hyperfine Structure 38931.5.2 Superfine Structure and Spontaneous Symmetry Breaking 39031.5.3 Extreme Molecular Symmetry Effects 392
31.1 DYNAMICS AND SPECTRA OFMOLECULAR ROTORS
Molecules are aggregates of two or more nuclei boundby at least one electron. The nuclei of most stablemolecules can be imagined to be points in a more or lessrigid body whose relative positions are constrained byan electronic bonding potential. This potential dependsstrongly upon the electronic state as described in Chap.30. For this discussion, we consider only stable moleculesin their electronic ground state.
Motions which stretch or compress the bonds arecalled vibrational motions, and give rise to spectralresonances in the infrared region of the spectrum. Typ-ical vibrational quanta (v0) lie between 80 cm−1 (the
37Atomic, Molecular, & Optical Physics Handbook, edit
lowest GeBr4 mode) and 3020 cm−1 (the highest CH4mode). (A wave number of 1000 cm−1 correspondsto a wavelength of 10 µm and a frequency of 29.9792458 THz.) Vibrational amplitudes are usually tiny, sincezero-point motions or vibrations involving one or twoquanta (v = 0, 1, 2, . . . ) are constrained by the steepbonding potential to less than a few percent of bondlengths.
Overall rotation of molecules in free space is un-constrained, and gives rise to far-infrared or microwavepure rotational transitions or sidebands on top of vibra-tional spectra. Typical rotational quanta lie between0.18 cm−1 (5.4 GHz) for SF6 and 10.6 cm−1 for CH4.Individual molecules are free to rotate or translate as awhole while undergoing their tiny but rapid vibrations.
8ed by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 379
A vibrating molecule can be thought of as a tumbling col-lection of masses held together by ‘springs’ (the electronicvibrational potential or force field) and is called a semi-rigid rotor . The coupling of rotational and vibrationalmotion is called rovibrational coupling, and includes cen-trifugal and Coriolis coupling discussed in Sect. 31.4.
This discussion of molecular dynamics and spectramainly involves molecular rotation and properties ofrotationally excited molecules, particularly those withhigh rotational quantum number J = 10–200. How-ever, the discussion also applies to molecules in ex-cited vibrational states, and even to certain cases ofmolecules in excited electronic states. The analysis of vi-bronic (vibrational-electronic), rovibrational (rotational-vibrational), or rovibronic (all three) types of excitationcan be very complicated [1–5] and is beyond the scope ofthis chapter, but consideration of these problems can allbenefit from the elementary treatment given here.
31.1.1 Rigid Rotors
As a first approximation, and for the purposes ofdiscussing basic molecular dynamics and spectra, onemay ignore vibrations and model stable molecules as‘stick-and-ball’ structures or rigid rotors . Then theHamiltonian just has the following three terms
H = AJ2x +BJ2
y + CJ2z . (31.1)
Here, Jx , Jy , Jz are rotational angular momentumoperators, and the rotational constants are
A = 12 (Ix), B = 1
2 (Iy), C = 12 (Iz) . (31.2)
where the Iα are the principal moments of inertia of thebody. This implies that the J-coordinate system beingused is a special one fixed to the rotor and aligned withits principal axes.
Many molecules, particularly all diatomic molecules,have two of these rotational constants equal, say A =B. Such rotors are called symmetric tops , and theirHamiltonian can be written in terms of the square of thetotal angular momentum J·J and one other componentJz as
H = BJ2x +BJ2
y + CJ2z = BJ·J + (C −B)J2
z .
(31.3)
This gives a simple formula for the symmetric toprotational energy levels in terms of the quantum numbersJ for total angular momentum and K for the body z-component
E(J,K) = BJ(J + 1) + (C −B)K2 . (31.4)
Atomic, Molecular, & Optical Physics Handbook, edit
However, this simple eigenvalue formula hides thestructure of the eigenstates or eigenfunctions. Indeed,the full Schrodinger angular differential equation basedupon Hamiltonian (31.1) is much more lengthy. It isimportant to remember that H is written in a rotatingbody coordinate system, and must be connected to a star-fixed or laboratory frame to get the full theory.
31.1.2 Molecular States Inside and Out
Rotor angular momentum eigenfunctions can be ex-pressed as a continuous linear combination of rotor an-gular position states |αβγ〉 defined by the Euler angles oflaboratory azimuth α, polar angle β of body z-axis, andbody azimuth or ‘gauge twist’ γ∣∣∣∣ J
MK
⟩=√
2J + 18π2
∫ 2π
0dα
∫ π
0sinβ dβ
×∫ 2π
0dγ DJ∗
MK(αβγ)|αβγ〉 , (31.5)
where the rotor wave functions DJ∗MK are just the conju-
gates of the Wigner rotation matrices described in Sect.2.4.1, and row and column indices M and K are the laband body components of the angular momentum, respec-tively [5–7].
An important feature of polyatomic molecules isthat their angular momentum states have two kinds ofazimuthal quantum numbers. In addition to the usuallab component M associated with the coordinate α (αand β are usually labeled φ and ϑ), there is a bodycomponent K associated with the Euler coordinate γ,the body azimuthal angle for the body z-axis relative tothe laboratory Z-axis.
The physics of atomic or diatomic angular momentumstates has no internal or ‘body’ structure, so that thequantum number K is always zero. Unless one setsK = 0, the energy formula (31.4) blows up for a pointparticle because z-inertia for a point is zero and C isinfinite. Also, the dimension of the angular momentumstate multiplet of a given J is larger than the usual(2J + 1) found in atomic or diatomic molecular physics.In polyatomic rotors, the number of states for each J is(2J + 1)2 since both M and K quantum numbers rangefrom −J to +J .
A futher important feature is that the molecular rotorwave functions contain as a special K = 0 case allthe usual atomic spherical harmonics Y `m complete withcorrect normalization and phase,
√4πY `m(φϑ) = D`∗
m0(φϑ)√
2`+ 1 . (31.6)
This is part of a powerful symmetry principle: grouprepresentations are quantum wave functions, and symmetry
ed by G. W. F. Drake, AIP Press, New York c© 1996.

380 / Atomic, Molecular, & Optical Physics Handbook
analysis is an extension of Fourier analysis — not just fortranslations as in standard Fourier transforms, but forany group of symmetry operations. The usual Fouriercoefficients exp(ikx) are replaced by the D functions inthe rotational Fourier transform embodied by Eq. (31.5).
Molecular rotational analysis displays another impor-tant aspect of symmetry analysis in general. For everygroup of symmetry operations, such as external lab-basedrotations, there is an independent dual group of internalor body-based operations. The external symmetry of theenvironment is independent of the internal symmetry ofthe molecular body, and all the operations of one com-mute with all those of the other. The molecular rotationgroup is written as an outer product R(3)lab ⊗R(3)bodyof the external and internal parts. Thus the degeneracyassociated with the group representations for a single Jis (2J + 1)2.
The inversion or parity operator I(r → −r) canbe defined to be the same for both lab and bodyframes. Including I with the rotational group R(3)gives the orthogonal group O(3) = R(3) ⊗ {1, I}. Ifparity is conserved (e.g., no weak neutral currents), thefundamental molecular orthogonal group is O(3)lab ⊗O(3)body.
How this symmetry breaks down and which levelssplit depends upon both the perturbative laboratoryenvironment and the internal molecular structure. Aspherical top Hamiltonian is Eq. (31.1) with A = B =C. This has a full O(3)body (spherical) symmetrysince it is just BJ·J. Given that the rotor is in anO(3)lab laboratory (empty space), the original symmetryO(3)lab ⊗ O(3)body remains intact, and the (2J + 1)2
degeneracy is to be expected. However, a symmetricrotor in a lab vacuum has its internal symmetry brokento O(2)body if A = B 6= C, and the energies given byEq. (31.4) result in internal quantum singlets for K = 0and ±K doublets for K 6= 0. But each of these levelsstill has a lab degeneracy of (2J + 1) if O(3)lab is stillin effect. So the (2J + 1)2 level degeneracies are eachsplit into multiplets of degeneracy (2J+1) and 2(2J+1)for K = 0 and K 6= 0 respectively. The resulting levelsare often labeled Σ, Π, ∆, Φ, Γ, . . . in analogy with theatomic s, p, d, f , g, . . . labels of Bohr model orbitals.
Only by perturbing the lab environment can one re-duce the O(3)lab symmetry and split the M degeneracies.For example, a uniform electric field would reduce theO(3)lab to O(2)lab, giving Stark splittings which consistof external quantum singlets for M = 0 and±M doubletsfor M 6= 0. A uniform magnetic field would reduce theO(3)lab to R(2)lab, giving Zeeman splittings into exter-nal quantum singlets for each M . The analogy betweenatomic external field splitting and internal molecular ro-tational structure is sometimes a useful one, as used inSects. 31.4 and 31.5.2.
Atomic, Molecular, & Optical Physics Handbook, edit
31.1.3 Rigid Asymmetric RotorEigensolutions and Dynamics
The general case for the rigid rotor Hamiltonian(31.1) has three unequal principal moments of inertia,so that A 6= B 6= C. This is called the rigid asymmetrictop Hamiltonian, and provides a first approximation formodeling rotation of low symmetry molecules such asH2O. Also, several properties of its eigensolutions areshared by more complicated systems. The dynamics ofan asymmetric top is quite remarkable, as demonstratedby tossing a tennis racquet in the air, flat side up. Thecorresponding behavior of molecules needs to be studiedcarefully in order to be well understood.
Given the total angular momentum J , a (2J + 1)-dimensional matrix representation of H may be con-structed using standard matrix elements of angular mo-mentum operators Jx, Jy and Jz as given in Chap. 2.The H matrix connects states with (2J + 1) differentbody quantum numbers K (−J ≤ K ≤ J), but the ma-trix is independent of the lab quantum numbers M , sothat there are (2J+1) identical H matrices, one for eachvalue of the lab quantum number M (−J ≤M ≤ J).
A plot of the 21 eigenvalues of (31.1) for J = 10is shown in Fig. 31.1. Here, the constants are setto A = 0.2 cm−1 and C = 0.6 cm−1 while B isvaried between B = A, which corresponds to a prolatesymmetric top (elongated cylindrical object) and B = C,which corresponds to an oblate symmetric top (flattenedcylindrical object). For all B values between those of Aand C, the object is asymmetric.
The left-hand end (A = B = 0.2, C = 0.6 cm−1 )of the plot in Fig. 31.1 corresponds to a prolate sym-metric top. The symmetric top level spectrum is givenby Eq. (31.4). It consists of a lowest singlet state cor-responding to K = 0 and an ascending quadratic ladderof doublets corresponding to K = ±1,±2, . . . ,±J . Theright-hand end (A = 0.2, B = C = 0.6 cm−1 ) of the plotcorresponds to an oblate symmetric top with a descend-ing quadratic ladder of levels, and theK = 0 level is high-est. Also, the internal K-axis of quantization switchesfrom the body z-axis for (A = B = 0.2, C = 0.6) tothe body x-axis for (A = 0.2, B = C = 0.6). The labM -degeneracy is invisible here, but exists nevertheless.
For intermediate values of B, one has an asymmetrictop level structure and, strictly speaking, no single axis ofquantization. As a result, the eigenlevel spectrum is quitedifferent. A detailed display of asymmetric top levels forthe case (A = 0.2, B = 0.4, C = 0.6) is given at thebottom of Fig. 31.2. They are shown to correspond tosemiclassical orbits in Sect. 31.2. This example is themost asymmetric top, since parameter B has a valuemidway between those of the symmetric top limits B = Aand B = C.
The twenty-one J = 10 asymmetric top levels are
ed by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 381
Figure 31.1. J = 10 eigenvalue plot for symmetric and asymmetric rigid rotors. (A = 0.2, C = 0.6 cm−1 ,A < B < C). Prolate and oblate RE surfaces are shown.
arranged into roughly ten doublets and one singlet.This is like the symmetric top spectrum except thatthe doublets are all split by varying amounts, andthe singlet is well separated from other levels in themiddle of the band instead of being crowded onto thetop or bottom. The doublet splittings are magnifiedin circles drawn next to the levels, and these indicatethat the splitting decreases quasi-exponentially with eachdoublet’s separation from the central singlet.
The rotor doublet splitting is called superfine struc-ture and can be viewed as the result of a dynamic tun-neling process in a semiclassical model of rotation [8–10].Such a model is useful for clarifying the classical-quantumcorrespondence for polyatomic rovibrational dynamics ingeneral. It can also help to derive simple approximationsfor eigenvalues and eigenvectors.
31.2 ROTATIONAL ENERGYSURFACES ANDSEMICLASSICAL ROTATIONALDYNAMICS
A semiclassical model of molecular rotation can bebased upon what is called a rotational energy surface(RE) [7–14]. Examples of RE surfaces for an asymmetrictop are shown in Fig. 31.2 and for prolate and oblate
Atomic, Molecular, & Optical Physics Handbook, edite
symmetric tops in Fig. 31.1. Each surface is a radialplot of the classical energy derived from the Hamiltonian(31.1) as a function of the polar directions of the classicalangular momentum J-vector in the body frame. Themagnitude of J is fixed for each surface. Note that theJ-vector in the lab frame is a classical constant of themotion if there are no external perturbations. However,J may gyrate considerably in the moving body frame,but its magnitude stays the same in all frames for freerotation.
The RE surface differs from what is called a constantenergy (CE) surface which is obtained by simply plottingE = H = const. in J-space using Eq. (31.1). A rigid rotorCE surface is an ellipsoid covering a range of |J| values ata single energy. On the other hand, an RE surface, is aspherical harmonic plot at a single |J|-value for a range ofenergies. The latter is more appropriate for spectroscopicstudies of fine structure, since one value of J correspondsto a multiplet of energy levels or transitions. An REsurface also shows loci of high and low energy rotations.Also, it has roughly the same shape as the body itrepresents, i.e., it is long in the direction in which thebody is long.
For a freely rotating molecule, the laboratory com-ponents of the classical total angular momentum J areconstant. If J is used to define the lab z-axis, then thedirection of the J-vector in the body frame is given by
d by G. W. F. Drake, AIP Press, New York c© 1996.

382 / Atomic, Molecular, & Optical Physics Handbook
Figure 31.2. J = 10 rotational energy surface and related level spectrum for an asymmetric rigid rotator(A = 0.2, B = 0.4, C = 0.6 cm−1 ).
Atomic, Molecular, & Optical Physics Handbook, edited by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 383
polar and body azimuthal coordinates β and γ, whichare the second and third Euler angles, respectively. (Itis conventional to use the negatives −β and −γ as polarcoordinates, but this will not be necessary here.) Thenthe body components of the J-vector are written as
Jx = |J | sinβ cos γ ,Jy = |J | sinβ sin γ , (31.7)Jz = |J | cosβ ,
where the magnitude is |J | =√J(J + 1) ∼= J + 1
2 .Substituting this into Eq. (31.1) gives an expression
E(β, γ) = 〈H〉 = J(J + 1)[
sin2 β(A cos2 γ +B sin2 γ
)+ C cos2 β
](31.8)
for the general rigid rotor RE surface radius in polarcoordinates. The prolate symmetric top (A = B < C)expression
E(β) = 〈H〉 = J(J + 1)[B + (C −B) cos2 β
](31.9)
is independent of azimuth γ. The 3-dimensional plots ofthese expressions are shown in Figs. 31.1 and 31.2.
The RE surfaces have topography lines of constantenergy (E = const.). These lines are the intersection ofthe RE surface (constant |J|) with spheres of constantenergy. The topography lines are allowed classical pathsof the J-vector in the body frame, since these pathsconserve both energy and momentum.
The trajectories in these figures are special ones.They are the quantizing trajectories for total angularmomentum J = 10. For the prolate symmetric top, thequantizing trajectories have integral values for the bodyz-component K. According to the Dirac vector model,angular momentum vectors trace out a cone of altitudeK and height |J | =
√J(J + 1). The quantizing polar
angles ΘJK are given by
ΘJK = cos−1 K√
J(J + 1), (K = J, J − 1, . . . ,−J)
(31.10)
and these are the latitude angles of the paths on the REsurfaces in Fig. 31.1 for K = 10, 9, . . . ,−10. (For theoblate RE surface, the angles are relative to the x-axis.)If β = ΘJ
K is substituted into the symmetric top REsurface equation (31.9), the result is
E(ΘJK) = J(J + 1)B + (C −B)K2 , (31.11)
which is precisely the symmetric top eigenvalue equation(31.4). The quantizing paths are circles which lie at theintersections of the Dirac angular momentum cones andthe RE surfaces. The angle ΘJ
K is a measure of theangular momentum uncertainty ∆Jx or ∆Jy transverse
Atomic, Molecular, & Optical Physics Handbook, edite
to the z-axis of quantization. Clearly, K = J states haveminimum uncertainty.
For the asymmetric top, the classical paths whichconserve both |J | and E fall into one of two types. First,there are those pairs of equal-energy orbits which goaround the hills on the plus or minus end of the bodyz-axis and correspond to the ±K pairs of levels in theupper half of the level spectrum drawn in Fig. 31.2. Thenthere are the pairs of levels belonging to the equal-energyorbits in either of the two valleys surrounding the bodyx-axis and associated with the pairs of levels in the lowerhalf of the level spectrum.
The upper pairs of paths are seen to be distortedversions of the prolate top orbits seen on the left ofFig. 31.1, while the lower pairs are distorted versions ofthe oblate top orbits seen on the right of Fig. 31.1. Thedistortion makes Jz deviate from a constantK-value, andcorresponds to K-mixing in the quantum states. Thisalso shows that more than one axis of quantization mustbe considered; the prolate-like paths are based on thez-axis, while the oblate-like paths belong to the bodyx-axis.
The two types of orbit are separated by what is calleda separatrix curve which crosses the saddle points oneither side of the body y-axis. In the example shown inFig. 31.2, the separatrix is associated with a single levelwhich separates the upper and lower energy doublets.The doublets that are closer to the separatrix level aresplit more than those which are further away. Apartfrom the splitting, the energy levels can be obtained bygeneralized Bohr quantization of the classical paths onthe RE surface. The quantization condition is∮
Jz dγ = K , (31.12)
where
Jz =
√J(J + 1)(C cos2 γ +B sin2 γ)−E
(C cos2 γ +B sin2 γ)−A(31.13)
follows from Eqs. (31.7) and (31.8). The resulting EK-values are obtained by iteration.
The doublet or superfine splitting is a quantum effectwhich may be associated with tunneling between orbitsthat would have had equal energies EK in the purelyclassical or semiclassical model. Most of the tunnelingcan be obtained from an integral over the saddle pointbetween each pair of equal-energy quantizing paths. Toa good approximation, the Kth tunneling amplitude isgiven by
SK = νK exp(−PK) (31.14)
where
PK = i
∫ γ+
γ−dγ
√J(J + 1)(C cos2 γ +B sin2 γ)−EK
(C cos2 γ +B sin2 γ)−A
(31.15)
d by G. W. F. Drake, AIP Press, New York c© 1996.

384 / Atomic, Molecular, & Optical Physics Handbook
Table 31.1.
Eigenvectors |z〉 |z〉 Eigenvalues
|A〉 1 1 EA(K) = EK + 2SK|B〉 1 –1 EB(K) = EK − 2SK
is the saddle path integral between closest approachpoints γ+ and γ−, and νK is the classical precessionfrequency or quantum level spacing around energy levelEK . Each amplitude SK is doubled, since there aretwo tunneling paths, and it appears in a tunnelingHamiltonian matrix for the Kth semiclassical doublet ofz and −z = z paths
〈H〉 =(
EK 2SK2SK EK
)|z〉|z〉 . (31.16)
The resulting tunneling energy eigensolutions are given inTable 31.1. A- or B-states correspond to symmetric andantisymmetric combinations of waves localized in the twosemiclassical paths. The symmetry of rotational states isconsidered in Sect. 31.3.
The total doublet splitting is 4SK , and decreasesexponentially with the saddle point integral (31.15). Thesuperfine A− B splittings in Fig. 31.2 are seen to rangefrom several GHz near the separatrix down to only26 kHz for the highest-K doublets at the band edges.
Meanwhile, the typical interdoublet level spacing orclassical precession frequency is about 150 GHz for theJ = 10 levels shown in Fig. 31.2. This K-level spacingis labeled rotational fine structure splitting, and wouldalso be present in the symmetric top case. The superfinesplitting of the symmetric top doublets goes exactly tozero, because the rotor has O(2)body symmetry if A = Bor B = C. Then all tunneling amplitudes cancel.
Finally, consider the spacing between adjacent J-levels called rotational structure in a spectrum. Thisspacing is
E(J,K)−E(J − 1,K) = 2BJ , (31.17)
according to the symmetric top energy formula (31.4).For the example just treated, this is about 10 cm−1 or300 GHz. It corresponds to the actual rotation frequencyof the body. It is the only kind of roatational dynamicsor spectrum possible for a diatomic rotor. However, adiatomic molecule can have internal electronic or nuclearspin rotation, which gives an additional fine structure[1, 6, 15].
To summarize, polyatomic molecules exhibit all threetypes of rotational motion (from faster to slower): rota-tional , precessional , and precessional tunneling . Theseare related to three kinds of spectral structure (fromcoarser to finer): rotational structure, fine structure, andsuperfine structure, respectively. Again, this neglects in-
Atomic, Molecular, & Optical Physics Handbook, edite
ternal rotational and spin effects which can have abnor-mally strong rotational resonance coupling due to the su-perfine structure [9, 16]. Examples of this are discussedin Sect. 31.5.2.
31.3 SYMMETRY OF MOLECULARROTORS
Molecular rotational symmetry is most easily intro-duced using examples of rigid rotors. Molecular rotorstructure may have more or less internal molecular sym-metry, depending on how the nuclei are positioned rel-ative to one another in the body frame. Molecular ro-tational symmetry is described by one of the elementaryrotational point symmetry groups. These are the n-foldaxial cyclic groups Cn and the polygonal dihedral groupsDn (n = 1, 2, . . . ), the tetrahedral group T , the cubic-octahedral group O, or the icosahedral group Y . Allother point groups such as Cnv, Td, and Oh are combina-tions of an elementary group with the inversion operationI(r→ −r). Each of these groups consists of operationswhich leave at least one point (the origin) of a structurefixed while mapping identical atoms or nuclei into eachother in such a way that the appearance of the structureis unchanged. The point groups are subgroups of thenuclear permutation groups [17].
In other words, molecular symmetry is based upon theabsolute identity of all atoms or, more precisely, nucleiof a given atomic number Z and mass number A. Itis the identity of the so-called ‘elementary’ electronicand nucleonic constituent particles which underlies thesymmetry.
The Pauli principle states that all half-integer spinparticles are antisymmetrized with every other one oftheir kind in the universe. The Pauli exclusion principleand the related Bose symmetrization principle determinemuch of molecular symmetry and dynamics, just as thePauli principle is fundamental to electronic structure.
31.3.1 Asymmetric Rotor SymmetryAnalysis
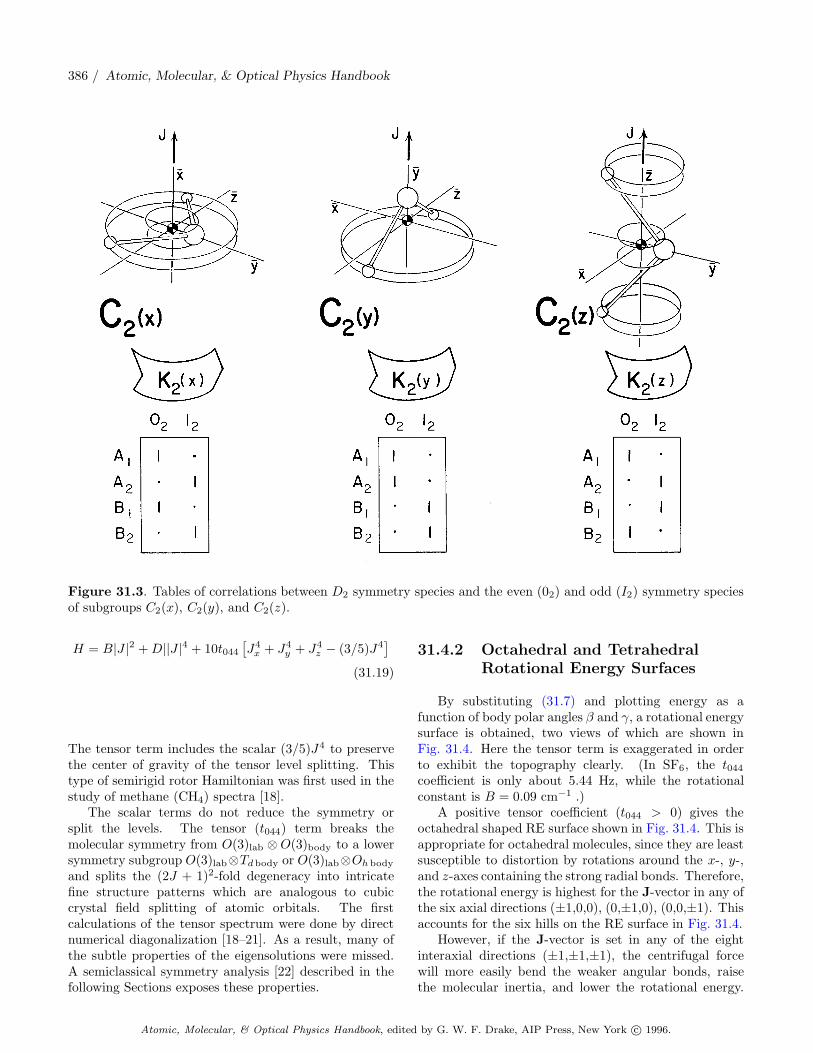
For an asymmetric rigid rotor, any rotation whichinterchanges x, y, or z-axes of the body cannot possiblybe a symmetry, since all three axes are assumed to havediffering inertial constants. This restricts considerationto 180◦ rotations about the body axes which are elementsof the groups C2 and D2.
The two symmetry types for C2 are even (denotedA or 02) and odd (denoted B or 12) with respect toa 180◦ rotation. For D2, which is just C2 ⊗ C2, thefour symmetry types are even-even (denoted A1), even-odd (denoted A2), odd-even (denoted B1), and odd-odd(denoted B2) with respect to 180◦ rotations about the y-
d by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 385
Table 31.2. Character table for symmetry group C2.
C2 1 R
A 1 1B 1 –1
and x-axes, repectively. (The z-symmetry is determinedby a product of the other two, since Rz = Rx Ry.)This is summarized in the character Tables 31.2 and31.3. The rotational energy surface for a rigid rotorshown in Fig. 31.2 is invariant under 180◦ rotationsabout each of the three body axes. Therefore, itsHamiltonian symmetry is D2 and its quantum eigenlevelsmust correspond to one of the four types in Table 31.3.The D2 symmetry labels are called rotational (or ingeneral rovibronic) species of the molecular state. Thespecies label the symmetry of a quantum wave functionassociated with a pair of C2 symmetric semiclassicalpaths.
The classical J-paths come in D2 symmetric pairs,but each individual classical J-path on the rigid rotorRE surface has a C2 symmetry which is a subgroupof D2. Each path in the valley around the x-axis isinvariant under just the 180◦ rotation around the x-axis. This is C2(x) symmetry. The other member ofits pair which goes around the negative x-axis also hasthis local C2(x) symmetry. The combined pair of pathshas full D2 symmetry, but classical mechanics does notpermit occupation of two separate paths. Multiple pathoccupation is a quantum effect.
Similarly, each individual J-path in the hill aroundthe z-axis is invariant under just the 180◦ rotationaround the z-axis, so it has C2(z) symmetry, as doesthe equivalent path around the negative z-axis. Only theseparatrix has the full D2 symmetry, since its pairs arelinked up on the y-axis to form the boundary betweenx and z paths. No J-paths encircle the unstable y-axis,since it is a saddle point.
Each classical J-path near the x- or z-axes belongs toa particular K-value through semiclassical quantizationconditions (31.12). Depending upon whether theK-valueis even (02) or odd (12), the corresponding K-doubletis correlated with a pair of D2 species as shown in the
Table 31.3. Character table for symmetry group D2.
D2 1 Rx Ry Rz
A1 1 1 1 1A2 1 –1 1 –1
B1 1 1 –1 –1B2 1 –1 –1 1
Atomic, Molecular, & Optical Physics Handbook, edite
correlation tables in Fig. 31.3. These three correlationtables give the axial 180◦ rotational symmetry of eachD2 species for rotation near each body axis x, y, and z,respectively. Only the stable rotation axes x and z needto be considered here.
For example, consider K = 10 paths which lie lowestin the x-axis valleys. Since K = 10 is even (02), it iscorrelated with an A1 and B1 superfine doublet [see the02 column in the C2(x) table]. On the high end near thez-axis hill top, K = 10 gives rise to an A1 and B2 doublet[see the 02 column in the C2(z) table]. All the doubletsin Fig. 31.2 may be assigned in this way.
31.4 TETRAHEDRAL-OCTAHEDRALROTATIONAL DYNAMICS ANDSPECTRA
The highest symmetry rigid rotor is the spherical topfor which the three inertial constants are equal (A = B =C). The spherical top Hamiltonian
H = BJ·J (31.18)
has the full R(3)lab⊗R(3)body symmetry. With inversionparity, the symmetry is O(3)lab ⊗O(3)body. In any case,the J-levels are (2J + 1)2-fold degenerate. The resultingBJ(J+1) energy expression is the first approximation formolecules which have the regular polyhedral symmetryof, for example, a tetrahedron (CF4), cube (C6H6),octahedron (SF6), dodecahedron or icosahedron (C20H20,B12H12, or C60). Rigid regular polyhedra have isotropicor equal inertial constants, and rotate as if their massdistributions were perfectly spherical.
However, no molecule can really have sphericalO(3)body symmetry. Even molecules of highest symme-try contain nuclear mass points, and therefore have afinite internal point symmetry. Evidence of octahedralor tetrahedral symmetry shows up in fine structure split-tings analogous to those discussed previously for asym-metric tops. However, spherical top fine structure is dueto symmetry breaking caused by anisotropic or tensor ro-tational distortion. To discuss this, one needs to considerwhat are called semirigid rotors.
31.4.1 Semirigid Octahedral Rotors andCentrifugal Tensor Hamiltonians
The lowest order tensor centrifugal distortion pertur-bation has the same form for both tetrahedral and octa-hedral molecules. It is simply a sum of fourth powers ofangular momentum operators given in the third term be-low. The first two terms are scalar rotor energy (31.18)and scalar centrifugal energy.
d by G. W. F. Drake, AIP Press, New York c© 1996.
386 / Atomic, Molecular, & Optical Physics Handbook
Figure 31.3. Tables of correlations between D2 symmetry species and the even (02) and odd (I2) symmetry speciesof subgroups C2(x), C2(y), and C2(z).
H = B|J |2 +D||J |4 + 10t044[J4x + J4
y + J4z − (3/5)J4]
(31.19)
The tensor term includes the scalar (3/5)J4 to preservethe center of gravity of the tensor level splitting. Thistype of semirigid rotor Hamiltonian was first used in thestudy of methane (CH4) spectra [18].
The scalar terms do not reduce the symmetry orsplit the levels. The tensor (t044) term breaks themolecular symmetry from O(3)lab ⊗O(3)body to a lowersymmetry subgroup O(3)lab⊗Tdbody or O(3)lab⊗Ohbodyand splits the (2J + 1)2-fold degeneracy into intricatefine structure patterns which are analogous to cubiccrystal field splitting of atomic orbitals. The firstcalculations of the tensor spectrum were done by directnumerical diagonalization [18–21]. As a result, many ofthe subtle properties of the eigensolutions were missed.A semiclassical symmetry analysis [22] described in thefollowing Sections exposes these properties.
Atomic, Molecular, & Optical Physics Handbook, edite
31.4.2 Octahedral and TetrahedralRotational Energy Surfaces
By substituting (31.7) and plotting energy as afunction of body polar angles β and γ, a rotational energysurface is obtained, two views of which are shown inFig. 31.4. Here the tensor term is exaggerated in orderto exhibit the topography clearly. (In SF6, the t044coefficient is only about 5.44 Hz, while the rotationalconstant is B = 0.09 cm−1 .)
A positive tensor coefficient (t044 > 0) gives theoctahedral shaped RE surface shown in Fig. 31.4. This isappropriate for octahedral molecules, since they are leastsusceptible to distortion by rotations around the x-, y-,and z-axes containing the strong radial bonds. Therefore,the rotational energy is highest for the J-vector in any ofthe six axial directions (±1,0,0), (0,±1,0), (0,0,±1). Thisaccounts for the six hills on the RE surface in Fig. 31.4.
However, if the J-vector is set in any of the eightinteraxial directions (±1,±1,±1), the centrifugal forcewill more easily bend the weaker angular bonds, raisethe molecular inertia, and lower the rotational energy.
d by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 387
Table 31.4. Character table for symmetry group O.
O 0◦ 120◦ 180◦ 90◦ 180◦
A1 1 1 1 1 1A2 1 1 1 –1 –1E 2 –1 2 0 0T1 3 0 –1 1 –1T2 3 0 –1 –1 1
This accounts for the eight valleys in Fig. 31.4.A negative tensor coefficient (t044 < 0) gives a cubic
shaped RE surface. Usually, this is appropriate forcubic and tetrahedral molecules, since they are mostsusceptible to distortion by rotations around the x-, y-,and z-axes which lie between the strong radial bonds onthe cube diagonals. Instead of six hills, one finds sixvalleys and eight hills on the cubic RE surface.
Note that a semirigid tetrahedral rotor may have thesame form of rotational Hamiltonian and RE surface asa cubic rotor. The four tetrahedral atomic sites are inthe same directions as four of the eight cubic sites. Theother four cubic sites form an inverted tetrahedron whichis indistinguishable from the original tetrahedron.
If only tetrahedral symmetry were required, theHamiltonian could contain a third order tensor of theform JxJyJz. However, pure rotational Hamiltoniansmust also satisfy time-reversal symmetry: the energy foreach J must be the same as for −J, and rotational senseshould not matter. This excludes all odd powers of J.Rotational energy surfaces must have inversion symmetryeven if the corresponding molecules do not. Hence thetetrahedral RE surface must have octahedral symmetry.
31.4.3 Octahedral and TetrahedralRotational Fine Structure
An example of rotational fine structure for angularmomentum quantum number J = 30 is shown inFig. 31.4. The levels are grouped mainly in clustersbelonging to the octahedral symmetry species A1, A2,E, T1, or T2. The characters of these species are given inTable 31.4, (which also applies to the group Td, whereT1 and T2 are sometimes labeled F1 and F2). Thefirst column gives the dimension or degeneracy of eachspecies; A1 and A2 are singlets, E is a doublet, whileT1 and T2 are triplets. These species form two clusters(A1, T1, T2, A2) and (T2, E, T1) in the low end ofthe spectrum and six clusters (T1, T2), (A2, T2, E),(T1, T2), (E, T1, A1), (T1, T2), and (A2, T2, E) in theupper part of the spectrum. (See the right-hand sideof Fig. 31.4). Note that the total dimension or (near)degeneracy for each of the two lower clusters is eight:(1+3+3+1) and (3+2+3), while the upper clusters eachhave sixfold (near) degeneracy: (3+3), (1+3+2), etc.
Each of the two lower eightfold clusters can beassociated with semiclassical quantizing paths in an REsurface valley as shown in Fig. 31.4. The eightfolddimension or (near) degeneracy occurs because eachquantizing path is repeated eight times; once in each ofthe eight identical valleys. Similarly, the sixfold clusterdimension occurs because there are six identical hills andeach quantizing path is repeated six times around thesurface.
The majority of the paths lie on the hills because the
Atomic, Molecular, & Optical Physics Handbook, edit
hills are bigger than the valleys. The hills subtend a halfangle of 35.3◦ at the separatrix, while the valleys onlyhave 19.5◦. To estimate the number of paths or clustersin hills or valleys, the angular momentum cone anglesfor J = 30 may be calculated using Eq. (31.10). Theresults are displayed in Fig. 31.5. The result is consistentwith the spectrum in Fig. 31.4. Only the two highest K-values of K = 29, 30 have cones small enough to fit inthe valleys, while the six states K = 25 to 30 can all fittogether on the hills.
The angular momentum cone formula also providesan estimate for each level cluster energy. The estimatesbecome more and more accurate as K increases andapproaches J and the uncertainty angle ΘJ
K decreases.Paths for higher K are more nearly circular and thereforemore nearly correspond to symmetric top quantum statesof pure K. The paths on octahedral surfaces are morenearly circular for a given K than are those on theasymmetric top RE surface, and so the octahedral rotorstates can be better approximated with those of thesymmetric top.
31.4.4 Octahedral Superfine Structure
The octahedral RE surface has many more localhills and valleys and corresponding types of semiclassicalpaths than are found on the rigid asymmetric top REsurface. The tunneling between multiple paths givesan octahedral surface superfine structure which can bemuch more complicated than the simple asymmetrictop doublet splittings. Still, the same basic symmetrycorrelations and tunneling mechanisms that work for theasymmetric top may be used.
First, the octahedral symmetry must be correlatedwith the local symmetry of the paths on the hills and inthe valleys. The hill paths have C4 symmetry, while thevalley paths have a local C3 symmetry. This is seen mostclearly for the low-K paths near the separatrix whichare least circular. The C3 and C4 correlations are givenin Fig. 31.6 along with a sketch of the correspondingmolecular rotation for each type of path.
To find the octahedral species associated with theK3 = 30 path in a C3 valley, one notes that 30 is 0 modulo3. Hence the desired species are found in the 03 column of
ed by G. W. F. Drake, AIP Press, New York c© 1996.

388 / Atomic, Molecular, & Optical Physics Handbook
Figure 31.4. J = 10 rotational energy surface and related level spectrum for a semirigid octahedral or tetrahedralrotor.
Atomic, Molecular, & Optical Physics Handbook, edited by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 389
Figure 31.5. J = 30 angular momentum cone halfangles and octahedral cutoffs.
the C3 correlation table: (A1, A2, T1, T2). This is whatappears (not necessarily in the same order) in the lowerleft corner of Fig. 31.4. Similarly, the species (A2, E, T2)for a K4 = 30 path on top of a C4 hill are found in the 24column of the C4 correlation table since 30 is 2 modulo4; these appear on the other side of Fig. 31.4. Clusters(T1, T2) for K4 = 29 and (A1, E, T1) for K4 = 28 arefound similarly.
A multiple path tunneling calculation analogous tothe one for rigid rotors can be applied to approximateoctahedral superfine splittings. Consider the cluster (A1,E, T1) for K4 = 28, for example. Six C4-symmetric pathslocated on octahedral vertices on opposite sides of thex-, y-, and z-axes may be labeled {|x〉, |x〉, |y〉, |y〉, |z〉,|z〉}. A tunneling matrix between the six paths is thefollowing:
〈H〉K4=28 =
H 0 S S S S0 H S S S SS S H 0 S SS S S S H 0S S S S 0 H
|x〉|x〉|y〉|y〉|z〉|z〉
(31.20)
where the tunneling amplitude between nearest neigh-bor octahedral vertices is S, but is assumed to be zerobetween antipodal vertices. The eigenvectors and eigen-values for this matrix are given in Table 31.5. This pre-dicts that the triplet (T1) level should fall between thesinglet (A1) and the doublet (E) levels and the singlet–triplet spacing (4S) should be twice the splitting (−2S)between triplet and doublet. This 2:1 ratio is observedin (E, T1, A1) and (A2, T2, E) clusters which can beresolved and also in numerical calculations [18–21].
The tunneling amplitudes can be calculated by aseparatrix path integral analogous to the asymmetrictop formula (31.14) [10, 11]. As shown in Fig. 31.4, thetunneling rates or superfine splittings near the separatrixare ∼ 1 MHz, which is only a little less than the classical
Atomic, Molecular, & Optical Physics Handbook, edit
precession frequency. But as K approaches J on the hilltops, the tunneling rate slows down to a few Hz.
31.5 HIGH RESOLUTIONROVIBRATIONAL STRUCTURE
A display of spectral hierarchy for higher and higherresolution is shown in Fig. 31.7 for the 630 cm−1 or 16 µmbands of CF4. This will serve to summarize the possiblerovibrational spectral structures and place them in alarger context. The v4 resonance in part (a) correspondsto a dipole-active n4 = 0→ 1 vibrational transition, andis just one of many vibrational structures to study. TheP (54) sideband resonance in part (b) corresponds to a(J = 54)→ (J − 1) rotational transition, and is just oneof a hundred or so rotational structures to study withinthe v4 bands.
Each band is something like a Russian doll; it containsstructure within structure within structure down to aresolution of a few tens of Hz. Examples of the rotationalfine and superfine structures discussed in Sect. 31.4 areshown in Fig. 31.7(c) and (d), but even more resolutionis needed to see the hyperfine structure in (e). Suchextremely high resolution has been reached with a CO2saturation absorption spectrometer [23, 24]. They havestudied the 10 µm bands of SF6 and SiF4, the latterbeing very simmilar to CF4 [25].
31.5.1 Tetrahedral Nuclear HyperfineStructure
High resolution spectral studies of SiF4 show unantic-ipated effects involving the four fluorine nuclear spins andmagnetic moments and their associated hyperfine states.First, the Pauli principle restricts the nuclear spin mul-tiplicity associated with each of the rotational symmetryspecies in much the same way that atomic L − S cou-pled states 2S+1L have certain spin multiplicities (2S+1)allowed for a given orbital L species involving two ormore equivalent electrons. Second, since superfine split-
Table 31.5. Eigenvectors and eigenvalues of the tunnel-ing matrix for the (A1, E, T1) cluster with K = 28
Eigenvector |x〉 |x〉 |y〉 |y〉 |z〉 |z〉 Eigenvalue√
6|A1〉 = 1 1 1 1 1 1 EA1 = H + 4S√
12|E, 1〉 = 2 2 –1 –1 –1 –1EE = H − 2S2|E, 2〉 = 0 0 1 1 –1 –1
√2|T1, 1〉 = 1 –1 0 0 0 0√2|T1, 2〉 = 0 0 1 –1 0 0 ET1 = H√2|T1, 3〉 = 0 0 0 0 1 –1
ed by G. W. F. Drake, AIP Press, New York c© 1996.

390 / Atomic, Molecular, & Optical Physics Handbook
Figure 31.6. Tables of correlations between O symmetry species and the cyclic axial symmetry species (Kp meansK mod p) of subgroups C3, C2 and C4.
tings can easily be tiny, different spin species can endup so close that hyperfine interactions, however small,can cause strongly resonant mixing of normally inviolatespecies. Finally, a very pure and simple form of sponta-neous symmetry breaking is observed in which otherwiseequivalent nuclei are separated into different subsets bythe rotational dynamics.
The nuclear spin to rotational species correlation iseasily found by correlating the full permutation symme-try (Sn for XYn molecules) with the full molecular rota-tion and parity symmetry [O(3)lab ⊗ Tdbody for CF4 orO(3)lab ⊗ Ohbody for SF6]. For spin- 1
2 nuclei, the Pauliprinciple allows a spin of I = 2 and a spin multiplicityof 2I + 1 = 5 for (J+, A2) or (J−, A1) species, but ex-cludes (J−, A2) or (J+, A1) species altogether. The Pauliallowed spin for (J+, T1) or (J−, T2) species is I = 1with multiplicity 3, but there are no allowed (J+, T2) or(J−, T1) species. Finally, both (J+, E) and (J−, E) be-long to singlet spin I = 0 and are singlet partners to aninversion doublet . (None of the other species can haveboth + and − parity.)
The E inversion doublet is analogous to the doubletin NH3 which is responsible for the ammonia maser.However, NH3-type inversion is not feasible in CF4 orSiF4, and so the splitting of the E doublet in thesemolecules is due to hyperfine resonance [9, 16, 29].
The Pauli analysis gives the number of hyperfine linesthat each species would exhibit if it were isolated andresolved, as shown in the center of Fig. 31.7(e). Therotational singlets A1 and A2 have 5 lines each, therotational triplets T1 and T2 are spin triplets, and therotational doublet E is a spin singlet but an inversion
Atomic, Molecular, & Optical Physics Handbook, edit
doublet. If the hyperfine structure of a given species A1,A2, T1, T2, or E is not resolved, then their line heightsare proportional to their total spin weights of 5, 5, 3, 3,and 2, respectively.
If the unresolved species are clustered, then thetotal spin weights of each add to give a characteris-tic cluster height. The line heights of the C4 clus-ters (T1,T2), (A2,T2,E), (T1,T2), (E,T1,A1) are 6, 10,6, 10, respectively. The line heights in the C3 clusters(A1,T1,T2,A2), (T1,E,T2), (T1,E,T2) are 16, 8, 8, respec-tively. This is roughly what is seen in the P (54) spectrumin Fig. 31.7(c).
31.5.2 Superfine Structure andSpontaneous Symmetry Breaking
The superfine cluster splittings (2S, 4S, etc.) are pro-portional to the J-precessional tunneling or ‘tumbling’rates bewteen equivalent C3 or C4 symmetry axes, andthey decrease with increasing K3 or K4. At some point,the superfine splittings decrease to less than the hyperfinesplittings which are actually increasing with K. The re-sulting collision of superfine and hyperfine structure hasbeen called superhyperfine structure or Case 2 clusters.The following is a rough sketch of the phenomenology ofthis very complex effect, using the results of Pfister [25].
As long as the tunneling rates are > 1 MHz, the nu-clear spins tend to average over spherical top motion.The spins couple into states of good total nuclear spin I,which in turn couple weakly with the overall angular mo-mentum and with well defined rovibrational speciesA1,
ed by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 391
Figure 31.7. Rovibrational structure in the 630 cm−1 or 16 µm bands of CF4 [16]. (a) Vibrational resonancesand band profiles. (Raman spectra from Ref. [26]). (b) Rotational P , Q, and R band structure corresponding toJ → J − 1, J → J , and J → J + 1 transitions. (FTIR spectra from Ref. [27]). (c) P (54) rotational fine structure dueto rotation-vibration coupling and angular momentum precessional motion. (Laser diode spectra from Ref. [28]). (d)Superfine structure due to precessional tunneling [25]. (e) Hyperfine structure due to nuclear spin precession [25].
Atomic, Molecular, & Optical Physics Handbook, edited by G. W. F. Drake, AIP Press, New York c© 1996.

392 / Atomic, Molecular, & Optical Physics Handbook
Figure 31.8. Stick sketches for examples of superfineand hyperfine spectral structure found by Pfister [25].(a),(b) Case 1 clusters (high tunneling amplitude S).(c),(d) Case 2 clusters (low tunneling amplitude S).
Table 31.6. Spin- 12 basis states for SiF4 rotating about
a C4 symmetry axis.
Iz = 2 Iz = 1 Iz = 0 Iz = −1 Iz = −2∣∣∣∣ ↑ ↑↓ ↓⟩
| ↑↓ ↑↓〉| ↑↓ ↑↑〉 | ↓↓ ↑↑〉 | ↓↓ ↑↓〉
| ↑↑ ↑↑〉 | ↑↑ ↑↓〉 | ↑↑ ↓↓〉 | ↑↓ ↓↓〉 | ↓↓ ↓↓〉
A2, T1, T2, or E as described above. The resulting iscalled Case 1 , and is analogous to LS coupling in atoms.
Stick figures for two examples of spectra observedby Pfister [25] are shown in Fig. 31.8(a) and (b). Thefirst Case 1 cluster in (a) is a C4-type (04) cluster(A1,T1,E) which was solved in Table 31.5. The otherCase 1 cluster in (b) is a C3-type (±13) cluster (T1,E,T2)(recall C3 correlations in Fig. 31.3). They are similar tothe corresponding sketches shown in Fig. 31.7(e). Onenotable difference is that the inversion doublet showslittle or no splitting in the (A1,T1,E) cluster, but doessplit in the (T1,E,T2) cluster.
When the tunneling rates fall below 10 or 20 kHz, theangular momentum can remain near a particular C3 orC4 symmetry axis for a time longer than the nuclear spinprecession periods. Spin precession rates and hyperfinesplittings are ∼ 50 kHz, and increase with K. Hencethere is plenty of time for each of the nuclear spins toalign or antialign with the C3 or C4 symmetry axes ofrotation. This is Case 2 coupling, and the resultingspectrum resembles that of an NMR scan of the nuclei,but the magnetic field here is provided by the moleculeitself through its own body frame rotation.
If SiF4 rotates uniformly about one C4 symmetryaxis, then all four F nuclei occupy equivalent positionsat the same average distance from the rotation axis andexperience the same local magnetic fields. The moleculecan be thought of as a pair of diatomic F-F rotorswith each one symmetrized or antisymmetrized so asto make the whole state symmetric. Table 31.6 showsthe spin- 1
2 base states arranged horizontally according
Atomic, Molecular, & Optical Physics Handbook, edite
to the total projection Iz of nuclear spins on the C4axis. Horizontal arrays (↑↓) of spins denote symmetricstates, while vertical arrays
(↑↓
)denote antisymmetric
spin states. The hyperfine energy is approximatelyproportional to the projection Iz . The resulting spectrumis a (1,2,4,2,1)-degenerate pyramid of equally spaced linesas shown in Fig. 31.8(c). Four spin- 1
2 states withoutsymmetry restrictions would give the standard binomial(1,4,6,4,1)-degeneracy seen in NMR spectra.
If the molecule settles upon C3 symmetry axes ofrotation, the situation is markedly different. The fournuclei no longer occupy equivalent positions. Onenucleus sits on the rotation axis, while the other threeoccupy equivalent off-axis positions. The off-axis nucleiexperience a different local magnetic field than the loneon-axis nucleus [see Fig. 31.8(d)]. From the spectrum, itappears that the spin-up to spin-down energy differenceis much greater for the on-axis nucleus than for thethree equatorial nuclei, whose spin states form an energyquartet {|↑↑↑〉, |↑↑↓〉, |↑↓↓〉, |↓↓↓〉}. The on-axis nucleushas an energy doublet with a large splitting, so that thefour nuclei together give a doublet of quartets as shownin the figure.
If the off-axis nuclei had experienced the greatestsplitting, then the spectrum would be a quartet ofdoublets instead of a doublet of quartets. This doesoccur in the SF6 superhyperfine structure, which showsa quintet of triplets for a Case 2 C4 symmetry cluster.For either one of these molecules, it is remarkable howdifferent the rovibrational ‘chemical shifts’ can becomefor equivalent symmetry sites. The result is a microscopicexample of spontaneous symmetry breaking.
31.5.3 Extreme Molecular SymmetryEffects
The most common high-symmetry molecules belongto either the tetrahedral Td or cubic-octahedralO groups.Until the recent discovery of fullerenes and of the struc-ture of virus coats, the occurrence of molecular pointgroups of icosahedral groups was thought to be rare ornonexistent in nature [30, 31].
d by G. W. F. Drake, AIP Press, New York c© 1996.

Molecular Symmetry and Dynamics / 393
For an extreme example of symmetry breaking effects,consider the buckminsterfullerene or Buckyball moleculeC60 which has the highest possible molecular point sym-metry Yh. A semiclassical approach to rotational sym-metry and dynamics is useful here, since the rotationalquantum constant is so small for fullerenes (for C60,2B = 0.0056 cm−1 or 168 MHz [32, 33]).
Since there are two isotopes 12C (nuclear spin0) and 13C (nuclear spin 1
2 ), it is possible to havea Bose-symmetric molecule 12C60, a Fermi-symmetricmolecule 13C60, or many broken-symmetry combinations12Cx 13C60−x. The most likely combination is 12C59
13C,which has no rotational symmetry at all, and only onereflection plane. This may be the most extreme exampleof molecular isotopic symmetry breaking; it goes fromthe highest possible symmetry Yh to the lowest Ch.
The Fermi-symmetric 13C60 molecule has ten timesas many rotating spin- 1
2 nuclei as SF6, and 210 timesas many hyperfine states, giving about 1.15 × 1018
spin states distributed among 10 symmetry species [34].In contrast, the Bose-symmetric 12C60 has only onespin symmetry species allowed by the Bose exclusionprinciple. It provides an even more extreme exampleof Bose exclusion than the Os16O4 molecule. In all,119 of the 120 Yh rovibrational symmetry states areBose excluded, giving 12C60 an extraordinarily sparserotational structure. However, it only takes the additionof one extra neutron to make 12C59
13C. Then all theexcluded rovibrational states must return!
REFERENCES
1. G.Herzberg, Molecular Spectra and Structure: Spectra ofDiatomic Molecules (Van Nostrand-Reinhold, New York,1950), Vol. I.
2. G.Herzberg, Molecular Spectra and Structure: Infraredand Raman Spectra of Polyatomic Molecules (VanNostrand-Reinhold, New York, 1945), Vol. II.
3. G.Herzberg, Molecular Spectra and Structure: Elec-tronic Structure of Polyatomic Molecules (Van Nostrand-Reinhold, New York, 1966), Vol. III.
4. E. B. Wilson, J. C. Decius, and P. C. Cross, MolecularVibrations (McGraw Hill, New York, 1955).
5. D. Papousek and M. R. Aliev, Studies in Physical andTheoretical Chemistry: Molecular Vibrational-RotationalSpectra (Elsevier, Amsterdam, 1982), Vol. 17.
6. R. N. Zare, Angular Momentum: Understanding SpatialAspects in Chemistry and Physics (Wiley Interscience,New York, 1988).
Atomic, Molecular, & Optical Physics Handbook, editeView publication statsView publication stats
7. W. G. Harter, Principles of Symmetry, Dynamics, andSpectroscopy (Wiley Interscience, New York, 1993).
8. W. G. Harter and C. W. Patterson, J. Math. Phys. 20,1453 (1979).
9. W. G. Harter, Phys. Rev. A24, 192 (1981).10. W. G. Harter and C. W. Patterson, J. Chem. Phys. 80,
4241 (1984).11. W. G. Harter, Comp. Phys. Rep. 8, 319 (1988).12. D. A. Sadovskiı and B. I. Zhilinskiı, Mol. Phys. 65, 109
(1988).13. D. A. Sadovskiı and B. I. Zhilinskiı, Phys. Rev. A47, 2653
(1993).14. L. M. Pavlichenkov, Phys. Rep. 226, 173 (1993).15. W. G. Harter, C. W. Patterson, and F. J. da Paixao, Rev.
Mod. Phys. 50, 37 (1978).16. W. G. Harter and C. W. Patterson, Phys. Rev. A 19,
2277 (1979).17. P. R. Bunker, Molecular Symmetry and Spectroscopy
(Academic Press, New York, 1979).18. K. T. Hecht, J. Mol. Spectrosc. 5, 535 (1960).19. K. R. Lea, M. J. M. Leask, and W. P. Wolf, J. Phys.
Chem. Solids 23, 1381 (1962).20. A. J. Dorney and J. K. G. Watson, J. Mol. Spectrosc. 42,
1 (1972).21. K. Fox, H. W. Galbraith, B. J. Krohn, and J. D. Louck,
Phys. Rev. A 15, 1363 (1977).22. W. G. Harter and C. W. Patterson, J. Chem. Phys. 66,
4872 (1977).23. J. Borde and Ch. J. Borde, J. Chem. Phys. 71, 417 (1982).24. Ch. Borde, J. Borde, Ch. Breant, Ch. Chardonnet, A.
Vanderberghe, and Ch. Salomon in Laser SpectroscopyVII 108, 95 (Springer, Berlin, 1985).
25. O. Pfister, thesis, Universite Paris-Nord, 1993.26. B. Montostari and A. Weber, J. Chem. Phys. 33, 1867
(1960).27. A. Stein, P. Rabinowitz, and A. Kaldor, Opt. Lett. 3, 97
(1978).28. R. S. McDowell, C. W. Patterson, C. R. Jones, M.
I. Buchwald, and J. M. Telle, Proc. Soc. Photo-Opt.Instrum. Eng. 190, 263 (1979).
29. R. J. Butcher, Ch. Chardonnet, and Ch. Borde, Phys.Rev. Lett. 70, 2698 (1993).
30. H. W. Kroto, J. R. Heath, S. C. O’Brian, R. F. Curl, andR. E. Smalley, Nature 318, 162 (1985).
31. W. Kratschmer, W. D. Lamb, K. Fostiropoulos, and D.R. Huffman, Nature 347, 354 (1990).
32. W. G. Harter and D. E. Weeks, Chem Phys. Lett. 132,187 (1986).
33. D. E. Weeks and W. G. Harter, Chem. Phys. Lett. 144,366 (1988); 176, 209 (1991).
34. W. G. Harter and T. C. Reimer, Chem. Phys. Lett. 194,230 (1992).
d by G. W. F. Drake, AIP Press, New York c© 1996.


















