
MOLECULAR SCAFFOLDS FOR PROTEIN SURFACE ... · Web viewProtein – protein interactions (PPIs)...
79
MOLECULAR SCAFFOLDS FOR PROTEIN SURFACE MIMICRY. NEW ERA IN PHARMACEUTICALS. Nikos Kyriakou Supervisor: Dr. J.M. van Marseveen
Transcript of MOLECULAR SCAFFOLDS FOR PROTEIN SURFACE ... · Web viewProtein – protein interactions (PPIs)...

MOLECULAR SCAFFOLDS FOR PROTEIN
SURFACE MIMICRY. NEW ERA IN
PHARMACEUTICALS.
Nikos Kyriakou
Supervisor: Dr. J.M. van Marseveen
Second Supervisor: Drs. L. Steemers

1 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Abstract Proteins are important for preserving life. Their highly complex surface imparts functionalities that are only found in proteins. Through specific sites on their surface they can interact with one another to carry out several biological functions, such as DNA replication, signal transduction, immune response and apoptosis. However, infectious proteins can also interact with physiological proteins inside a living organism. This results in harming the organism which can even lead to death. Nevertheless, by administrating a molecule that generates therapeutic antibodies can save the day. The problem is the easiest way that can be done is by using a natural protein, however, proteins are known as bad drugs candidates, basically due to degradation that exhibit inside the body. A novel solution would be to re-create the “hot spots” of a protein in a considerable smaller molecule, i.e. to mimic the protein.
A good way to do that is to connect linear peptides, which bear the antigenic sites of a protein, on an organic molecule (scaffold). This molecular scaffold has to induce certain conformation on the peptides and stability. Moreover, reactions that are applicable to unprotected peptides (“Click Chemistry”) are needed so that protective groups are absent in the synthesis.
2 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

ContentsCOVER PAGE....................................................................................................................................................... 0
ABSTRACT........................................................................................................................................................... 2
ABBREVIATIONS.................................................................................................................................................. 4
1. INTRODUCTION.......................................................................................................................................... 5
PROTEIN – PROTEIN INTERACTIONS (PPIS) AND THEIR IMPORTANCE.............................................................................5 ARTIFICIAL PROTEINS AND THE EXPERTISE OF MIMICKING.............................................................................................6
2. DISCRIMINATION: THE ABILITY TO DISTINGUISH MINOR DIFFERENCES – SELECTIVITY IN REACTIONS...........9
3. RE-CREATING NATURE IN A FLASK............................................................................................................. 14
DIVERGENT APPROACH FOR MIMICKING PROTEIN BINDING SITES..........................................................................................143.1 SCAFFOLD-INDUCED CONFORMATION TO PROTEIN MIMICS.....................................................................................15
3.1.2 Mimicking α-Helix secondary structure................................................................................................163.1.3 Mimicking β-Turn and β-Sheet Secondary Structures...........................................................................183.1.4 Mimicking Looped conformations........................................................................................................213.1.5 Mimicking Discontinuous Protein Binding Sites....................................................................................22
4. STARTING FROM SIMPLE ORGANIC MOLECULES TOWARDS DRUG CANDIDATES....................................26
DISCONTINUOUS PROTEIN BINDING SITE MIMICS.............................................................................................................264.1 THE TRIAZACYCLOPHANE (TAC) – SCAFFOLD......................................................................................................264.2 FUNCTIONALIZED CYCLIC PEPTIDES AS SCAFFOLDS.................................................................................................294.3 CLIPSTM - TECHNOLOGY.................................................................................................................................32
4.3.1 Peptide ligation via Copper(II)-catalyzed Azide-alkyne Cycloaddition..................................................354.3.2 Peptide ligation via Strain-promoted Alkyne-Azide Cycloaddition........................................................364.3.3 Peptide ligation via Thiol-ene Reaction................................................................................................364.3.4 Peptide Ligation via Oxime Formation.................................................................................................374.3.5 Constrain-induced via Single and Double Disulfide Bond.....................................................................384.3.6 Double-CLIPS Technology in the Mimicry of Discontinuous Binding Sites.............................................39
4.4 OTHER MULTIFUNCTIONAL SCAFFOLDS..............................................................................................................41
REFERENCES...................................................................................................................................................... 48
3.1.1
3 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Abbreviations
AAPH 2,2'-Azobisisobutyramidinium chlorideAcm AcetamidomethylAFB2 Aflatoxin B2AIDS Acquired immunodeficiency syndromeAla AlanineAlloc AllyloxycarbonylArg ArginineAsp Aspartic acidBn BenzylBoc Di-t-butyl dicarbonateBSA Bovine Serum
albumin Cbz Benzyl chloroformateCD3 Cluster of differentiation 3CH3CN Acetonitrile Cu CopperCuAAC Copper(I)-catalyzed azide-alkyne
cycloadditionCuSO4 Copper(II) sulfateCys CysteineDAVE Asp -Ala-Val-GluDNA Deoxyribonucleic acidDPAP 2,2-Dimethoxy-2-Phenyl AcetophenoneDTT DithiothreitolELISA Enzyme-linked immunosorbent assayEVH Ena/Vasp homology proteinsFAKL Phenylalanine-Alanine-Lysine-LeucineFRET Förster resonance energy transferFmoc Chloroformic acid 9H-fluoren-9-
ylmethyl esterGlu Glutamic acidGly Glycinegp120 Envelope glycoprotein GP120
HIV Human immunodeficiency virusH2O WaterHPLC High-performance liquid
chromatographyI2 IodineIgG Immunoglobulin GKLA Lys-Leu-Ala LC-MS Liquid chromatography–mass
spectrometryLeu LeucineLys LysineMBP Maltose-Binding ProteinMeOH MethanolNaHCO3 Sodium hydrogen carbonateNa2S4O6 Sodium tetrathionateOKT 3 Muromonab-CD3/ Orthoclone OKT3PBS Phosphate-buffered salinePPIs Protein-protein interactionsRAFT Regioselectively addressable
functionalized templatesRGD Arg-Gly-Aspr.t. Room temperatureSN2 Bi-molecular nucleophilic substitutionSPAAC Strain-promoted azide-alkyne
cycloadditiontBuOH 2-methyl-2-propanolTCEP Tris(2-carboxyethyl)phosphineTFA Trifluoroacetic acidTris tris-(Hydroxymethyl)-aminomethaneVal ValineVEGF-A Vascular endothelial growth factor AVP1 Polyomavirus Capsid Protein
4 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

1.Introduction Life on Earth emerged when highly complex molecules were formed; these were given the name macromolecules (from the Greek macro which means long). These make up the backbone of life; the cells (1). In living cells, macromolecules are found in four different types: proteins, nucleic acids, carbohydrates and lipids. These are essential for all living organisms, as they control all life processes. However, proteins by themselves hold a key role in life’s cycle, as they are the most abundant macromolecules found in living cells; the diversity of functional proteins in cells and tissues is incomparably larger than of any other class of macromolecules and they are involved in practically all life processes (1).
However, the question is “why a synthetic chemist should trouble his/her mind with developing artificial proteins”. Some scientists find it challenging to devise and synthesize new molecules, but, the most valid answer could be, the need of constructing molecules (usually smaller than those found in nature) that bear comparable activity with nature’s macromolecules such as proteins.
Protein – Protein Interactions (PPIs) and their Importance. Protein – protein interactions (PPIs) govern most of biological processes in a living organisms (2). Such interactions take part in the signal transduction, immune response, DNA replication and apoptosis (3; 4;5). Hence, even the slightest alternation of these interactions can be fatal to an organism (source of HIV,
cancer, leukemia, malaria and other auto-immune or infectious diseases) (6; 7; 8; 9). This fact unveils the significance of vaccines and drugs.
Although, the idea of using proteins as drugs is tempting, proteins in general are poor drug candidates due to bioavailability problems that arise from conformational instability (proteins readily lose their three-dimensional structure under a variety of conditions), susceptibility to proteolytic degradation, poor membrane penetration and low cellular uptake, and unfavorable pharmacokinetics due to rapid clearance or instability of peptide bonds to degradation by peptidases inside the human body (10).
The old fashioned view that a protein must keep its native morphology (primary, secondary, tertiary and quaternary structure) to be functional, is wrong; recent studies have shown that about 30% of eukaryotic proteins are composed of proteins that do not have a well-defined structure prior to interaction with their binding partner (11; 12; 13). Moreover, their biological activity is not globally extended on all protein surface and only small regions of their folded surfaces exhibit any activity (14), hence, their functionality can be reproduced in smaller molecules (protein mimics) that retain these bioactive surfaces (10).
5 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Scheme 1.1: Protein - Protein Interactions.Figure 1.1: Two proteins forming a complex. The binding site is denoted with yellow color.

6 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Artificial Proteins and the expertise of mimicking. Protein mimics have been used in the past and have proved to be highly selective and, therefore, successful in the treatment of several life-threatening diseases (15). In the literature, several cases of linear peptides being used as drugs have been documented (16). However, further research on the subject indicates that certain structural fixation is required for biological activity (17). It is well known nowadays among scientists in the peptide field that proteins recognize (interact) one another via defined parts on their surfaces, these constitute the antigenic sites of a protein and are defined as epitopes (18). A protein mimic is usually a smaller compound than a protein, which bears enough information to reproduce the functionalities of a protein or peptide (19).
Epitopes are distinguished in two types; the “continuous epitopes”, which are seldom in proteins and the “discontinuous epitopes”, which are most abundant antigens in natural proteins. A contiguous epitope is defined by consecutive residues directly linked in the sequence (primary structure) by peptide bonds, whereas, a discontinuous epitope is defined as spatially adjacent group of surface amino acid residues that are
partially or totally without any peptide linkage directly connected (20).
Since, epitopes cover a small part of a protein’s surface, an artificial and, at the same time, active drug requires no more than the antigen sites of the molecule. Chemists try to achieve that goal, the past few decades, by binding only the active parts of a protein on a small organic molecule, i.e. “scaffold” (or “template”). Scaffolds are useful especially for mimicking discontinuous epitopes. The latter can only be reproduced effectively when their three-dimensional topology is retained in the artificial protein (21).
The origins of immunotherapy are along with the discovery of the structure of antibodies and the development of hybridoma technology, with which the first monoclonal antibody became reality (22). The latter, was approved by the FDA (Food and Drug Administration) in 1986 and was a murine IgG2a CD3 specific transplant rejection drug, OKT3 (brand name muromonab). This drug found use in solid organ transplant recipients who became steroid resistant (23).
It has not being a while since, artificial proteins were synthesized via long and repetitive routes, utilizing the properties of orthogonal protective groups (: groups that are selectively removed). Those routes were characterized by several limitations
7 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 1.2: Homer 1a EVH1 Domain bound to oligoproline (left). β-Hairpin mimicking the EVH1 domain (right).
Figure 1.3: Representation of an Antibody and its Antigen (an epitope is the part an antigen) (171).

spotted in a divergent approach; according to a divergent method, a central molecule (or scaffold) bearing orthogonally protected functional groups is used, to which multiple different peptides are being attached. A divergent approach suffers from limitations regarding deprotection reactions and purification steps. Moreover, organic molecules such as those exhibit strongly hydrophobic properties(21). To overcome the aforementioned limitations a convergent approach must be used instead.
A convergent approach, unlike to any divergent, relies on orthogonal ligation of pre-functionalized peptide fragments that simultaneously constitute the central scaffold. Moreover, protection or pre-activation of the functionalities is not required. Usually fewer steps are involved in convergent synthesis. (21). The introduction of the concept was made by Velluz et al in 1967 (24). According to him, in a convergent procedure various parts of the target molecule are assembled separately and independently and at the final steps are linked together. A convergent procedure has the effect of lowering the path length of the main line, which in turn makes the procedure more economical than a simple linear procedure (25).
Scheme 1.3: Convergent synthesis.
For the successful mimicry of all epitopes, three categories (or levels) must be ascribed (21): 1) “Continuous (Linear) Epitope”; it can be reproduced usually by a linear peptide, 2) “Continuous (Conformational) Epitope”; is necessary to have a spectacular structure to bind on the corresponding protein and, 3) “Discontinuous (Conformational) Epitope”; successful mimic of the epitope requires more than one separate peptide fragment on the scaffold and by forming the right structure (Scheme 1.3).
8 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Scheme 1.4: Three levels of protein mimicry.
The scope of this project is to present the latest fashion in protein mimics. The focus will be upon orthogonal ligation utilizing scaffolds and also different approaches will be mentioned. Several examples from the recent literature will be demonstrated with the advantages and the disadvantages of each methodology.
In the final chapter it will be attempted to show the best possible way, if it’s feasible, for the synthesis of complex proteins and their mimics.
2.
9 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

2.Discrimination: The ability to distinguish minor Differences – Selectivity in Reactions
The problem with the old fashion syntheses towards protein mimics, was the large number of reactive centers present in a single reaction. To bypass this obstacles, chemists thought of deactivating certain groups selectively. This was made possible by the introduction of protective groups and especially orthogonal protective groups (26). However, it has been proved in practice that using protective groups creates some additional problems; i) incomplete coupling and deprotection reactions lead to truncated and deletion sequences, ii) accumulation of side products from incomplete reactions, impurities from reagents, solvents and protected amino acids, and iii) aggregation of growing peptides (27). Hence, strategies employing protective groups are limited in the number of reactive groups present in one reaction, i.e. small peptides had to be used with no exceptions.
The only solution is to rely on convergent approaches that are fully chemoselective and render protective groups obsolete. Those kind of strategies use reagents with unique functionalities and usually are facile and robust. But what do chemist imply when they refer to a reaction as chemoselective?
In 1983 Trost set it as follows, “The ability to discriminate among the reactive sites is referred to as chemoselectivity” (28). In simple words, single reaction of one functional group in the presence of others renders the reaction chemoselective. For example, the preferential reduction of reduction of a ketone in the presence of an ester and a carbon-carbon double bond, as stated in a recent publication by the group of Procter, is highly chemoselective (29).
Chemists were able to use convergent strategies for mimicking nature after the introduction of several relatively new chemical reactions, these belong to the more general group of reactions called Chemical Ligation. With chemical ligation approach large peptide fragments are joined together chemoselectively through the formation of an amide or non-amide linkage (26). Moreover, a chemoselective approach is usually based on thiol and imine functionalities for forming an amidic bond or by employing hydrazine, oxime, thioester and thioether functionalities, which results in a non-amidic bond formation (30; 31; 32; 33). However, a more recent approach, coined by the Nobel laureate in Chemistry in 2001 (34) Sharpless and co-workers, promises fast, facile and robust reactions towards the desire products (35). The new approach is termed Click Chemistry and is defined by a set of stringent criteria that a process must meet to belong in this context. A reaction must be i) modular, ii) wide in scope, iii) high in yields, iv) no by-products or produce harmless by-products that can be removed by nonchromatographic methods, v) stereospecific but not necessarily enantioselective, vi) simple reaction conditions, vii) readily available starting materials and reagents, viii) no solvent or a solvent that is
benign or easily removed, and ix) simple product isolation. A reaction that fits perfect in this category is the azide-alkyne Huisgen cycloaddition or Click reaction. It is a 1,3-dipolar cycloaddition between an azide and an alkyne to give a 1,2,3-
10 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals. Figure 2.1: Azide-alkyne Huisgen 1,3-dipolar cycloaddition.

triazole (36). However, a variation to the Huisgen cycloaddition has shown better results; though, Huisgen reaction is devoid of side products, usually the outcome is a mixture of 1,4 and 1,5 regioisomers (Figure 2.1) and usually proceeds in elevated temperatures (37). This variant, on the other hand, is copper(I) catalyzed and proceeds in room temperature in a variety of solvents (including water with no organic co-solvent). It is better termed as Copper(I)-catalyzed Azide-Alkyne Cycloaddition or CuAAC. However, click chemistry includes several other chemical reactions such as the famous Diels-Alder reaction, nucleophilic substitution and particulary ring-opening of strained heterocyclic electrophiles, carbonyl chemistry of the non-aldol type (oxime ethers, urea formation, thiourea etc.) and additions to carbon-carbon multiple bonds (37; 38).
CuAAC reactions is perfect in every aspect except of one, the mandatory use of copper as catalyst renders the reaction useless, due to the toxicity of copper, in both bacterial and mammalian cells (39). Bertozzi et al. thought of a different route to promote [3 +2] cycloadditions (40). By using strain were able to promote the reaction; the best candidate was already known since 1961, when Wittig and Krebs publish their findings, of a simple and facile reaction between cyclooctyne (the smallest stable cycloalkyne) and an azide, which proceed fast under physiological conditions in the absence of catalysts or other reagents to give one single product, the triazole. The reaction was studied further, from the Bertozzi group, in living cells with positive results and no harmful effects on the cells were observed.
A different and chemoselective route of connecting peptides on scaffolds is Native Chemical Ligation (NCL). Following Kemp’s “priop thiol capture” (41) approach and Kent’s chemical ligation method (33), Dawson et al. developed a procedure that is completely entropically driven, overcoming
the problems associated with enthalpy driven approaches, which are due to low reactant concentrations and entropy barrier imposed by high molecular masses, especially when side-chain protection is extensively used (42). . NCL relies on the unique properties of sulfur chemistry; in a certain environment thiol groups can act as extreme nucleophiles and react chemoselectively. NCL employs two fully unprotected peptide fragments, on the C-terminal there is a thioester group and on the N-terminal is always a Cys moiety.
Firstly, the sulfhydryl residue on the Cys attacks the thioester group on the other peptide fragment to give the chemoselective reaction, then an intramolecular acyl transfer results in the final product bearing a native amide bond in every case of NCL.
However, NCL has a Cys requirement at the N-terminal, which is quite a problem due to scarcity of Cys amino acids in proteins. A solution to this problem is by utilizing serine or threonine ligation methods along with a (removable) salicylaldehyde ester as scaffold (43). This procedure relies on the properties of
11 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 2.2: Native Chemical Ligation method (167).

salicylaldehyde ester, which renders the ester completely inert to amine nucleophiles. The first step involves imine ligation; nucleophilic attack of the amine moiety of the N-terminal fragment, followed by a reversible oxazolidine formation. Then a non-reversible 1,5 O/S → N acyl transfer forms an amide bond, then if it’s desirable the scaffold can be removed under acidic conditions. The reaction proceeds smoothly in pyridine/acetic acid (1:1) and usually takes 4 – 24 hours to completion in room temperature.
Though, is a purely chemoselective ligation procedure due requirement of mainly organic solvents (pyridine – acetic acid buffer) the method is limited until it is applicable to physiological conditions.
All these approach have being applied in the synthesis of protein mimics.
Figure 2.2: 1) CLIPS scaffolds developed by Timmerman et al. 2) 2nd generation CLIPS scaffold. 2) Copper(I)-Catalyzed Azide-Alkyne cycloaddition. 3) Strain-Promoted Azide-Alkyne cycloaddition.
A novel method towards protein mimics was developed by Timmerman et al. of Pepscan Therapeutics in 2005, this is called CLIPS-technology (Chemical Linkage of Peptides onto Scaffolds) (44). The reaction has been found to be chemoselective to side chain unprotected dithiol-containing peptides. It was developed mainly for mimicking looped protein functionalities CLIPS reactions employ readily available materials such as 1,3-dibromoxylene or m-T2 (m stand for meta bromide positions, T for Template and 2 for the number of bromides), 1,3,5-tribromomesitylene or T3 and 1,2,4,5-tetrabromodurene or T4 (3 Figure 2.2). However, further development, under the supervision of Timmerman, in the subject resulted in a second generation of scaffolds (4 Figure 2.2), where a second exquisite functionality was added to the scaffold. By differentiating the central molecules of a protein
12 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

mimic they were able to add more peptide fragments on the central molecule and imitate protein functions more effectively (45).
In this point, a reference to a second concept slightly deviating from chemoselectivity is imposed by the scope of this project. The newly introduced (in this text) concept is termed as orthogonality. It was first devised by Barany and Merrifield for a completely independent classes of protecting groups (46). Since then, orthogonality became wider in context. Orthogonality refers to multiple (more than one otherwise is simply chemoselective) chemoselective reaction operating with no interference from one another(47). However, in the literature this two concepts appear interchangeable several times.
Scheme 2.1: Ligation reaction utilizing a salicylaldehyde ester scaffold.
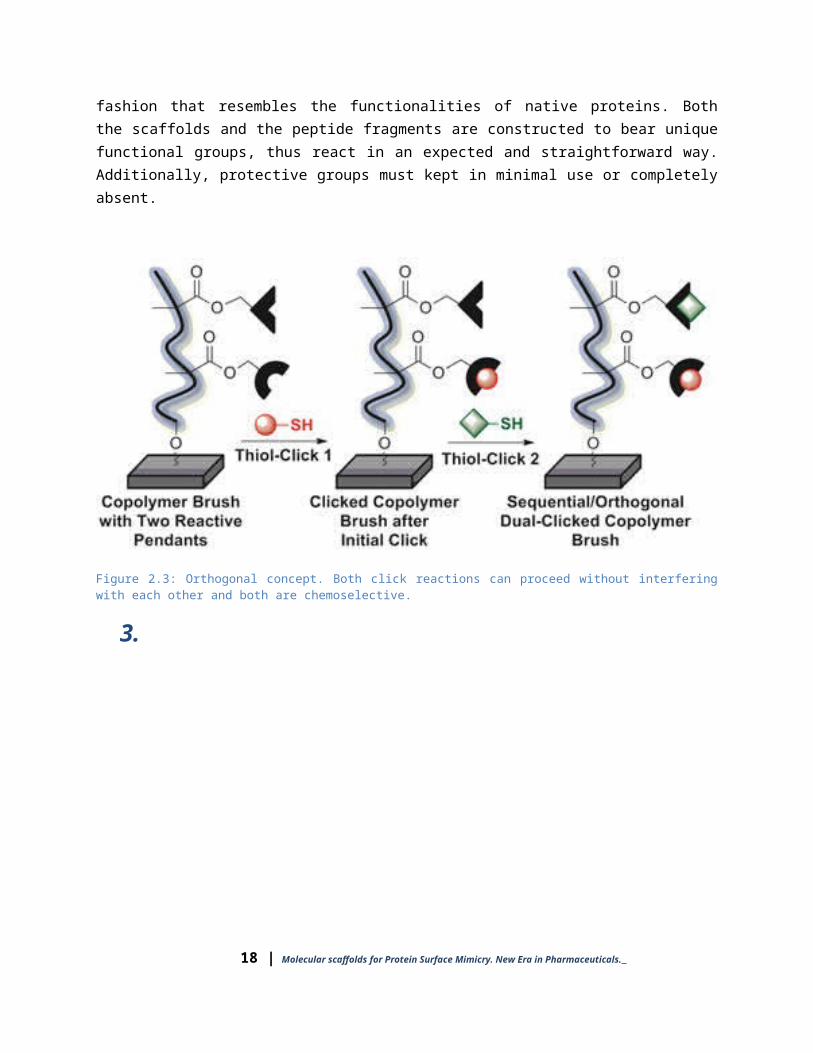
In the following sections, chemoselective and orthogonal reactions towards epitope mimics will be given. All the mimics referred here are consist of a small central organic molecule (here we will call them scaffolds) on which multiple peptides are anchored in a fashion that resembles the functionalities of native proteins. Both the scaffolds and the peptide fragments are constructed to bear unique functional groups, thus react in an expected and straightforward way. Additionally, protective groups must kept in minimal use or completely absent.
13 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Figure 2.3: Orthogonal concept. Both click reactions can proceed without interfering with each other and both are chemoselective.
3.
14 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

3.Re-creating Nature in a Flask One can think of nature as the perfect and wiser chemist. Every single reaction in nature results in a desirable product with no or minor by-products. On the other hand, humans face several obstacles when they try to develop new techniques to address a problem. This along with others, are the reasons for chemists attempting to recreate nature in flasks.
The past 20 years chemists have discovered how protein-protein interactions (see Introduction) and by manipulating them can preserve life. With this in mind, mimicking protein functionalities is of great importance for humans to survive. However, due to the complexity of the chemistry used in nature chemists have found a back door towards mimicking protein binding sites; via utilizing organic molecules that bear the desired protein functionalities. This molecules are named templates or scaffolds.
All the past efforts to synthesize protein mimics follow divergent approaches and they will be discussed briefly in this chapter as a historical review.
Divergent Approach for mimicking protein binding sites. A divergent approach (for convergent see introduction) utilizes a scaffold, on which peptides or amino acids are bound to via covalent bond, usually, or metallic bond, which depends on the nature of the template. A divergent strategy generally involves long and repetitive multi-step reactions to furnish the desire product (Scheme 2.1). In this kind of reactions the use of protective groups was inevitable due to the presence of multiple reactive groups in one reaction. This limits the procedure in utilizing only small peptide fragments. Any divergent approach is an alternative route to a convergent one (21; 49).
The primary attempts for generating neutralizing antibodies were made with short linear peptides. First, Anderer et al, who conceived the idea of employing short C-terminal peptides derived from a viral coat protein to generate antibodies against the protein (50). Nevertheless, his novel work did not get the appropriate attention because of the use in plants and therefore was considered irrelevant to humans by many. No long after Anderer and in 1981 two independent publications by the Audibert (51) and Beachy (52) groups confirmed the principle of utilizing linear peptides for neutralizing toxins derived from bacteria. The first group showed that by administrating a 14-residue peptide in guinea pigs antibodies could be generated for diphtheria toxin. Beachy, on the other hand, used a 35-residue peptide for generating protective antibodies against the bacteria infection Streptococcus pyogenes.
In 1989, a group in the UK defined the three-dimensional structure of the Foot-and-mouth disease virus (FMDV) protein VP1 via X-ray crystallography (53). With the structure of VP1 known and its binding site, first Brittle (54) and then Dimarchi (55) used a three-residue peptide (Arg-Gly-Asp) to neutralize the virus, however, the use of inactivated virus was more efficient.
15 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

The first successful synthetic vaccine was made by Langeveld et al (16) in 1994; they used a linear peptide based on the continuous N-terminus binding site of the VP2 protein from the canine parvovirus. It was confirmed to induce protective antibodies when administrated in animals. After Langeveld, several synthetic vaccines were developed utilizing the properties of neutralizing virus with linear peptides (56; 57; 58). Although some linear synthetic peptides were able to generate protective antibodies against virus and toxins it was obvious that protein structure influences protein function.
Scheme 3.1: Convergent approach towards protein mimicry; an organic scaffold with orthogonal protective groups, is first deprotected and then a peptide fragment is inserted to the central scaffold.
3.1Scaffold-induced conformation to protein mimics. Veber et al. in 1976 (59) first speculated that conformation is more important than primary structures (sequence of amino acids). Replacement of somatostatin sulfur atoms (forming disulfide bridge and constrain the peptide in a looped conformation) by one or two methylene groups showed that, the analogue with both sulfur atoms replaced (i.e. the lesser metabolic analogue) could be more potent
16 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Figure 3.1: The native structure of somatostatin (Left) and the analogue with the two sulfur atoms replaced by two methylene groups (Right). Disulfide bonds are more easily hydrolyzed than carbon-carbon bonds, thus the structure with no disulfide bridge will be more stable.
biologically than the rest. Moreover, a publication by the same group in 1979 (60), in which an analogue of somatostatin bearing a D-tryptophan in position 8 instead of L-tryptophan (increased the stability of βΙΙ’-turn), not only retained its bioactivity but also a slight increase in biological activity was observed.
Due to the importance of conformation in inhibiting protein-protein interactions linear peptides cannot be used effectively. However, conformation can be induced to continuous peptides by employing a scaffold (or template). Scaffolds retain the property of forcing linear peptides to arrange in an appropriate conformation necessary for mimicking the structure and, therefore, the functionality of a protein.
In nature there are several secondary structures which are essential in protein function; α-helix, β- and γ-turn and, β-sheets. Herein, a reference to divergent approaches toward conformational constrained mimics is handed. In every case mentioned below, a divergent approach have been used to address the problem; i.e. use of multiple (orthogonal) protective groups (Fmoc, Boc, Bn, Alloc etc.), isolation and purification after each step due to possible multiple side-products, complex synthetic routes and coupling reagents were necessary. In addition, this types of synthetic strategies were limited in the size of peptides amenable to use and usually the final products were highly hydrophobic.
3.1.2 Mimicking α -Helix secondary structure. α-Helices are the most prevalent secondary structures in proteins (61), hence, their fundamental role in PPIs (62). Due to difficulties predicting secondary structures of a polypeptide from its primary sequence, scaffolds had to be incorporated for achieving the right 3D structure.
17 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Sasaki and Kaiser used a tetrasubstituted coproporphyrin (see below) as scaffold to synthesize an artificial hemeprotein, bearing a four helix bundle. They used four fully protected peptides, which coupled on each carboxylic residue on the porphyrin scaffold by segment condensation reaction. This artificial protein mimic, which was named “helichrome”, was designed to mimic the hydrolase activity of cytochrome P-450 whose porphyrin-based active site is surrounded by several α-helices which serve as a hydrophobic substrate binding pocket. The peptide sequence was carefully chosen and the four helices make up a hydrophobic cavity or pocket above the porphyrin scaffold; the latter serves as the binding site for substrates of the hydroxylase reaction. CD studies on the product and the single peptide had shown that the product is high in α-helices content, whereas, the peptide adopted a disordered conformation. Therefore, it was concluded that the scaffold provides great stability to α-helices (63). However, because fully protected peptides had to be used the procedure suffers from the problems regarding divergent approaches; i.e. multistep procedure, isolation and purification after each step etc.
Figure 3.2: Coproporphyrin scaffold (Left). Artificial hemeprotein or Helichrome, displaying the cavity, with a substrate inside, that the 4 helices create (Right).
Tetraphenylporphyrins were used for the same scope by DeGrado and co-workers. By functionalizing the meta position of the phenyl groups with amphiphilic peptides. The final product was
used as ion channel. They found that, meta substitution provides optimal interhelical spacing for ion transport and the amino sequence was based on model peptides developed by DeGrado et al. which self-assemble into proton-selective ion channels in membranes (64).
18 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

An entirely different approach to induce α-helices utilizing a scaffold, was conceived by Kemp (65). Their scaffold contained three carbonyl moieties oriented in such fashion that an α-helix would result featuring the right spacing and gradient of a right-handed α-helix. In this paradigm, the authors utilize the properties of hydrogen bonding; the carbonyl groups of the scaffold (Figure 3.3) were oriented to form hydrogen bonds with the amide NH protons of the attached peptide, and hence, inducing an α-helix.
In 2000, Verdine and co-workers (66) thought of a different approach towards α-helices. They developed the scaffold seen in the margin, in which they used the double bonds via ruthenium catalyzed ring closing metathesis to induce and stabilize helical structures on peptides. These kind of peptides are called ‘stapled peptides’ and they bear spectacular properties; they retain specificity and natural multi-target recognition capabilities of native therapeutic proteins with minor limitations in their ability to address extracellular and intracellular targets. Due to the fact that stapled peptides are been forced to retain their structure, proteases cannot recognize and disassemble them (67; 68).
Figure 3.4: Stapled peptides.
Other methods found in the literature to stabilize α-helices include metal chelates (69), disulfide bridges (70), salt bridges (71), lactam bridges (72) and amide bonds (73) to connect i and i+4 position within an α-helix.
3.1.3 Mimicking β -Turn and β -Sheet Secondary Structures. β-Sheets are entirely different from α-helices, not only in the way they fold but also in the facility of adopting their secondary structure. According to studies, the thermodynamic propensity of a given residue to adopt a β-sheet conformation is more context dependent than analogous α-helical propensities (74; 75; 76). This shows that the long range interactions are more important in β-sheet folding than α-helix formation. Moreover, biophysical studies on proteins confirm that for a given protein to adopt a β-sheet formation is relatively slower than folding in an α-helical structure, hence, the former is rate limiting in tertiary structure formation (77; 78; 79; 80). All these indicate that β-sheets and α-helices fold differently. Due to difficulties of protein folding in sheet structures, scaffolds that induce these secondary structures are necessary in developing protein mimics.
19 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 3.3: Kemp and co-workers scaffold (in blue color) for inducing α-helix.

N
NO
O
H
H
N
N
NH
N NN
HN
NNN
N
O O
HO
H
Ph
O
H O
H
N
OO
OO
HO
H
Ph
Pro-D-Ala
Pro-D-Ala
Epidolindine Scaffold
N
NO
O
H
H
N
N
NH
N NN
HN
NN
N
O O
HO
H
Ph
O
H O
H
N
OO
OO
HO
H
Ph
N
The properties of rigidity to construct a desire conformation was conceived first by Hirschmann, who utilized the rigid γ-lactam to induce a single conformation to a peptide (59). In a second publication from the same group, in an attempt to develop a bioactive mimic of Luteinizing Hormone-Releasing Hormone (LH-RH) they used a γ-lactam to induce a β-turn in the final product. The introduction of the lactam group in the backbone of the protein stabilizes the β-turn conformation due to the restriction of
movement granted by the scaffolds’ rigidity. Model studies conducted in vivo and in vitro showed that their product not only mimics’ the activity of the native hormone but is even more potent, which is ascribed to better binding on the receptor than resistant increase to
proteases (81).
Many other examples of nucleators prompting β-turns can be found in the literature and some of them can be seen in the figure below.
Figure 3.6: The top three scaffolds are the simplest examples for inducing β-turn (82; 83; 84). The bottom two scaffolds illustrate more complex tetrapeptide-based mimics that highlight the potential to incorporate both the natural peptides as well as several amine or carbonyl functionalities already within the template (85; 86; 87).
However, the most challenging structure to mimic between the two is the β-sheet this is due to the fact that β-sheet is more context dependent than an analogous turn (21). Kemp and co-workers displayed in 1988 the first induced β-sheet mimic. By utilizing hydrogen bonding and the rigidity of proline it was possible to artificially construct a β-sheet structure. They incorporate an epindolidione template, in which hydrogen bond donors and acceptors are well oriented to mimic β-sheet structures. However, due to planarity of the central scaffold, no strand-strand interactions are possible; strand-strand interactions are capable of stabilizing β-sheet structures in nature. To overcome this problem, Kemp’s group used amino acid sequences, such as Pro-D-Ala (Figure 3.7a), bound to the scaffold to favor the reverse turn conformation as shown in the figure above (88). To induce antiparallel β-sheet folding, the Kemp group replaced the Pro-D-Ala sequence with two urea moieties as shown in Figure 3.7b.
20 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 3.5: γ-lactam template.

Another good example for β-sheet formation was introduced by Kelly’s group. In their method, via incorporating a β-turn nucleator, they were able to replace the i+1 and i+2 residues of β-turn. When a linear 13-residue peptide was attached on the benzofuran-based scaffold, the former was folded into a β-sheet structure, even though at first it was estimated that repulsion between positively charged moieties would disfavor the resulted structure. The stability of the final product stems from the hydrogen bonding mainly, which is promoted from the scaffold. Moreover, the tertiary clustering of hydrophobic side chains in the early stages of protein folding and the rigid structure of the scaffold results in the product’s final conformation (89).
Bipyridine-based scaffolds have also been used as promoters of β-sheet nucleation. In this case the scaffold lacks of rigidity, hence, the final product can adopt two conformations; the transoid, favored due to repulsions between the two peptides, and the cisoid conformation, which is the desire one. The cisoid conformation is mediated by ligating within the
peptides a copper(II) atom in the final product as shown below (90).
Scheme 3.2: Transoid to cisoid shift mediated by Cu(II) ligation between the peptides; R = a-amino acid side chain.
3.1.4 Mimicking Looped conformations. Any loop conformation consists of a combination of β-sheets and β-turns, or “sheet-turn-sheet” loops (21). Studies conducted on linear peptides whether these can generate antibodies capable of neutralizing virus showed that in some cases cyclization of these peptides was required for the mimic to show immunogenic properties and generate antibodies (91; 92; 93; 94).
Looped structures are hard to mimic due to complex spatial arrangement. An appropriate scaffold has to direct the attached peptides to adopt the correct three dimensional structure (tertiary structure). Moreover, for a protein to function certain secondary structure is required (10), hence for mimicking a loop conformation the scaffold must be chosen wisely.
To mimic the Ω-loop of interleukin-1α (a cytokine responsible for triggering inflammatory and immunological responses by interacting with the receptor on lymphocytes), Sarabu and
21 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 3.7b: Antiparallel β-sheet.
Figure 3.7a: Parallel β-turn mimic.
Figure 3.8: Ω-loop mimic induced by a napthalene-based scaffold (highlighted).

co-workers utilized a naphthalene-based scaffold (Figure 3.8). The apparent loop is believed to hold the biological activity of interleukin-1α. The scaffold they had chosen kept the peptide’s termini in right distance so the peptide fragment attached to the scaffold could fold in the desired conformation (confirmed by NMR experiments). However, testing the final product revealed that the Ω-loop was not enough for receptor recognition and activation (95).
Peluso et al., utilized the so-called Template-Assembled Synthetic Protein scaffold (TASP or RAFT; Regioselective Addressable Functionalized Template), developed by Mutter and co-workers (96), to mimic the complementary-determining region of the monoclonal phosphorylcholine-binding myeloma antibody McPC603 (97). By employing orthogonal protective groups they were able to fashion a three-loop molecule as shown in the scheme below. The template is stabilized by hydrogen intramolecular interactions (the template has a β-sheet secondary structure), therefore, all the binding sites are facing the right direction during the peptide condensation reaction, which is consistent with high cyclisation rates. No side reactions were observed, however, due to simultaneous condensation of the N- and C-terminal chain ends of each loop peptide onto the template resulted in two orientational isomers in each step, i.e. eight isomers were observed of identical molecular mass.
Scheme 3.3: Synthesis of three-loop mimic on the TASP scaffold.
22 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 3.8: Ω-loop mimic induced by a napthalene-based scaffold (highlighted).

Other approaches towards loop mimics include backbone-to-backbone or side-to-side chain cyclizations (98) for which reactions like olefin metathesis (99), macrocyclization by conventional lactamization chemistry (100) and click reaction (101) were used, however, this approaches do not serve the scope of this project, hence no further discussion on the topics will be given.
3.1.5 Mimicking Discontinuous Protein Binding Sites. Discontinuous epitopes are the most challenging to reproduce; the majority of protein epitopes are discontinuous (102). A discontinuous binding site or epitope, consists of several separated strands bearing unique binding moieties that are brought in close proximity by protein folding (tertiary structure) (103). Discontinuous epitopes cannot be mimicked by linear peptides of an antigen, due to their complexity. Therefore, by attaching several peptides on a scaffold a tertiary protein structure can be imitated with great success.
Mutter and co-workers coined a way to make this possible; by developing the TASP scaffold (see section 1.1.3) they were able to attach five different peptide fragments (orthogonal protective groups) on the scaffold and, therefore, reproduce a large surface area of a discontinuous protein binding site. The result was interesting, the scaffold forced each element to adopt the three-dimensional structure of the native protein (96). Since, its introduction to the scientific community TASP scaffold has found wide range of use; Rau and Haehnel used the TASP scaffold for the synthesis of a water-soluble cytochrome b model (104), Grouzmann and co-workers synthesized the very first potent and selective Y2 antagonist, via exploiting the concept of template-assembled synthetic proteins. They were able to anchor four peptides on every lysine residue on the scaffold and reproduce an Y2 antagonist for the first time (105).
Figure 3.9: Tamplate-Assembled Synthetic Protein (TASP) concept by Mutter and co-workers.
After the introduction of the TASP-concept, several groups around the globe focused their research interest around mimicking discontinuous epitopes via attaching several peptide fragments on one central scaffold. Liskamp et al. (106), for example, employed a triazacyclophane (TAC) scaffold for constructing artificial receptors. The TAC-scaffold is an organic molecule bearing three secondary protected amines on one end and an aromatic carboxylic acid on the other end. Due to the orthogonality of the protected groups used, they were able to anchor multiple different peptides. TAC-scaffold
23 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 3.10: A triazacyclophane (TAC) scaffold with three different protective groups bound on the amines. The scaffold is anchored on a solid support from the carboxylic acid's side.

has been used for mimicking papain inhibitory protein (107), constructing of synthetic receptors (108), developing an artificial vaccine for pertussis (109) and for developing a mimic of the copper-binding site of a protein (110).
The rigid character of the TAC scaffold in several cases has shown good propensity for the right placing of a discontinuous epitope, or peptide fragments can be positioned on a certain way that a large area is covered. Adding to that, the synthesis of TAC scaffold is convenient and robust, making it as an ideal choice for the syntheses of protein and peptide mimics (111).
Fairlie and co-workers on the other hand, attempted and succeeded on mimicking the two inter-helical loops of cytochrome b562 (112). They developed a macrocyclic scaffold constrained by oxazoles and thiazoles (4 FIgure 3.11) to support two peptide loops projecting orthogonally from the same face of the scaffold deciphered by NMR analysis. Comparison with the crystal structure of cytochrome b 562
indicated good matching of the artificial molecule with the native one.
Goodman (113) and after him Liskamp (114), used scaffolds bearing carboxylic acid functionalities for replicating the triple helix of collagen (7 Figure 3.11). Due to the characteristic preorganization of the scaffold and its rigid structure, both groups succeeded in mimicking the triple helix. Moreover, the scaffold clearly induced some sort of stability on the helix compared to non-assembled peptides. However, these constructs are relatively small and may have limitations when large surface areas in proteins need to be covered.
In 1997, the group of Hamilton (115) presented the synthesis of an antibody mimic that can bind on the surface of cytochrome c in a similar way to the natural protein partners. Calix[4]arene ( 8 Figure 3.11) was chosen as the central scaffold; it’s readily available and adopts a semirigid cone conformation when the phenol groups are alkylated. This cone conformation as outcome projects the para-substituents on the same side of the ring to form a potential binding site (116). Their final product was able to bind strongly to a complementary surface on cytochrome c receptor and disturb the approach of reducing agents to the active site of the protein, as with cytochrome c peroxidase (115). Later Spadaro employed calix[4]arene in his synthesis of a fully synthetic cancer vaccine candidate (117). In this case, both faces of the scaffold were used; via sequential amide coupling, four S-Tn glycomimetic antigens were attached on the wide upper rim and one P3CS immunoadjuvant residue on the lower side. It was the first example of a vaccine candidate built on a nonpeptidic platform. Clinical trials on mice revealed that the candidate drug enhances production of Tn specific IgG antibodies compared to an analogous monovalent reference compound.
Several other scaffolds were used from time to time, like D-glucose (6 Figure 3.11); Hirschmann et al used it to mimic the functionalities of somatostatin hormone and proved that peptidic scaffolds are not necessary for reproducing protein functionalities (118). Eichler (119), in an attempt to build scaffolds that cover a large protein surface area developed scaffold such as 9 (Figure 3.11). In 2007, he employed this scaffold to mimic the CD4 binding site of HIV-1 gp120 (120). This mimic has been found to generate antibodies that recognize the parent protein gp120, as well as to compete with the broadly neutralizing antibody mAb b12 whose binding site overlaps with the CD4-binding site for binding to gp120.
24 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

In a diverse approach, Hamilton et al. (121) used non-covalently stabilized macrocyclic scaffolds to which peptides were attached. By employing a peptide-oligonucleotide click conjugation methodology, they developed a self-organizing structure that positions two peptide loops on one surface, which in turn gives rise to the formation of both homo- and hetero-combinations. The supramolecular approach is limited to that formation of mixtures of homo- and hetero-dimers can never be prevented, due to the nature of this approach.
As stated before in this chapter, all the synthetic approaches are based on divergent approaches. In the rest of this report only convergent synthetic strategies will be discussed, as these are the main scope of this project.
25 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Figure 3.11: Scaffolds mimicking protein tertiary structures.
26 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

4.Starting from Simple Organic Molecules towards Drug Candidates
Ever since scientists comprehend (even in the slightest degree that we understand today) how vital for living organisms PPIs are, they focused their research interests in replicating protein “hot spots” (the most important residues or areas of a protein associated in biding).
The first thought was that the use of simple linear peptides could do the job, however, this has been proven to be an erroneous conception. Studies on PPIs have shown that primary structure is not as important as the secondary and tertiary structures (122). With this in mind synthetic chemists attempt to construct small molecules which have the ability to bind on certain proteins.
Although ideally proteins could be used as drugs, due to the complex and usually unstable structures, unpredictable bioavailability, sometimes unknown action, inability of manufacturing proteins ex vivo (due to their nature) and low pharmacokinetics, proteins are poor drug candidates (21; 123). So, as Jacob Bronowski (124) once said “Science, like art, is not a copy of nature but a re-creation of her”, chemists nowadays realize that the most appropriate solution in drug manufacturing is to recreate the way proteins interact with one another.
In this chapter, a review of recent ways for mimicking nature toward possible drug candidates will be handed. This mimics are constructed via simple procedures utilizing a central scaffold on which several peptides are bound to express desirable protein functions.
Discontinuous Protein Binding Site Mimics. Proteins have gained their unique structures after billions of years of evolution. Nature was slow but effective, humans, on the other hand, devise different routes to address the problem. In this case, small organic molecules that bear protein functionalities, are able to disrupt PPIs. These molecules are based on templates or scaffolds. These are molecules that can be used as the backbone of bigger compounds that one or more peptide fragments are attached to, for inhibiting or disturbing certain PPIs.
A keen supporter of this approach is Prof. Liskamp of Utrecht University. His name has long been connected with triazacyclophane scaffold or TAC-scaffold (see also Chapter 2).
27 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

4.1The Triazacyclophane (TAC) – Scaffold. The TAC-scaffold bears a relatively rigid 14-member heterocyclic ring with three tertiary amines well distributed and hence able to cover large protein surfaces (125). Although, the TAC-scaffold was synthesized to bear three orthogonal protective groups (106) hence, selective deprotection is possible, in the last few years they alternate their approach by utilizing Click chemistry (126). One year ago, the Liskamp group described a fast and convenient approach
towards “smart libraries” (combinatorial chemistry) of discontinuous epitope mimics of HIV gp120 using a TAC scaffold. To mimic the interaction of HIV gp120 with the CD4 receptor (successful mimicry of the gp120 discontinuous epitope can possibly lead to synthetic vaccines against AIDS), several groups have determined that three loop constrained peptide fragments are required (127; 128; 129). To do so, they devised a derivative of the fully protected TAC scaffold (Scheme 3.11), with three alkyne residues projecting from the molecule. With the scaffold in hand click chemistry can be applied; via regular solid-phase synthesis they were able to manufacture the necessary
Scheme 4.1: Combinatorial approach toward epitope mimics libraries, utilizing Click reaction or CuAAC. Microwave radiation have proven to accelerate the process (citation needed).
loops with an azidolysine residue projecting toward the exterior site of the cyclic peptide fragments. Then, by utilizing the CuAAC reaction (Click reaction - see Chapter 1) an equimolar library of gp120 mimics was observed, with no undesirable side products. LC-MS studies revealed that all combinations of cyclic peptide loops on the TAC-scaffold could be identified from their mass value. They were able to separate the products easily and up to mg quantities of each pure epitope mimic was obtained. The procedure was highly reproducible and identical libraries were easily prepared.
28 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.1: Conserved epitopes in the CD4 binding site on gp120.

The final products were tested in a gp120-capture ELISA experiment; interestingly two out of all, were observed to highly compete with the native protein for binding on the CD4 receptor. It was noted by the authors that, the current convergent approach compared to earlier divergent methods (non-stop solid phase approach) accelerated the process towards smart libraries of protein mimics. Moreover, the un-scaffolded peptides showed no binding at all, which implies that pre-organization of a protein’s ”hot spots” is of great importance in PPIs.
To further investigate the properties of a good protein mimic, the Liskamp group, tested along with the TAC-scaffold three more scaffolds, all of them functionalized with alkyne residues for developing smart libraries of epitope mimics for the interaction between gp120 and CD4 via CuAAC protocol (130). They employed a triamine scaffold (1 Scheme 4.2); it can be considered as a flexible analogue of the TAC-scaffold, a dendrimer building block (3 Scheme 4.2); which diverges from the rest in organizing the peptide loops in roughly one plane and, a cyclotriveratrylene or CTV-scaffold (5 Scheme 4.2); the most pre-organized scaffold usually used for stabilizing triple helical mimics (114). Again, competitive ELISA experiment was chosen to evaluate the affinity of the gp120 mimics. They run two screening tests; at concentrations of 250 μg/mL more than 60% inhibition of gp120 was observed by the most artificial mimics, whereas, at 125 μg/mL of concentration the triamine-based mimics showed significant drop of inhibition while all CTV-based mimics showed almost complete inhibition. In fact, a specific CTV-based mimic compared to the most potent TAC-based protein mimic was ten times more active; this reveals the crucial importance of orientation in constructing a successful protein mimic.
29 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Scheme 4.2: Synthesis of smart libraries of three versatile scaffolds.
The approach developed by the group is rapid, versatile and highly reproducible toward libraries of discontinuous epitope mimics. The CuAAC reaction is fully chemoselective reaction and usually gives products in high yields (see Chapter 1). Microwave irradiation enhances the reaction (131; 132).
The TAC-scaffold is a good tool for mimicking protein binding sites and the above text proves it. The fact that the CTV-based mimic was more effective shows only that mimicry of protein binding sites is a very complex task and that different scaffolds can be used in different cases depending on the topology of the protein binding site.
30 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

4.2Functionalized cyclic peptides as scaffolds.
Figure 4.2: The TASP concept.
In 1989, Mutter developed the so called Template Assembled Synthetic Protein (TASP) approach (for more details see Chapter 2). This utilize Lys residues for attaching several peptide fragments to construct protein mimics. The usual procedures toward protein mimics rely on orthogonally protected groups, however, in the literature can be found procedures following convergent approaches (133; 134; 135; 136; 137).
The very first convergent TASP approach was attempted in 1993 by Dawson and Kent (137). The goal was to synthesize a four helical TASP molecule via chemoselective ligation. To do so, they manufactured a peptide, the scaffold, bearing four bromoacetyl residues. For the reaction to be fully chemoselective the peptide fragments had be fashioned in a way that no protective groups were needed. Chemical ligation (42) procedure (which was introduced by the same group) was followed by introducing on the peptide C-terminal a thioacid
moiety, which can selectively displace bromide via SN2 reaction and attach the peptide fragment on the scaffold covalently.
Chemical ligation reaction proceeds cleanly at ambient temperature in aqueous buffer at pH 5 to furnish the desire product after just 70 min. However, some by-products detected but the main product was easily isolated and purified. This procedure utilizes the advantages of the chemical ligation approaches; products are readily purified, due to the absence of protective groups, high yielding reactions and the method is of general applicability because the chemistry is compatible with all functional groups found in peptides and proteins. Moreover, if necessary other chemical ligation approaches are applicable (see Chapter 1), therefore, four different, if is desirable, peptide fragments can be attached on the scaffold.
31 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.3: TASP-approach via chemical ligation reaction.

Tuchscherer, on the other hand, used oxime ligation to bind four peptide fragments on a TASP-scaffold (133). The ligation reaction proceeded under mild conditions in sodium acetate buffer at pH 5 with threefold excess of the peptide fragment with respect to the functional groups on the template and was found to be quantitative by analytical RP-HPLC. Separation was necessary to isolate the final product from the unreacted peptide fragments.
A combinatorial approach was used by Plé et al. employing a RAFT scaffold (136). As before, chemical ligation strategy was used for anchoring four peptide fragments on the scaffold. Again oxime ligation was preferred. In this case though, four peptide fragments with different primary structures were used to furnish a library of RAFT molecules. The group was able to optimize the reaction conditions of the oxime ligation to consume all the amount of RAFT present and hence, simplify the purification of the mixture after the reaction is complete; an ammonium acetate buffer was chosen and the scaffold was completely transform to products after 24 h at room temperature. They succeeded in synthesizing 256 different protein mimics, which they tested on affinity chromatography. By utilizing an avidin - agarose bead, they were able to see which were recognized, due to a dabsyl dye anchored on the RAFT molecules (gives red color to the bead).
The TASP-scaffolds are limited in the protein surface area that can cover, mainly because all the peptide fragments are projected in the upper face of the scaffold (Figure 4.2). To further wide the use of cyclic peptides, the bottom side has to be functional as well. A group from Universitè Joseph Fourier in France, Grenoble developed a trifunctional TASP-scaffold (138). Their approach relies completely on orthogonal chemoselective ligation techniques; on the upper side four Arg-Gly-Asp (RGD) groups were anchored by employing the “Click reaction” (CuAAC) and on the bottom face a carbohydrate residue via oxime or thioether ligation along with a molecular fragment that can be used for diagnostics (fluorescent probe) or for therapy ( peptide or nucleic acid residues).
To demonstrate the capabilities of chemoselective ligation techniques, they decided to prepare three different products via one-pot sequential chemoselective ligation of different biomolecules. In the first synthetic route by utilizing oxime ligation (under acidic conditions and after 1 hour) they incorporate in the bottom face a carbohydrate residue, then by simple increase of pH to neutral the KLA peptide fragment was added to the scaffold. Rapid thiol addition was observed at ambient conditions within 30 min. Moreover, no reaction between the remaining carbohydrate reagent and the maleimide moiety was detected. In the last step via “click reaction” furnished the desired compound in 55% yield after purification. However, during the cycloaddition reaction, the KLA peptide residues formed dimers, probably due to oxidation of the residual thiopeptide, as the only by-product. The second route followed the previous successful way with the functional groups on the scaffold slightly different ( b Scheme 4.3). Also, a fluorescent probe was successfully added to the scaffold. The third route was mainly to show how versatile this approach can be by anchoring a nucleonic acid on the scaffold’s bottom face with yields of about 60%.
32 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Scheme 4.3: i) Reagents and conditions for the synthesis of compounds 2-4: 1) R1-ONH2 (3 eq.), CH3CN/PBS (pH 2.2 40:60), 1h; 2) R2-SH (1.3 eq.), pH adjusted to 7 (NaHCO3), 30 min; 3) R3-CH2-N3 (6 eq.), CuO (5 eq.), tBuOH. ii) Reagents and conditions for the synthesis of compounds 6-8: 4) lactose (5 eq.), CH3CN/ammonium acetate buffer (pH 4.5, 40:60), 50oC, 5h; 5) Alexa Fluor 647 C2-maleimide (1.3 eq.), pH adjusted to 7 (NaHCO3), 30 min; 6) R3-CH2-N3 (6 eq.), CuO (5 eq.), tBuOH. iii) Reagents and conditions for the synthesis of compounds 10-12: 6) R3-CHO (5 eq.), TFA/CH3CN/H2O (5:45:50), 1h; 7) pH adjusted to 7 (NaHCO3), R1-SH (2 eq.), 30 min; 8) R6-C≡CH (0.8 eq.), CuO (5 eq.), tBuOH, 50oC, 5h. PBS = phosphate-buffered saline, TFA = trifluoroacetic acid.
All in all, the TASP-scaffolds constitute a versatile way of manufacturing protein biding sites. Utilizing chemoselective ligation techniques makes it a powerful tool for protein mimicry. It is applicable to combinatorial approaches but also for constructing specific compounds toward drug discovery.
33 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Moreover, the fact that it’s structure consists purely of amino acids renders the compounds fully water-soluble and applicable to in vivo approaches. The conditions of each reaction are mild enough that makes the procedure easy to handle.
4.3CLIPS TM - Technology. In 2005 Timmerman et al. coined a simple and reliable way, called CLIPS chemistry, for inducing loop conformations onto unprotected peptides bearing Cys residues (45). CLIPS stands for Chemical Linkage of Peptides onto Scaffolds and the reaction has proven to be completely chemoselective towards
unprotected dithiol-contained peptides (21).In this route, readily available organic synthetic molecules are been used as scaffolds bearing two, three or four bromides attached on a benzene ring. The reaction was characterized as “unusually fast and entirely selective” for Cys under aqueous solutions (45) Reaction of a 14-mer peptide with 1.05 equiv of m-T2 (see Scheme 4.5) in a 1:7 mixture of ACN/NH4HCO3 (pH 7.8) results in the corresponding constrained looped compound in > 80% yield within 15 min at ambient conditions, deprived of any by-product (< 5%). Oxidative cyclization is not a competitive reaction under these conditions. Therefore, CLIPS reactions are orthogonal and fully compatible with oxidative disulfide bond formation,
hence, double looped conformation can be applied to a peptide containing four sulfhydryl moieties by using only a T2 scaffold and subsequently disulfide oxidation, although, possible protective groups might be needed (139). Moreover, an independent group (of J. W. Szostak at Harvard Medical School and a Nobel Prize winner for Physiology or Medicine) characterize the two thioether bonds of a cyclic CLIPS product as “stable, apolar and may improve bioavailability”, uncovering even more the possibilities of CLIPS chemistry (140; 141).
34 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.4: CLIPS-Technology. A T3 scaffold constrains a linear peptide into a double-looped mimic.

Scheme 4.4: CLIPS reaction. The polymer does not form (or forms in less than 5%) with the intramolecular reaction been favored.
The CLIPS reaction depends highly on the nature of the scaffold and less on the size of the linear peptides. For example p-T2 scaffold reacts much slower than the other two T2 analogues (o- and m-T2); HPLC studies showed that, the less stable scaffolds (i.e. o- and p-T4) can be observed only for large ring sizes, whereas, the m-T4 molecule (significantly more stable than its analogues) can be and it was observed in significant amounts (15%) for most peptides. An explanation is that the first SN2 reaction is the rate determining step (slower) and the subsequent intramolecular reaction is activated by a stabilizing resonance effect and proceeds fast to the formation of the final product, since the second step is less energetically demanding reaction (142).To prove that peptide mimics based on CLIPS-technology exhibit any immunological properties (and thus can be used as artificial therapeutic drugs), Timmerman and his colleagues devised an experiment, in which peptide mimics were manufactured via the CLIPS reaction. Based on epitope mapping studies, they designed two constrained compounds (a single- and a double-constrained) that showed improved mAb-binding and generate high-titred antisera in rats, which in turn neutralize the biological activity of Follicle Stimulating Hormone (hFSH) in vitro and bind to hFSH in ELISA. In simple words, the experiment was more than successful (139).
Several other groups used CLIPS technology in different tasks, such as simultaneous cyclization and peptide labeling (143), in phage display libraries for the selection of ligands based on bicyclic peptides (144) for macrocyclization of peptides (145; 146; 147; 148).
35 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

However, this procedure relies only on cysteine-contained peptides, something that limits its use. To further extend its applicability, Smeenk et al. (46) designed a more versatile generation of CLIPS scaffolds. These still follow the same efficient pathway of CLIPS technology towards mimicry, though, by adding a third orthogonal functional group on the scaffold different peptides can be attached on and therefore, carry more information thus be more efficient.
The 1st generation of CLIPS scaffolds was inspired by the work of Hartman et al. (143). Though, the syntheses were not hard and the yields were sufficient enough (≈ 70%), due to their highly hydrophobic nature, a 2nd generation of water-soluble molecular scaffolds had to be fashioned (the insolubility of the scaffold
disturbs the water-solubility of the CLIPS-cyclized peptides when used in the CuAAC reaction) (21).
The 2nd generation of scaffolds (named as oS2, “o” stands for “ortho” and denotes the relative positions of the bromides and “S” for “soluble”) have found to be easily made ( Figure 4.5), chemically very stable and highly water-soluble due to the quaternary ammonium ion. The newly introduced functionality follows the Click chemistry protocol (see Chapter 2), i.e. straightforward and completely chemoselective reactions, mild conditions, no by-products etc. hence, four different chemistry were chosen for this task, the Copper(II) Azide-Alkyne cycloaddition (CuAAC), the thiol-ene reaction, the oxime formation reaction and the strain-promoted azide-alkyne cycloaddition (SPAAC). By employing these kind of chemistries, along with disulfide bond formation, two separate linear peptides can be constrained into loops and be ligated together in close proximity to mimic a unique peptide surface.After synthesizing all the possible scaffolds the most appropriate route had to be found for constructing the desired mimics.
Figure 4.6: Retrosynthetic analysis of the 2nd generation of CLIPS-scaffolds.
36 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.5: 1st generation CLIPS-scaffolds

4.3.1 Peptide ligation via Copper(II)-catalyzed Azide-alkyne Cycloaddition.
Scheme 4.4: CuAAC optimized reaction with 1st generation of Double-CLIPS scaffolds.
A trial reaction to test the efficacy of the CuAAC reaction was made using 1 st generation CLIPS scaffolds. First, the peptide fragments bearing two Cys residues were anchored via the regular CLIPS reaction. Then several trial reactions were investigated with different conditions to see if CuAAC it’s applicable reaction.The first successful reaction was made with CLIPSed peptides in concentration of 50 mM, 0.05 equiv. of copper and 0.5 mM of sodium ascorbate in room temperature. Within 15 min the reaction was completed with full conversion of the reagents. However, reducing the starting concentration of the CLIPSed peptides to 1 mM, was found that either quantitative amounts of copper were needed for full conversion or increased temperature, or by using THTA as ligand. Moreover, microwave irradiation was used to enhance the CuAAC reaction which did in some extent. Yet, the results were far from sufficient for ligating CLIPSed peptides even when the 2nd generation CLIPS scaffolds were used. The failure of this approach was mainly ascribed to oxidation of the peptides by relatively large amounts of Cu(I) present in combination with the aerobic reaction conditions used, as well as decomposition of the peptides due to the high temperatures been used in ligation reactions.
37 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

Scheme 4.5: CuAAC reaction with 2nd generation of Double-CLIPS scaffolds. The reaction resulted in insignificant amounts of product.
4.3.2 Peptide ligation via Strain-promoted Alkyne-Azide Cycloaddition. To overcome the problems associated with the use of catalyst, the so called “Strain-Promoted Click reaction” was employed. An oS2 scaffold bearing a fluoro-substituted cyclooctyne was utilized for this reaction. But, due to the hydrophobic nature of the cyclooctyne, the observed low reactivity (never full conversion of the CLIPSed peptides was observed) and the complex synthesis of the cyclooctyne fragment the thermal Click reaction was no further
exploited.
4.3.3 Peptide ligation via Thiol-ene Reaction. This route involves a photo-induced radical mechanism between a thiol and alkene group. Advantage of this reaction is that, it proceeds with complete absence of catalyst and under aqueous conditions. It can be initiated thermally or via UV-irradiation at room temperature, however, it has been proved in the past that the photo-induced route is more efficient (149; 150).
To test the reaction, several reaction conditions were tested using Fmoc-Cys and an alkene-functionalized Cbz-piperazine scaffold as model compounds. As photo-initiator DPAP was used, along with TCEP to prevent disulfide dimer formation, which in turn resulted in higher conversions while the conditions with no TCEP present were not altered. The optimized conditions were found to be with much more use of DPAP resulting in heterogeneous conditions, since DPAP was not fully soluble.
38 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.7: Double-CLIPS scaffolds for SPAAC.
Figure 4.8: Scaffolds been used for the "Thiol-ene" reaction.

However, when this conditions were employed to a reaction between alkene and thiol-functionalized RGD-peptides, no product was observed, mainly due to high concentrations of the initiator the starting materials were decomposed.Finally, by replacing DPAP with the water-soluble analog AAPH, the model reaction did not proceed with equimolar quantities of the thiol and the alkene. Full conversion was possible only with excess of either the alkene or the initiator, indicating for one more time that the thiol-ene reaction is not applicable for generate mimics using 2nd generation CLIPS scaffolds.
4.3.4 Peptide Ligation via Oxime Formation. Again a model system was tested first similar to the thiol-ene approach. This was found to be extremely successful with more than 99% conversion in every trial reaction (Table 4.1). It was observed that the reaction was optimized in the presence of 100 mM of aniline as catalyst of the reaction. Then a model reaction with RGD peptides was attempted successfully, since the reaction proceeded fast to completion (15 min) in 77% yield of the expected double-CLIPS product and no by-products were observed.
Table 4.1: Oxime bond formation in a model system with optimized conditions.
Buffer (mM) Concentration (mM) Time (h) Conversion (%)0 10 5 > 99%100 10 0.5 > 99%0 1 24 > 99%1 1 8 > 99%10 1 1.5 > 99%100 1 0.5 > 99%
With these results in hand oxime ligation was the most appropriate reaction for the ligation of unprotected CLIPSed peptides.
39 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

ONH2
N N
O
O
N
NO
N
NO
N
N N
OH
O
SS
= Amino Acid
= Cystein Residue
100 mM citric acid/aniline
buffer
r.t., 15 min
S S
Scheme 4.6: Oxime ligation towards Double-constrained CLIPS-mimic.
4.3.5 Constrain-induced via Single and Double Disulfide Bond. Preorganization in protein mimicry is more than important. Ligation of two constrained peptide fragments sometimes is not enough for a synthetic molecule to reproduce natural functions found in proteins.To further constrain the peptides disulfide
bonds consist of a facile and fast way. There two ways, via intra- and inter-
molecular disulfide bond formation.Formation of an intramolecular disulfide bond between two oxime-ligated loops, was achieved by utilizing iodine to cleave the Acm protecting groups from the Cys residues and at the same time facilitate the formation of a disulfide bond. Though, an efficient method, several by-products were observed limiting the procedure. To overcome this problem a different approach was used; fully unprotected ligated loops were first reduced with DTT in excess and purified by HPLC and subsequently re-oxidized using a Tris-HCl buffer pH 8.0. This way showed increase in overall yields and allowed to monitor the SS-oxidation reaction.
40 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.9: Intramolecular disulfide bond formation.

Different route towards double constrained peptides can be done by first forming a disulfide bond and then ligate the two peptides together. To do so, one of the thiols has to be activated first in order to exclude the formation of the symmetrical disulfide products, as shown below.
Scheme 4.7: Asymmetric disulfide bond formation.
This way consists of an alternative way to produce mimics bearing a disulfide bond. Even the non-optimized CuAAC reaction could be facilitated with better results; intramolecular CuAAC reaction of two peptides brought together by disulfide bond, proceeded to completion within 10 min in the presence of CuSO4 (5 equiv) and sodium ascorbate (2.5 equiv). However, immediate purification was necessary to prevent slow reduction of the disulfide bond by the reducing agent.Additionally, two disulfide bonds can be introduced in a CLIPS mimic to further constrain the peptides for better mimicry. This is done by simple use of a Tris-HCl buffer and waiting time of about 24-48 h. However, due to unclear route of formation of both disulfide bonds there are difficulties in controlling these reactions.Having in hand an optimized way towards protein surface mimics the next step is to actually mimic a protein binding site.
4.3.6 Double-CLIPS Technology in the Mimicry of Discontinuous Binding Sites. Three different protein binding sites were investigated within the context of double-CLIPS technology; i) the human Follicle Stimulating Hormone Beta (hFSH-β), ii) the human Chorionic Gonadotropin Beta (hCG-β) and iii) the human Vascular Endothelial Growth Factor (hVEGF).
This proteins contain highly discontinuous and conformational topology of two assembled loop structures within the same binding area, the β1- and β3-loop. The hVEGF protein was chosen due to bevacizumab (AvastinTM), which was the first ever therapeutic antibody, developed by Roche®, that produces angiogenesis (an angiogenesis inhibitor is a substance that inhibits the growth of new blood vessels, this are used to treat several form of cancer) inhibition by inhibiting VEGF-A (151). The main difference of this protein from the other two is
41 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.10: The human Follicle Stimulating Hormone Beta (hFSH-β) bound on the FSH-receptor (in red) (168).

that lacks of an additional disulfide bond that could help stabilizing the structural organization of both loops.
Three different techniques were chosen for studying the double-CLIPS compounds in detail. The first involves a peptide array technology, which made it possible to study the amino acid sequence in β1-loop, the position of the CLIPS-scaffolds at the β1- and β3-loop and the loop lengths. The most successful candidates were then further tested for antibody binding properties in a binding ELISA setup (with this experiment the influence of various structural parameters were investigated, such as the linkage between the constrained loops, the position of the scaffold, loop lengths and the structural organization of loops induced by disulfide bonds). Ultimately, the mimics were tested in competitive ELISA experiments to exclude the influence of surface immobilization effects on the binding of the mimics and to verify if the results of the previous experiments apply in solution.
Previous studies showed that a CLIPS mimic of the β3-loop could generate polyclonal antisera with strong cross-reactivity in ELISA to native hFSH and neutralizing activity in the Y1-cell assay. To further investigate if double-CLIPS technology can be even more efficient, a CLIPS-constrained mimic containing the β3- and β1-loop in close proximity by a disulfide bond was tested. Peptide array screening studies were able to demonstrate the importance of the β1-loop in the mimic,
which was observed to bind more efficient even at low antibody concentrations (10-100 ng/ml). Studies in binding ELISA showed that both loops are important for binding on the native receptor, however, the IC50
values (concentration of inhibitor producing 50 per cent inhibition, i.e. it shows the effectiveness of a compound in inhibiting biological or biochemical function) (152) obtained were not near the native protein values, indicating that more information must be included in an hFSH mimic or a different loop than β1 must be the key for sufficient competition with the native protein (such as α2-loop).Next, double-CLIPS technology was applied towards hCG mimicry. Based on the findings for hFSH, which is consider to be a ‘sister’ protein, they investigated a certain number of mimics. They observed that β1-loop was essential for binding on the receptor (via peptide screening studies) and the loop lengths were important for successful binding, since mimics with longer
loops showed enhanced competition with the native protein in competitive ELISA studies. Finally, sheep immunization studies with the best hCG CLIPS-mimic did not show significant cross-reactivity with hCG-receptor, which was attributed to incorrect overall topology of the loops within the mimic. A possible solution could be utilizing longer peptide loops constrained with an extra disulfide bond, or by alternating the already developed mimic.The third and final test was to use double-CLIPS mimics for improving an already existing and FDA approved epitope mimic, the bevacizumab. The findings were promising for double-CLIPS technology,
42 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Figure 4.11: The human Chorionic Gonadotropin Beta (hCG-β) (169).
Figure 4.22: the human Vascular Endothelial Growth Factor (hVEGF) (170).

nevertheless, more investigation is required for manufacturing effective protein surface mimics which eventually will find their way to the drug market.
All in all, CLIPS technology is a novel tool towards developing compounds bearing functionalities that can only be found to nature. Moreover, its capabilities can be seen if one only considers how young this method is.
4.4Other Multifunctional Scaffolds. Though several different scaffolds are reported in the recent literature, due to insufficient data a comprehensive references cannot be given. Therefore, a small reference to most interesting scaffolds will be handed here.
To start with, a trifunctional scaffold bearing three completely orthogonal reactive groups was presented by Clavè et al. (153). Though, their synthesis was mostly for different biomolecules than proteins or peptides, in the communication they mentioned the possibility of introducing peptides as well as with other biomolecules. The scaffold or tripod, as the authors named it, bears an azido group for Staudinger ligation or CuAAC/SPAAC, oxime group for oxime ligation and a cysteine residue for basically thiol addition (also native chemical ligation approach could be possible).
Figure 4.13: Scaffold with three orthogonal functional groups.
For testing the bioorthogonality of the tripod, a reliable and high yielding sequence of reaction had to be chosen careful. So, the reaction scheme followed was namely, thiol alkylation through Michael addition or SN2 reaction, oxime ligation (these two reactions do not interfere one another) and finally the famous “Click reaction”. Moreover, they selected “wet chemistry” (chemistry done in liquid phase excluding quantitative analytical chemistry done instrumentally) with the mentioned sequence due to the fact that, sulfhydryl groups are oxidatively damaged by copper and sodium ascorbate, the main reagents for generating the active copper(I) catalyst essential for the click reaction, especially with the generation of reactive oxygen species, and copper salts are capable of catalyzing the N-O bond cleavage and hence, lead to degradation of the free aminooxy moieties.
To illustrate the possibility of utilizing a trifunctional scaffold in this way, ligation of fluorescent organic dyes, or FRET cassette, bio-probes and immunosensors were selected. The group picked molecules that are relatively sensitive to work with (they mentioned even more sensitive than peptides) to prove the applicability of their approach towards bio-conjugation. So, a decision was made and the mycotoxin AFB2 was selected, as a previous attempt lead to failure for conjugating the AFB2 to a protected analog
43 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

of the current tripod, due to highly nucleophile-sensitive lactone constituted in the AFB2 molecule to the deprotection conditions.
First, the free thiol was reacted with a fluorescent compound bearing an iodine residue (SN2). The reaction was found to be complete within 1 hour and purification by PR-HPLC gave quantitative yield. Then, oxime ligation yielded the desired product exclusively after 3 hours (the product structures confirmed by ESI mass spectrometry). Finally, the mycotoxin was added via CuAAC route and the final product was obtained bearing the AFB2 moiety with no alternations on the cyanine and xanthene dyes. Therefore, the group demonstrated successfully the applicability of a trifunctional scaffold which can be used for labelling studies (however, may be with small deviations can be applied to other areas also). The reaction scheme they have followed was efficient and reproducible and resulted in product with high wield. Nevertheless, more research must be done for speeding up the procedure and if its application can be as wide as mentioned in the communication.
Oxime ligation is consider to be among the best ligation ways used for bio-conjugation of biomolecules. However, it suffers of long waiting times, often reagents in excess are required and depends on pH (154; 155; 156). An alternative approach which is fast and reliable is the copper catalyzed dipolar cycloaddition or CuAAC. Yet, this also has the problems associated with an active copper(I) catalyst (see above). A novel free of catalysts azide-alkyne cycloaddition was introduced by the Bertozzi group (157).This relies on strain to “catalyze” the reaction, as in the case of the SPAAC reaction, though, instead of preparing a cyclooctyne (which is a hard synthesis) they employed a commercially obtainable dibenzoazacyclooctyne or DIBAC for labelling procedures and for protein fusion. So, the Bertozzi group constructed four different scaffolds all bearing two different functionalities; one aminooxy group and an azide or a DIBAC group, as seen in figure below.
Figure 4.15: Scaffolds used in Strain-Promoted cycloaddition (SPAAC).
The labeling experiments showed good results except of the linker 2, which was the least efficient labelling reagent. To exclude the possibility of side reaction with free thiols (noted with other cyclooctynes) a test reaction of the DIBAC-488 with aldehyde-tagged human serum albumin (HAS), containing a cysteine residue, gave no significant labeling, thus, providing sufficient evidence of no side reactions of the DIBAC reagent with free thiol groups (158; 159).To further test the applicability of this procedure, they applied the method on protein-peptide conjugations by conjugating full-length hIgG protein with hGH or MBP protein (the products are relevant
44 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

to increase of the serum halflife of protein therapeutics, hGH-hIgG) (160; 161). To do so, a longer bifunctional linker was constructed bearing an aminooxy moiety and the DIBAC group. Then, they treated both 1 and 4 with hGH, MBP and hIgG separately, then subsequently with the labeling compounds, to observe that the oxime-conjugated proteins were efficiently labeled with dye and formed the expected homo and heterodimers.
All in all, the Bertozzi group demonstrated a sufficient Cu-free click approach for conjugating protein and peptides of unprecedented size and complexity. The procedure chosen by the group opens new avenues for constructing possible drugs even in large scale (experiments made in large scale show that is applicable).
An article published this year, describes a trifunctional scaffold bearing completely orthogonal functionalities. As the other groups, the presented herein, scaffold has been constructed for oxime ligation, thiol addition and CuAAC or Staudinger ligation. This is a benzene-based molecule built up in an efficient six step procedure. Again, labeling experiments were conducted that display the versatile character of the three most widely used bioorthogonal groups; the azido, thiol and aminooxy group. Their results were relatively good showing the applicability of such a scaffold to a variety of areas.
Beal and co-workers (162) based on the work of Aucagne (163) and Kele (164), on sequential CuAAC reactions and combination of both CuAAC and SPAAC, respectively, devised a trifanctional scaffold capable for bio-conjugation in one-pot synthesis. Moreover, a cysteine could be used as the third functional moiety, however, due to lack of suitable commercial starting materials and concerns over transfer of the acyl group from sulfur to the β-amino functionality, as observed in NCL, alternatives to cysteine were chosen, such as aspartic acid, glutamic acid and lysine.The use of a cylcooctyne to promote 1,3-dipolar addition was expected to result into mixtures of regioisomers, however, a successful reaction even with unwanted products would be enough proof for further studies. Moreover, the authors speculated that the resulted isomers could be more likely to adopt a closer conformation to the native protein. Initial experiments with low template concentrations showed slow conversion (24 h and 89% conversion) with both starting materials remaining after completion. Increase in concentrations (from 1 to 10 mg/ml), helped the reaction to complete within 6 h. Subsequent CuAAC reaction was successful under classic “click” condition after 2 h and required no optimization. To validate the applicability of the template and the subsequent strain- and catalyzed promoted dipolar reactions with complex biomolecules, trial reactions with different peptides were investigated, such as the cyclic RGD peptide, the DAVE-N3 and the FAKL-N3 peptides. Furthermore, to demonstrate potential application in the synthesis of immunogens, a commercial maleimide-activated BSA was employed as starting material.
45 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

The tag experiments revealed that various functional groups are tolerated by the template and the conditions required for the syntheses, though, some isomers were believed that have been also produced during the reactions.The results lead to the conclusion that SPAAC and CuAAC are completely orthogonal to one another and no by-products result from a one-pot synthesis using both reactions. But, due to several by-products observed, optimization of the approach is necessary.
In a communication by Yim and co-workers (165), a series of dimeric [Tyr3]octreotate peptides, with different spacers were synthesized through click chemistry to evaluate the binding on somatostatin receptors (SSTRs). The compounds under investigation were ligated via CuAAC reaction on a benzene-based molecular scaffold bearing two alkyne groups.
Firstly, the different [Tyr3]octreotate analogues were evaluated in competitive binding experiments, which pointed towards the largest dimer.
Next, the strongest binder (i.e. the largest dimer) along with the smallest dimer and the monomeric analog were selected for further DOTA-functonalization, radiolabeling and in vivo evaluation. The DOTA molecule was anchored by thio acid/sulfonylazide amidation or “sulfo click” reaction.
46 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.
Scheme 4.8: Sequential bioconjugation on Click-enabled template.
Figure 4.16: Molecular structure of the dimeric alkyne building block.

The dimer with the longest spacer between the two [Tyr3]octreotate peptides was found to result in unfavorable tumor accumulation, which was ascribed to a combination of rapid wash-out from circulation, high kidney accumulation and a relatively internalization rate. Whereas, the shortest dimer showed activity in the tumor after 4 h post-injection.The observed results indicate the importance of small differences in similar compounds on tumor targeting and pharmacokinetics (relevant for therapeutic drugs as well).
5.
47 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

5. Summary & Outlook This project aims to describe synthetic organic molecules that have been used for binding biomolecules, mainly peptides and proteins, with the ultimate goal of mimicking nature. Chemists usually refer to this molecules as scaffolds or templates.
An organic molecule is a good candidate as central scaffold if is stable and inert to peptides, affects the stability of the peptides bound on it in a way that further stabilize them, bears orthogonal functional groups (thus only specific reactions can proceed), no protective groups are necessary, the synthesis of both the scaffold and the final mimic are feasible in mild conditions (no degradation of peptides or side reaction are involved in the procedure), the reagents used preferably are not toxic, the scaffolds are water-soluble and lastly, the induced conformation is the desirable. Though, most of the above requirements are universally important for manufacturing efficient protein surface mimics, yet, others are of minor importance. All these facts will be used for evaluating the scaffolds mentioned in Chapter 4.
At this point I would like to clarify that in every single case mentioned herein (Chapter 4 as well) protection/deprotection schemes were avoided and exclusively orthogonal reactions were chosen. Moreover, scaffolds which have been mentioned but the information are inadequate for comparison will be left out or will be mentioned where is appropriate.
Figure 5.1: Scaffolds that have been used widely in the literature.
The most exploited scaffolds in the literature are three; the triazacyclophane (TAC)-scaffold, the cyclic peptides or TASP/RAFT scaffolds and the CLIPS-scaffolds. These three have been tried out in
48 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

several experiments as mimics and have been found success in some of them.
In almost every case the mimic that gave the best results, bore the more rigid structure (59). For example the experiment conducted by the Liskamp group, where four scaffolds (including the TAC-scaffold) were tested after synthesizing a library of mimics, showed that most successful was a construct that had less degrees of freedom. The same result was found by Smeenk and co-workers for CLIPS-mimics; the mimic which was fashioned to be less resilient yielded the best results.
Scaffolds that are constructed of amino acids have the advantage of being already friendly to aqueous solutions. It has been proved long ago before convergent procedures were introduced in synthesis of protein mimics that cyclopeptide-based mimics can be used well as backbones for mimicking protein surfaces. Galibert and co-workers by introducing three orthogonal functional groups in the cyclic decapeptide scaffold showed the versatile character of this construct. They were able to introduce three different molecules in one-pot synthesis by only changing the conditions.
CLIPS-technology, on the other hand, has the advantages of easily accessible starting materials, i.e. the scaffold is synthesized in only few simple steps with good yields (71% for oS2-CHO, 97% for oS2-ONH2). The oxime ligation reaction is a facile and robust route towards mimic which proceeds in mild conditions (100 Mm citric acid/aniline Buffer, room temperature) with high yields and conversions that exceeded 99% in all the examples. Moreover, double-CLIPS methods have proved to increase the efficacy of the mimics, mainly due to larger coverage of a protein surfaces, i.e. greater input of information into a mimic. Additionally, disulfide bonds which do not interfere with the other reactions, yield even more constrained molecules that can bind easier on a receptor, if the protein binding site is already known, otherwise this might result in some problems since certain proteins are believed to change, on some degree, their structure when they interact with other proteins (166). However, if our knowledge in PPIs goes beyond a satisfactory level then double-CLIPS technology will be one of the most powerful tools in mimicking protein functions towards synthetic drugs.
As been mentioned before, several groups in the last few years focused their research interests towards this type of chemistry; i.e. utilizing a central scaffold and click chemistry to create large constructs for mainly therapeutic purposes. Though, most of them attained good results, yet a lot of work needs to be done. Each scaffold presented here has its own advantages and disadvantages, some follow relatively easy synthetic schemes and others more complex. However, all the cases display how powerful selective reactions are and that nature can be reproduced. Synthetic chemists have as main target the development of completely selective reactions, where protective groups and complex purification protocols are unnecessary. If this is accessible then complex molecules can be synthesized in the laboratories with reliable and reproducible routes. As a result, chemistry enters a new era where everything is possible, we just need to use our most powerful weapon, the mind. After all, nature evolved in this point after millions of years of “trial and error”, one cannot assume that humans can do it after one night.
49 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

References1. A. Kessel, N. Ben-Tal. Introduction to Proteins: Structure, Function and Motion. s.l. : CRC Press , 2011.
2. Tinnefeld, Philip. 2011, Nature, Vol. 473, pp. 461-462.
3. M. W. Peczuh, A. D. Hamilton. 2000, Chem. Rev., Vol. 100, pp. 2479-2493.
4. S. Fletcher, A. D. Hamilton. 2006, J. R. Soc. Interface, Vol. 3, pp. 215-233.
5. F. J. Dekker, N. J. de Mol, J. van Ameijde, M. J. E. Fischer, R. Ruijtenbeek, F. A. M. Redegeld, R. M. J. Liskamp. 2002, Chembiochem, Vol. 3, pp. 238-242.
6. F. Lecaille, J. Kaleta, D. Bromme. 2002, Chem. Rev., Vol. 102, pp. 4459-4488.
7. Hausen, H. zur. 2002, Nat. Rev. Cancer, Vol. 2, pp. 342-350.
8. P. Harjes, E. E. Wanker. 2003, Trends Biochem. Sci., Vol. 28, pp. 425-433.
9. T. Wen, L. Zhang, S. K. P. Kung, T. J. Molina, R. G. Miller, T. W. Mak. 1995, Eur. J. Immunol., Vol. 25, pp. 3155-3159.
10. David P. Fairlie, Micheal L. West, Allan K. Wong. 1998, Curr. Med. Chem., Vol. 5, pp. 29-62.
11. K. Sugase, H. J. Dyson, P. E. Wright. 2007, Nature, Vol. 447, p. 1021.
12. J. Wang, Q. Lu,H. P. Lu. 2006, PLOS Comput. Biol., Vol. 2, p. No. e78.
13. B. A. Shoemaker, J. J. Portman, P. G. Wolynes. 2000, Pro. Natl. Acad. Sci. U.S.A, Vol. 41, p. 415.
14. Daniel F. Veber, Roger M. Freidinger. 1985, Trends Neurosci., Vol. 8, pp. 392-396.
15. N. Ferrara, K. J. Hillan, H. P. Gerber, W. Novotny. 2004, Nat. Rev. Drug. Discov., Vol. 3, pp. 391-400.
16. J. Langeveld, J. I. Casal, A. Osterhaus, E Corte,R. De Swart, C. Vela, K. Dalsgaard, W. C. Puijk, W. M. M. Schaaper, R. H. Meloen. 1994, J. Virol., Vol. 68, pp. 4506-4513.
17. T. Lehner, C. Doyle, Y. Wang, K. Babaahmady, T. Whittall, L. Tao, L. Bergmeier, C.Kelly, , 2001, 166, 7446-7455. 2001, J. Immunol., Vol. 166, pp. 7446-7455.
18. Jiri Novotny, Jürgen Bajorath. 1996, Advances in Protein Chemistry, Vol. 49, pp. 149-180.
19. Stephen J. Lippard, Jeremy M. Berg. Principles of Bioinorganic Chemistry. s.l. : University Science Books, 1994.
20. M. Zouhair Atassi, John A. Smith. 1978, Immunochemistry, Vol. 15, pp. 609-610.
50 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

21. Smeenk, L. Double-CLIPS Technlogy for the Mimicry of Structurally Complex Antibody Binding Sites on Proteins. 2013.
22. G. Köhler, C. Milstein. 1975, Nature, Vol. 256, pp. 495-497.
23. Michael A. Hooks, Catherine S. Wade, William J. Millikan. 1991, Pharmacotherapy, Vol. 11, pp. 26-37.
24. L. Velluz, J. Valls, J. Mathieu. 1967, Angew. Chem. Int. Ed. Engl, Vol. 6, pp. 778-789.
25. Hendrickson, James B. 1977, J. Am. Chem. Soc., Vol. 99, pp. 5439-5450.
26. T. Kimmerlin, D. Seebach. 2005, J. Peptide Res., Vol. 65, pp. 229-260.
27. Hosahalli P. Hemantha, Narasimhamurthy Narendra, Vommina V. Sureshbabu. 2012, Tetrahedron, Vol. 68, pp. 9491-9537.
28. Trost, Barry M. 1983, Science, Vol. 219, pp. 245-250.
29. Michal Szostak, Malcolm Spain, David J. Procter. 2013, Chem. Soc. Rev., Vol. 42, pp. 9155-9183.
30. Anouk Dirksen, Tilman M. Hackeng, Philip E. Dawson. 2006, Angew. Chem. Int. Ed., Vol. 118, pp. 7743-7746.
31. Florence M. Brunel, Philip E. Dawson. 2005, Chem. Commun., pp. 2552-2554.
32. Kieran F. Geoghegan, Justin G. Stroh. 1992, Bioconjugate Chem., Vol. 3, pp. 138-146.
33. M. Schnolzer, S. B. Kent. 1992, Science, Vol. 256, pp. 221-225.
34. www.nobelprize.org. [Online] Nobel Foundation, 2001.
35. Hartmuth C. Kolb, M. G. Finn, K. Barry Sharpless. 2001, Angew. Chem. Int. Ed., Vol. 40, pp. 2004-2021.
36. Huisgen, Rolf. 1961, Proceeding of the Chemical Society of London, p. 357.
37. Vsevolod V. Rostovtsev, Luke G. Green, Valery V. Fokin, K. Barry Sharpless. 2002, Angew. Chem. Int. Ed., Vol. 41, pp. 2596-2599.
38. Hartmuth C. Kolb, K.Barry Sharpless. 2003, Drug Discovery Today, Vol. 8, pp. 1128-1137.
39. Matthew Lukeman, Peter Wan. 2003, J. Am. Chem. Soc., Vol. 125, pp. 1164-1165.
40. icholas J. Agard, Jennifer A. Prescher, Carolyn R. Bertozzi. 2004, J. Am. Chem. Soc., Vol. 126, pp. 15046-15047.
41. D. S. Kemp, Nicholas George Galakatos. 1986, J. Org. Chem., Vol. 51, pp. 1821-1829.
51 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

42. Martina Schnölzer, Stephen B. H. Kent. 1992, Science, Vol. 256, pp. 221-225.
43. Philip E. Dawson, Tom W. Muir, Ian Clark-Lewis, Stephen B. H. Kent. 1994, Science, Vol. 266, pp. 776-779.
44. Yinfeng Zhang, Ci Xu, Hiu Yung Lam, Chi Lung Lee, Xuechen Li. 2013, Proc. Natl. Acad. Sci. U.S.A., Vol. 110, pp. 6657-6662.
45. Peter Timmerman, Joris Beld, Wouter C. Puijk, Rob H. Meloen. 2005, ChemBioChem, Vol. 6, pp. 821-824.
46. Linde E. J. Smeenk, Nicolas Dailly, Henk Hiemstra, Jan H. van Maarseveen, Peter Timmerman. 2012, Org. Lett., Vol. 14, pp. 1194-1197.
47. George Barany, Robert B. Merrifield. 1977, J. Am. Chem. Soc., Vol. 99, pp. 7363-7365.
48. Chun-Ho Wong, Steven C. Zimmerman. 2013, Chem. Commun., Vol. 49, pp. 1679-1696.
49. R. A. Gossage, E. Munoz-Martinez, H. Frey, A. Burgath, M. Lutz, A. L. Spek, G. van Koten. 1995, Chem. Eur. J., Vol. 5, pp. 2191-2197.
50. Anderer, F. A. 1963, Adv. Protein Chem., Vol. 18, pp. 1-35.
51. F. Audibert, M. Jolivet, L. Chedid, J. Alouf, P. Boquet, P. Rivaille, O. Siffert. 1981, Nature, Vol. 289, pp. 593-594.
52. E. H. Beachy, J. M. Seyer, J. B.Dale, W. A. Simpson, A. H. Kang. 1981, Nature, Vol. 292, pp. 457-459.
53. R. Acharya, E. Fry, D. Stuart, G. Fox, D. Rowlands, F. Brown. 1989, Nature, Vol. 337, pp. 709-716.
54. J. L. Brittle, R. A. Houghten, H. Alexander, T. M. Shinnick, J. G. Sutcliffe, R. A. Lerner, D. J. Rowlands, F. Brown. 1982, Nature, Vol. 298, pp. 30-33.
55. R. Dimarchi, G. Brooke, C. Gale, V. Cracknell, T. Doel, N. Mowat. 1986, Science, Vol. 232, pp. 639-641.
56. A. Meola, P. Delmastro, P. Monaci, A. Luzzago, A. Nicosia, F. Felici, R. Cortese, G. Galfre. 1995, J. Immunol., Vol. 154, pp. 3162-3172.
57. Talwar, G. P. 1997, Immunol. Cell Biol., Vol. 75, pp. 184-189.
58. P. Sarobe, C. Pendleton, T. Akatsuka, D. Lau, V. Engelhard, S. Feinstone, J. Berzofsky. 1998, J. Clin. Invest., Vol. 102, pp. 1239-1248.
59. Daniel F. Veber, Robert G. Strachan , Susan J. Bergstrand , Frederick W. Holly , Carl F. Homnick , Ralph Hirschmann , Mary L. Torchiana , Richard Saperstein. 1976, J. Am. Chem. Soc., Vol. 98, pp. 2367-2369.
52 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

60. Daniel F. Veber, Frederick W. Holly, Ruth F. Nutt, Susan J. Bergstrand , Stephen J. Bergstrand , Stephen F. Brady, Ralph Hirschmann , Monroe S. Glitzer, Richard Saperstein. 1979, Nature, Vol. 280, pp. 512-514.
61. Stickle, D. F., L. G. Presta, K. A. Dill, and G. D. Rose. 1992, J. Mol. Biol., Vol. 226, pp. 1143-1159.
62. Thomas A. Edwards, Andrew J. Wilson. 2011, Amino Acids, Vol. 41, pp. 743-754.
63. Tomikazu Sasaki, Emil T. Kaiser. 1989, J. Am. Chem. Soc., Vol. 111, pp. 381-383.
64. Karin S. Ikkerfeldt, Ronald M. Kim, Daniel Camac, John T. Groves, Jim D. Lear, William F. DeGrado. 1992, J. Am. Chme. Soc., Vol. 114, pp. 9656-9657.
65. Kemp, Daniel S. 1990, Trends Biotechnol., Vol. 8, pp. 249-255.
66. Christian E. Schafmeister, Julia Po, Gregory L. Verdine. 2000, J. Am. Chem. Soc., Vol. 122, pp. 5891-5892.
67. L. D.Walwnsky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine, S. J. Korsmeyer. 2004, Science, Vol. 305, pp. 1466-1470.
68. F. Bernal, A. F. Tyler, L. D. Walensky, G. L. Verdine. 2007, J. Am. Chem. Soc., Vol. 129, p. 2456.
69. M. R. Ghadiri, A. K. Fernholz. 1990, J. Am. Chem. Soc., Vol. 112, pp. 9633-9635.
70. D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh, P. G. Shcultz. 1991, J. Am. Chem. Soc., Vol. 113, pp. 9391-9392.
71. S. Marquese, R. L. Baldwin. 1987, Proc. Natl. Acad. Sci. U.S.A., Vol. 84, pp. 8898-8902.
72. C. Bracken, J. Gulyas, J. W. Taylor, J. Baum. 1994, J. Am. Chem. Soc., Vol. 116, pp. 6431-6432.
73. M. Chorev, E. Roubini, R. L. McKee, S. W. Gibbons, M. E. Goldman, M. P.Caulifield, M Rosenblatt. 1991, Biochemistry, Vol. 30, pp. 5968-5974.
74. Chongwoo A. Kim, Jeremy M. Berg. 1993, Nature, Vol. 362, pp. 267-270.
75. Daniel L. Minor Jr, Peter S. Kim. 1994, Nature, Vol. 367, pp. 660-663.
76. Daniel L. Minor Jr, Peter S. Kim. 1994, Nature, Vol. 371, pp. 264-267.
77. H.Jane Dyson, James R. Sayre, Gene Merutka, Hang-Cheol Shin, Richard A. Lerner, Peter E. Wright. 1992, J. Mol. Biol., Vol. 226, pp. 819-835.
78. Yuji Goto, Kozo Hamaguchi. 1982, J. Mol. Biol., Vol. 156, pp. 891-910.
79. Sakae Tsuda, Kenji Ogura , Yasushi Hasegawa , Koichi Yagi , Kunio Hikichi. 1990, Biochemistry, Vol. 29, pp. 4951-4958.
53 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

80. P. Varley, A. M. Gronenborn, H. Christensen, P. T. Wingfield, R. H. Pain, G. M. Clore. 1993, Science, Vol. 260, pp. 1110-1113.
81. Roger M. Freidinger, Daniel F. Veber, Debra Schwenk Perlow, Jerry R. Brooks Richard Saperstein. 1980, Science, Vol. 210, pp. 656-658.
82. K. Sato, M. Hott, M. H. Dong, H. Y. Hu, J. P. Taulene, M. Goodman, U. Nagai, N. Lingi. 1991, Int. J. Pept. Protein Res., Vol. 38, pp. 340-345.
83. Feigel, M. 1986, J. Am. Chem. Soc., Vol. 108, pp. 181-182.
84. M. Kahn, S. Wilke, B. Chen, K. Fujita. 1988, J. Am. Chem. Soc., Vol. 110, pp. 1638-1639.
85. M. Kahn, S. Wilke, B. Chen, K. Fujita, Y. H. Lee, M. E. Johnson. 1988, J. Mol. Recognit., Vol. 1, pp. 75-79.
86. W. C. Ripka, G. V. De Lucca, A. C. Bach, R. S. Pottorf, J. M. Blaney. 1993, Tetrahedron, Vol. 49, pp. 3609-3628.
87. W. C. Ripka, G. V. De Lucca, A. C. Bach, R. S. Pottorf, J. M. Blaney. 1993, Tetrahedron, Vol. 49, pp. 3593-3608.
88. Daniel S. Kemp, Benjamin R. Bowen. 1988, Tetrahedron Lett., Vol. 29, pp. 5077-5080.
89. H. Diaz, J. W. Kelly. 1991, Tetrahedron Lett., Vol. 32, pp. 5725-5728.
90. Joel P. Schneider, Jeffety W. Kelly. 1995, Chem. Rev., Vol. 95, pp. 2169-2187.
91. S. van der Werf, J. P. Briand, S. Plaue J. Burckard, M. Girard, M. H. V. Van Regenmorte. 1994, Res. Virol., Vol. 145, pp. 349-359.
92. Gordon R. Dreesman, Yanuario Sanchez, Irina Ionescu-Matiu, James T. Sparrow, Howard R. Six, Darrell L. Peterson, F. Blaine Hollinger, Joseph L. Melnick. 1982, Nature, Vol. 295, pp. 158-160.
93. M. Ferguson, D.M.A. Evans, D.I. Magrath, P.D. Minor, J.W. Almond, G.C. Schild. 1985, Virology, Vol. 143, pp. 505-515.
94. Steven W. Millward, Stephen Fiacco, Ryan J. Austin, Richard W. Roberts. 2007, ACS Chem. Biol., Vol. 2, pp. 625-634.
95. Ramakanth Sarabu, Kathleen Lovey, Vincent S. Madison, David C. Fry, David N. Greeley, Charles M. Cook, Gary L. Olson. 1993, Tetrahedron, Vol. 49, pp. 3629-3640.
96. M. Mutter, S. Vuilleumier. 1989, Angew. Chem. Int. Edit. , Vol. 28, pp. 535-554.
97. Stephane Peluso, Pascal Dumy, Ian M. Eggleston, Patrick Garrouste, Manfred Mutter. 1997, Tetrahedron, Vol. 53, pp. 7231-7236.
54 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

98. C. J. White, A. K. Yudin. 2011, Nat. Chem., Vol. 3, pp. 509-524.
99. J. F. Reichwein. B. Wels, J. A. Kruijtzer, C. Versluis, R. M. Liskamp. 1999, Angew. Chem. Int. Ed., Vol. 38, pp. 3684-3687.
100. J. Illesinghe, C. X. Guo, R. Garland, A. Ahmed, B. van Lierop, J. Elaridi, W. R. Jacksonb, A. J. Robinson. 3, 2009, Chem. Commun., pp. 295-297.
101. V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra, J. H. van Maarseveen. 2006, Org. Lett., Vol. 8, pp. 919-922.
102. L. Jin, B. M. Fendly, J. A. Wells. 1992, J. Mol. Biol., Vol. 226, pp. 851-865.
103. D.J. Barlow, M.S. Edwards, J.M. Thornton,. 1986, Nature, Vol. 322, pp. 747-748.
104. Harald K. Rau, Wolfgang Haehnel. 1998, J. Am. Chem. Soc., Vol. 120, pp. 468-476.
105. Eric Grouzmann, Thierry Buclin, Maria Martire, Clara Cannizzaro, Barbara Dörner, Alain Razaname, Manfred Mutter. 1997, J. Biol. Chem., Vol. 272, pp. 7699-7706.
106. Till Opatz, Rob M. J. Liskamp. 2001, Org. Lett., Vol. 3, pp. 3499-3502.
107. Dirk-Jan van Zoelen, Maarten R. Egmond, Roland J. Pieters, Rob M. J. Liskamp. 2007, ChemBiochem, Vol. 8, pp. 1950-1956.
108. Till Opatz, Rob M. J. Liskamp. 2002, J. Comb. Chem., Vol. 4, pp. 275-284.
109. Marcel Hijnena, Dirk J. van Zoelenc, Cristina Chamorroc, Pieter van Gageldonka, Frits R. Mooia, Guy Berbersa, Rob M.J. Liskamp. 2007, Vaccine, Vol. 25, pp. 6807-6817.
110. H. Bauke Albada, Fouad Soulimani, Bert M. Weckhuysen, Rob M. J. Liskamp. 46, 2007, Chem. Commun., pp. 4895-4897.
111. H. Bauke Albada, F. Soulimani, H. J. F. Jacobs, C. Versluis, B. M. Weckhuysenb, R. M. J. Liskamp. 2012, Org. Biomol. Chem. , Vol. 10, pp. 1088-1092.
112. Y. Singh, M. J. Stoermer, A. J. Lucke, T. Guthrie, D. P. Fairlie. 2005, J. Am. Chem. Soc., Vol. 127, pp. 6563-6572.
113. Yangbo Feng, Giuseppe Melacini, Joseph P. Taulane, Murray Goodman. 1996, J. Am. Chem. Soc., Vol. 118, pp. 10351-10358.
114. Erik T. Rump, Dirk T. S. Rijkers, Hans W. Hilbers, Philip G. de Groot, Rob M. J. Liskamp. 2002, Chem. Eur. J., Vol. 8, pp. 4613-4621.
115. Yoshitomo Hamuro, Mercedes Crego Calama, Hyung Soon Park, Andrew D. Hamilton. 1997, Angew. Chem. Int. Ed., Vol. 36, pp. 2680-2683.
55 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

116. Alberto Marra, Alessandro Dondoni, Francesco Sansone. 1996, J. Org. Chem., Vol. 61, pp. 5155-5158.
117. Corrada Geraci, Grazia M. L. Consoli, Eva Galante, Ennio Bousquet, Maria Pappalardo, Angelo Spadaro. 2008, Bioconjugate Chem., Vol. 19, pp. 751-758.
118. Ralph Hirschmann, K. C. Nicolaou , Sherrie Pietranico , Ellen M. Leahy , Joseph Salvino , Byron Arison , Maria A. Cichy , P. Grant Spoors , William C. Shakespeare. 1993, J. Am. Chem. Soc., Vol. 115, pp. 12550-12568.
119. Raimo Franke, Christian Doll, Victor Wray, Jutta Eichler. 2004, Org. Biomol. Chem., Vol. 2, pp. 2847-2851.
120. Raimo Franke, Tatjana Hirsch, Heike Overwin, Jutta Eichler Priv.-Doz. 2007, Angew. Chem. Int. Ed., Vol. 46, pp. 1253-1255.
121. Partha S. Ghosh, Andrew D. Hamilton. 2012, J. Am. Chem. Soc., Vol. 134, pp. 13208-13211.
122. Brooke N. Bullock, Andrea L. Joachim, Paramjit S. Arora. 2011, J. Am. Chem. Soc., Vol. 133, pp. 14220-14223.
123. Tomlinson, Ian M. 2004, Nat. Biotech., Vol. 22, pp. 521-522.
124. Bronowski, Jacob. 1956, Higher Education Quarterly, Vol. 11, pp. 26-42.
125. Zoelen, Dirk-Johan van. Mimicry of Discontinuous Epitopes Employing the Triazacyclophane (TAC) Scaffold for Modulation of Protein-Protein Interactions. Utrecht : University of Utrecht, 2008.
126. Gwenn E. Mulder, John A. W. Kruijtzer, Rob M. J. Liskamp. 2012, ChemComm, Vol. 48, pp. 10007-10009.
127. Peter D. Kwong, Richard Wyatt, James Robinson, Raymond W. Sweet, Joseph Sodroski, Wayne A. Hendrickson. 1998, Nature, Vol. 393, pp. 648-659.
128. Cristina Chamorro, John A. W. Kruijtzer, Marina Farsaraki, Jan Balzarini, Rob M. J. Liskamp. 2009, Chem. Commun. , pp. 821-823.
129. Raimo Franke, Tatjana Hirsch, Heike Overwin, Jutta Eichler. 2007, Angew. Chem. Int. Ed., Vol. 46, pp. 1253-1255.
130. Gwenn E. Mulder, H (Linda). C. Quarles van Ufford, Jeroen van Ameijde,Arwin J. Brouwer, John A. W. Kruijtzer, Rob M. J. Liskamp. 2013, Org. Biomol. Chem., Vol. 11, pp. 2676-2684.
131. Choon Young Lee, Rich Held , Ajit Sharma , Rom Baral , Cyprien Nanah , Dan Dumas , Shannon Jenkins , Samik Upadhaya , Wenjun Du. 2013, J. Org. Chem., Vol. 78, pp. 11221-11228.
56 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

132. Prasad Appukkuttan, Wim Dehaen , Valery V. Fokin , Erik Van der Eycken. 2004, Org. Lett., Vol. 6, pp. 4223-4225.
133. Tuchscherer, Gabriel. 1993, Tetrahedron Lett., Vol. 34, pp. 8419-8422.
134. Manfred Mutter, Pascal Dumy, Patrick Garrouste, Christian Lehmann, Marc Mathieu, Cristina Peggion, Stephane Peluso, Alain Razaname, Gabriele Tuchscherer. 1996, Angew. Chem. Int. Ed., Vol. 35, pp. 1481-1485.
135. Shiroh Futaki, Mika Aoki, Tomoko Ishikawa, Fusayo Kondo, Tomoko Asahara, Mineo Niwa, Yutaka Nakaya, Takeshi Yagamica, Kouki Kitagawa. 1999, Bioorg. Med. Chem., Vol. 7, pp. 187-192.
136. Sophie Plé, Mélanie Figuet, Pascal Dumy. 2005, C. R. Chimie, Vol. 8, pp. 833-839.
137. Philip E. Dawson, Stephen B. H. Kent. 1993, J. Am. Chem. Soc., Vol. 115, pp. 7263-7266.
138. Mathieu Galibert, Olivier Renaudet, Pascal Dumy, Didier Boturyn. 2011, Angew. Chem. Int. Ed., Vol. 50, pp. 1901-1904.
139. Peter Timmerman, Wouter C. Puijk, Rob H. Meloen. 2007, J. Mol. Recognit., Vol. 20, pp. 283-299.
140. Florian P. Seebeck, Jack W. Szostak. 2006, J. Am. Chem. Soc., Vol. 128, pp. 7150-7151.
141. Nobelprize.org. http://www.nobelprize.org/nobel_prizes/medicine/laureates/2009/szostak-facts.html. [Online] 2013.
142. Gunther Hennrich, Vincent M. Lynch, Eric V. Anslyn. 2001, Chem. Commun. , pp. 2436-2437.
143. Gajanan K. Dewkar, Pedro B. Carneiro, Matthew C. T. Hartman. 2009, Org. Lett., Vol. 11, pp. 4708-4711.
144. Christian Heinis, Trevor Rutherford, Stephan Freund, Greg Winte. 2009, Nat. Chem. Biol., Vol. 5, pp. 502-507.
145. Alessandro Angelini, Philippe Diderich, Julia Morales-Sanfrutos, Sarah Thurnheer, David Hacker, Laure Menin, Christian Heinis. 2012, Bioconjugate Chem., Vol. 23, pp. 1856-1863.
146. Shiyu Chen, Julia Morales-Sanfrutos, Alessandro Angelini, Brian Cutting, Christian Heinis. 2012, ChemBioChem, Vol. 13, pp. 1032-1038.
147. Alessandro Angelini, Julia Morales-Sanfrutos, Philippe Diderich, Shiyu Chen, Christian Heinis. 2012, J. Med.Chem., Vol. 55, pp. 10187-10197.
148. Alessandro Angelini, Laura Cendron, Shiyu Chen, Jeremy Touati, Greg Winter, Giuseppe Zanotti, Christian Heinis. 2012, ACS Chem. Biol., Vol. 7, pp. 817-821.
57 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

149. L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore, C. J. Hawker. 2008, Macromolecules, Vol. 41, pp. 7063-7070.
150. A. Dondoni, A. Massi, P. Nanni, A. Roda. 2009, Chem. Eur. J., Vol. 15, pp. 11444-11449.
151. Hayden, Erika Check. 2009, Nature, Vol. 458, pp. 686-687.
152. Cheng Yung-Chi. 1973, Biochem. Pharma., Vol. 22, pp. 3099-3108.
153. Guillaume Clave, Herve Volland, Melanie Flaender, Didier Gasparutto, Anthony Romieu, Pierre-Yves Renard. 2010, Org. Biomol. Chem., Vol. 8, pp. 4329-4335.
154. I. P. Decostaire, D. Lelievre, H. Zhang, A. F. Delmas. 2006, Tetrahedron Lett., Vol. 47, p. 7057.
155. J. M. Chalker, G. J. L. Bernardes, B. G. Davis. 2011, Acc. Chem. Res., Vol. 44, pp. 730-741.
156. R. K. V. Lim, Q. Lin. 2010, Chem. Comm., Vol. 46, p. 1589.
157. Jason E. Hudak, Robyn M. Barfield, Gregory W. de Hart, Patricia Grob, Eva Nogales, Carolyn R. Bertozzi, David Rabuka. 2012, Angew. Chem. Int. Ed., Vol. 51, pp. 4161-4165.
158. Mauro Lo Conte, Samuele Staderini, Alberto Marra, Macarena Sanchez-Navarro, Benjamin G. Davis, Alessandro Dondoni. 2011, Chem. Commun., Vol. 47, pp. 11086-11088.
159. Benjamin D. Fairbanks, Evan A. Sims, Kristi S. Anseth, Christopher N. Bowman. 2010, Macromolecules, Vol. 43, pp. 4113-4119.
160. Ian R Wilkinson, Eric Ferrandis, Peter J Artymiuk, Marc Teillot, Chantal Soulard, Caroline Touvay, Sarbendra L Pradhananga, Sue Justice, Zida Wu, Kin C Leung, Christian J Strasburger, Jon R Sayers, Richard J Ross. 2007, Nat. Med., Vol. 13, pp. 1108-1113.
161. Blaire L Osborn, Les Sekut, Marta Corcoran, Carol Poortman, Bonnie Sturm, Guoxian Chen, Donald Mather, Hsiu Ling Lin, Tom J Parry. 2002, Eur. J. Pharmacol., Vol. 456, pp. 149-158.
162. David M. Beal, Victoria E. Albrow, George Burslem, Louisa Hitchen, Carla Fernandes, Cris Lapthorn, Lee R. Roberts, Matthew D. Selby, Lyn H. Jones. 2012, Org. Biomol. Chem., Vol. 10, pp. 548-554.
163. Ibai E. Valverde, Agnès F. Delmas, Vincent Aucagne. 2009, Tetrahedron, Vol. 65, pp. 7597-7602.
164. Péter Kele, Gábor Mezö, Daniela Achatz and Otto S. Wolfbeis. 2009, Angew. Chem. Int. Ed., Vol. 48, pp. 344-347.
165. Cheng-Bin Yim, Annemarie Eek, Berend van der Wildt, Cees Versluis, Ingrid Dijkgraaf, Dirk T. S. Rijkers, Lieke Joosten, Otto C. Boerman, Rob M. J. Liskamp. 2011, ChemBioChem, Vol. 12, pp. 750-760.
166. Dror Tobi, Ivet Bahar. 2005, Proc. Natl. Acad. Sci. U.S.A., Vol. 102, pp. 18908-18913.
58 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.

167. Vijaya R. Pattabiraman, Jeffrey W. Bode. 2011, Nature, Vol. 480, pp. 471-479.
168. Qing R. Fan, Wayne A. Hendrickson. 2005, Nature, Vol. 433, pp. 269-277.
169. Hao Wu, Joyce W. Lustbader, Yee Liu, Robert E. Canfield, Wayne A. Hendrickson. 1994, Structure, Vol. 2, pp. 545-558.
170. Yves A. Muller, Hans W. Christinger, Bruce A. Keyt, Abraham M. de Vos. 1997, Structure, Vol. 5, pp. 1325-1338.
171. Goldsby RA, Kindt TJ, Kuby J, Osborne BA. Immunology. 5. New York : W.H. Freeman and Company, 2002.
59 | Molecular scaffolds for Protein Surface Mimicry. New Era in Pharmaceuticals.


















