
Molecular Modeling Using HPC and Gaussian: Density-Functional Theory and Noncovalent Interactions
21
Molecular Modeling Using HPC and Gaussian: Density-Functional Theory and Noncovalent Interactions Gino A. DiLabio National Institute for Nanotechnology 11421 Saskatchewan Drive Edmonton, Alberta T5T 5A6 [email protected] www.ualberta.ca/~gdilabio Westgrid Seminar Series University of Alberta
description
Molecular Modeling Using HPC and Gaussian: Density-Functional Theory and Noncovalent Interactions. Gino A. DiLabio National Institute for Nanotechnology 11421 Saskatchewan Drive Edmonton, Alberta T5T 5A6 [email protected] www.ualberta.ca/~gdilabio. Westgrid Seminar Series - PowerPoint PPT Presentation
Transcript of Molecular Modeling Using HPC and Gaussian: Density-Functional Theory and Noncovalent Interactions

Molecular Modeling Using HPC and Gaussian:Density-Functional Theory and Noncovalent Interactions
Gino A. DiLabioNational Institute for Nanotechnology11421 Saskatchewan DriveEdmonton, AlbertaT5T [email protected]/~gdilabio
Westgrid Seminar SeriesUniversity of Alberta
Feb. 5, 2014

12252–12256 PNAS September 17, 2002 vol. 99 no. 19 www.pnas.orgcgidoi10.1073pnas.192252799
Geckos have evolved one of the most versatile and effective adhesives known. The mechanism of dry adhesion in the millions of setae on the toes of geckos has been the focus of scientific studyfor over a century. We provide the first direct experimental evidence for dry adhesion of gecko setae by noncovalent interactions (also called van der Waals forces)
Gino DiLabio
You certainly care about weak interactions if you're a gecko. Gecko feet have little structures called setae which allow them to grip surfaces. There are about 14400 setae /mm2 which generate a 0.5 MN/m2 of adhesive force, which is ca. 83 psi.

Why do we care about weak interactions?
Formation of 1D and 2D organic lines on Si
Nature, 2005, 435, 658-661.

Why do we care about weak interactions?
Heavy oil/bitumen upgrading
Mackie and DiLabio, Energy and Fuels, 2010, 24, 6468-6475.

Why do we care about weak interactions?
Organic electronic material
J. Phys. Chem. C, 2010, 114. 10952.

How do we model these systems?
• Large systems necessitate the use of density-functional theory (DFT)
• B3LYP was one of the first hybrid DFT that was implemented in most computational chemistry programs. It has since found general acceptance and use and works well for thermochemistry.
• However, most conventional DFT methods, including B3LYP, cannot accurately treat weak, non-covalently bonded systems – specifically “dispersion” interactions.

How bad are conventional DFT methods for weak interactions?
Chem. Phys. Lett. 2006, 419, 333-339
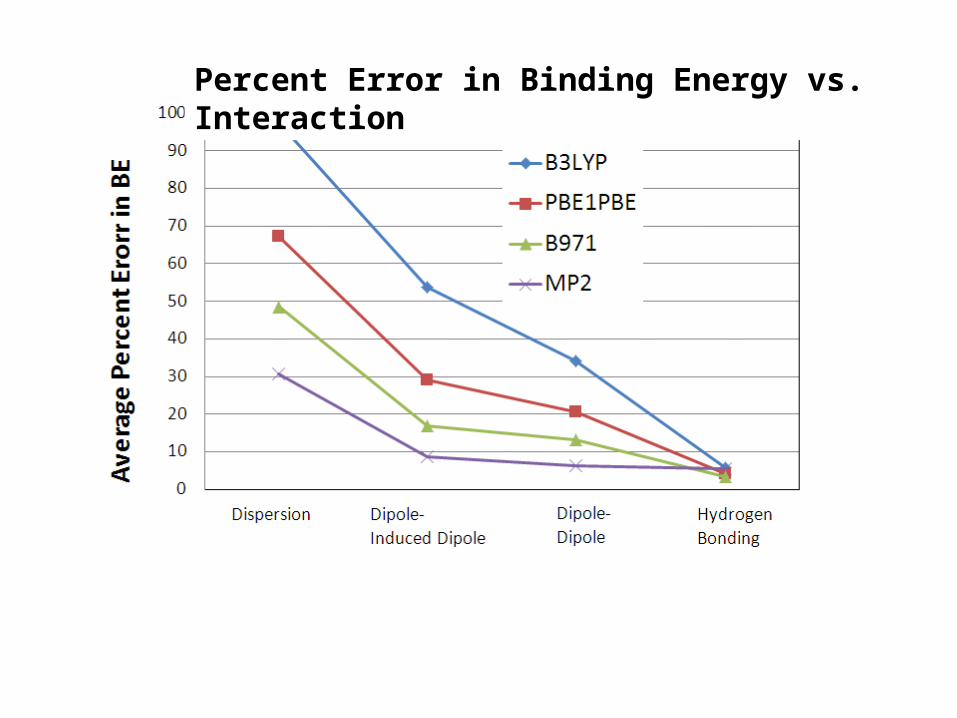
Percent Error in Binding Energy vs. Interaction

•DFT-D/D3: Add-on to the DFT energy an empirical van der Waals term. Developed for use with many functionals. (Grimme, J. Chem. Phys. 2010, 132, 154104)
•M06-2X: A DFT method parameterized to reproduce dispersion binding, among other properties. (Zhao and Truhlar, Theor. Chim. Acta, 2008, 120, 215.)
•DCP: Dispersion correcting potentials correct the erroneous noncovalent behaviour of a small number of DFT methods. (Torres and DiLabio, J. Phys. Chem. Lett. 2012, 3, 1738) – Compute Canada RAC supported work.
Improving the performance of DFT methods: Dispersion-corrected DFT in Gaussian
Review: J. Phys. Org. Chem. 2009, 22, 1127-1135.See also: 2014 version of Reviews in Computational Chemistry
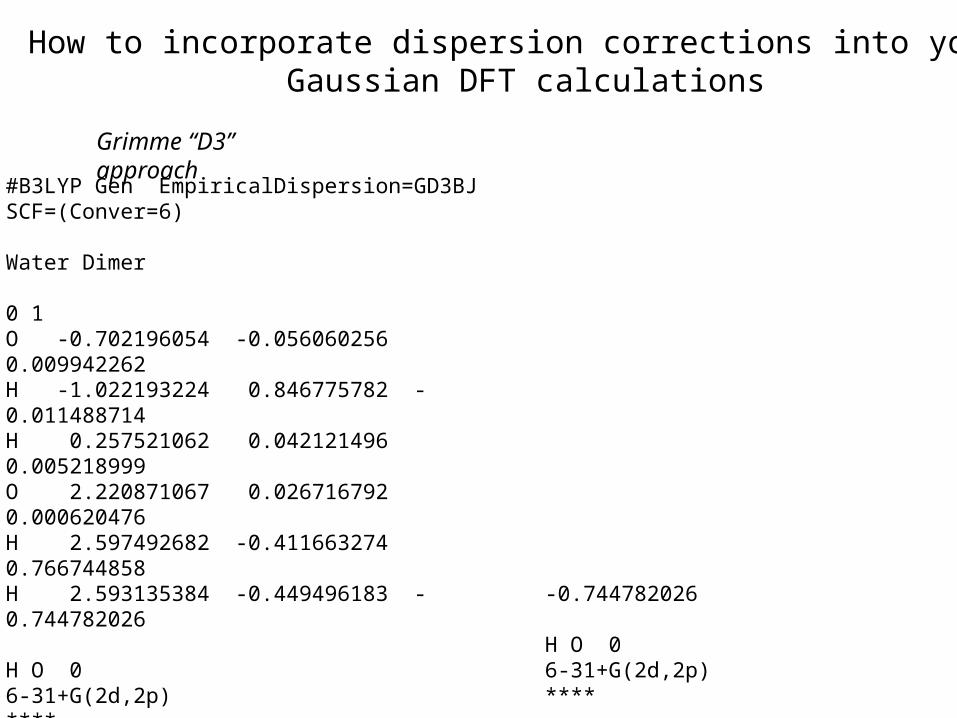
How to incorporate dispersion corrections into your Gaussian DFT calculations
#B3LYP Gen EmpiricalDispersion=GD3BJ SCF=(Conver=6)
Water Dimer
0 1O -0.702196054 -0.056060256 0.009942262H -1.022193224 0.846775782 -0.011488714H 0.257521062 0.042121496 0.005218999O 2.220871067 0.026716792 0.000620476H 2.597492682 -0.411663274 0.766744858H 2.593135384 -0.449496183 -0.744782026
H O 06-31+G(2d,2p)****
#M062X Gen SCF=(Conver=6)
Water Dimer
0 1O -0.702196054 -0.056060256 0.009942262H -1.022193224 0.846775782 -0.011488714H 0.257521062 0.042121496 0.005218999O 2.220871067 0.026716792 0.000620476H 2.597492682 -0.411663274 0.766744858H 2.593135384 -0.449496183 -0.744782026
H O 06-31+G(2d,2p)****
Grimme “D3” approach Truhlar M06-2X approach
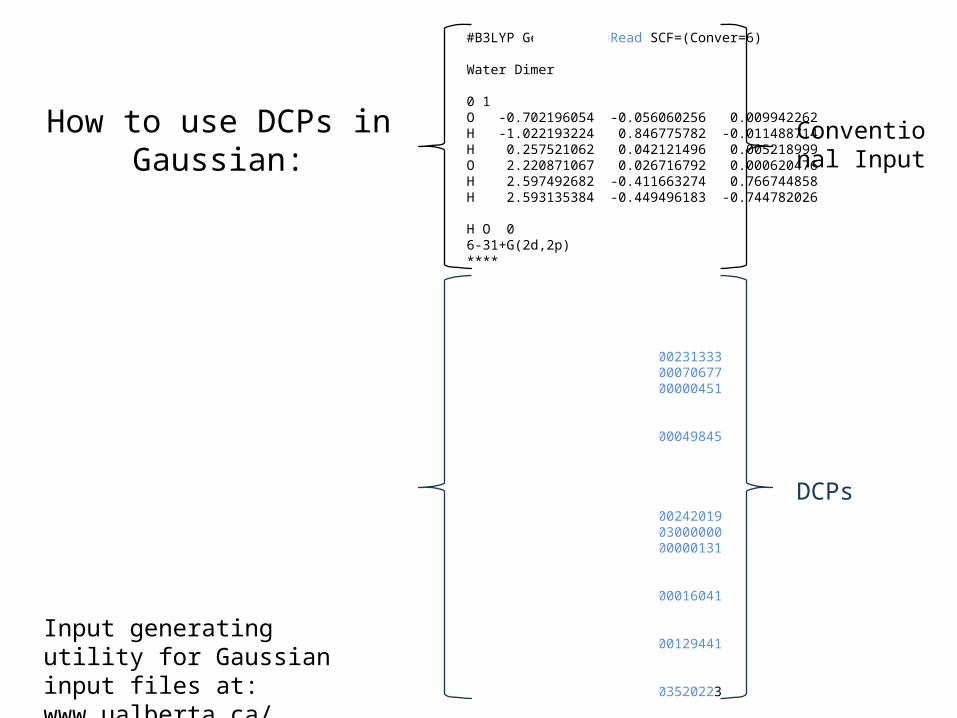
How to use DCPs in Gaussian:
#B3LYP Gen Pseudo=Read SCF=(Conver=6)
Water Dimer
0 1O -0.702196054 -0.056060256 0.009942262H -1.022193224 0.846775782 -0.011488714H 0.257521062 0.042121496 0.005218999O 2.220871067 0.026716792 0.000620476H 2.597492682 -0.411663274 0.766744858H 2.593135384 -0.449496183 -0.744782026
H O 06-31+G(2d,2p)****
H 0H 1 0P an up 3 2 0.120883601 0.000231333 2 0.044528578 -0.000070677 2 0.005658790 -0.000000451S-P 1 2 0.174740501 -0.000049845O 0O 3 0F an up 3 2 0.192168931 0.000242019 2 0.166560549 0.003000000 2 0.016734867 -0.000000131S-F 1 2 0.039337457 0.000016041P-F 1 2 0.123780982 -0.000129441D-F 1 2 0.061418151 -0.003520223
Conventional Input
DCPs
Input generating utility for Gaussian input files at:www.ualberta.ca/~gdilabio

How DCPs work
Based on (old) ECP technology:
e.g. Iodine = 1s22s22p6…5s24d105p5 [Potential] 5s24d105p5
In the case of DCPs, the potentials don’t replace any core electrons but instead are constructed such that the long-range behaviour of a DFT is corrected.
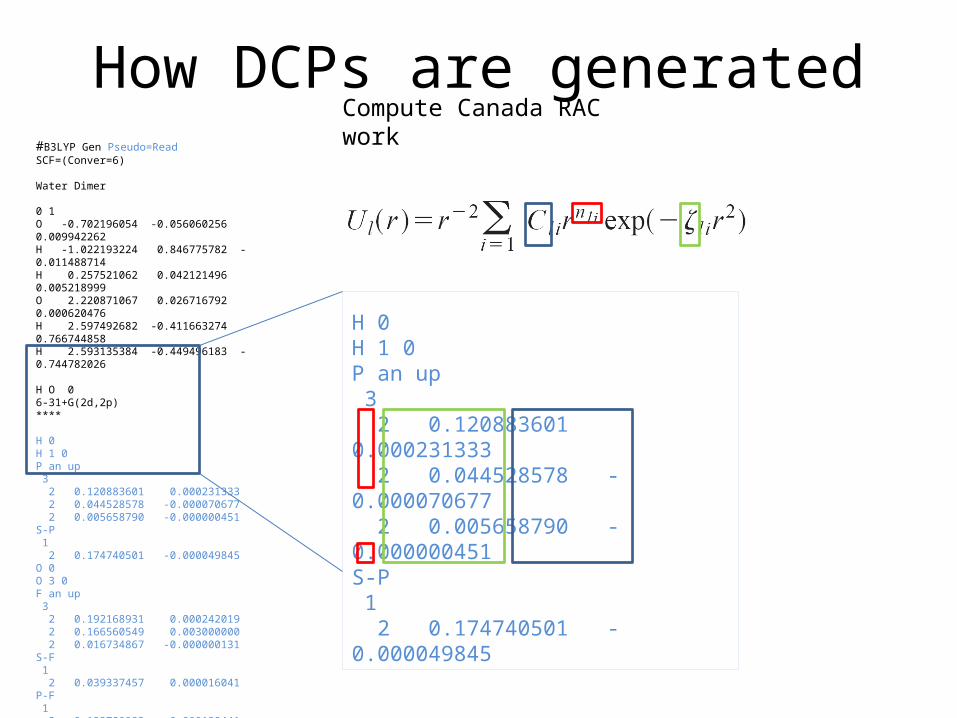
How DCPs are generated#B3LYP Gen Pseudo=Read SCF=(Conver=6)
Water Dimer
0 1O -0.702196054 -0.056060256 0.009942262H -1.022193224 0.846775782 -0.011488714H 0.257521062 0.042121496 0.005218999O 2.220871067 0.026716792 0.000620476H 2.597492682 -0.411663274 0.766744858H 2.593135384 -0.449496183 -0.744782026
H O 06-31+G(2d,2p)****
H 0H 1 0P an up 3 2 0.120883601 0.000231333 2 0.044528578 -0.000070677 2 0.005658790 -0.000000451S-P 1 2 0.174740501 -0.000049845O 0O 3 0F an up 3 2 0.192168931 0.000242019 2 0.166560549 0.003000000 2 0.016734867 -0.000000131S-F 1 2 0.039337457 0.000016041P-F 1 2 0.123780982 -0.000129441D-F 1 2 0.061418151 -0.003520223
H 0H 1 0P an up 3 2 0.120883601 0.000231333 2 0.044528578 -0.000070677 2 0.005658790 -0.000000451S-P 1 2 0.174740501 -0.000049845
Compute Canada RAC work

How DCPs are generatedCompute Canada RAC work
Steps:
1. Develop an initial guess for a DCP for an atom/DFT method/basis set combination.
2. Select the fitting data to which DCPs will be optimized.
3. Submit optimization script to Grex. The script:a) Builds Gaussian input files based on the information in 1&2.b) Builds queue files for Gaussian runsc) Submits queue files to the queued) Monitors status of runse) Extracts energies of noncovalently bonded systems and their monomersf) Adjusts values of Ci and ζi according to some optimization scheme
4. Select next atom and repeat from step 2.

-30 -20 -15 -10 -5 0 5 10 15 20 30 40 500
5
10
15
20
25Functional/6-31+G(2d,2p)
M06-2X
Performance on the S66 benchmark set: Occurrences in %error in BE
Past SuccessesB3LYP-DCP versus other dispersion-corrected DFT methods

-30 -20 -15 -10 -5 0 5 10 15 20 30 40 500
5
10
15
20
25Functional/6-31+G(2d,2p)
M06-2X ωB97XD
Performance on the S66 benchmark set: Occurrences in %error in BE
Past SuccessesB3LYP-DCP versus other dispersion-corrected DFT methods
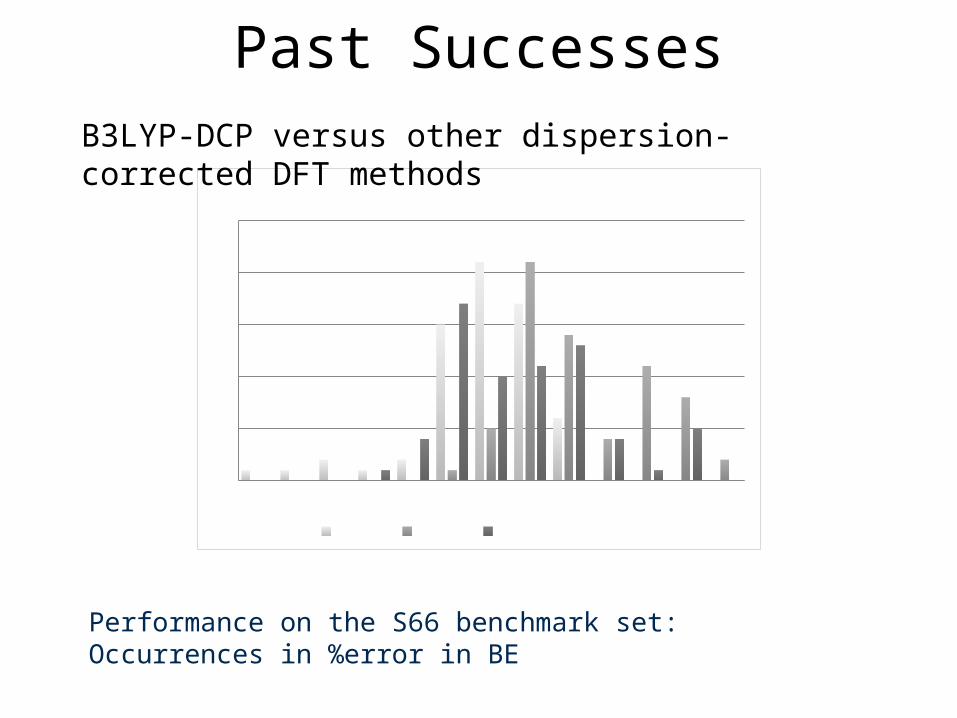
-30 -20 -15 -10 -5 0 5 10 15 20 30 40 500
5
10
15
20
25Functional/6-31+G(2d,2p)
M06-2X ωB97XD B97D
Performance on the S66 benchmark set: Occurrences in %error in BE
Past SuccessesB3LYP-DCP versus other dispersion-corrected DFT methods
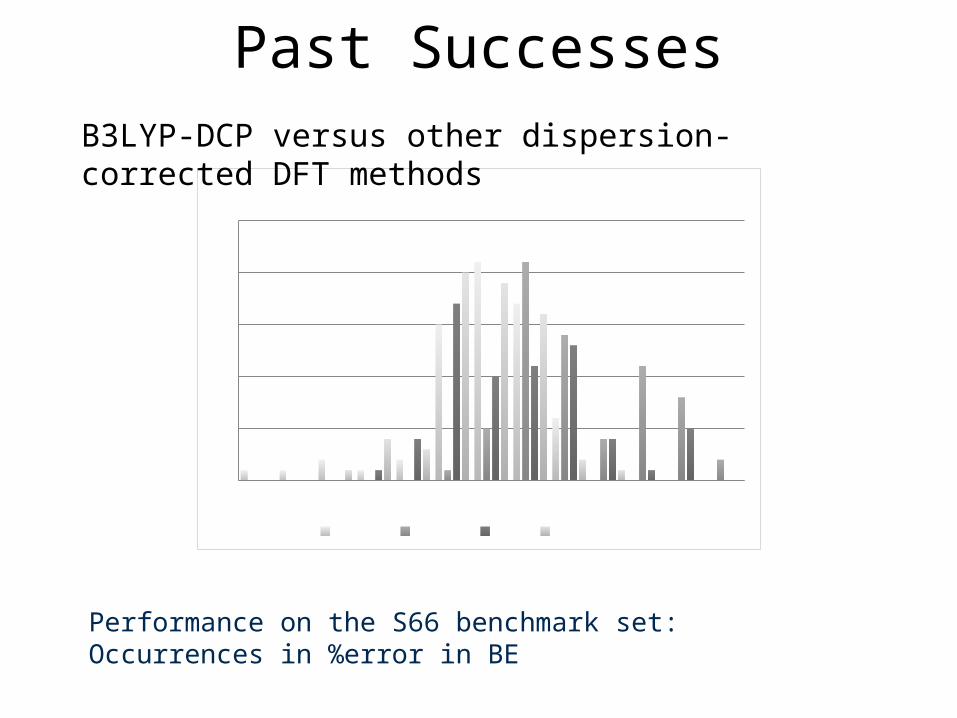
-30 -20 -15 -10 -5 0 5 10 15 20 30 40 500
5
10
15
20
25Functional/6-31+G(2d,2p)
M06-2X ωB97XD B97D B3LYP-DCP
Performance on the S66 benchmark set: Occurrences in %error in BE
Past SuccessesB3LYP-DCP versus other dispersion-corrected DFT methods
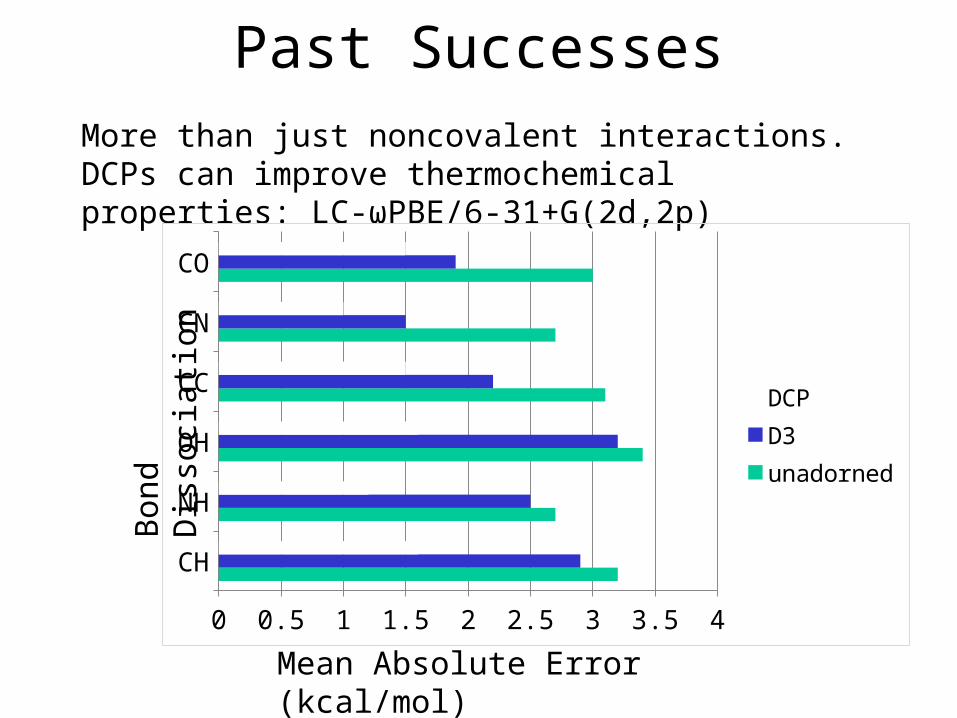
Past SuccessesMore than just noncovalent interactions. DCPs can improve thermochemical properties: LC-ωPBE/6-31+G(2d,2p)
CH
NH
OH
CC
CN
CO
0 0.5 1 1.5 2 2.5 3 3.5 4
DCPD3unadorned
Bond
Diss
ocia
tion
Mean Absolute Error (kcal/mol)

Future Efforts on DCPs
1. “Low cost” Density-functional theory methods + various computational chemistry/physics codes.
2. Thermochemistry, kinetics and noncovalent interactions.
3. Develop a deeper understanding of how DCPs work.
Challenges
1. Optimization scripts are complicated and platform dependent.
2. Conventional resource allocation scheme is not ideal for this work.

Thanks to:
Dr. Edmanuel Torres - NINT
Dr. Iain D. Mackie - NINT
Prof. Erin R. Johnson – UC Merced


















