
Molecular Modeling - Bioinformatics Institute · Molecular Modeling. ... [email protected]....
47
Computational Biology 1 Computational Biology 1 Molecular Modeling Molecular Modeling Guna Rajagopal, Bioinformatics Institute, [email protected] References References : Molecular Modeling Molecular Modeling – – Principles and Applications, Principles and Applications, Andrew Leach, 2 Andrew Leach, 2 nd nd Ed., Prentice Hall,2001 Ed., Prentice Hall,2001
Transcript of Molecular Modeling - Bioinformatics Institute · Molecular Modeling. ... [email protected]....

Computational Biology 1Computational Biology 1
Molecular ModelingMolecular Modeling
Guna Rajagopal,Bioinformatics Institute, [email protected]
ReferencesReferences : Molecular Modeling Molecular Modeling –– Principles and Applications, Principles and Applications, Andrew Leach, 2Andrew Leach, 2ndnd Ed., Prentice Hall,2001Ed., Prentice Hall,2001

Why Model?Why Model?•• To To understandunderstand biological/chemical data ? (But what does this mean?)biological/chemical data ? (But what does this mean?)
•• To To extract knowledgeextract knowledge from data we need to be able to search, from data we need to be able to search, merge, & check data via models.merge, & check data via models.
•• IntegratingIntegrating diverse data types can reduce random & systematic errors.diverse data types can reduce random & systematic errors.
•• By building By building simple modelssimple models through appropriate approximations, we can through appropriate approximations, we can get a handle on the phenomena at hand. We then refine/tune the mget a handle on the phenomena at hand. We then refine/tune the model odel via validation through experiments, to fit as close as possible via validation through experiments, to fit as close as possible to reality!to reality!
•• To understand the To understand the behavior of complex systemsbehavior of complex systems so that we can so that we can ultimately reultimately re--engineer them for our purposes.engineer them for our purposes.

Why do simulations?Why do simulations?
• Simulations are the onlyonly general method for “solving” many-body problems. Other methods involve approximations and experts.
• Experiment is limited and expensive. Simulations can complementcomplement the experiment.
• Simulations are relatively easyrelatively easy even for complex systems.
• They usually scale upscale up with the computer power.

Definition of SimulationDefinition of Simulation•• What is a simulation?What is a simulation?
An internal state “S” (e.g. position and momentum)A rule for changing the state SSnn+1 +1 = T (= T (SSnn))
We repeat the iteration many times: n⇒∞
SS00⇒⇒ SS11⇒⇒ SS22⇒⇒ SS33⇒⇒ SS44⇒⇒ SS55⇒⇒…….. .. SSnn⇒⇒ SSnn+1+1⇒⇒
•• The iteration index “n” is called “time.”The iteration index “n” is called “time.”• Simulations can be
– Deterministic (e.g. Newton’s equations=MD)– Stochastic (Monte Carlo, Brownian motion,…)– You analyze the errors the same way.
•• Similar to an experiment: rules simple but output unpredictable.Similar to an experiment: rules simple but output unpredictable.

ErgodicityErgodicity• Typically simulations are ergodicergodic: There is a correlation time T.
For times much longer than that, all non-conserved properties are close to their average value. Used for:– Warm up period at the beginning (equilibration) of MD/MC – To get independent samples for computing errors.
•• No matter what the initial state, one can characterize the stateNo matter what the initial state, one can characterize the stateof the system for t>T by a unique probability distribution of the system for t>T by a unique probability distribution function F(S) (if function F(S) (if ergodicergodic).).
• The typical example is the Boltzmann Boltzmann distributiondistribution:
• One goal is to compute averages over S. Another is to compute dynamics.
( )E SeZ
β−

Problems with estimating errorsProblems with estimating errors•• Any good simulation quotes systematic and statistical Any good simulation quotes systematic and statistical
errors for anything important. errors for anything important. • The error and mean are simultaneously determined from
the same data. HOW?•• Central limit theoremCentral limit theorem: the distribution of an average
approaches a normal distribution (if the variance is finite). One standard deviation means ~2/3 of the time the correct answer is within σ of the sample average.
• Problem in simulations is that data is correlated in timedata is correlated in time. It takes a “correlation” time to be “ergodic”
• We must throw away the initial transient

Two Simulation ModesTwo Simulation Modes
A. Give us the phenomena and invent a Give us the phenomena and invent a model to mimic the problem.model to mimic the problem. The semiThe semi--empirical approach.empirical approach. But one cannot reliably extrapolate the model away from the empirical data.
B. Maxwell, Boltzmann and Maxwell, Boltzmann and SchrSchröödingerdingergave us the model based on fundamental gave us the model based on fundamental laws of physicslaws of physics. All we must do is numerically solve the mathematical problem and determine the properties. (first principles first principles or ab initio methodsor ab initio methods).

Molecular DynamicsMolecular Dynamics

Molecular Dynamics (MD) SimulationsMolecular Dynamics (MD) Simulations
In biology, macroscopic properties are often determined by In biology, macroscopic properties are often determined by moleculemolecule--level behavior.level behavior.
Quantitative and/or qualitative informationQuantitative and/or qualitative information about macroscopic behavior of macromolecules can be obtained from simulation of a system at atomistic level.
Classical Molecular DynamicsClassical Molecular Dynamics simulations calculate the motion of the atoms in a molecular assembly using Newtonian dynamics to determine the net force and acceleration experienced by each atom. Each atom i at position ri, is treated as a point with a mass mi and a fixed charge qi. (In quantum molecular dynamicsquantum molecular dynamics, we use Schrodinger’s equation etc.)

Equations of Motion
• Closed system of N-interacting particles• Equations governing dynamical evolution of the
system (classical equations of motion):• Newton’s Law - 2
2 ( )d Xm F Xdt
=uur uur
1 2( , ,..., )NX r r r= Position of N-particles of systemuur ur ur uur
( ) ( )F X V X= −∇ ≡uur uur
Generalized force

Steps in MD SimulationsSteps in MD Simulations
1) Build realistic atomistic model of the system
2) Simulate the behavior of the system over time using specific conditions (temperature, pressure, volume, etc)
3) Analyze the results obtained from MD and relate to macroscopic level properties

What is a What is a ForcefieldForcefield??In molecular dynamics a molecule is described as a series of charged points (atoms) linked by springs (bonds).
To describe the time evolution of bond lengths, bond angles and torsions, also the non-bond van der Waals and electrostatic interactions between atoms, one uses a forcefield.The forcefield is a collection of equations and associated constants designed to reproduce molecular geometry and selected propertiesof tested structures.

A typical PE function (force field)A typical PE function (force field) Harmonic oscillators
Prevent collapse

Empirical force fields (EFF)Empirical force fields (EFF)
• Translation of protein sequence to structure rests upon a central dogma of biology: proteins adopt their lowest free energy proteins adopt their lowest free energy conformation as their functional state.conformation as their functional state.
• Force fields used to represent biological systems must yield the functional structure of the known proteins as its lowest free lowest free energy statesenergy states.

Limitations of existing EFFLimitations of existing EFF
EFF’sEFF’s without inclusion of without inclusion of electrostatic electrostatic polarizationpolarization cannot accurately describe the cannot accurately describe the solvationsolvation of highly charged of highly charged biomoleculesbiomolecules..
EFF are also EFF are also unableunable to treat bondto treat bond--breaking and breaking and bondbond--making processes in biochemical making processes in biochemical reactions (DNA, protein synthesis, reactions (DNA, protein synthesis, allosteric allosteric enzyme regulation etc.enzyme regulation etc.
Need for quantum mechanical techniques.Need for quantum mechanical techniques.

The need for quantum mechanicsThe need for quantum mechanics
With quantum mechanics, we can address problems such as:With quantum mechanics, we can address problems such as:
• Hydration structure of the DNA nucleoside bases• Energetic factors leading to DNA base pairing• Hydration of the DNA backbone and basic sites• Role of polarization in the stability of proteins,
alpha helices etc.• Catalytic mechanisms at the active sites of
enzymes,• Interaction of proteins with light, …

Minimization StepMinimization Step
Conformational change
Energy
The energy of the system can be calculated using the forcefield. The conformation of the system can be altered to find lower energy conformations through a process called minimizationminimization.
Minimization algorithms:Minimization algorithms:• steepest descent (slowly converging – use for highly restrained systems• conjugate gradient (efficient, uses intelligent choices of search direction – use for large systems)• BFGS (quasi-newton variable metric method)• Newton-Raphson (calculates both slope of energy and rate of change)

SolvationSolvation
Biological activity is the result of interactions between molecules and occurs at the interfaces between molecules (protein-protein, protein-DNA, protein-solvent, DNA-solvent, etc).
Why model Why model solvationsolvation??• many biological processes occur in aqueous solution• solvation effects play a crucial role in determining molecular conformation, electronic properties, binding energies, etc
How to model How to model solvationsolvation??• explicit treatment: solvent molecules are added to the molecular system • implicit treatment: solvent is modeled as a continuum dielectric

MD:MD: VerletVerlet MethodMethodEnergy function:
used to determine the force on each atom:
Newton’s equation represents a set of N second order differential equations which are solved numerically at discrete time stepssolved numerically at discrete time stepsto determine the trajectory of each atom.
Advantage of the Verlet Method: requires only one force evaluation per timestep

Overview of MD Overview of MD
What you have up to now:• pdb file, • topology file• parameter file
What you need next: a program capable of reading and manipulating this information
Programs:X-PLOR, CHARMm, NAMD2, AMBER, GROMACS, EGO, etc

Example: MD Simulations of Example: MD Simulations of the K+ Channel Proteinthe K+ Channel Protein
Ion channels are membrane -spanning proteins that form a pathway for the flux of inorganic ions across cell membranes.
Potassium channels are a particularly interesting class of ion channels, managing to distinguish with impressive fidelity between K+ and Na+ ions while maintaining a very high throughput of K+ ions when gated.
Klaus Schluten, UIUC

Setting up the system (1)Setting up the system (1)
• retrieve the PDB (coordinates) file from the Protein Data Bank
• use topology and parameter files to set up the structure
• add hydrogen atoms using X-PLOR
• minimize the protein structure using NAMD2
Klaus Schluten, UIUC
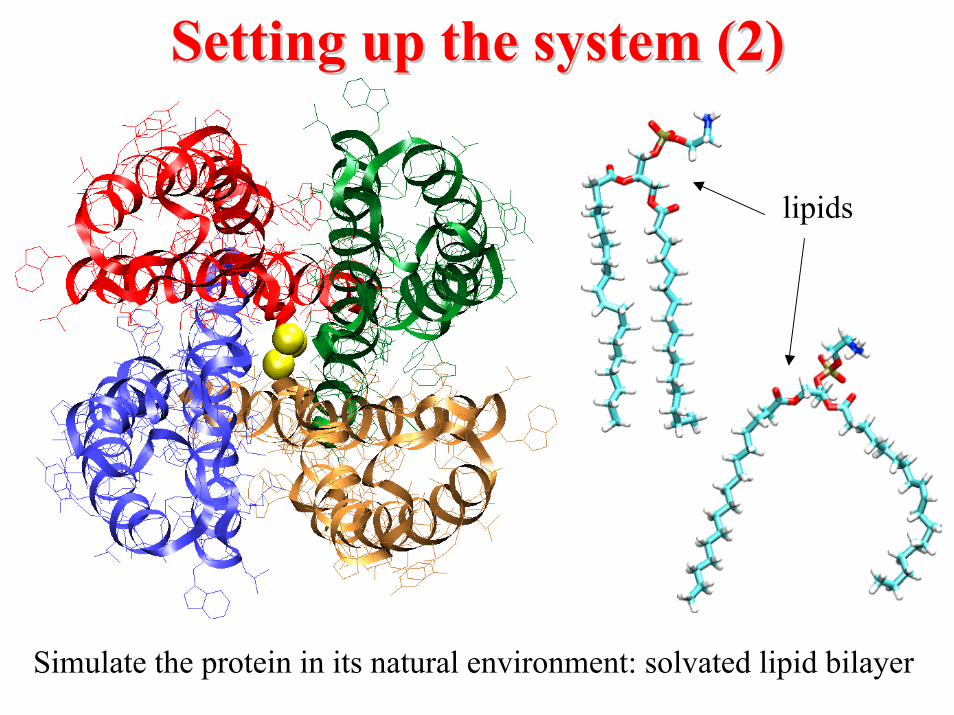
Setting up the system (2)Setting up the system (2)
lipids
Simulate the protein in its natural environment: solvated lipid bilayer

Setting up the system (3)Setting up the system (3)Inserting the protein in the lipid bilayer
gaps
Automatic insertion into the lipid bilayer leads to big gaps between the protein and the membrane => long equilibration time required to fill the gaps.Solution: manually adjust the position of lipids around the protein
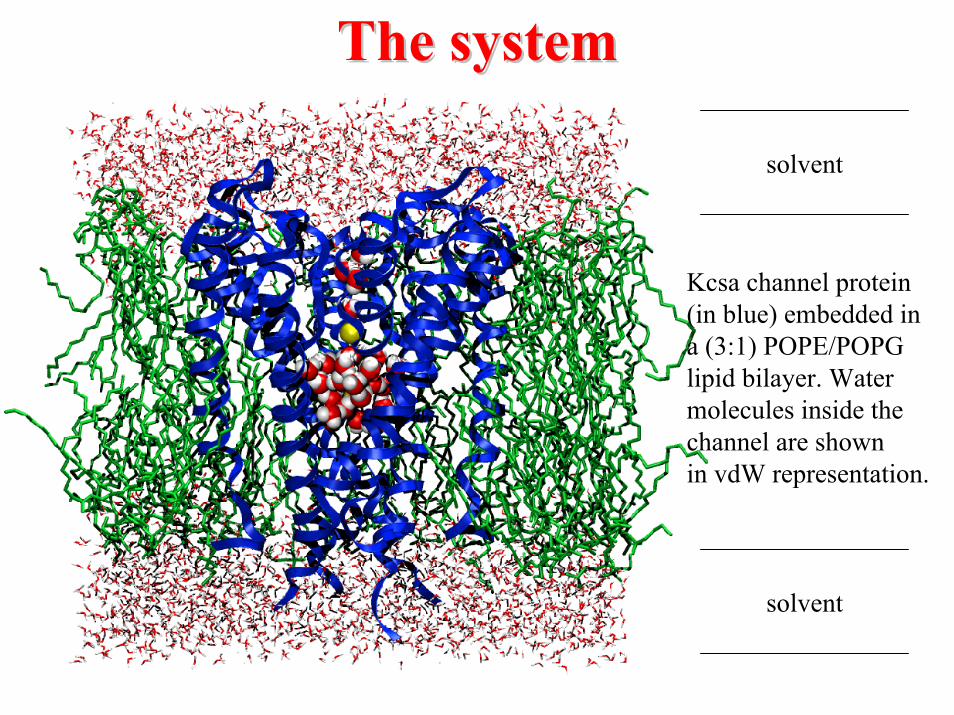
The systemThe system
Kcsa channel protein(in blue) embedded in a (3:1) POPE/POPGlipid bilayer. Watermolecules inside thechannel are shownin vdW representation.
solvent
solvent

Simulating the systemSimulating the system
Summary of simulations:Summary of simulations:• protein/membrane system contains 38,112 atoms, including 5117 water molecules, 100 POPE and 34 POPG lipids, plus K+ counterions• CHARMM26 forcefield• periodic boundary conditions, PME electrostatics• 1 ns equilibration at 310K, NpT• 2 ns dynamics, NPT
Program:Program: NAMD2
Platform:Platform: Cray T3E (Pittsburgh Supercomputer Center)Klaus Schluten, UIUC

MD ResultsMD Results
RMS deviations for the KcsA protein and its selectivity filer indicate that the protein is stable during the simulation with the selectivity filter the most stable part of the system.
Temperature factors for individual residues in the four monomers of the KcsA channel protein indicate that the most flexible parts of the protein are the N and C terminal ends, residues 52-60 and residues 84-90. Residues 74-80 in the selectivity filter have low temperature factors and are very stable during the simulation.

Dr. C Varma
Haemoglobin Porphyrin/Oxygen

oxygen
histidine
porphyrin
Oxygen buried
Need fluctuationsof protein to get in
Dr. C Varma

MD MD ––ShortcomingsShortcomings• Quality of the forcefield
• Size and TimeSize and Time – atomistic simulations can be performed only for systems of a few tenths of angstroms on the length scale and for a few nanoseconds on the time scale
• Conformational freedom of the moleculeConformational freedom of the molecule – the number of possible conformations a molecule can adopt is enormous, growingexponentially with the number or rotatable bonds.
• Only applicable to systems that have been parameterizedOnly applicable to systems that have been parameterized
• Connectivity of atoms cannot change during dynamicsConnectivity of atoms cannot change during dynamics – no bond making/breaking so no chemical reactions

Monte Carlo MethodsMonte Carlo Methods

Monte Carlo MethodsMonte Carlo Methods
•• What is the Monte Carlo method?What is the Monte Carlo method?
– Any method which uses random numbersrandom numbers as an essential part of the algorithm
– Often a method for doing high dimensional high dimensional integralsintegrals by sampling the integrand,
– error independent independent of the dimensionality and proportional to 1/1/SqrtSqrt(computing effort)(computing effort)
– Often a Markov chain, called Metropolis MC–– Ideal for parallel computersIdeal for parallel computers

Monte Carlo AlgorithmsMonte Carlo Algorithms• MC methods used widely to explore explore
conformation efficientlyconformation efficiently, and like in many other optimization problems, to search for the minimum. Simple minimization methods based on moving “downhill” in energy fail as they get trapped in a trapped in a local minimumlocal minimum far from the native state.
• The MC method allows for escaping from these local minimum.
• Has proved extremely effective in many applications especially in the calculations of calculations of thermodynamic quantitiesthermodynamic quantities such as the free energy, entropy etc.

Metropolis AlgorithmMetropolis AlgorithmSuppose the configuration of a system is specified by a variableSuppose the configuration of a system is specified by a variable XX.
1. Generate a randomrandom set of values for X to provide a starting conformation. Calculate the energy of this conformation E=E(X).
2.2. PerturbPerturb the variables X to X’ to generate a new conformation. (One usually changes the coordinates by a small amount).
3. Calculate the energy of the new conformation, E’ = E(X’). (Usually the most expensive part of the calculation.) We use the same force fieldssame force fields as in the MD calculations above.

4. Decide whether to accept or rejectaccept or reject the move. • If the energy has decreased, i.e. we went
downhill in the move, always accept italways accept it. Then, set X’ as the new conformation and E’ as the new energy.
• If the energy has increased or stayed the same, sometimes accept the movesometimes accept the move i.e. if D=E’ – E, accept the step with probability exp(exp(--D/KT), D/KT), where K is Boltzmann’s constant and T is the effective temperature. (This has the potential to This has the potential to get over barriers, out of traps in local minima. get over barriers, out of traps in local minima. The effective temperature T controls the The effective temperature T controls the chance that a uphill move will be accepted.)chance that a uphill move will be accepted.)
5. Return to step 2.

Protein Structure PredictionProtein Structure Prediction

Knowledge based methodsKnowledge based methods• Based on assembling clues to the structure of a
target sequence by finding similaritiessimilarities to known structures.
• We are coming closer to saturating the set of possible folds with known structures. This is the stated goal of structural structural genomics genomics projects.projects.
• Once we have a complete set of folds and sequences, and methods for relating them, empirical methods can provide pragmatic solutions of many problems.

Structure Prediction MethodsStructure Prediction Methods
•• Secondary Structure PredictionSecondary Structure Prediction: Prediction of secondary structure from AA sequence without attempting to assemble these regions in 3D space. The results are lists of regionslists of regions of the sequence predicted to form alpha helices and beta strands.
•• Homology modelingHomology modeling: prediction of the 3D structure of a protein from the known structures known structures of one or more related proteinsof one or more related proteins. The results are a complete coordinate set for mainchain and sidechains, intended to be a high quality model of the structure (comparable to at least a low resolution X-ray structure).

•• Fold RecognitionFold Recognition: given a library of known structures, determine which of them shares a folding pattern with a query protein of known sequence but unknown structure.The result could be the nomination of a known structure that has the same fold as the query protein, or a statement that no protein in the library has the same fold as the query protein.
•• Critical Assessment of Structure Prediction Critical Assessment of Structure Prediction (CASP(CASP) – Experimentalists make public the AA sequences of the proteins they are investigating, and keep their experimental structures secret until predictors have had a chance to submit their models. CASP runs on a two-year cycle.

The protein folding problemThe protein folding problem

The protein folding problemThe protein folding problemTwo facets of the protein folding problem:Two facets of the protein folding problem:
• Prediction of the three dimensional structure from the amino-acid sequence
• Understanding the mechanisms and pathways whereby the 3D structure forms within biologically relevant timescales.
Note: The importance of protein (protein (mismis)folding)folding e.g. in Alzheimer’sdisease and mad cow disease.

Simple model of protein foldingSimple model of protein folding

Example : Protein Example : Protein DenaturationDenaturation
•Proteins have a native statenative state. (Actually, they tend to have a tight cluster of native states.)
•Denaturation occurs when heat or denaturantsheat or denaturants such as guanidine, urea or detergent are added to solution. Also, the pH can affect folding.
•When performing a denaturation process nonnon--covalent interactions are brokencovalent interactions are brokeni.e.Ionic, van der-Waals, dipolar, hydrogen bonding, etc.
•Solvent is reorganized.

Protein folding mechanismsProtein folding mechanismsA simplisticsimplistic view of the folding mechanism:
We know that the amino acid R groups fall into charges, hydrophobic, and hydrophilic catagories.
Therefore the folded state of the polypeptide chain is stablizedstablized primarily by the sequestration of sequestration of much of the hydrophobic groups into the core much of the hydrophobic groups into the core of the proteinof the protein (out of contact with water) while the hydrophilic and charged groups remain in contact with water.

Computational Protein FoldingComputational Protein FoldingAb initio methods to fold AA sequences to 3D native conformation based on several assumptions:
•• AssumedAssumed that the equilibrium conformation is the global global free energy minimumfree energy minimum on a folding pathway. (No one knows for sure!)
•• AssumedAssumed that current interaction models (force fields) are sufficiently accurate.
• Many globular proteins are oligomericoligomeric and many may not fold as monomers; but only monomeric proteins treated by these methods.
• Problem of recognizing that a given sequence will produce a dimeric or tetrameric protein, and how to treat oligomerization in computational approaches to folding has not even been properly addressedhas not even been properly addressed.

BioComputingBioComputing : Range of Computational Methods: Range of Computational Methods
HomologyHomology--basedbasedStructure Prediction
Molecular DynamicsMolecular Dynamicsand Molecular Mechanics
First PrinciplesFirst PrinciplesQuantum MechanicsStructure Prediction and Molecular Mechanics Quantum Mechanics
Overlapping Continuum of Methods
Qualitative properties and Qualitative properties and classical trajectories using classical trajectories using
empirical potentialsempirical potentials
Static properties from exact Static properties from exact quantum mechanicsquantum mechanics
• Protein structures• Structure-based homologies
• Dynamic structural data• Solvent distributions• Qualitative energetics• Docking
• Interaction energies• Molecular structures• Reaction energies• Spectroscopic parameters• Solvation energies• Reaction rates
Predict structure based onPredict structure based onanalogy with known structuresanalogy with known structures

What we are aiming for with large scale computing!What we are aiming for with large scale computing!


















