
Modeling human endothelial cell transformation in...
14
dmm.biologists.org 1066 Introduction Vascular neoplasias are a spectrum of disorders that include rare, aggressive malignancies such as angiosarcoma, as well as commonly occurring infantile hemangiomas that are usually indolent but can give rise to life-threatening complications when vital organs are involved (Drolet et al., 1999; Szajerka and Jablecki, 2007; Koch et al., 2008; Young et al., 2010). The unifying pathological feature of these conditions is perturbation of endothelial cell (EC) proliferation and disorganization of endothelial tissue architecture. In a non- pathological state, mature ECs have limited proliferative ability and form an organized monolayer along the inner surface of blood and lymphatic vessels. These cells are derived from a hierarchy of highly proliferative progenitors that are thought to reside within the bone marrow and are ontologically related to hematopoietic stem cells (Asahara et al., 1997; Asahara et al., 1999). An array of cytokines and chemokines direct the recruitment and homing of endothelial progenitors from the bone marrow, via the peripheral circulation, to the inner walls of blood and lymph vessels where they differentiate and contribute to many normal physiological processes, such as wound healing, inflammation, coagulation, cell migration and hematopoiesis (Shi et al., 1998; Asahara et al., 1999; Krishnaswamy et al., 1999; Takahashi et al., 1999). The mitogenic signaling pathways that regulate the proliferation of normal ECs have been intensively studied and are reviewed elsewhere (Carmeliet and Jain, 2011). However, relatively few investigations have focused on the molecular mechanisms that promote unregulated proliferation and transformation of ECs during the development of vascular neoplasia. This is in part due to the rarity of EC-derived malignancies as well as the focus of most investigations on end-stage disease, which precludes study of the molecular events involved in the initiation and progression of disease. Consequently, little progress has been made in the development of novel therapeutics for the treatment of endothelial malignancies over the past few decades. By describing the molecular features of vascular neoplasias, as well as the cell lines and in vivo models that are available to model EC transformation, the aim of this review is to inform and motivate preclinical studies of new therapeutic approaches to the treatment of specific EC neoplasias. Clinical and molecular features of EC-derived neoplasias Hemangioma Hemangioma is the most common tumor of infancy, affecting ~10% of Caucasian neonates (Table 1) (Drolet et al., 1999). This neoplasia develops as a rapidly growing disorganized mass of ECs. Most hemangiomas appear within the first few weeks of birth and are characterized by an initial period of rapid tumor growth that is driven by the proliferation of immature ECs. This is followed by a ‘rest phase’, in which there is little change in the appearance of the tumor. A subsequent slow involuting phase associated with EC differentiation and apoptosis marks regression of the tumor in almost all cases. The involuted tumor is replaced by fibrous and fatty tissue. Although histologically benign, non-invasive and unlikely to progress to a malignancy, visceral hemangiomas can result in substantial morbidity and mortality by causing obstruction, hemorrhage or by diverting blood flow. Currently available treatments for life-threatening or otherwise complicated cases of hemangioma have limited efficacy and commonly result in treatment-associated toxicity. REVIEW Disease Models & Mechanisms 6, 1066-1079 (2013) doi:10.1242/dmm.012674 1 Cancer Cell Development Group, Children’s Cancer Institute Australia for Medical Research, Lowy Cancer Research Centre, University of New South Wales, Randwick, NSW, Australia *Author for correspondence ([email protected]) © 2013. Published by The Company of Biologists Ltd This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed. Endothelial cell (EC)-derived neoplasias range from benign hemangioma to aggressive metastatic angiosarcoma, which responds poorly to current treatments and has a very high mortality rate. The development of treatments that are more effective for these disorders will be expedited by insight into the processes that promote abnormal proliferation and malignant transformation of human ECs. The study of primary endothelial malignancy has been limited by the rarity of the disease; however, there is potential for carefully characterized EC lines and animal models to play a central role in the discovery, development and testing of molecular targeted therapies for vascular neoplasias. This review describes molecular alterations that have been identified in EC-derived neoplasias, as well as the processes that underpin the immortalization and tumorigenic conversion of ECs. Human EC lines, established through the introduction of defined genetic elements or by culture of primary tumor tissue, are catalogued and discussed in relation to their relevance as models of vascular neoplasia. Modeling human endothelial cell transformation in vascular neoplasias Victoria W. Wen 1 and Karen L. MacKenzie 1, * Disease Models & Mechanisms DMM
-
Upload
phungthien -
Category
Documents
-
view
213 -
download
0
Transcript of Modeling human endothelial cell transformation in...

dmm.biologists.org1066
IntroductionVascular neoplasias are a spectrum of disorders that include rare,aggressive malignancies such as angiosarcoma, as well as commonlyoccurring infantile hemangiomas that are usually indolent but cangive rise to life-threatening complications when vital organs areinvolved (Drolet et al., 1999; Szajerka and Jablecki, 2007; Koch et al.,2008; Young et al., 2010). The unifying pathological feature of theseconditions is perturbation of endothelial cell (EC) proliferation anddisorganization of endothelial tissue architecture. In a non-pathological state, mature ECs have limited proliferative ability andform an organized monolayer along the inner surface of blood andlymphatic vessels. These cells are derived from a hierarchy of highlyproliferative progenitors that are thought to reside within the bonemarrow and are ontologically related to hematopoietic stem cells(Asahara et al., 1997; Asahara et al., 1999). An array of cytokines andchemokines direct the recruitment and homing of endothelialprogenitors from the bone marrow, via the peripheral circulation,to the inner walls of blood and lymph vessels where they differentiateand contribute to many normal physiological processes, such aswound healing, inflammation, coagulation, cell migration andhematopoiesis (Shi et al., 1998; Asahara et al., 1999; Krishnaswamyet al., 1999; Takahashi et al., 1999).
The mitogenic signaling pathways that regulate the proliferationof normal ECs have been intensively studied and are reviewedelsewhere (Carmeliet and Jain, 2011). However, relatively fewinvestigations have focused on the molecular mechanisms that
promote unregulated proliferation and transformation of ECsduring the development of vascular neoplasia. This is in part dueto the rarity of EC-derived malignancies as well as the focus ofmost investigations on end-stage disease, which precludes studyof the molecular events involved in the initiation and progressionof disease. Consequently, little progress has been made in thedevelopment of novel therapeutics for the treatment of endothelialmalignancies over the past few decades. By describing themolecular features of vascular neoplasias, as well as the cell linesand in vivo models that are available to model EC transformation,the aim of this review is to inform and motivate preclinical studiesof new therapeutic approaches to the treatment of specific ECneoplasias.
Clinical and molecular features of EC-derivedneoplasiasHemangiomaHemangioma is the most common tumor of infancy, affecting ~10%of Caucasian neonates (Table 1) (Drolet et al., 1999). This neoplasiadevelops as a rapidly growing disorganized mass of ECs. Mosthemangiomas appear within the first few weeks of birth and arecharacterized by an initial period of rapid tumor growth that isdriven by the proliferation of immature ECs. This is followed by a‘rest phase’, in which there is little change in the appearance of thetumor. A subsequent slow involuting phase associated with ECdifferentiation and apoptosis marks regression of the tumor inalmost all cases. The involuted tumor is replaced by fibrous andfatty tissue. Although histologically benign, non-invasive andunlikely to progress to a malignancy, visceral hemangiomas canresult in substantial morbidity and mortality by causing obstruction,hemorrhage or by diverting blood flow. Currently availabletreatments for life-threatening or otherwise complicated cases ofhemangioma have limited efficacy and commonly result intreatment-associated toxicity.
REVIEW Disease Models & Mechanisms 6, 1066-1079 (2013) doi:10.1242/dmm.012674
1Cancer Cell Development Group, Children’s Cancer Institute Australia for MedicalResearch, Lowy Cancer Research Centre, University of New South Wales, Randwick,NSW, Australia*Author for correspondence ([email protected])
© 2013. Published by The Company of Biologists LtdThis is an Open Access article distributed under the terms of the Creative Commons AttributionLicense (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distributionand reproduction in any medium provided that the original work is properly attributed.
Endothelial cell (EC)-derived neoplasias range from benign hemangioma to aggressive metastatic angiosarcoma, whichresponds poorly to current treatments and has a very high mortality rate. The development of treatments that aremore effective for these disorders will be expedited by insight into the processes that promote abnormal proliferationand malignant transformation of human ECs. The study of primary endothelial malignancy has been limited by therarity of the disease; however, there is potential for carefully characterized EC lines and animal models to play a centralrole in the discovery, development and testing of molecular targeted therapies for vascular neoplasias. This reviewdescribes molecular alterations that have been identified in EC-derived neoplasias, as well as the processes thatunderpin the immortalization and tumorigenic conversion of ECs. Human EC lines, established through theintroduction of defined genetic elements or by culture of primary tumor tissue, are catalogued and discussed inrelation to their relevance as models of vascular neoplasia.
Modeling human endothelial cell transformation invascular neoplasiasVictoria W. Wen1 and Karen L. MacKenzie1,*
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
Disease Models & Mechanisms 1067
Modeling endothelial cell neoplasia REVIEW
Hemangiomas are composed of morphologically normal,immature ECs that are clonally derived and exhibit enhancedproliferation and migration (Mulliken et al., 1982; Boye etal., 2001; Dadras et al., 2004). Evidence has recently emergedto suggest that the excessive proliferation of ECs inhemangiomas is driven by an imbalance in angiogenicsignaling factors and activation of nuclear factor-κB (NFκB)(Jinnin et al., 2008; Greenberger et al., 2010). Notably, arecent study of genetic risk factors for hemangioma identifiedgermline mutations in the genes encoding vascularendothelial growth factor (VEGF) receptor 2 (VEGFR2) andanthrax toxin receptor 1 (also known as tumor endothelialmarker 8) that perturb VEGF signaling and contribute todisorganized angiogenesis (Jinnin et al., 2008). Othermolecular events resulting from the cytogeneticabnormalities that have been identified in hemangiomascould also play a role in this disorder. Chromosomalaberrations that have been observed in hemangioma includeloss of heterozygosity at chromosome regions 5q, 13q14 and17p13, as well as loss of the Y chromosome and amplificationof the cyclin D1 gene on chromosome 11 (Berg et al., 2001;Domfeh et al., 2006; Mohamed et al., 2009). Insights into themolecular regulation of different phases of hemangioma willbe valuable for the development of improved therapies forthese disorders.
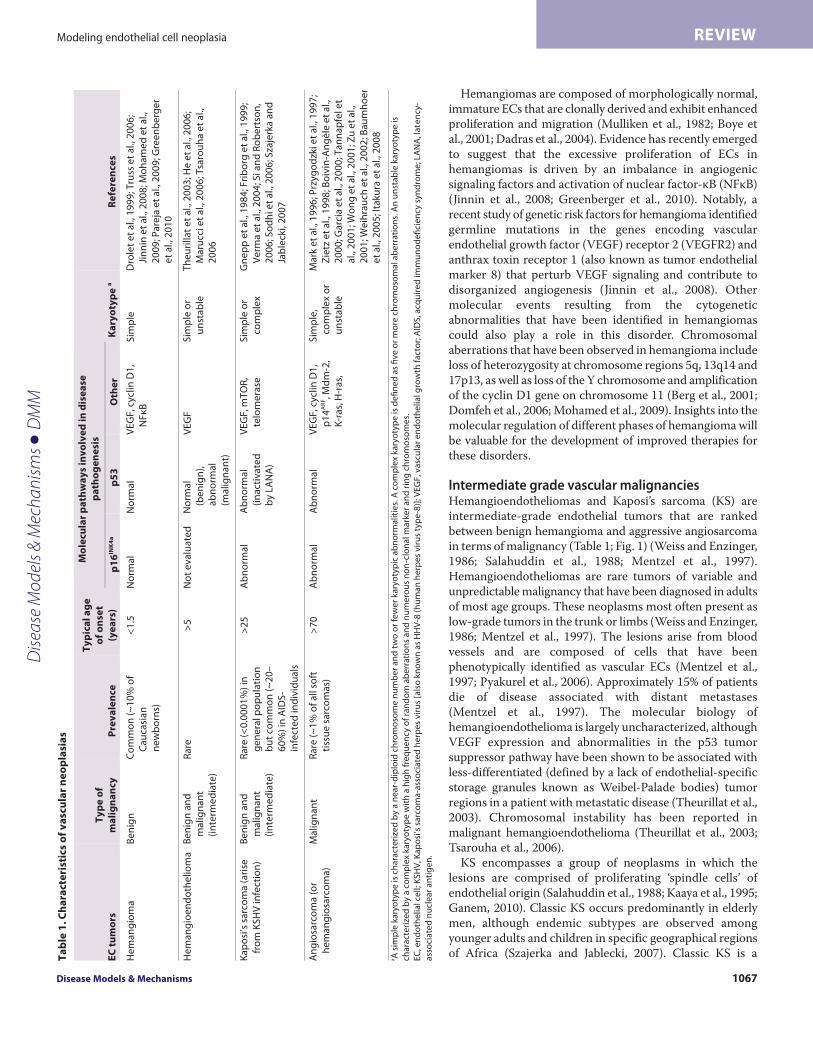
Intermediate grade vascular malignanciesHemangioendotheliomas and Kaposi’s sarcoma (KS) areintermediate-grade endothelial tumors that are rankedbetween benign hemangioma and aggressive angiosarcomain terms of malignancy (Table 1; Fig. 1) (Weiss and Enzinger,1986; Salahuddin et al., 1988; Mentzel et al., 1997).Hemangioendotheliomas are rare tumors of variable andunpredictable malignancy that have been diagnosed in adultsof most age groups. These neoplasms most often present aslow-grade tumors in the trunk or limbs (Weiss and Enzinger,1986; Mentzel et al., 1997). The lesions arise from bloodvessels and are composed of cells that have beenphenotypically identified as vascular ECs (Mentzel et al.,1997; Pyakurel et al., 2006). Approximately 15% of patientsdie of disease associated with distant metastases (Mentzel et al., 1997). The molecular biology ofhemangioendothelioma is largely uncharacterized, althoughVEGF expression and abnormalities in the p53 tumorsuppressor pathway have been shown to be associated withless-differentiated (defined by a lack of endothelial-specificstorage granules known as Weibel-Palade bodies) tumorregions in a patient with metastatic disease (Theurillat et al.,2003). Chromosomal instability has been reported inmalignant hemangioendothelioma (Theurillat et al., 2003;Tsarouha et al., 2006).
KS encompasses a group of neoplasms in which thelesions are comprised of proliferating ‘spindle cells’ ofendothelial origin (Salahuddin et al., 1988; Kaaya et al., 1995;Ganem, 2010). Classic KS occurs predominantly in elderlymen, although endemic subtypes are observed amongyounger adults and children in specific geographical regionsof Africa (Szajerka and Jablecki, 2007). Classic KS is aT
ab
le 1
. Ch
ara
cte
rist
ics
of
va
scu
lar
ne
op
lasi
as
EC
tu
mo
rs
Ty
pe
of
ma
lig
na
ncy
P
rev
ale
nce
Ty
pic
al
ag
e
of
on
set
(ye
ars
)
Mo
lecu
lar
pa
thw
ay
s in
vo
lve
d i
n d
ise
ase
pa
tho
ge
ne
sis
Ka
ryo
typ
e a
R
efe
ren
ces
p1
6IN
K4
a
p5
3
Oth
er
He
ma
ng
iom
a
Be
nig
n
Co
mm
on
(~
10
% o
f
Ca
uca
sia
n
ne
wb
orn
s)
<1
.5
No
rma
l N
orm
al
VE
GF
, cyc
lin D
1,
NF
κB
S
imp
le
Dro
let
et
al.,
19
99
; Tru
ss e
t a
l., 2
00
6;
Jin
nin
et
al.,
20
08
; Mo
ha
me
d e
t a
l.,
20
09
; Pa
reja
et
al.,
20
09
; Gre
en
be
rge
r e
t a
l., 2
01
0
He
ma
ng
ioe
nd
oth
elio
ma
B
en
ign
an
d
ma
lign
an
t (i
nte
rme
dia
te)
Ra
re
>5
N
ot
ev
alu
ate
d
No
rma
l
(be
nig
n),
a
bn
orm
al
(ma
lign
an
t)
VE
GF
S
imp
le o
r
un
sta
ble
T
he
uri
llat
et
al.,
20
03
; He
et
al.,
20
06
;
Ma
rucc
i et
al.,
20
06
; Tsa
rou
ha
et
al.,
2
00
6
Ka
po
si’s
sa
rco
ma
(a
rise
fro
m K
SH
V in
fect
ion
) B
en
ign
an
d
ma
lign
an
t (i
nte
rme
dia
te)
Ra
re (
<0
.00
01
%)
in
ge
ne
ral p
op
ula
tio
n
bu
t co
mm
on
(~
20
–
60
%)
in A
IDS
-in
fect
ed
ind
ivid
ua
ls
>2
5
Ab
no
rma
l A
bn
orm
al
(in
act
iva
ted
b
y LA
NA
)
VE
GF
, mT
OR
,
telo
me
rase
S
imp
le o
r
com
ple
x G
ne
pp
et
al.,
19
84
; Fri
bo
rg e
t a
l., 1
99
9;
Ve
rma
et
al.,
20
04
; Si a
nd
Ro
be
rtso
n,
20
06
; So
dh
i et
al.,
20
06
; Sza
jerk
a a
nd
Jab
leck
i, 2
00
7
An
gio
sarc
om
a (
or
he
ma
ng
iosa
rco
ma
) M
alig
na
nt
Ra
re (
~1
% o
f a
ll so
ft
tiss
ue
sa
rco
ma
s)
>7
0
Ab
no
rma
l A
bn
orm
al
VE
GF
, cyc
lin D
1,
p1
4A
RF, M
dm
-2,
K-r
as,
H-r
as,
Sim
ple
,
com
ple
x o
r u
nst
ab
le
Ma
rk e
t a
l., 1
99
6; P
rzyg
od
zki e
t a
l., 1
99
7;
Zie
tz e
t a
l., 1
99
8; B
oiv
in-A
ng
èle
et
al.,
2
00
0; G
arc
ia e
t a
l., 2
00
0; T
an
na
pfe
l et
al.,
20
01
; Wo
ng
et
al.,
20
01
; Zu
et
al.,
20
01
; We
ihra
uch
et
al.,
20
02
; Ba
um
ho
er
et
al.,
20
05
; Ita
kura
et
al.,
20
08
aA
sim
ple
ka
ryo
typ
e is
ch
ara
cte
rize
d b
y a
ne
ar-
dip
loid
ch
rom
oso
me
nu
mb
er
an
d t
wo
or
few
er
kary
oty
pic
ab
no
rma
litie
s. A
co
mp
lex
kary
oty
pe
is d
efi
ne
d a
s fi
ve
or
mo
re c
hro
mo
som
al a
be
rra
tio
ns.
An
un
sta
ble
ka
ryo
typ
e is
cha
ract
eri
zed
by
a c
om
ple
x ka
ryo
typ
e w
ith
a h
igh
fre
qu
en
cy o
f ra
nd
om
ab
err
ati
on
s a
nd
nu
me
rou
s n
on
-clo
na
l ma
rke
r a
nd
rin
g c
hro
mo
som
es.
EC
, en
do
the
lial c
ell;
KS
HV
, Ka
po
si’s
sa
rco
ma
-ass
oci
ate
d h
erp
es
vir
us
[als
o k
no
wn
as
HH
V-8
(h
um
an
he
rpe
s v
iru
s ty
pe
-8)]
; VE
GF
, va
scu
lar
en
do
the
lial g
row
th f
act
or;
AID
S, a
cqu
ire
d im
mu
no
de
fici
en
cy s
ynd
rom
e; L
AN
A, l
ate
ncy
-
ass
oci
ate
d n
ucl
ea
r a
nti
ge
n.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

dmm.biologists.org1068
Modeling endothelial cell neoplasiaREVIEW
relatively indolent disease that is rarely life-threatening and is oftenleft untreated. Indeed, spontaneous remissions have been reported(Brooks, 1986). Cases of KS that do progress usually involve localinvasion, whereas distant dissemination is extremely rare. KS alsomanifests in association with immune suppression in individualswith AIDS and organ-transplant recipients (Gnepp et al., 1984). Incontrast to classic KS, AIDS-associated KS presents as a morewidespread, multifocal disorder where life-threateningcomplications can result from visceral involvement.
KS arises in individuals infected with human herpes virus 8 [alsoknown as KS-associated herpes virus (KSHV)] (Chang et al., 1994).Since the discovery of KSHV, there has been considerable
investigation of the genetics and oncogenic potential of this virus(reviewed by Ganem, 2010). The KSHV genome encodes viralhomologs of cellular genes that promote the cell cycle and cellularimmortalization, inhibit apoptosis, stimulate angiogenesis, andenable infected cells to evade the immune system (Boshoff et al.,1995; Bais et al., 1998; Knight et al., 2001; Bais et al., 2003; Sodhiet al., 2006; Chang et al., 2009). Consequently, activation of cellularoncogenes and mutation of tumor suppressor genes might play aless substantial role in the initiation of KS relative to other sarcomas.Although TP53 (encoding p53) mutations seem to be rare in KS,p53 protein function can be suppressed through direct interactionwith the KSHV-encoded latency-associated nuclear antigen
Classic KS
AIDS- and transplant-associatedKS
Angiosarcoma VEGF pathway dysfunction p53 mutation p14ARF inactivation Mdm-2 overexpression p16INK4a silencing Ras mutation ALT Complex or unstable karyotype
Malignant hemangioendothelioma
VEGF pathway dysfunction p53 dysfunction Unstable karyotype
Spontaneous regression
Spontaneous regression Local
invasion VEGF pathway dysfunction NFκB pathway dysfunction Cyclin D1 amplification Simple karyotype
Hemangioma
Hemangioendothelioma Wild-type p53 Simple karyotype
VEGF pathway dysfunction p53 dysfunction (LANA inactivated) mTOR activation (KSHV activated) Telomerase activation (KSHV activated) Simple or complex karyotype p53-p21Cip1 activation
p16INK4a -pRb activation Aneuploidy
Senescent ECs
KSHV infection
Telomere shortening
A Senescence
B Benign
C Intermediate
D Malignant
Normal ECs
Fig. 1. Molecular alterations underlying vascular neoplasias. (A)Normal ECs (illustrated in the center) have a finite replicative lifespan that is dictated by theshortening of telomeres with each cell division. Critical shortening of telomeres leads to activation of p53 and pRb pathways, and the onset of senescence. (B-D)Abnormal proliferation of ECs is a common feature of vascular neoplasias. Abnormalities in the VEGF pathway are common across the spectrum of vascularneoplasias. Other mechanisms implicated in oncogenesis are listed, and chromosomal characteristics are described in blue. A simple karyotype is characterizedby a near-diploid chromosome number and two or fewer karyotypic abnormalities. A complex karyotype is defined as five or more chromosomal aberrations.(B)Hemangioma is a benign condition in which hyperproliferation of immature ECs is driven by constitutive activation of NFκB and VEGF signaling pathways.(B,C)Infection by KSHV gives rise to classic Kaposi’s sarcoma (KS), which is relatively indolent and can either undergo spontaneous regression or can becomelocally invasive. p53 is inactivated, whereas mTOR and telomerase are activated by the KSHV-encoded protein LANA and the G-protein coupled receptor,respectively. (C)In immune-compromised individuals, KSHV often progresses to a multifocal tumor. (C,D)Hemangioendothelioma also presents in variousgrades, with defects in the p53 pathway found in malignant hemangioendothelioma, but not in intermediate-grade hemangioendothelioma. Chromosomalabnormalities are observed in intermediate-grade and malignant EC tumors. (D)In addition to p53 dysfunction, malignant hemangioendothelioma andangiosarcoma often exhibit chromosomal instability. Activation of oncogenes such as Ras is also common in angiosarcoma. EC, endothelial cell; NFκB, nuclearfactor-κB; ALT, alternate telomere lengthening; KS, Kaposi’s sarcoma; KSHV, Kaposi’s sarcoma-associated herpes virus [also known as HHV-8 (human herpes virustype-8)]; VEGF, vascular endothelial growth factor; LANA, latency-associated nuclear antigen; AIDS, acquired immune deficiency syndrome; pRb, retinoblastomatumor suppressor protein; mTOR, mammalian target of rapamycin.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Disease Models & Mechanisms 1069
Modeling endothelial cell neoplasia REVIEW
(LANA) (Friborg et al., 1999). Insights into the role of KSHV inthe pathogenesis of KS have provided a platform for several earlyphase clinical trials of new therapeutic strategies, which pave theway for rapid progress towards the more effective treatment ofAIDS- and transplant-related KS. Promising novel therapeuticsinclude inhibitors of tyrosine kinase signaling and compounds thattarget the mTOR (mammalian target of rapamycin) pathway, whichare activated by the KSHV G-protein coupled receptor (Koon etal., 2005; Sodhi et al., 2006).
AngiosarcomaAngiosarcoma is a rare malignancy that makes up less than 1% ofall sarcomas. In contrast to other EC-derived neoplasias,angiosarcomas are highly malignant (Table 1; Fig. 1) (Young et al.,2010). This malignancy occurs with a peak incidence in the seventhdecade of life and tends to arise in the viscera, skin and soft tissue.Angiosarcomas have a propensity to recur locally and spread widely,exhibiting a high rate of lymph node and systemic metastases. Thedisease is associated with a high mortality rate as a result of poorresponse to treatment (Mark et al., 1996; Lezama-del Valle et al.,1998). With a reported 5-year survival rate of ~20% and very littlechange in treatment options over the past decades, innovativetherapies are urgently needed. Tumor resection with adjuvantradiotherapy is currently the main treatment approach toangiosarcoma, although a few groups have recently begun topursue biological and molecular targeted approaches (Koontz etal., 2008; George et al., 2009; Maki et al., 2009; Young et al., 2010).A more detailed understanding of the molecular pathways involvedin malignant transformation of ECs and more extensive preclinicalstudies will be invaluable for the validation of these therapies andthe optimization of effective therapeutic strategies.
Most cases of angiosarcoma arise sporadically; however, previousirradiation, exposure to toxic chemicals and chronic lymphedemaare known risk factors (Mark et al., 1996; Lezama-del Valle et al.,1998). Activation of the oncogene K-ras through acquisition ofspecific point mutations is a common event in angiosarcoma,reportedly occurring in 29% of hepatic angiosarcomas (7 out of 24)and 60% of cardiac angiosarcomas (3 out of 5) (Przygodzki et al.,1997; Boivin-Angèle et al., 2000; Garcia et al., 2000). The incidenceof K-ras mutations seems to be highest among angiosarcomasassociated with exposure to vinyl chloride, where mutations havebeen detected in ~80% of cases (5 out of 6 cases tested) (Boivin-Angèle et al., 2000). Ras mutations are likely to contribute to thehighly metastatic nature of angiosarcomas, given that they aregenerally not found in non-malignant vascular neoplasias (Table 1).The malignant growth of angiosarcoma is driven by theproliferation of pleomorphic cells that exhibit varying degrees ofEC differentiation and give rise to a chaotic tissue architecture(Young et al., 2010). VEGF is expressed at high levels inangiosarcoma and the expression of VEGF receptors is associatedwith a more differentiated tumor phenotype and favorableprognosis (Zietz et al., 1998; Itakura et al., 2008). Consistent withthose observations, inhibition of VEGF receptor 1 (VEGFR1) hasbeen shown to increase the proliferation of canine angiosarcomacells in vitro (Tamburini et al., 2009).
Abnormalities in the TP53 pathway – including p53 mutations,Mdm-2 overexpression and inactivation of p14ARF – are commonmolecular aberrations in angiosarcoma (Hollstein et al., 1994; Zietz
et al., 1998; Weihrauch et al., 2002). For example, mutations in theTP53 gene were detected in ~30% of liver angiosarcomas(Przygodzki et al., 1997; Weihrauch et al., 2002). Consistent withthe high frequency of p53 dysfunction, complex karyotypes andchromosomal instability are often observed in angiosarcomas.Chromosomal aberrations reported in angiosarcomas includedeletions within the CDKN2A locus at chromosome region 9p21,which encodes tumor suppressors p16INK4a, p15INK4b and p14ARF.Inactivation of p16INK4a by promoter methylation is anotherfrequent event in liver angiosarcoma (Tannapfel et al., 2001;Weihrauch et al., 2002). Studies of liver angiosarcoma showed thatp16INK4a was repressed by promoter methylation in 63% (12 out of19) of tumor samples, whereas 5% (1 out of 19) exhibitedhomozygous deletion and 10% (2 of 19) exhibited loss ofheterozygosity at the CDKN2A locus. Overall, inactivation ofp16INK4a was detected in 74% (14 out of 19) of angiosarcomas. Theimportance of p16INK4a repression in angiosarcoma was also evidentfrom canine studies that showed sustained expression of p16INK4a
in 100% of benign hemangiomas (a total of ten were examined),and p16INK4a suppression in 82% (32 out of 39) angiosarcomas(Yonemaru et al., 2007). Together, these studies implicate Rasactivation plus aberrations in the p53 pathway and silencing ofp16INK4a as key events in the pathogenesis of advanced, but notbenign, vascular tumors (Table 1, Fig. 1).
Immortalization and tumorigenic conversion of human ECsThe limited replicative potential of normal human ECsA fundamental step in the development of endothelial neoplasiasis the deregulation of cell proliferation. Normal human cells havea finite replicative ability, yet cancer cells are capable of unlimitedreplication, i.e. they are immortal. Cellular immortality isconsidered to be a requirement for malignant transformation andis a defining property of the subset of tumor cells that drive tumorgrowth and recurrence (Zhou et al., 2009). Previous studies haverevealed that the in vitro replicative potential of normal humancells, including ECs, is restricted by both intrinsically andextrinsically initiated stresses that induce senescence (e.g. telomereshortening and an oxidative environment, respectively) (Hayflickand Moorhead, 1961; Johnson and Longenecker, 1982; Yuan et al.,2008). Senescent ECs typically adopt a flattened morphology anddemonstrate cytoplasmic spreading, increased granularity,vacuolization and multi-nucleation. They are resistant to mitogenicstimulation and exhibit pH-dependent β-galactosidase activity(Johnson and Longenecker, 1982; Dimri et al., 1995).
It is well established that the shortening of chromosomal-endstructures, referred to as telomeres, plays a central role in the onsetof senescence (Artandi and DePinho, 2010). Telomeres normallyfunction to prevent chromosomal end-to-end fusions and tomaintain genomic integrity during cell division. However, telomericDNA is eroded during consecutive cell divisions as a consequenceof the inability of DNA polymerases to synthesize the 5� terminusof linear DNA. Exposure to oxidizing agents accelerates telomereshortening and the onset of EC senescence; conversely, free radicalscavengers reduce the rate of telomere shortening (von Zglinickiet al., 1995; Xu et al., 2000; Kurz et al., 2004; Napier et al., 2010).The progressive shortening of telomeres has been demonstratedin serially passaged cultures of human ECs derived from the
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

dmm.biologists.org1070
Modeling endothelial cell neoplasiaREVIEW
umbilical vein, iliac arteries and veins, abdominal aorta, and bonemarrow (Chang and Harley, 1995; Aviv et al., 2001; MacKenzie etal., 2002). Telomere shortening has also been demonstrated inarterial ECs in vivo in association with increasing age (Okuda etal., 2000).
When telomeres reach a critically short length, they are proneto fusion and promote chromosome rearrangements. Hence,primary ECs that are maintained in culture for long periods of timefrequently harbor aberrant chromosomes (Wagner et al., 2001). Inaddition to structural rearrangements, changes in wholechromosome number (aneuploidy) tend to occur as cultured ECsage and approach senescence in vitro (Nichols et al., 1987; Johnsonet al., 1992; Zhang et al., 2000; Aviv et al., 2001). In normal humancells, chromosome fusions and rearrangements initiate a DNAdamage response that culminates in activation of tumor suppressorp53 and transcriptional upregulation of p21Cip1/Waf1, which haltscell cycle progression and initiates senescence (Stein et al., 1999).Tumor suppressor p16INK4a is subsequently upregulated, resultingin hypo-phosphorylation of the retinoblastoma tumor suppressorprotein (pRb) and a sustained growth arrest (Munro et al., 1999;Wagner et al., 2001; Freedman, 2005). The activation of tumorsuppressor pathways and induction of senescence plays a crucialrole in preventing replication of aged endothelial cells harboringcytogenetic aberrations. Disruption of these mechanisms enablescells to continue to proliferate and initiate neoplastic growth, asoutlined below.
Lifespan extension, immortality and genetic evolution ofECsDuring neoplastic transformation, the replicative lifespan of humancells is extended by either the activation of a telomere maintenancemechanism (described below) or inactivation of a tumor suppressorpathway (reviewed by Reddel, 2000). Inactivation of tumorsuppressor pathways enables cells to progress through the cell cyclewith critically short telomeres. However, short, dysfunctionaltelomeres that are present in proliferating cells promotechromosomal fusions and genomic instability (Ray et al., 1990;Counter et al., 1992). Telomere dysfunction in pre-malignant andcancerous cells is typically evidenced by dicentric chromosomes,unbalanced translocations, gene amplifications, deletions and ringchromosomes (Ducray et al., 1999; Gisselsson et al., 2001; O’Haganet al., 2002). These types of chromosomal aberrations are frequentlyobserved in KS and angiosarcoma, and have also been identifiedin immortalized EC lines, particularly those with defective tumorsuppressor pathways (Schuborg et al., 1998; Wong et al., 2001; Wenet al., 2006). Chromosomal instability accelerates the rate andaccumulation of molecular changes that facilitate malignanttransformation and tumor progression (Rudolph et al., 2001),including activation of a telomere maintenance mechanism,oncogene activation or silencing of tumor suppressor genes.
Under standard culture conditions, ECs that bypass senescenceusually succumb to a proliferative ‘crisis’ that is characterized byan increased rate of cell death and reduced cell expansion(MacKenzie et al., 2002; Gu et al., 2003; Nisato et al., 2004; Wenet al., 2006). Crisis is thought to be initiated by telomeredysfunction, genetic catastrophe and/or fatal oxidative damage.Cells that are driven into crisis by the deactivation of tumorsuppressor pathways resume proliferation if they undergo a
spontaneous molecular event that activates a telomere maintenancemechanism: either activation of the enzyme telomerase or analternate telomere lengthening mechanism (ALT) (Counter et al.,1992; Bryan et al., 1995). Telomere maintenance stabilizeschromosomes and thereby enables both escape from crisis andunlimited proliferation.
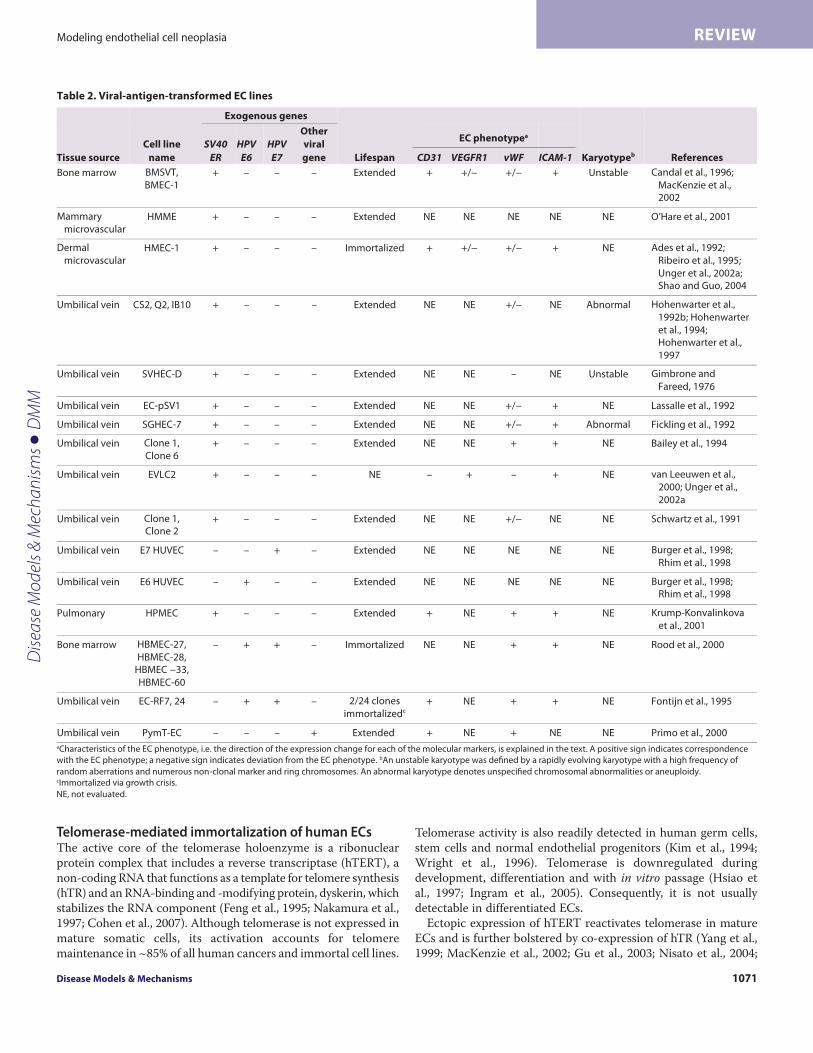
Extension of replicative lifespan and immortalization ofECs following transfection with viral antigensThe extended replicative lifespan that results from the inactivationof tumor suppressor pathways is thought to represent a pre-malignant stage of the multistep process of cancer development.Several investigations have demonstrated that the replicativelifespan of normal human ECs derived from bone marrowmicrovasculature, the umbilical vein and the iliac vein is extendedby transfection with Simian virus 40 (SV40) T antigen or humanpapilloma virus (HPV)-encoded E6 and E7 oncogenes (Table 2).The large T antigen of SV40 and the HPV E6 and E7 oncogenesfunctionally inactivate the p53 and pRb tumor suppressor pathwaysand thereby enable transfected cells to bypass senescence (Girardiet al., 1965; Werness et al., 1990; Shay et al., 1991). Viral-antigen-transfected ECs have been shown to replicate for up to 42 passagesbeyond senescence (Table 2) (Gimbrone and Fareed, 1976; Schwartzet al., 1991; Fickling et al., 1992; Hohenwarter et al., 1992b; Lassalleet al., 1992; Bailey et al., 1994; Rhim et al., 1998; Krump-Konvalinkova et al., 2001; O’Hare et al., 2001; MacKenzie et al.,2002).
A number of studies also reported that viral transformation leadsto immortalization of ECs (Ades et al., 1992; Fontijn et al., 1995;Burger et al., 1998; Rhim et al., 1998; Rood et al., 2000). However, agrowth crisis prior to immortalization was noted in several studies(Ades et al., 1992; Fontijn et al., 1995; Rood et al., 2000; MacKenzieet al., 2002) and two studies showed that telomerase wasspontaneously activated in the immortalized ECs (Rhim et al., 1998;Gagnon et al., 2002). Ectopic activation of telomerase was shown tocooperate with SV40 transformation to overcome crisis andefficiently promote immortalization of bone-marrow- andmammary-derived ECs (O’Hare et al., 2001; MacKenzie et al., 2002).Together, these studies suggest that inactivation of tumor suppressorpathways and activation of a telomere maintenance mechanism isnecessary and sufficient for in vitro immortalization of human ECs.
Immortalization of ECs by transfection with viral oncogenes andactivation of telomerase is not sufficient for malignant conversionof ECs (Burger et al., 1998; Rhim et al., 1998; MacKenzie et al.,2002). Nevertheless, viral-antigen-transfected ECs do exhibit somecharacteristics of transformed cells, including the ability toproliferate independently of exogenous VEGF, and reducedexpression of proteins that define the endothelial phenotype,including VEGFR2, platelet endothelial cell adhesion molecular-1(CD31; also known as PECAM-1), VE cadherin and von Willebrandfactor (vWF) (Table 2) (Gimbrone and Fareed, 1976; Hohenwarteret al., 1992b; Rood et al., 2000; MacKenzie et al., 2002; Shao andGuo, 2004). These features parallel the abnormal expression ofVEGF receptors and tendency for downregulation of vWF andCD31 in late-stage hemangiomas (Takahashi et al., 1994). Viral-oncogene-transformed ECs also accumulate genomicrearrangements that are similar to those observed in ECmalignancies (Gimbrone and Fareed, 1976; MacKenzie et al., 2002).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
Disease Models & Mechanisms 1071
Modeling endothelial cell neoplasia REVIEW
Telomerase-mediated immortalization of human ECsThe active core of the telomerase holoenzyme is a ribonuclearprotein complex that includes a reverse transcriptase (hTERT), anon-coding RNA that functions as a template for telomere synthesis(hTR) and an RNA-binding and -modifying protein, dyskerin, whichstabilizes the RNA component (Feng et al., 1995; Nakamura et al.,1997; Cohen et al., 2007). Although telomerase is not expressed inmature somatic cells, its activation accounts for telomeremaintenance in ~85% of all human cancers and immortal cell lines.
Telomerase activity is also readily detected in human germ cells,stem cells and normal endothelial progenitors (Kim et al., 1994;Wright et al., 1996). Telomerase is downregulated duringdevelopment, differentiation and with in vitro passage (Hsiao etal., 1997; Ingram et al., 2005). Consequently, it is not usuallydetectable in differentiated ECs.
Ectopic expression of hTERT reactivates telomerase in matureECs and is further bolstered by co-expression of hTR (Yang et al.,1999; MacKenzie et al., 2002; Gu et al., 2003; Nisato et al., 2004;
Table 2. Viral-antigen-transformed EC lines
Exogenous genes
Tissue source
Cell line
name
SV40 ER
HPV E6
HPV E7
Other
viral
gene Lifespan CD31 VEGFR1 vWF ICAM-1 Karyotypeb References
Bone marrow BMSVT,
BMEC-1 + – – – Extended + +/− +/− + Unstable Candal et al., 1996;
MacKenzie et al., 2002
Mammary
microvascular HMME + – – – Extended NE NE NE NE NE O’Hare et al., 2001
Dermal
microvascular HMEC-1 + – – – Immortalized + +/− +/− + NE Ades et al., 1992;
Ribeiro et al., 1995;
Unger et al., 2002a; Shao and Guo, 2004
Umbilical vein CS2, Q2, IB10 + – – – Extended NE NE +/− NE Abnormal Hohenwarter et al.,
1992b; Hohenwarter
et al., 1994; Hohenwarter et al.,
1997
Umbilical vein SVHEC-D + – – – Extended NE NE – NE Unstable Gimbrone and
Fareed, 1976
Umbilical vein EC-pSV1 + – – – Extended NE NE +/− + NE Lassalle et al., 1992
Umbilical vein SGHEC-7 + – – – Extended NE NE +/− + Abnormal Fickling et al., 1992
Umbilical vein Clone 1,
Clone 6 + – – – Extended NE NE + + NE Bailey et al., 1994
Umbilical vein EVLC2 + – – – NE – + – + NE van Leeuwen et al.,
2000; Unger et al.,
2002a
Umbilical vein Clone 1, Clone 2
+ – – – Extended NE NE +/− NE NE Schwartz et al., 1991
Umbilical vein E7 HUVEC – – + – Extended NE NE NE NE NE Burger et al., 1998;
Rhim et al., 1998
Umbilical vein E6 HUVEC – + – – Extended NE NE NE NE NE Burger et al., 1998; Rhim et al., 1998
Pulmonary HPMEC + – – – Extended + NE + + NE Krump-Konvalinkova
et al., 2001
Bone marrow HBMEC-27,
HBMEC-28, HBMEC −33,
HBMEC-60
– + + – Immortalized NE NE + + NE Rood et al., 2000
Umbilical vein EC-RF7, 24 – + + – 2/24 clones
immortalizedc + NE + + NE Fontijn et al., 1995
Umbilical vein PymT-EC – – – + Extended + NE + NE NE Primo et al., 2000 aCharacteristics of the EC phenotype, i.e. the direction of the expression change for each of the molecular markers, is explained in the text. A positive sign indicates correspondence
with the EC phenotype; a negative sign indicates deviation from the EC phenotype. bAn unstable karyotype was defined by a rapidly evolving karyotype with a high frequency of
random aberrations and numerous non-clonal marker and ring chromosomes. An abnormal karyotype denotes unspecified chromosomal abnormalities or aneuploidy.
cImmortalized via growth crisis.
NE, not evaluated.
EC phenotypea
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
dmm.biologists.org1072
Modeling endothelial cell neoplasiaREVIEW
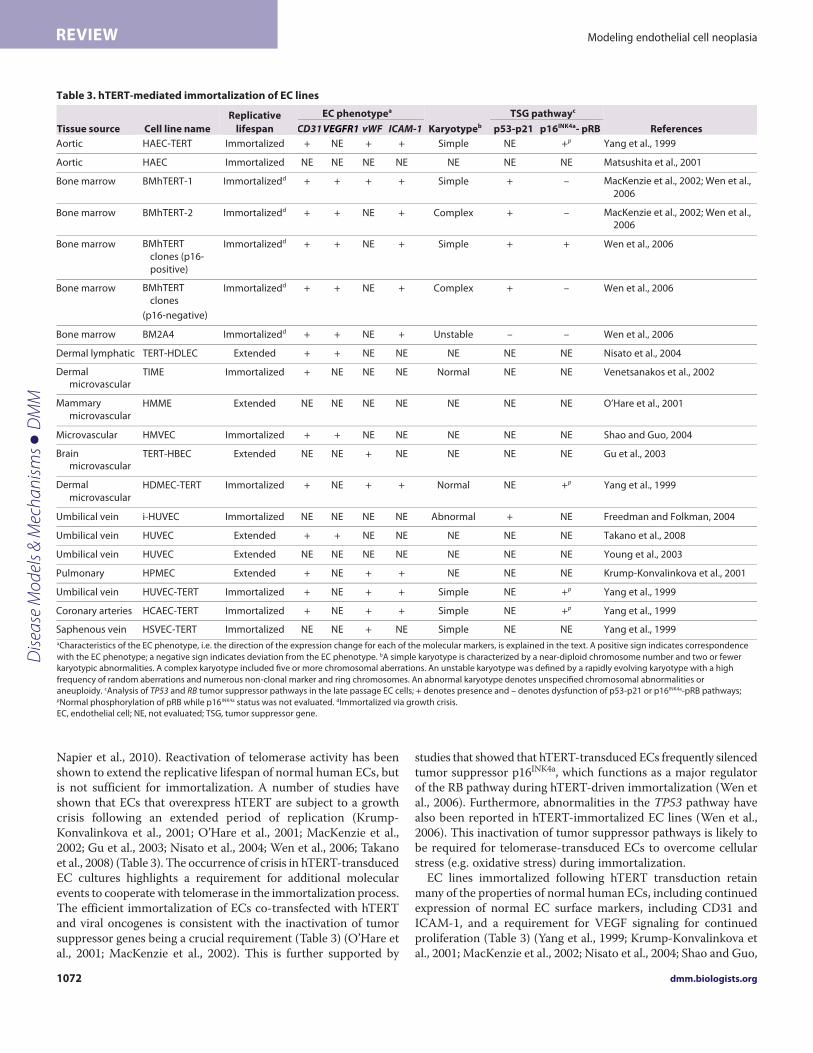
Napier et al., 2010). Reactivation of telomerase activity has beenshown to extend the replicative lifespan of normal human ECs, butis not sufficient for immortalization. A number of studies haveshown that ECs that overexpress hTERT are subject to a growthcrisis following an extended period of replication (Krump-Konvalinkova et al., 2001; O’Hare et al., 2001; MacKenzie et al.,2002; Gu et al., 2003; Nisato et al., 2004; Wen et al., 2006; Takanoet al., 2008) (Table 3). The occurrence of crisis in hTERT-transducedEC cultures highlights a requirement for additional molecularevents to cooperate with telomerase in the immortalization process.The efficient immortalization of ECs co-transfected with hTERTand viral oncogenes is consistent with the inactivation of tumorsuppressor genes being a crucial requirement (Table 3) (O’Hare etal., 2001; MacKenzie et al., 2002). This is further supported by
studies that showed that hTERT-transduced ECs frequently silencedtumor suppressor p16INK4a, which functions as a major regulatorof the RB pathway during hTERT-driven immortalization (Wen etal., 2006). Furthermore, abnormalities in the TP53 pathway havealso been reported in hTERT-immortalized EC lines (Wen et al.,2006). This inactivation of tumor suppressor pathways is likely tobe required for telomerase-transduced ECs to overcome cellularstress (e.g. oxidative stress) during immortalization.
EC lines immortalized following hTERT transduction retainmany of the properties of normal human ECs, including continuedexpression of normal EC surface markers, including CD31 andICAM-1, and a requirement for VEGF signaling for continuedproliferation (Table 3) (Yang et al., 1999; Krump-Konvalinkova etal., 2001; MacKenzie et al., 2002; Nisato et al., 2004; Shao and Guo,
Table 3. hTERT-mediated immortalization of EC lines
Tissue source Cell line name Replicative
lifespan
EC phenotypea
Karyotypeb
TSG pathwayc
References CD31 vWF ICAM-1 p53-p21 p16INK4a- pRB
Aortic HAEC-TERT Immortalized + NE + + Simple NE +p Yang et al., 1999
Aortic HAEC Immortalized NE NE NE NE NE NE NE Matsushita et al., 2001
Bone marrow BMhTERT-1 Immortalizedd + + + + Simple + – MacKenzie et al., 2002; Wen et al.,
2006
Bone marrow BMhTERT-2 Immortalizedd + + NE + Complex + – MacKenzie et al., 2002; Wen et al., 2006
Bone marrow BMhTERT
clones (p16-
positive)
Immortalizedd + + NE + Simple + + Wen et al., 2006
Bone marrow BMhTERT
clones
(p16-negative)
Immortalizedd + + NE + Complex + – Wen et al., 2006
Bone marrow BM2A4 Immortalizedd + + NE + Unstable – – Wen et al., 2006
Dermal lymphatic TERT-HDLEC Extended + + NE NE NE NE NE Nisato et al., 2004
Dermal microvascular
TIME Immortalized + NE NE NE Normal NE NE Venetsanakos et al., 2002
Mammary
microvascular HMME Extended NE NE NE NE NE NE NE O’Hare et al., 2001
Microvascular HMVEC Immortalized + + NE NE NE NE NE Shao and Guo, 2004
Brain microvascular
TERT-HBEC Extended NE NE + NE NE NE NE Gu et al., 2003
Dermal
microvascular HDMEC-TERT Immortalized + NE + + Normal NE +p Yang et al., 1999
Umbilical vein i-HUVEC Immortalized NE NE NE NE Abnormal + NE Freedman and Folkman, 2004
Umbilical vein HUVEC Extended + + NE NE NE NE NE Takano et al., 2008
Umbilical vein HUVEC Extended NE NE NE NE NE NE NE Young et al., 2003
Pulmonary HPMEC Extended + NE + + NE NE NE Krump-Konvalinkova et al., 2001
Umbilical vein HUVEC-TERT Immortalized + NE + + Simple NE +p Yang et al., 1999
Coronary arteries HCAEC-TERT Immortalized + NE + + Simple NE +p Yang et al., 1999
Saphenous vein HSVEC-TERT Immortalized NE NE + NE Simple NE NE Yang et al., 1999 aCharacteristics of the EC phenotype, i.e. the direction of the expression change for each of the molecular markers, is explained in the text. A positive sign indicates correspondence
with the EC phenotype; a negative sign indicates deviation from the EC phenotype. bA simple karyotype is characterized by a near-diploid chromosome number and two or fewer
karyotypic abnormalities. A complex karyotype included five or more chromosomal aberrations. An unstable karyotype was defined by a rapidly evolving karyotype with a high
frequency of random aberrations and numerous non-clonal marker and ring chromosomes. An abnormal karyotype denotes unspecified chromosomal abnormalities or
aneuploidy. cAnalysis of TP53 and RB tumor suppressor pathways in the late passage EC cells; + denotes presence and – denotes dysfunction of p53-p21 or p16INK4a-pRB pathways; pNormal phosphorylation of pRB while p16INK4a status was not evaluated. dImmortalized via growth crisis.
EC, endothelial cell; NE, not evaluated; TSG, tumor suppressor gene.
VEGFR1
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Disease Models & Mechanisms 1073
Modeling endothelial cell neoplasia REVIEW
2004; Takano et al., 2008; Dill et al., 2012). Interestingly, somehTERT-transduced EC lines were also shown to form functionalvessels in vivo and in vitro (Yang et al., 2001; MacKenzie et al., 2002;Freedman and Folkman, 2004). Thus, despite their deregulatedreplicative lifespan, hTERT-immortalized ECs generally seem toretain more accurately the properties of normal, untransformedECs than do viral-antigen-transformed ECs. Nevertheless, it isimportant to note that spontaneous molecular alterations that occurduring hTERT-driven immortalization can confer neoplasticproperties. For instance, our recent investigations revealed thatrepression of p16INK4a during immortalization of ECs is associatedwith cytoskeletal changes, altered motility and defects inmorphogenesis in vitro (Kan et al., 2012). These changes are likelyto reflect some of the phenotypic changes required for theacquisition of metastatic ability in vivo.
Repression of p16INK4a by promoter methylation in hTERT-immortalized ECs is a molecular event that has also been observedin angiosarcoma (Fig. 1) (Tannapfel et al., 2001; Weihrauch et al.,2002). In our study of hTERT-transduced bone marrow ECs(BMECs), the subset of clones that repressed p16INK4a all developedcomplex karyotypes (Wen et al., 2006). The cytogeneticheterogeneity and abnormalities detected in the p16INK4a-negativeBMECs were typical of those that have been documented in EC-derived tumors. Specific aberrations that recur in p16INK4a-repressed cell lines and are also observed in angiosarcoma includemonosomy 13, loss of the Y chromosome, rearrangements involving17p, and trisomies 20 and 11 (Mandahl et al., 1990; Kindblom etal., 1991; Gil-Benso et al., 1994; Cerilli et al., 1998; Schuborg et al.,1998; Wong et al., 2001; Zu et al., 2001; Baumhoer et al., 2005;Tsarouha et al., 2006; Wen et al., 2006). Repression of p16INK4a andthe concurrent development of karyotypic complexity is a feasibledepiction of the molecular alterations that permit the accumulationof genomic imbalances in EC neoplasias.
Relatively little is known about the mechanism(s) that supporttelomere maintenance in the various types of human vascularneoplasias in patients. However, one study demonstratedtelomerase activity in 22 out of 22 KS lesions (Chen et al., 2001),and others have shown that KSHV-encoded LANA directlyactivates Sp1-mediated transcription of hTERT (Knight et al., 2001;Verma et al., 2004). In contrast, an assessment of telomerase activityin canine angiosarcomas showed no detectable telomerase enzymeactivity in 6 out of 7 cases (Yazawa et al., 1999; Chandler et al.,2009). Telomerase activity was also undetectable in two single-clone-derived angiosarcoma cell lines, ISO-HAS.1 and AS-M.5(Krump-Konvalinkova et al., 2003). These studies suggest that ALTcould be a common mechanism of telomere length maintenancein angiosarcoma. However, this possibility remains to be confirmedby demonstration of the specific molecular characteristics of ALT-immortalized cells, such as very long heterogeneous telomerelengths, DNA c-circles and PML bodies in angiosarcoma tissue(Bryan et al., 1997; Henson et al., 2005; Fasching et al., 2007).
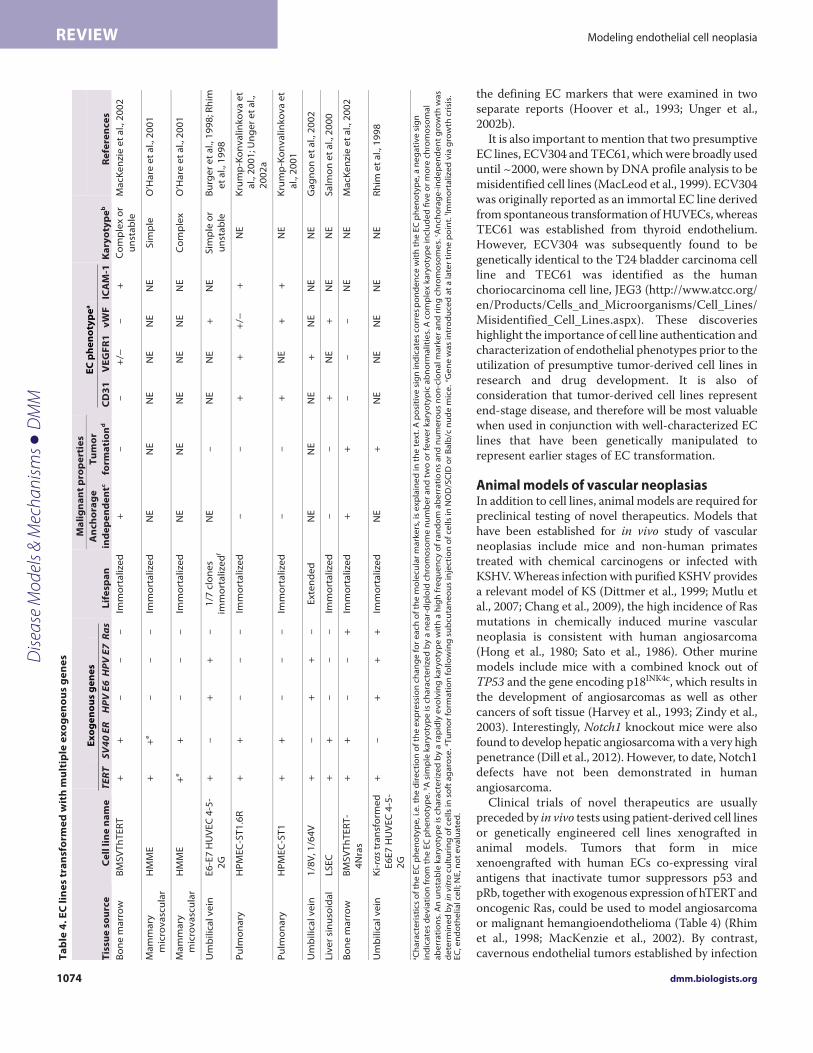
Tumorigenic conversion of human EC linesImmortalized EC lines established by co-expression of hTERT andSV40 T antigen exhibit a partially transformed phenotype (Table 4)(Salmon et al., 2000; O’Hare et al., 2001; MacKenzie et al., 2002;Krump-Konvalinkova et al., 2003). BMECs that co-expressed SV40T antigen and hTERT were shown to be capable of robust
anchorage-independent growth in soft-agarose, but were notovertly tumorigenic, because implantation in immune-compromised mice resulted in very small subcutaneous nodulesthat regressed within 3 weeks (MacKenzie et al., 2002). Pulmonaryand sinusoidal ECs transformed with hTERT and SV40 ER weresimilarly non-tumorigenic (Salmon et al., 2000; Krump-Konvalinkova et al., 2001). In addition to the downregulation ofphenotypic markers, ECs immortalized by SV40 T antigen grewindependently of exogenous VEGF (Tables 2, 4), which is consistentwith the constitutive activation of VEGF signaling andheterogeneous phenotype observed in human vascular neoplasias(Ohsawa et al., 1995; MacKenzie et al., 2002; Bais et al., 2003; Itakuraet al., 2008; Young et al., 2010).
Overexpression of oncogenic N-Ras mediated tumorigenicconversion of immortalized BMECs (MacKenzie et al., 2002).Upon subcutaneous injection into immunocompromised mice,BMECs that co-expressed hTERT, SV40 T antigen and N-Rasgenerated large, rapidly growing tumors that were organized intolumen-like structures and in some mice metastasized to lymphnodes (MacKenzie et al., 2002). When injected via the tail vein, theECs lodged within the lungs, where they formed tumors that causedmorbidity within 3 weeks of implantation. Similarly, humanumbilical vein endothelial cells (HUVECs) transfected with HPVE6 and E7 oncogenes and immortalized via spontaneous activationof telomerase were vulnerable to tumorigenic conversion bytransfection with activated K-ras (Rhim et al., 1998). Thedemonstration of K-ras mutations in angiosarcoma underscores therelevance of these models to human vascular neoplasias (Boivin-Angèle et al., 2000; Hong et al., 2000).
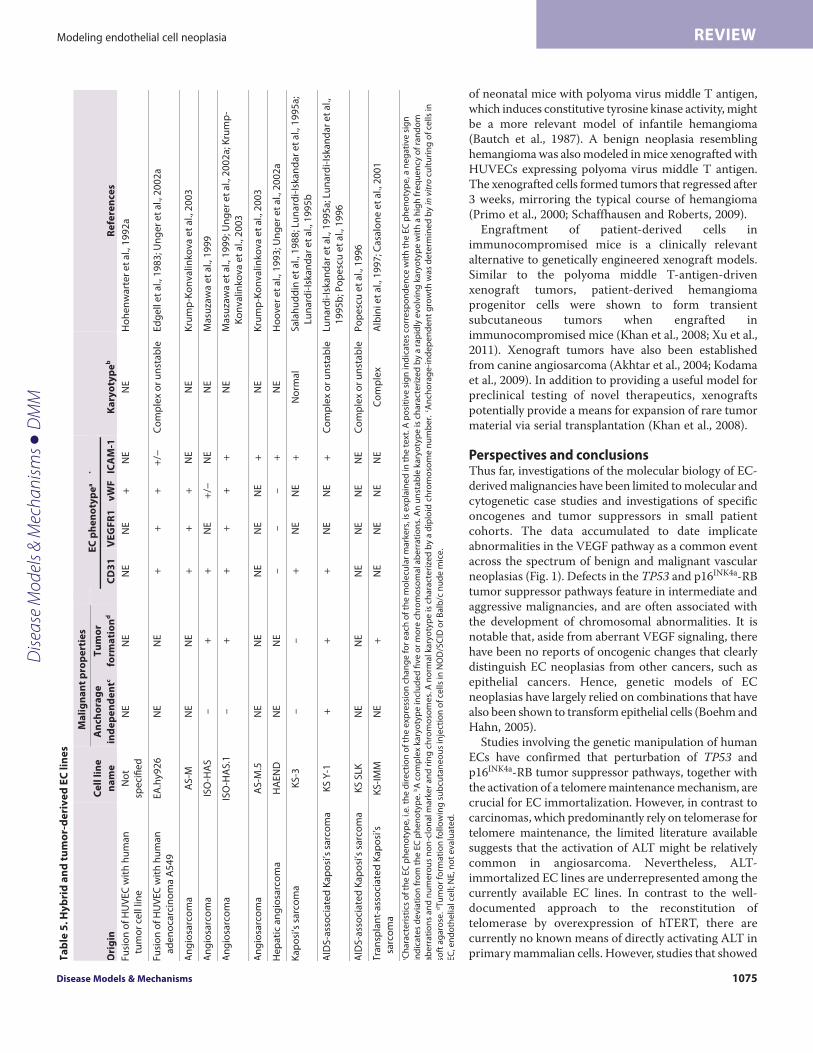
EC hybrid and vascular tumor-derived cell linesIn addition to genetically engineered ECs, immortalized EC lineshave been generated by fusion of HUVECs with human tumor celllines, such as the adenocarcinoma A549 cell line (Table 5) (Edgellet al., 1983; Hohenwarter et al., 1992a; Unger et al., 2002b). Hybridendothelial tumor cell lines have unlimited replicative potential andretain some, but not all, EC characteristics (Unger et al., 2002a).In addition, because the hybrid EC A549 cell line displays karyotypicchanges that have also been identified in A549 cells, it does notseem an ideal representation of the molecular biology of endothelialtumor cells (Edgell et al., 1983).
Authentic EC lines isolated directly from vascular tumors areclearly valuable for studies of the molecular biology of the disease.Cell lines established by ex vivo culture of human tumor tissuesinclude the angiosarcoma-derived cell lines ISO-HAS, AS-M andHAEND (Hoover et al., 1993; Masuzawa et al., 1999; Unger et al.,2002a; Krump-Konvalinkova et al., 2003). Aberrant p53 expression,a lack of telomerase activity and an ability to form tumors in vivo(evaluated in ISO-HAS and ISO-HAS.1) are some of thecharacteristics that angiosarcoma-derived clonal EC lines have incommon with primary angiosarcomas. At least four KS-derivedEC lines have been established from AIDS- and non-AIDS-associated KS patients (Table 5) (Salahuddin et al., 1988; Lunardi-Iskandar et al., 1995b; Popescu et al., 1996; Albini et al., 1997;Casalone et al., 2001). Most of the reported EC-tumor-derived celllines retained typical endothelial characteristics, includingexpression of CD31, VEGFR1, vWF and I-CAM. One exceptionwas the HAEND cell line, which suppressed expression of all of
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
dmm.biologists.org1074
Modeling endothelial cell neoplasiaREVIEW
the defining EC markers that were examined in twoseparate reports (Hoover et al., 1993; Unger et al.,2002b).
It is also important to mention that two presumptiveEC lines, ECV304 and TEC61, which were broadly useduntil ~2000, were shown by DNA profile analysis to bemisidentified cell lines (MacLeod et al., 1999). ECV304was originally reported as an immortal EC line derivedfrom spontaneous transformation of HUVECs, whereasTEC61 was established from thyroid endothelium.However, ECV304 was subsequently found to begenetically identical to the T24 bladder carcinoma cellline and TEC61 was identified as the humanchoriocarcinoma cell line, JEG3 (http://www.atcc.org/en/Products/Cells_and_Microorganisms/Cell_Lines/Misidentified_Cell_Lines.aspx). These discoverieshighlight the importance of cell line authentication andcharacterization of endothelial phenotypes prior to theutilization of presumptive tumor-derived cell lines inresearch and drug development. It is also ofconsideration that tumor-derived cell lines representend-stage disease, and therefore will be most valuablewhen used in conjunction with well-characterized EClines that have been genetically manipulated torepresent earlier stages of EC transformation.
Animal models of vascular neoplasiasIn addition to cell lines, animal models are required forpreclinical testing of novel therapeutics. Models thathave been established for in vivo study of vascularneoplasias include mice and non-human primatestreated with chemical carcinogens or infected withKSHV. Whereas infection with purified KSHV providesa relevant model of KS (Dittmer et al., 1999; Mutlu etal., 2007; Chang et al., 2009), the high incidence of Rasmutations in chemically induced murine vascularneoplasia is consistent with human angiosarcoma(Hong et al., 1980; Sato et al., 1986). Other murinemodels include mice with a combined knock out ofTP53 and the gene encoding p18INK4c, which results inthe development of angiosarcomas as well as othercancers of soft tissue (Harvey et al., 1993; Zindy et al.,2003). Interestingly, Notch1 knockout mice were alsofound to develop hepatic angiosarcoma with a very highpenetrance (Dill et al., 2012). However, to date, Notch1defects have not been demonstrated in humanangiosarcoma.
Clinical trials of novel therapeutics are usuallypreceded by in vivo tests using patient-derived cell linesor genetically engineered cell lines xenografted inanimal models. Tumors that form in micexenoengrafted with human ECs co-expressing viralantigens that inactivate tumor suppressors p53 andpRb, together with exogenous expression of hTERT andoncogenic Ras, could be used to model angiosarcomaor malignant hemangioendothelioma (Table 4) (Rhimet al., 1998; MacKenzie et al., 2002). By contrast,cavernous endothelial tumors established by infectionT
ab
le 4
. E
C l
ine
s tr
an
sfo
rme
d w
ith
mu
ltip
le e
xo
ge
no
us
ge
ne
s
Tis
sue
so
urc
e
Ce
ll l
ine
na
me
Lif
esp
an
Ma
lig
na
nt
pro
pe
rtie
s
Ka
ryo
typ
eb
TER
T SV
40 E
R
A
nc
ho
rag
e
ind
ep
en
de
ntc
Tu
mo
r
form
ati
on
d
CD
31
V
EG
FR
1
vW
F
ICA
M-1
R
efe
ren
ce
s
Bo
ne
ma
rro
w
BM
SV
Th
TE
RT
+
+
–
–
–
Im
mo
rta
lize
d
+
–
–
+/−
–
+
C
om
ple
x o
r
un
sta
ble
M
acK
en
zie
et
al.,
20
02
Ma
mm
ary
mic
rov
asc
ula
r H
MM
E
+
+e
–
–
–
Imm
ort
ali
zed
N
E
NE
N
E
NE
N
E
NE
S
imp
le
O’H
are
et
al.,
20
01
Ma
mm
ary
m
icro
va
scu
lar
HM
ME
+
e
+
–
–
–
Imm
ort
ali
zed
N
E
NE
N
E
NE
N
E
NE
C
om
ple
x
O’H
are
et
al.,
20
01
Um
bil
ica
l ve
in
E6
-E7
HU
VE
C 4
-5-
2G
+
–
+
+
–
1
/7 c
lon
es
imm
ort
ali
zed
f N
E
–
NE
N
E
+
NE
S
imp
le o
r
un
sta
ble
Bu
rge
r e
t a
l., 1
99
8; R
him
et
al.,
19
98
Pu
lmo
na
ry
HP
ME
C-S
T1
.6R
+
+
–
–
–
Im
mo
rta
lize
d
–
–
+
+
+/−
+
N
E
Kru
mp
-Ko
nv
ali
nk
ov
a e
t a
l., 2
00
1; U
ng
er
et
al.,
20
02
a
Pu
lmo
na
ry
HP
ME
C-S
T1
+
+
–
–
–
Im
mo
rta
lize
d
–
–
+
NE
+
+
N
E
Kru
mp
-Ko
nv
ali
nk
ov
a e
t
al.,
20
01
Um
bil
ica
l ve
in
1/8
V, 1
/64
V
+
–
+
+
–
Ex
ten
de
d
NE
N
E
NE
+
N
E
NE
N
E
Ga
gn
on
et
al.,
20
02
Liv
er
sin
uso
ida
l L
SE
C
+
+
–
–
–
Imm
ort
ali
zed
–
–
+
N
E
+
NE
N
E
Sa
lmo
n e
t a
l., 2
00
0
Bo
ne
ma
rro
w
BM
SV
Th
TE
RT
-
4N
ras
+
+
–
–
+
Imm
ort
ali
zed
+
+
–
–
–
N
E
NE
M
acK
en
zie
et
al.,
20
02
Um
bil
ica
l ve
in
Ki-
ras
tra
nsf
orm
ed
E6
E7
HU
VE
C 4
-5-
2G
+
–
+
+
+
Imm
ort
ali
zed
N
E
+
NE
N
E
NE
N
E
NE
R
him
et
al.,
19
98
aC
ha
ract
eri
stic
s o
f th
e E
C p
he
no
typ
e, i
.e. t
he
dir
ect
ion
of
the
ex
pre
ssio
n c
ha
ng
e f
or
ea
ch o
f th
e m
ole
cula
r m
ark
ers
, is
ex
pla
ine
d in
th
e t
ex
t. A
po
siti
ve
sig
n in
dic
ate
s co
rre
sp
on
de
nce
wit
h t
he
EC
ph
en
oty
pe
, a n
eg
ati
ve
sig
n
ind
ica
tes
de
via
tio
n f
rom
th
e E
C p
he
no
typ
e. b
A s
imp
le k
ary
oty
pe
is c
ha
ract
eri
zed
by
a n
ea
r-d
iplo
id c
hro
mo
som
e n
um
be
r a
nd
tw
o o
r fe
we
r k
ary
oty
pic
ab
no
rma
liti
es.
A c
om
ple
x k
ary
oty
pe
incl
ud
ed
fiv
e o
r m
ore
ch
rom
oso
ma
l
ab
err
ati
on
s. A
n u
nst
ab
le k
ary
oty
pe
is c
ha
ract
eri
zed
by
a r
ap
idly
ev
olv
ing
ka
ryo
typ
e w
ith
a h
igh
fre
qu
en
cy o
f ra
nd
om
ab
err
ati
on
s a
nd
nu
me
rou
s n
on
-clo
na
l ma
rke
r a
nd
rin
g c
hro
mo
som
es.
cA
nch
ora
ge
-in
de
pe
nd
en
t g
row
th w
as
de
term
ine
d b
y in
vitr
o cu
ltu
rin
g o
f ce
lls
in s
oft
ag
aro
se. d
Tu
mo
r fo
rma
tio
n f
oll
ow
ing
su
bcu
tan
eo
us
inje
ctio
n o
f ce
lls
in N
OD
/SC
ID o
r B
alb
/c n
ud
e m
ice
. eG
en
e w
as
intr
od
uce
d a
t a
late
r ti
me
po
int.
f Imm
ort
ali
zed
via
gro
wth
cri
sis.
E
C, e
nd
oth
eli
al c
ell
; NE
, no
t e
va
lua
ted
.
Ex
og
en
ou
s g
en
es
HPV
E6
HPV
E7
Ras
EC
ph
en
oty
pe
a
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
Disease Models & Mechanisms 1075
Modeling endothelial cell neoplasia REVIEW
of neonatal mice with polyoma virus middle T antigen,which induces constitutive tyrosine kinase activity, mightbe a more relevant model of infantile hemangioma(Bautch et al., 1987). A benign neoplasia resemblinghemangioma was also modeled in mice xenografted withHUVECs expressing polyoma virus middle T antigen.The xenografted cells formed tumors that regressed after3 weeks, mirroring the typical course of hemangioma(Primo et al., 2000; Schaffhausen and Roberts, 2009).
Engraftment of patient-derived cells inimmunocompromised mice is a clinically relevantalternative to genetically engineered xenograft models.Similar to the polyoma middle T-antigen-drivenxenograft tumors, patient-derived hemangiomaprogenitor cells were shown to form transientsubcutaneous tumors when engrafted inimmunocompromised mice (Khan et al., 2008; Xu et al.,2011). Xenograft tumors have also been establishedfrom canine angiosarcoma (Akhtar et al., 2004; Kodamaet al., 2009). In addition to providing a useful model forpreclinical testing of novel therapeutics, xenograftspotentially provide a means for expansion of rare tumormaterial via serial transplantation (Khan et al., 2008).
Perspectives and conclusionsThus far, investigations of the molecular biology of EC-derived malignancies have been limited to molecular andcytogenetic case studies and investigations of specificoncogenes and tumor suppressors in small patientcohorts. The data accumulated to date implicateabnormalities in the VEGF pathway as a common eventacross the spectrum of benign and malignant vascularneoplasias (Fig. 1). Defects in the TP53 and p16INK4a-RBtumor suppressor pathways feature in intermediate andaggressive malignancies, and are often associated withthe development of chromosomal abnormalities. It isnotable that, aside from aberrant VEGF signaling, therehave been no reports of oncogenic changes that clearlydistinguish EC neoplasias from other cancers, such asepithelial cancers. Hence, genetic models of ECneoplasias have largely relied on combinations that havealso been shown to transform epithelial cells (Boehm andHahn, 2005).
Studies involving the genetic manipulation of humanECs have confirmed that perturbation of TP53 andp16INK4a-RB tumor suppressor pathways, together withthe activation of a telomere maintenance mechanism, arecrucial for EC immortalization. However, in contrast tocarcinomas, which predominantly rely on telomerase fortelomere maintenance, the limited literature availablesuggests that the activation of ALT might be relativelycommon in angiosarcoma. Nevertheless, ALT-immortalized EC lines are underrepresented among thecurrently available EC lines. In contrast to the well-documented approach to the reconstitution oftelomerase by overexpression of hTERT, there arecurrently no known means of directly activating ALT inprimary mammalian cells. However, studies that showedT
ab
le 5
. Hy
bri
d a
nd
tu
mo
r-d
eri
ve
d E
C l
ine
s
Ori
gin
C
ell
lin
e
na
me
Ma
lig
na
nt
pro
pe
rtie
s
Ka
ryo
typ
eb
R
efe
ren
ces
An
cho
rag
e
ind
ep
en
de
ntc
Tu
mo
r
form
ati
on
d
CD
31
V
EG
FR
1
vW
F
ICA
M-1
Fu
sio
n o
f H
UV
EC
wit
h h
um
an
tum
or
cell
line
No
t
spe
cifi
ed
N
E
NE
NE
N
E
+
NE
N
E
Ho
he
nw
art
er
et
al.,
19
92
a
Fu
sio
n o
f H
UV
EC
wit
h h
um
an
ad
en
oca
rcin
om
a A
54
9
EA
.hy9
26
N
E
NE
+
+
+
+/−
C
om
ple
x o
r u
nst
ab
le
Ed
ge
ll e
t a
l., 1
98
3; U
ng
er
et
al.,
20
02
a
An
gio
sarc
om
a
AS
-M
NE
N
E
+
+
+
N
E
NE
K
rum
p-K
on
va
linko
va
et
al.,
20
03
An
gio
sarc
om
a
ISO
-HA
S
–
+
+
N
E
+/−
N
E
NE
M
asu
zaw
a e
t a
l., 1
99
9
An
gio
sarc
om
a
ISO
-HA
S.1
–
+
+
+
+
+
NE
M
asu
zaw
a e
t a
l., 1
99
9; U
ng
er
et
al.,
20
02
a; K
rum
p-
Ko
nv
alin
kov
a e
t a
l., 2
00
3
An
gio
sarc
om
a
AS
-M.5
N
E
NE
NE
N
E
NE
+
N
E
Kru
mp
-Ko
nv
alin
kov
a e
t a
l., 2
00
3
He
pa
tic
an
gio
sarc
om
a
HA
EN
D
NE
N
E
–
–
–
+
N
E
Ho
ov
er
et
al.,
19
93
; Un
ge
r e
t a
l., 2
00
2a
Ka
po
si’s
sa
rco
ma
K
S-3
–
–
+
NE
N
E
+
No
rma
l S
ala
hu
dd
in e
t a
l., 1
98
8; L
un
ard
i-Is
kan
da
r e
t a
l., 1
99
5a
;
Lun
ard
i-Is
kan
da
r e
t a
l., 1
99
5b
AID
S-a
sso
cia
ted
Ka
po
si’s
sa
rco
ma
K
S Y
-1
+
+
+
N
E
NE
+
C
om
ple
x o
r u
nst
ab
le
Lun
ard
i-Is
kan
da
r e
t a
l., 1
99
5a
; Lu
na
rdi-
Iska
nd
ar
et
al.,
19
95
b; P
op
esc
u e
t a
l., 1
99
6
AID
S-a
sso
cia
ted
Ka
po
si’s
sa
rco
ma
K
S S
LK
NE
N
E
N
E
NE
N
E
NE
C
om
ple
x o
r u
nst
ab
le
Po
pe
scu
et
al.,
19
96
Tra
nsp
lan
t-a
sso
cia
ted
Ka
po
si’s
sarc
om
a
KS
-IM
M
NE
+
NE
N
E
NE
N
E
Co
mp
lex
Alb
ini e
t a
l., 1
99
7; C
asa
lon
e e
t a
l., 2
00
1
aC
ha
ract
eri
stic
s o
f th
e E
C p
he
no
typ
e, i
.e. t
he
dir
ect
ion
of
the
exp
ress
ion
ch
an
ge
fo
r e
ach
of
the
mo
lecu
lar
ma
rke
rs, i
s e
xpla
ine
d in
th
e t
ext
. A p
osi
tiv
e s
ign
ind
ica
tes
corr
esp
on
de
nce
wit
h t
he
EC
ph
en
oty
pe
, a n
eg
ati
ve
sig
n
ind
ica
tes
de
via
tio
n f
rom
th
e E
C p
he
no
typ
e. b
A c
om
ple
x ka
ryo
typ
e in
clu
de
d fi
ve
or
mo
re c
hro
mo
som
al a
be
rra
tio
ns.
An
un
sta
ble
ka
ryo
typ
e is
ch
ara
cte
rize
d b
y a
ra
pid
ly e
vo
lvin
g k
ary
oty
pe
wit
h a
hig
h f
req
ue
ncy
of
ran
do
m
ab
err
ati
on
s a
nd
nu
me
rou
s n
on
-clo
na
l ma
rke
r a
nd
rin
g c
hro
mo
som
es.
A n
orm
al k
ary
oty
pe
is c
ha
ract
eri
zed
by
a d
iplo
id c
hro
mo
som
e n
um
be
r. c
An
cho
rag
e-i
nd
ep
en
de
nt
gro
wth
wa
s d
ete
rmin
ed
by
in v
itro
cult
uri
ng
of
cells
in
soft
ag
aro
se. d
Tu
mo
r fo
rma
tio
n f
ollo
win
g s
ub
cuta
ne
ou
s in
ject
ion
of
cells
in N
OD
/SC
ID o
r B
alb
/c n
ud
e m
ice
.
EC
, en
do
the
lial c
ell;
NE
, no
t e
va
lua
ted
.
EC
ph
en
oty
pe
a
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

dmm.biologists.org1076
Modeling endothelial cell neoplasiaREVIEW
spontaneous activation of ALT in SV40-transformed epithelial andfibroblastic cell lines allude to an indirect means of establishingALT-immortalized EC lines (Bryan et al., 1995).
The currently available genetic models might also be made moreclinically relevant by substituting viral oncogenes with short hairpinRNA (shRNA) targeting the specific tumor suppressor genesimplicated in EC malignancies, and by constitutive activation ofVEGF signaling. Conditional deletion of tumor suppressor genesspecifically within ECs would also provide a valuable alternativemurine model of endothelial neoplasia. Additional refinement ofgenetic models will then be contingent upon furthercharacterization of the specific molecular defects that underpin theinitiation and progression of specific vascular neoplasias. Althoughhampered by sample numbers, genome-wide mapping and next-generation sequencing might be applied toward this goal.Information from broad-platform technologies such as these willbe valuable for improving diagnosis, identifying therapeutic targets,and establishing new in vitro and in vivo models that accuratelydepict the molecular biology of specific EC-derived neoplasias.Such models will then provide an avenue for preclinical tests ofnew therapeutics and for ultimately improving outcomes forpatients with these disorders.COMPETING INTERESTSThe authors declare that they do not have any competing or financial interests.
FUNDINGThe Children’s Cancer Institute Australia is affiliated with the University of NewSouth Wales and the Sydney Children’s Hospital Network. K.L.M. received researchgrants from National Health and Medical Research Council Career DevelopmentAward (510378), NSW Cancer Council and Cancer Institute NSW.
REFERENCESAdes, E. W., Candal, F. J., Swerlick, R. A., George, V. G., Summers, S., Bosse, D. C.
and Lawley, T. J. (1992). HMEC-1: establishment of an immortalized humanmicrovascular endothelial cell line. J. Invest. Dermatol. 99, 683-690.
Akhtar, N., Padilla, M. L., Dickerson, E. B., Steinberg, H., Breen, M., Auerbach, R.and Helfand, S. C. (2004). Interleukin-12 inhibits tumor growth in a novelangiogenesis canine hemangiosarcoma xenograft model. Neoplasia 6, 106-116.
Albini, A., Paglieri, I., Orengo, G., Carlone, S., Aluigi, M. G., DeMarchi, R.,Matteucci, C., Mantovani, A., Carozzi, F., Donini, S. et al. (1997). The beta-corefragment of human chorionic gonadotrophin inhibits growth of Kaposi’s sarcoma-derived cells and a new immortalized Kaposi’s sarcoma cell line. AIDS 11, 713-721.
Artandi, S. E. and DePinho, R. A. (2010). Telomeres and telomerase in cancer.Carcinogenesis 31, 9-18.
Asahara, T., Murohara, T., Sullivan, A., Silver, M., van der Zee, R., Li, T.,Witzenbichler, B., Schatteman, G. and Isner, J. M. (1997). Isolation of putativeprogenitor endothelial cells for angiogenesis. Science 275, 964-967.
Asahara, T., Masuda, H., Takahashi, T., Kalka, C., Pastore, C., Silver, M., Kearne, M.,Magner, M. and Isner, J. M. (1999). Bone marrow origin of endothelial progenitorcells responsible for postnatal vasculogenesis in physiological and pathologicalneovascularization. Circ. Res. 85, 221-228.
Aviv, H., Khan, M. Y., Skurnick, J., Okuda, K., Kimura, M., Gardner, J., Priolo, L. andAviv, A. (2001). Age dependent aneuploidy and telomere length of the humanvascular endothelium. Atherosclerosis 159, 281-287.
Bailey, T. S., Auersperg, N. and Myrdal, S. E. (1994). Recloning of SV40 early genetransfected human endothelial cells repeatedly recovers subpopulations with lowpassage characteristics and morphologies. Biochem. Cell Biol. 72, 117-125.
Bais, C., Santomasso, B., Coso, O., Arvanitakis, L., Raaka, E. G., Gutkind, J. S., Asch,A. S., Cesarman, E., Gershengorn, M. C. and Mesri, E. A. (1998). G-protein-coupledreceptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene andangiogenesis activator. Nature 391, 86-89.
Bais, C., Van Geelen, A., Eroles, P., Mutlu, A., Chiozzini, C., Dias, S., Silverstein, R.L., Rafii, S. and Mesri, E. A. (2003). Kaposi’s sarcoma associated herpesvirus Gprotein-coupled receptor immortalizes human endothelial cells by activation of theVEGF receptor-2/ KDR. Cancer Cell 3, 131-143.
Baumhoer, D., Gunawan, B., Becker, H. and Füzesi, L. (2005). Comparative genomichybridization in four angiosarcomas of the female breast. Gynecol. Oncol. 97, 348-352.
Bautch, V. L., Toda, S., Hassell, J. A. and Hanahan, D. (1987). Endothelial cell tumorsdevelop in transgenic mice carrying polyoma virus middle T oncogene. Cell 51, 529-537.
Berg, J. N., Walter, J. W., Thisanagayam, U., Evans, M., Blei, F., Waner, M.,Diamond, A. G., Marchuk, D. A. and Porteous, M. E. (2001). Evidence for loss ofheterozygosity of 5q in sporadic haemangiomas: are somatic mutations involved inhaemangioma formation? J. Clin. Pathol. 54, 249-252.
Boehm, J. S. and Hahn, W. C. (2005). Cancer genetics: Finding the right mix. Eur. J.
Hum. Genet. 13, 1099-1100.Boivin-Angèle, S., Lefrançois, L., Froment, O., Spiethoff, A., Bogdanffy, M. S.,
Wegener, K., Wesch, H., Barbin, A., Bancel, B., Trépo, C. et al. (2000). Ras genemutations in vinyl chloride-induced liver tumours are carcinogen-specific but varywith cell type and species. Int. J. Cancer 85, 223-227.
Boshoff, C., Schulz, T. F., Kennedy, M. M., Graham, A. K., Fisher, C., Thomas, A.,McGee, J. O., Weiss, R. A. and O’Leary, J. J. (1995). Kaposi’s sarcoma-associatedherpesvirus infects endothelial and spindle cells. Nat. Med. 1, 1274-1278.
Boye, E., Yu, Y., Paranya, G., Mulliken, J. B., Olsen, B. R. and Bischoff, J. (2001).Clonality and altered behavior of endothelial cells from hemangiomas. J. Clin. Invest.
107, 745-752.Brooks, J. J. (1986). Kaposi’s sarcoma: a reversible hyperplasia. Lancet 328, 1309-1311.Bryan, T. M., Englezou, A., Gupta, J., Bacchetti, S. and Reddel, R. R. (1995).
Telomere elongation in immortal human cells without detectable telomeraseactivity. EMBO J. 14, 4240-4248.
Bryan, T. M., Englezou, A., Dalla-Pozza, L., Dunham, M. A. and Reddel, R. R. (1997).Evidence for an alternative mechanism for maintaining telomere length in humantumors and tumor-derived cell lines. Nat. Med. 3, 1271-1274.
Burger, A. M., Fiebig, H. H., Kuettel, M. R., Lautenberger, J. A., Kung, H. F. andRhim, J. S. (1998). Effect of oncogene expression on telomerase activation andtelomere length in human endothelial, fibroblast and prostate epithelial cells. Int. J.
Oncol. 13, 1043-1048.Candal, F. J., Rafii, S., Parker, J. T., Ades, E. W., Ferris, B., Nachman, R. L. and Kellar,
K. L. (1996). BMEC-1: a human bone marrow microvascular endothelial cell line withprimary cell characteristics. Microvasc. Res. 52, 221-234.
Carmeliet, P. and Jain, R. K. (2011). Molecular mechanisms and clinical applications ofangiogenesis. Nature 473, 298-307.
Casalone, R., Albini, A., Righi, R., Granata, P. and Toniolo, A. (2001). Nonrandomchromosome changes in Kaposi sarcoma: cytogenetic and FISH results in a new cellline (KS-IMM) and literature review. Cancer Genet. Cytogenet. 124, 16-19.
Cerilli, L. A., Huffman, H. T. and Anand, A. (1998). Primary renal angiosarcoma: a casereport with immunohistochemical, ultrastructural, and cytogenetic features andreview of the literature. Arch. Pathol. Lab. Med. 122, 929-935.
Chandler, H. L., Newkirk, K. M., Kusewitt, D. F., Dubielzig, R. R. and Colitz, C. M.(2009). Immunohistochemical analysis of ocular hemangiomas andhemangiosarcomas in dogs. Vet. Ophthalmol. 12, 83-90.
Chang, E. and Harley, C. B. (1995). Telomere length and replicative aging in humanvascular tissues. Proc. Natl. Acad. Sci. USA 92, 11190-11194.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M. andMoore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865-1869.
Chang, H., Wachtman, L. M., Pearson, C. B., Lee, J. S., Lee, H. R., Lee, S. H., Vieira, J.,Mansfield, K. G. and Jung, J. U. (2009). Non-human primate model of Kaposi’ssarcoma-associated herpesvirus infection. PLoS Pathog. 5, e1000606.
Chen, Z., Smith, K. J., Skelton, H. G., 3rd, Barrett, T. L., Greenway, H. T., Jr and Lo,S. C. (2001). Telomerase activity in Kaposi’s sarcoma, squamous cell carcinoma, andbasal cell carcinoma. Exp. Biol. Med. (Maywood) 226, 753-757.
Cohen, S. B., Graham, M. E., Lovrecz, G. O., Bache, N., Robinson, P. J. and Reddel,R. R. (2007). Protein composition of catalytically active human telomerase fromimmortal cells. Science 315, 1850-1853.
Counter, C. M., Avilion, A. A., LeFeuvre, C. E., Stewart, N. G., Greider, C. W., Harley,C. B. and Bacchetti, S. (1992). Telomere shortening associated with chromosomeinstability is arrested in immortal cells which express telomerase activity. EMBO J. 11,1921-1929.
Dadras, S. S., North, P. E., Bertoncini, J., Mihm, M. C. and Detmar, M. (2004).Infantile hemangiomas are arrested in an early developmental vasculardifferentiation state. Mod. Pathol. 17, 1068-1079.
Dill, M. T., Rothweiler, S., Djonov, V., Hlushchuk, R., Tornillo, L., Terracciano, L.,Meili-Butz, S., Radtke, F., Heim, M. H. and Semela, D. (2012). Disruption of Notch1induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas inlivers of mice. Gastroenterology 142, 967-977, e2.
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E. E.,Linskens, M., Rubelj, I., Pereira-Smith, O. et al. (1995). A biomarker that identifiessenescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA
92, 9363-9367.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Disease Models & Mechanisms 1077
Modeling endothelial cell neoplasia REVIEW
Dittmer, D., Stoddart, C., Renne, R., Linquist-Stepps, V., Moreno, M. E., Bare, C.,McCune, J. M. and Ganem, D. (1999). Experimental transmission of Kaposi’ssarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med.
190, 1857-1868.Domfeh, A. B., Fichera, M. and Hunt, J. L. (2006). Allelic loss of 3 different tumor
suppressor gene loci in benign and malignant endothelial tumors of the head andneck. Arch. Pathol. Lab. Med. 130, 1184-1187.
Drolet, B. A., Esterly, N. B. and Frieden, I. J. (1999). Hemangiomas in children. N. Engl.
J. Med. 341, 173-181.Ducray, C., Pommier, J. P., Martins, L., Boussin, F. D. and Sabatier, L. (1999).
Telomere dynamics, end-to-end fusions and telomerase activation during thehuman fibroblast immortalization process. Oncogene 18, 4211-4223.
Edgell, C. J., McDonald, C. C. and Graham, J. B. (1983). Permanent cell lineexpressing human factor VIII-related antigen established by hybridization. Proc. Natl.
Acad. Sci. USA 80, 3734-3737.Fasching, C. L., Neumann, A. A., Muntoni, A., Yeager, T. R. and Reddel, R. R. (2007).
DNA damage induces alternative lengthening of telomeres (ALT) associatedpromyelocytic leukemia bodies that preferentially associate with linear telomericDNA. Cancer Res. 67, 7072-7077.
Feng, J., Funk, W. D., Wang, S.-S., Weinrich, S. L., Avilion, A. A., Chiu, C.-P., Adams,R. R., Chang, E., Allsopp, R. C., Yu, J. et al. (1995). The RNA component of humantelomerase. Science 269, 1236-1241.
Fickling, S. A., Tooze, J. A. and Whitley, G. S. (1992). Characterization of humanumbilical vein endothelial cell lines produced by transfection with the early regionof SV40. Exp. Cell Res. 201, 517-521.
Fontijn, R., Hop, C., Brinkman, H. J., Slater, R., Westerveld, A., van Mourik, J. A.and Pannekoek, H. (1995). Maintenance of vascular endothelial cell-specificproperties after immortalization with an amphotrophic replication-deficientretrovirus containing human papilloma virus 16 E6/E7 DNA. Exp. Cell Res. 216, 199-207.
Freedman, D. A. (2005). Senescence and its bypass in the vascular endothelium. Front.
Biosci. 10, 940-950.Freedman, D. A. and Folkman, J. (2004). Maintenance of G1 checkpoint controls in
telomerase-immortalized endothelial cells. Cell Cycle 3, 811-816.Friborg, J., Jr, Kong, W., Hottiger, M. O. and Nabel, G. J. (1999). p53 inhibition by the
LANA protein of KSHV protects against cell death. Nature 402, 889-894.Gagnon, E., Cattaruzzi, P., Griffith, M., Muzakare, L., LeFlao, K., Faure, R., Béliveau,
R., Hussain, S. N., Koutsilieris, M. and Doillon, C. J. (2002). Human vascularendothelial cells with extended life spans: in vitro cell response, protein expression,and angiogenesis. Angiogenesis 5, 21-33.
Ganem, D. (2010). KSHV and the pathogenesis of Kaposi sarcoma: listening to humanbiology and medicine. J. Clin. Invest. 120, 939-949.
Garcia, J. M., Gonzalez, R., Silva, J. M., Dominguez, G., Vegazo, I. S., Gamallo, C.,Provencio, M., España, P. and Bonilla, F. (2000). Mutational status of K-ras andTP53 genes in primary sarcomas of the heart. Br. J. Cancer 82, 1183-1185.
George, S., Merriam, P., Maki, R. G., Van den Abbeele, A. D., Yap, J. T., Akhurst, T.,Harmon, D. C., Bhuchar, G., O’Mara, M. M., D’Adamo, D. R. et al. (2009).Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromaltumor sarcomas. J. Clin. Oncol. 27, 3154-3160.
Gil-Benso, R., López-Ginés, C., Soriano, P., Almenar, S., Vazquez, C. and Llombart-Bosch, A. (1994). Cytogenetic study of angiosarcoma of the breast. Genes
Chromosomes Cancer 10, 210-212.Gimbrone, M. A., Jr and Fareed, G. C. (1976). Transformation of cultured human
vascular endothelium by SV40 DNA. Cell 9, 685-693.Girardi, A. J., Jensen, F. C. and Koprowski, H. (1965). Sv40-Induced Tranformation of
Human Diploid Cells: Crisis and Recovery. J. Cell. Physiol. 65, 69-83.Gisselsson, D., Jonson, T., Petersén, A., Strömbeck, B., Dal Cin, P., Höglund, M.,
Mitelman, F., Mertens, F. and Mandahl, N. (2001). Telomere dysfunction triggersextensive DNA fragmentation and evolution of complex chromosome abnormalitiesin human malignant tumors. Proc. Natl. Acad. Sci. USA 98, 12683-12688.
Gnepp, D. R., Chandler, W. and Hyams, V. (1984). Primary Kaposi’s sarcoma of thehead and neck. Ann. Intern. Med. 100, 107-114.
Greenberger, S., Adini, I., Boscolo, E., Mulliken, J. B. and Bischoff, J. (2010).Targeting NF-κB in infantile hemangioma-derived stem cells reduces VEGF-Aexpression. Angiogenesis 13, 327-335.
Gu, X., Zhang, J., Brann, D. W. and Yu, F.-S. X. (2003). Brain and retinal vascularendothelial cells with extended life span established by ectopic expression oftelomerase. Invest. Ophthalmol. Vis. Sci. 44, 3219-3225.
Harvey, M., McArthur, M. J., Montgomery, C. A., Jr, Butel, J. S., Bradley, A. andDonehower, L. A. (1993). Spontaneous and carcinogen-induced tumorigenesis inp53-deficient mice. Nat. Genet. 5, 225-229.
Hayflick, L. and Moorhead, P. S. (1961). The serial cultivation of human diploid cellstrains. Exp. Cell Res. 25, 585-621.
He, M., Das, K., Blacksin, M., Benevenia, J. and Hameed, M. (2006). A translocationinvolving the placental growth factor gene is identified in an epithelioidhemangioendothelioma. Cancer Genet. Cytogenet. 168, 150-154.
Henson, J. D., Hannay, J. A., McCarthy, S. W., Royds, J. A., Yeager, T. R., Robinson,R. A., Wharton, S. B., Jellinek, D. A., Arbuckle, S. M., Yoo, J. et al. (2005). A robustassay for alternative lengthening of telomeres in tumors shows the significance ofalternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res.
11, 217-225.Hohenwarter, O., Schmatz, C. and Katinger, H. (1992a). Stability of von Willebrand
factor secretion in different human endothelial hybrid cell lines. Cytotechnology 8,31-37.
Hohenwarter, O., Zinser, E., Schmatz, C., Rüker, F. and Katinger, H. (1992b).Influence of transfected SV40 early region on growth and differentiation of humanendothelial cells. J. Biotechnol. 25, 349-356.
Hohenwarter, O., Jakoubek, A., Schmatz, C. and Katinger, H. (1994). Expression ofSV40 tumour antigens enables human endothelial cells to grow independently fromfoetal calf serum and exogenous growth factors. J. Biotechnol. 34, 205-211.
Hohenwarter, O., Waltenberger, A., Strutzenberger, K. and Katinger, H. (1997).Human endothelial cell lines established by mutated forms of the simian virus 40large T oncogene. J. Biotechnol. 54, 131-137.
Hollstein, M., Marion, M. J., Lehman, T., Welsh, J., Harris, C. C., Martel-Planche, G.,Kusters, I. and Montesano, R. (1994). p53 mutations at A:T base pairs inangiosarcomas of vinyl chloride-exposed factory workers. Carcinogenesis 15, 1-3.
Hong, C. B., Winston, J. M. and Lee, C. C. (1980). Hepatic angiosarcoma. Animalmodel: angiosarcoma of rats and mice induced by vinyl chloride. Am. J. Pathol. 101,737-740.
Hong, H. H., Devereux, T. R., Melnick, R. L., Moomaw, C. R., Boorman, G. A. andSills, R. C. (2000). Mutations of ras protooncogenes and p53 tumor suppressor genein cardiac hemangiosarcomas from B6C3F1 mice exposed to 1,3-butadiene for 2years. Toxicol. Pathol. 28, 529-534.
Hoover, M. L., Vĕtvicka, V., Hoffpauir, J. M. and Tamburro, C. H. (1993). Humanendothelial cell line from an angiosarcoma. In Vitro Cell Dev. Biol. 29, 199-202.
Hsiao, R., Sharma, H. W., Ramakrishnan, S., Keith, E. and Narayanan, R. (1997).Telomerase activity in normal human endothelial cells. Anticancer Res. 17 2A, 827-832.
Ingram, D. A., Mead, L. E., Moore, D. B., Woodard, W., Fenoglio, A. and Yoder, M. C.(2005). Vessel wall-derived endothelial cells rapidly proliferate because they containa complete hierarchy of endothelial progenitor cells. Blood 105, 2783-2786.
Itakura, E., Yamamoto, H., Oda, Y. and Tsuneyoshi, M. (2008). Detection andcharacterization of vascular endothelial growth factors and their receptors in a seriesof angiosarcomas. J. Surg. Oncol. 97, 74-81.
Jinnin, M., Medici, D., Park, L., Limaye, N., Liu, Y., Boscolo, E., Bischoff, J., Vikkula,M., Boye, E. and Olsen, B. R. (2008). Suppressed NFAT-dependent VEGFR1expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 14,1236-1246.
Johnson, L. K. and Longenecker, J. P. (1982). Senescence of aortic endothelial cells invitro: influence of culture conditions and preliminary characterization of thesenescent phenotype. Mech. Ageing Dev. 18, 1-18.
Johnson, T. E., Umbenhauer, D. R., Hill, R., Bradt, C., Mueller, S. N., Levine, E. M.and Nichols, W. W. (1992). Karyotypic and phenotypic changes during in vitro agingof human endothelial cells. J. Cell. Physiol. 150, 17-27.
Kaaya, E. E., Parravicini, C., Ordonez, C., Gendelman, R., Berti, E., Gallo, R. C. andBiberfeld, P. (1995). Heterogeneity of spindle cells in Kaposi’s sarcoma: comparisonof cells in lesions and in culture. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10,295-305.
Kan, C.-Y., Wen, V. W., Pasquier, E., Jankowski, K., Chang, M., Richards, L. A.,Kavallaris, M. and MacKenzie, K. L. (2012). Endothelial cell dysfunction andcytoskeletal changes associated with repression of p16(INK4a) duringimmortalization. Oncogene 31, 4815-4827.
Khan, Z. A., Boscolo, E., Picard, A., Psutka, S., Melero-Martin, J. M., Bartch, T. C.,Mulliken, J. B. and Bischoff, J. (2008). Multipotential stem cells recapitulate humaninfantile hemangioma in immunodeficient mice. J. Clin. Invest. 118, 2592-2599.
Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L.,Coviello, G. M., Wright, W. E., Weinrich, S. L. and Shay, J. W. (1994). Specificassociation of human telomerase activity with immortal cells and cancer. Science
266, 2011-2015.Kindblom, L. G., Stenman, G. and Angervall, L. (1991). Morphological and
cytogenetic studies of angiosarcoma in Stewart-Treves syndrome. Virchows Arch. A
Pathol. Anat. Histopathol. 419, 439-445.Knight, J. S., Cotter, M. A., 2nd and Robertson, E. S. (2001). The latency-associated
nuclear antigen of Kaposi’s sarcoma-associated herpesvirus transactivates thetelomerase reverse transcriptase promoter. J. Biol. Chem. 276, 22971-22978.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

dmm.biologists.org1078
Modeling endothelial cell neoplasiaREVIEW
Koch, M., Nielsen, G. P. and Yoon, S. S. (2008). Malignant tumors of blood vessels:angiosarcomas, hemangioendotheliomas, and hemangioperictyomas. J. Surg. Oncol.
97, 321-329.Kodama, A., Sakai, H., Matsuura, S., Murakami, M., Murai, A., Mori, T., Maruo, K.,
Kimura, T., Masegi, T. and Yanai, T. (2009). Establishment of caninehemangiosarcoma xenograft models expressing endothelial growth factors, theirreceptors, and angiogenesis-associated homeobox genes. BMC Cancer 9, 363.
Koon, H. B., Bubley, G. J., Pantanowitz, L., Masiello, D., Smith, B., Crosby, K.,Proper, J., Weeden, W., Miller, T. E., Chatis, P. et al. (2005). Imatinib-inducedregression of AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 23, 982-989.
Koontz, B. F., Miles, E. F., Rubio, M. A., Madden, J. F., Fisher, S. R., Scher, R. L. andBrizel, D. M. (2008). Preoperative radiotherapy and bevacizumab for angiosarcomaof the head and neck: two case studies. Head Neck 30, 262-266.
Krishnaswamy, G., Kelley, J., Yerra, L., Smith, J. K. and Chi, D. S. (1999). Humanendothelium as a source of multifunctional cytokines: molecular regulation andpossible role in human disease. J. Interferon Cytokine Res. 19, 91-104.
Krump-Konvalinkova, V., Bittinger, F., Unger, R. E., Peters, K., Lehr, H. A. andKirkpatrick, C. J. (2001). Generation of human pulmonary microvascular endothelialcell lines. Lab. Invest. 81, 1717-1727.
Krump-Konvalinkova, V., Kleideiter, E., Friedrich, U., Klotz, U. and Kirkpatrick, C. J.(2003). Tumorigenic conversion of endothelial cells. Exp. Mol. Pathol. 75, 154-159.
Kurz, D. J., Decary, S., Hong, Y., Trivier, E., Akhmedov, A. and Erusalimsky, J. D.(2004). Chronic oxidative stress compromises telomere integrity and accelerates theonset of senescence in human endothelial cells. J. Cell Sci. 117, 2417-2426.
Lassalle, P., LaGrou, C., Delneste, Y., Sanceau, J., Coll, J., Torpier, G., Wietzerbin, J.,Stehelin, D., Tonnel, A.-B. and Capron, A. (1992). Human endothelial cellstransfected by SV40 T antigens: characterization and potential use as a source ofnormal endothelial factors. Eur. J. Immunol. 22, 425-431.
Lezama-del Valle, P., Gerald, W. L., Tsai, J., Meyers, P. and La Quaglia, M. P. (1998).Malignant vascular tumors in young patients. Cancer 83, 1634-1639.
Lunardi-Iskandar, Y., Bryant, J. L., Zeman, R. A., Lam, V. H., Samaniego, F., Besnier,J. M., Hermans, P., Thierry, A. R., Gill, P. and Gallo, R. C. (1995a). Tumorigenesisand metastasis of neoplastic Kaposi’s sarcoma cell line in immunodeficient miceblocked by a human pregnancy hormone. Nature 375, 64-68.
Lunardi-Iskandar, Y., Gill, P., Lam, V. H., Zeman, R. A., Michaels, F., Mann, D. L.,Reitz, M. S., Jr, Kaplan, M., Berneman, Z. N., Carter, D. et al. (1995b). Isolation andcharacterization of an immortal neoplastic cell line (KS Y-1) from AIDS-associatedKaposi’s sarcoma. J. Natl. Cancer Inst. 87, 974-981.
MacKenzie, K. L., Franco, S., Naiyer, A. J., May, C., Sadelain, M., Rafii, S. andMoore, M. A. (2002). Multiple stages of malignant transformation of humanendothelial cells modelled by co-expression of telomerase reverse transcriptase,SV40 T antigen and oncogenic N-ras. Oncogene 21, 4200-4211.
MacLeod, R. A., Dirks, W. G., Matsuo, Y., Kaufmann, M., Milch, H. and Drexler, H. G.(1999). Widespread intraspecies cross-contamination of human tumor cell linesarising at source. Int J Cancer 83, 555-563.
Maki, R. G., D’Adamo, D. R., Keohan, M. L., Saulle, M., Schuetze, S. M., Undevia, S.D., Livingston, M. B., Cooney, M. M., Hensley, M. L., Mita, M. M. et al. (2009).Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin.
Oncol. 27, 3133-3140.Mandahl, N., Jin, Y. S., Heim, S., Willén, H., Wennerberg, J., Biörklund, A. and
Mitelman, F. (1990). Trisomy 5 and loss of the Y chromosome as the solecytogenetic anomalies in a cavernous hemangioma/angiosarcoma. Genes
Chromosomes Cancer 1, 315-316.Mark, R. J., Poen, J. C., Tran, L. M., Fu, Y. S. and Juillard, G. F. (1996). Angiosarcoma.
A report of 67 patients and a review of the literature. Cancer 77, 2400-2406.Marucci, G., Barbanera, A., Tosi, A. L., Andreoli, A., Simonetti, L., Magrini, E.,
Farnedi, A. and Foschini, M. P. (2006). Epithelioid hemangioendothelioma of thespinal cord: Description of a case with cytogenetic analysis. Int. J. Surg. Pathol. 14,340-343.
Masuzawa, M., Fujimura, T., Hamada, Y., Fujita, Y., Hara, H., Nishiyama, S.,Katsuoka, K., Tamauchi, H. and Sakurai, Y. (1999). Establishment of a humanhemangiosarcoma cell line (ISO-HAS). Int. J. Cancer 81, 305-308.
Matsushita, H., Chang, E., Glassford, A. J., Cooke, J. P., Chiu, C. P. and Tsao, P. S.(2001). eNOS activity is reduced in senescent human endothelial cells: Preservationby hTERT immortalization. Circ. Res. 89, 793-798.
Mentzel, T., Beham, A., Calonje, E., Katenkamp, D. and Fletcher, C. D. (1997).Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic andimmunohistochemical study of 30 cases. Am. J. Surg. Pathol. 21, 363-374.
Mohamed, A. M., Elwakil, T. F., Taher, I. M., Elbarbary, M. M., Kayed, H. F., Hussein,H. A. and Eid, O. M. (2009). Cyclin D1 gene amplification in proliferatinghaemangioma. Cell Tissue Res. 338, 107-115.
Mulliken, J. B., Zetter, B. R. and Folkman, J. (1982). In vitro characteristics ofendothelium from hemangiomas and vascular malformations. Surgery 92, 348-353.
Munro, J., Stott, F. J., Vousden, K. H., Peters, G. and Parkinson, E. K. (1999). Role ofthe alternative INK4A proteins in human keratinocyte senescence: evidence for thespecific inactivation of p16INK4A upon immortalization. Cancer Res. 59, 2516-2521.
Mutlu, A. D., Cavallin, L. E., Vincent, L., Chiozzini, C., Eroles, P., Duran, E. M.,Asgari, Z., Hooper, A. T., La Perle, K. M., Hilsher, C. et al. (2007). In vivo-restrictedand reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animalmodel of virally induced Kaposi’s sarcoma. Cancer Cell 11, 245-258.
Nakamura, T. M., Morin, G. B., Chapman, K. B., Weinrich, S. L., Andrews, W. H.,Lingner, J., Harley, C. B. and Cech, T. R. (1997). Telomerase catalytic subunithomologs from fission yeast and human. Science 277, 955-959.
Napier, C. E., Veas, L. A., Kan, C. Y., Taylor, L. M., Yuan, J., Wen, V. W., James, A.,O’Brien, T. A., Lock, R. B. and MacKenzie, K. L. (2010). Mild hyperoxia limits hTRlevels, telomerase activity, and telomere length maintenance in hTERT-transducedbone marrow endothelial cells. Biochim. Biophys. Acta 1803, 1142-1153.
Nichols, W. W., Buynak, E. B., Bradt, C., Hill, R., Aronson, M., Jarrell, B. E., Mueller,S. N. and Levine, E. M. (1987). Cytogenetic evaluation of human endothelial cellcultures. J. Cell. Physiol. 132, 453-462.
Nisato, R. E., Harrison, J. A., Buser, R., Orci, L., Rinsch, C., Montesano, R., Dupraz, P.and Pepper, M. S. (2004). Generation and characterization of telomerase-transfected human lymphatic endothelial cells with an extended life span. Am. J.Pathol. 165, 11-24.
O’Hagan, R. C., Chang, S., Maser, R. S., Mohan, R., Artandi, S. E., Chin, L. andDePinho, R. A. (2002). Telomere dysfunction provokes regional amplification anddeletion in cancer genomes. Cancer Cell 2, 149-155.
O’Hare, M. J., Bond, J., Clarke, C., Takeuchi, Y., Atherton, A. J., Berry, C., Moody, J.,Silver, A. R. J., Davies, D. C., Alsop, A. E. et al. (2001). Conditional immortalizationof freshly isolated human mammary fibroblasts and endothelial cells. Proc. Natl.Acad. Sci. USA 98, 646-651.
Ohsawa, M., Naka, N., Tomita, Y., Kawamori, D., Kanno, H. and Aozasa, K. (1995).Use of immunohistochemical procedures in diagnosing angiosarcoma. Evaluation of98 cases. Cancer 75, 2867-2874.
Okuda, K., Khan, M. Y., Skurnick, J., Kimura, M., Aviv, H. and Aviv, A. (2000).Telomere attrition of the human abdominal aorta: relationships with age andatherosclerosis. Atherosclerosis 152, 391-398.
Pareja, M. J., Vargas, M. T., Sánchez, A., Ibáñez, J. and González-Cámpora, R.(2009). Cytogenetic study of a pulmonary sclerosing hemangioma. Cancer Genet.Cytogenet. 195, 80-84.
Popescu, N. C., Zimonjic, D. B., Leventon-Kriss, S., Bryant, J. L., Lunardi-Iskandar,Y. and Gallo, R. C. (1996). Deletion and translocation involving chromosome 3 (p14)in two tumorigenic Kaposi’s sarcoma cell lines. J. Natl. Cancer Inst. 88, 450-455.
Primo, L., Roca, C., Ferrandi, C., Lanfrancone, L. and Bussolino, F. (2000). Humanendothelial cells expressing polyoma middle T induce tumors. Oncogene 19, 3632-3641.
Przygodzki, R. M., Finkelstein, S. D., Keohavong, P., Zhu, D., Bakker, A., Swalsky, P.A., Soini, Y., Ishak, K. G. and Bennett, W. P. (1997). Sporadic and Thorotrast-induced angiosarcomas of the liver manifest frequent and multiple point mutationsin K-ras-2. Lab. Invest. 76, 153-159.
Pyakurel, P., Pak, F., Mwakigonja, A. R., Kaaya, E., Heiden, T. and Biberfeld, P.(2006). Lymphatic and vascular origin of Kaposi’s sarcoma spindle cells during tumordevelopment. Int. J. Cancer 119, 1262-1267.
Ray, F. A., Peabody, D. S., Cooper, J. L., Cram, L. S. and Kraemer, P. M. (1990). SV40 Tantigen alone drives karyotype instability that precedes neoplastic transformation ofhuman diploid fibroblasts. J. Cell. Biochem. 42, 13-31.
Reddel, R. R. (2000). The role of senescence and immortalization in carcinogenesis.Carcinogenesis 21, 477-484.
Rhim, J. S., Tsai, W. P., Chen, Z. Q., Chen, Z., Van Waes, C., Burger, A. M. andLautenberger, J. A. (1998). A human vascular endothelial cell model to studyangiogenesis and tumorigenesis. Carcinogenesis 19, 673-681.
Ribeiro, M. J., Phillips, D. J., Benson, J. M., Evatt, B. L., Ades, E. W. and Hooper, W.C. (1995). Hemostatic properties of the SV-40 transfected human microvascularendothelial cell line (HMEC-1). A representative in vitro model for microvascularendothelium. Thromb. Res. 79, 153-161.
Rood, P. M., Calafat, J., von dem Borne, A. E., Gerritsen, W. R. and van der Schoot,C. E. (2000). Immortalisation of human bone marrow endothelial cells:characterisation of new cell lines. Eur. J. Clin. Invest. 30, 618-629.
Rudolph, K. L., Millard, M., Bosenberg, M. W. and DePinho, R. A. (2001). Telomeredysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet.28, 155-159.
Salahuddin, S. Z., Nakamura, S., Biberfeld, P., Kaplan, M. H., Markham, P. D.,Larsson, L. and Gallo, R. C. (1988). Angiogenic properties of Kaposi’s sarcoma-derived cells after long-term culture in vitro. Science 242, 430-433.
Salmon, P., Oberholzer, J., Occhiodoro, T., Morel, P., Lou, J. and Trono, D. (2000).Reversible immortalization of human primary cells by lentivector-mediated transferof specific genes. Mol. Ther. 2, 404-414.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Disease Models & Mechanisms 1079
Modeling endothelial cell neoplasia REVIEW
Sato, N., Sato, T., Takahashi, S. and Kikuchi, K. (1986). Establishment of murineendothelial cell lines that develop angiosarcomas in vivo: brief demonstration of aproposed animal model for Kaposi’s sarcoma. Cancer Res. 46, 362-366.
Schaffhausen, B. S. and Roberts, T. M. (2009). Lessons from polyoma middle Tantigen on signaling and transformation: A DNA tumor virus contribution to the waron cancer. Virology 384, 304-316.
Schuborg, C., Mertens, F., Rydholm, A., Brosjö, O., Dictor, M., Mitelman, F. andMandahl, N. (1998). Cytogenetic analysis of four angiosarcomas from deep andsuperficial soft tissue. Cancer Genet. Cytogenet. 100, 52-56.
Schwartz, B., Vicart, P., Delouis, C. and Paulin, D. (1991). Mammalian cell lines canbe efficiently established in vitro upon expression of the SV40 large T antigen drivenby a promoter sequence derived from the human vimentin gene. Biol. Cell 73, 7-14.
Shao, R. and Guo, X. (2004). Human microvascular endothelial cells immortalized withhuman telomerase catalytic protein: a model for the study of in vitro angiogenesis.Biochem. Biophys. Res. Commun. 321, 788-794.
Shay, J. W., Pereira-Smith, O. M. and Wright, W. E. (1991). A role for both RB and p53in the regulation of human cellular senescence. Exp. Cell Res. 196, 33-39.
Shi, Q., Rafii, S., Wu, M. H., Wijelath, E. S., Yu, C., Ishida, A., Fujita, Y., Kothari, S.,Mohle, R., Sauvage, L. R. et al. (1998). Evidence for circulating bone marrow-derived endothelial cells. Blood 92, 362-367.
Si, H. and Robertson, E. S. (2006). Kaposi’s sarcoma-associated herpesvirus-encodedlatency-associated nuclear antigen induces chromosomal instability throughinhibition of p53 function. J. Virol. 80, 697-709.
Sodhi, A., Chaisuparat, R., Hu, J., Ramsdell, A. K., Manning, B. D., Sausville, E. A.,Sawai, E. T., Molinolo, A., Gutkind, J. S. and Montaner, S. (2006). The TSC2/mTORpathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 10, 133-143.
Stein, G. H., Drullinger, L. F., Soulard, A. and Dulić, V. (1999). Differential roles forcyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescenceand differentiation in human fibroblasts. Mol. Cell. Biol. 19, 2109-2117.
Szajerka, T. and Jablecki, J. (2007). Kaposi’s sarcoma revisited. AIDS Rev. 9, 230-236.Takahashi, K., Mulliken, J. B., Kozakewich, H. P. W., Rogers, R. A., Folkman, J. and
Ezekowitz, R. A. B. (1994). Cellular markers that distinguish the phases ofhemangioma during infancy and childhood. J. Clin. Invest. 93, 2357-2364.
Takahashi, T., Kalka, C., Masuda, H., Chen, D., Silver, M., Kearney, M., Magner, M.,Isner, J. M. and Asahara, T. (1999). Ischemia- and cytokine-induced mobilization ofbone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med.5, 434-438.
Takano, H., Murasawa, S. and Asahara, T. (2008). Functional and gene expressionanalysis of hTERT overexpressed endothelial cells. Biologics 2, 547-554.
Tamburini, B. A., Trapp, S., Phang, T. L., Schappa, J. T., Hunter, L. E. and Modiano,J. F. (2009). Gene expression profiles of sporadic canine hemangiosarcoma areuniquely associated with breed. PLoS ONE 4, e5549.
Tannapfel, A., Weihrauch, M., Benicke, M., Uhlmann, D., Hauss, J., Wrbitzky, R. andWittekind, C. (2001). p16INK4A-alterations in primary angiosarcoma of the liver. J.Hepatol. 35, 62-67.
Theurillat, J. P., Vavricka, S. R., Went, P., Weishaupt, D., Perren, A., Leonard-Meier,C. and Bachli, E. B. (2003). Morphologic changes and altered gene expression in anepithelioid hemangioendothelioma during a ten-year course of disease. Pathol. Res.Pract. 199, 165-170.
Truss, L., Dobin, S. M. and Donner, L. R. (2006). Deletion (21)(q21.2q22.12) as a soleclonal cytogenetic abnormality in a lobular capillary hemangioma of the nasalcavity. Cancer Genet. Cytogenet. 170, 69-70.
Tsarouha, H., Kyriazoglou, A. I., Ribeiro, F. R., Teixeira, M. R., Agnantis, N. andPandis, N. (2006). Chromosome analysis and molecular cytogenetic investigations ofan epithelioid hemangioendothelioma. Cancer Genet. Cytogenet. 169, 164-168.
Unger, R. E., Krump-Konvalinkova, V., Peters, K. and Kirkpatrick, C. J. (2002a). Invitro expression of the endothelial phenotype: comparative study of primaryisolated cells and cell lines, including the novel cell line HPMEC-ST1.6R. Microvasc.Res. 64, 384-397.
Unger, R. E., Oltrogge, J. B., von Briesen, H., Engelhardt, B., Woelki, U., Schlote, W.,Lorenz, R., Bratzke, H. and Kirkpatrick, C. J. (2002b). Isolation and molecularcharacterization of brain microvascular endothelial cells from human brain tumors.In Vitro Cell. Dev. Biol. Anim. 38, 273-281.
van Leeuwen, E. B., Veenstra, R., van Wijk, R., Molema, G., Hoekstra, A., Ruiters,M. H. and van der Meer, J. (2000). Characterization of immortalized humanumbilical and iliac vein endothelial cell lines after transfection with SV40 large T-antigen. Blood Coagul. Fibrinolysis 11, 15-25.
Venetsanakos, E., Mirza, A., Fanton, C., Romanov, S. R., Tlsty, T. and McMahon, M.(2002). Induction of tubulogenesis in telomerase-immortalized human microvascularendothelial cells by glioblastoma cells. Exp. Cell Res. 273, 21-33.
Verma, S. C., Borah, S. and Robertson, E. S. (2004). Latency-associated nuclearantigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription ofhuman telomerase reverse transcriptase promoter through interaction withtranscription factor Sp1. J. Virol. 78, 10348-10359.
von Zglinicki, T., Saretzki, G., Döcke, W. and Lotze, C. (1995). Mild hyperoxiashortens telomeres and inhibits proliferation of fibroblasts: a model for senescence?Exp. Cell Res. 220, 186-193.
Wagner, M., Hampel, B., Bernhard, D., Hala, M., Zwerschke, W. and Jansen-Dürr, P.(2001). Replicative senescence of human endothelial cells in vitro involves G1 arrest,polyploidization and senescence-associated apoptosis. Exp. Gerontol. 36, 1327-1347.
Weihrauch, M., Markwarth, A., Lehnert, G., Wittekind, C., Wrbitzky, R. andTannapfel, A. (2002). Abnormalities of the ARF-p53 pathway in primaryangiosarcomas of the liver. Hum. Pathol. 33, 884-892.
Weiss, S. W. and Enzinger, F. M. (1986). Spindle cell hemangioendothelioma. A low-grade angiosarcoma resembling a cavernous hemangioma and Kaposi’s sarcoma.Am. J. Surg. Pathol. 10, 521-530.
Wen, V. W., Wu, K., Baksh, S., Hinshelwood, R. A., Lock, R. B., Clark, S. J., Moore, M.A. and Mackenzie, K. L. (2006). Telomere-driven karyotypic complexity concurswith p16INK4a inactivation in TP53-competent immortal endothelial cells. CancerRes. 66, 10691-10700.
Werness, B. A., Levine, A. J. and Howley, P. M. (1990). Association of humanpapillomavirus types 16 and 18 E6 proteins with p53. Science 248, 76-79.
Wong, K. F., So, C. C., Wong, N., Siu, L. L., Kwong, Y. L. and Chan, J. K. (2001).Sinonasal angiosarcoma with marrow involvement at presentation mimickingmalignant lymphoma: cytogenetic analysis using multiple techniques. Cancer Genet.Cytogenet. 129, 64-68.
Wright, W. E., Piatyszek, M. A., Rainey, W. E., Byrd, W. and Shay, J. W. (1996).Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet.18, 173-179.
Xu, D., Neville, R. and Finkel, T. (2000). Homocysteine accelerates endothelial cellsenescence. FEBS Lett. 470, 20-24.
Xu, D., O, T. M., Shartava, A., Fowles, T. C., Yang, J., Fink, L. M., Ward, D. C., Mihm,M. C., Waner, M. and Ma, Y. (2011). Isolation, characterization, and in vitropropagation of infantile hemangioma stem cells and an in vivo mouse model. J.Hematol. Oncol. 4, 54.
Yang, J., Chang, E., Cherry, A. M., Bangs, C. D., Oei, Y., Bodnar, A., Bronstein, A.,Chiu, C. P. and Herron, G. S. (1999). Human endothelial cell life extension bytelomerase expression. J. Biol. Chem. 274, 26141-26148.
Yang, J., Nagavarapu, U., Relloma, K., Sjaastad, M. D., Moss, W. C., Passaniti, A.and Herron, G. S. (2001). Telomerized human microvasculature is functional in vivo.Nat. Biotechnol. 19, 219-224.
Yazawa, M., Okuda, M., Setoguchi, A., Nishimura, R., Sasaki, N., Hasegawa, A.,Watari, T. and Tsujimoto, H. (1999). Measurement of telomerase activity in dogtumors. J. Vet. Med. Sci. 61, 1125-1129.
Yonemaru, K., Sakai, H., Murakami, M., Kodama, A., Mori, T., Yanai, T., Maruo, K.and Masegi, T. (2007). The significance of p53 and retinoblastoma pathways incanine hemangiosarcoma. J. Vet. Med. Sci. 69, 271-278.
Young, A. T., Lakey, J. R., Murray, A. G., Mullen, J. C. and Moore, R. B. (2003). In vitrosenescence occurring in normal human endothelial cells can be rescued by ectopictelomerase activity. Transplant. Proc. 35, 2483-2485.
Young, R. J., Brown, N. J., Reed, M. W., Hughes, D. and Woll, P. J. (2010).Angiosarcoma. Lancet Oncol. 11, 983-991.
Yuan, J., Wen, V. W., Schuller, C. E. and MacKenzie, K. L. (2008). Molecularmechanisms that regulate the replicative lifespan of endothelial cells. In CellApoptosis Research Progress (ed. R. H. Fenton and C. V. Burnside), pp. 83-113. NewYork, NY: Nova Science Publishers.
Zhang, L., Aviv, H., Gardner, J. P., Okuda, K., Patel, S., Kimura, M., Bardeguez, A.and Aviv, A. (2000). Loss of chromosome 13 in cultured human vascular endothelialcells. Exp. Cell Res. 260, 357-364.
Zhou, B. B., Zhang, H., Damelin, M., Geles, K. G., Grindley, J. C. and Dirks, P. B.(2009). Tumour-initiating cells: challenges and opportunities for anticancer drugdiscovery. Nat. Rev. Drug Discov. 8, 806-823.
Zietz, C., Rössle, M., Haas, C., Sendelhofert, A., Hirschmann, A., Stürzl, M. andLöhrs, U. (1998). MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGFup-regulation in angiosarcomas. Am. J. Pathol. 153, 1425-1433.
Zindy, F., Nilsson, L. M., Nguyen, L., Meunier, C., Smeyne, R. J., Rehg, J. E., Eberhart,C., Sherr, C. J. and Roussel, M. F. (2003). Hemangiosarcomas, medulloblastomas, andother tumors in Ink4c/p53-null mice. Cancer Res. 63, 5420-5427.
Zu, Y., Perle, M. A., Yan, Z., Liu, J., Kumar, A. and Waisman, J. (2001). Chromosomalabnormalities and p53 gene mutation in a cardiac angiosarcoma. Appl.Immunohistochem. Mol. Morphol. 9, 24-28.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM


















