
Quality Systems Compliance 1 FDA Medical Device Quality Systems Compliance.
Upload
sterlingmedicaldevicesCategory
view
511download
6description
Mobile Medical Device Compliance
Vector Software Webinar March 27, 2013
Daniel Sterling, President

What we do:
o System development and test
Software and Electronics Experts
Any Phase
o Risk planning and hazard identification
o DHF Remediation
o Project Rescue
o Quality System Consulting
300+ Projects with 100 Clients
Who is Sterling?
ISO 13485
FM 543438
Registered
IEC 62304 Compliant
Your Partner in Medical Device Development
There when you need us!

Is Your App a Device? … and if so??
In the United States
LAW (FD&C Act)
Regulation (21CFRxxx)
FDA Guidance
ANSI / AAMI / ISO / IEC Standards

Medical Device Defined
Section 201(h) of the FD&C Act:
“…an instrument, apparatus, implement, machine, contrivance, implant, in vitro
reagent…..”, that is “…intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man…” or
“…intended to affect the structure or any function of the body of man or other animals…”

Intended Use
21 CFR 801.4:
… may, for example, be shown by labeling claims, advertising matter, or oral or written statements by such persons or their representatives.
… by the circumstances that the article is, with the knowledge of such persons or their representatives, offered and used for a purpose for which it is neither labeled nor advertised.

FDA Guidance for Mobile Medical Applications:
Draft Guidance for Industry and Food and Drug Administration Staff , Mobile Medical Applications, DRAFT GUIDANCE , July 21, 2011
http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatorinformation/ Guidances/ default.htm
IMHO, nice summary - not much new, Just Another Computing Platform
Why? Because all the previous guidance covers mobile
Some clarification on types of apps which have not usually been considered devices (e.g. office automation, EMR accessories)
Distinguishes between manufacturers and distributors (e.g. iTunes)
If the intended use makes the mobile device into a medical device or a medical device accessory, then it is a medical device.

Risk Risk Risk Assess, Mitigate, Test & Trace … and Repeat How Much Determined by Class and LOC
What if my App is a Device?

FDA Guidance for Mobile Medical Applications:
Guidance for Industry, Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software , January 14, 2005
http://www.fda.gov/cdrh/comp/guidance/1553.pdf
Risk considerations for networked devices – vulnerable to altered behavior
Plan for control of the device, updates/patches, subsequent validation
IMHO, drives requirements for mobile devices (e.g. control of OS updates)

Underlying FDA Guidance for Software:
• Guidance for Industry and FDA Staff -- Guidance for the Content of Premarket
Submissions for Software Contained in Medical Device, May 11, 2005
http://www.fda.gov/cdrh/ode/guidance/337.html
What Documents to Submit to FDA
Based on Level of Concern
• General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 11, 2002
http://www.fda.gov/cdrh/comp/guidance/938.html
Name is deceiving; outlines “good content” for all/most design output
“Validation” is based on a preponderance of evidence that good practices were used.

Underlying FDA Guidance for Software:
Guidance for Industry, FDA Reviewers and Compliance on Off-The-Shelf Software Use in Medical Device, September 9, 1999
http://www.fda.gov/cdrh/ode/guidance/585.html
Defines OTS Software
What you need to do with OTS as part of Validation and Risk Assessment

What is IEC 62304 and Why do We Care?
What is the Impact of Implementing This Standard?
Two Questions We Will Discuss:
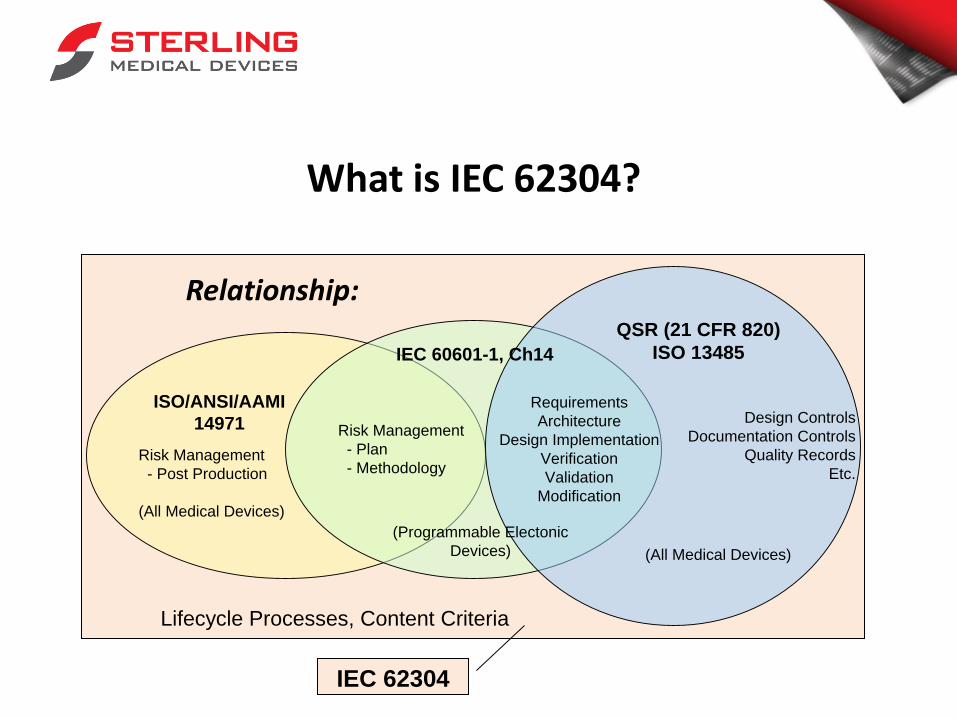
What is IEC 62304?
Relationship:
Risk Management
- Post Production
(All Medical Devices)
Risk Management
- Plan
- Methodology
Design Controls
Documentation Controls
Quality Records
Etc.
Requirements
Architecture
Design Implementation
Verification
Validation
Modification
IEC 60601-1, Ch14
ISO/ANSI/AAMI
14971
(All Medical Devices)
(Programmable Electonic
Devices)
QSR (21 CFR 820)
ISO 13485
IEC 62304
Lifecycle Processes, Content Criteria

What is IEC 62304?
1. Specifies activities and tasks for the development and
maintenance of software
2. A “how-to” for software compliance
3. What constitutes ‘good’ design output (in conjunction
with Guidance)
4. Where is review appropriate

What is IEC 62304?
5. What practices support the quality system and risk
management
6. Maps to PMA Guidance
7. Designed within context of 13485/QSR and 14971

FDA Standard Acceptance:
A standard from any standards body may be
acceptable to some extent; check at:
Standards Search (for recognition): http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/se
arch.cfm

… and Why Do We Care?
IEC 62304
…
Conformance to this standard provides
evidence that a software development
process is in place and fulfills the document
recommendation of the Software
Development Environment Description
section of the "Guidance for the Content of
Premarket Submissions for Software
Contained in Medical Devices".
(http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/detail.cfm?id=22436)
FDA: Recognized

Risk Management Standards
ISO 14971
FDA: Hot
Complete standard. A declaration of
conformity…
A declaration of conformity to ISO
14971 may be used to satisfy the risk
management needs for a Special
510(k). …
2007 version is accepted by FDA
2009 and 2012 version are NOT
(http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/Detail.cfm?ID=5188)

Impact of 62304
1. Quality System Alterations, Audits
2. Document/Template Content Revisions
3. Iterative Development Required
4. Focus on Risk Management
a) Architectural Decomposition
b) Risk Associated with Each Item
c) Safety Class Determines Process

Impact of 62304 Alterations…
Verification at Every Stage
E.g. At Unit Level
Verification Process including acceptance criteria
Based on Safety Class
Costly May affect economics of Tools & Automation

Impact of 62304 Iterative Development Required
IEC 80002-1, Conclusion
Plan for Risk (Re) Evaluations
Plan for Requirement, Design,
Implementation, and Test
Updates as a Result

Impact of 62304 Risk Management: Architectural Decomposition
Class A, B, C ~ LOC Minor, Moderate, Major
Independence Rationale Required

Impact of 62304
Risk Management: SFMEA Risk of Each Item/Unit
Why P1 and P2??
ID Class Unit Class Sub-Unit Class Sub-Unit 2
Potential Failure Mode
Potential Cause(s)/ Mechanism(s) of Failure
Potential Effect(s) of Failure (Harm)
Sev
P1
P2
Risk SRS ID
Mitigations
Sev
P1
P
2 Res
. Risk
C Main Control
Module
C User
Interface
C Main Panel Failure to display warning and notification messages
Failure to display correct images
Failure to display correct patient information
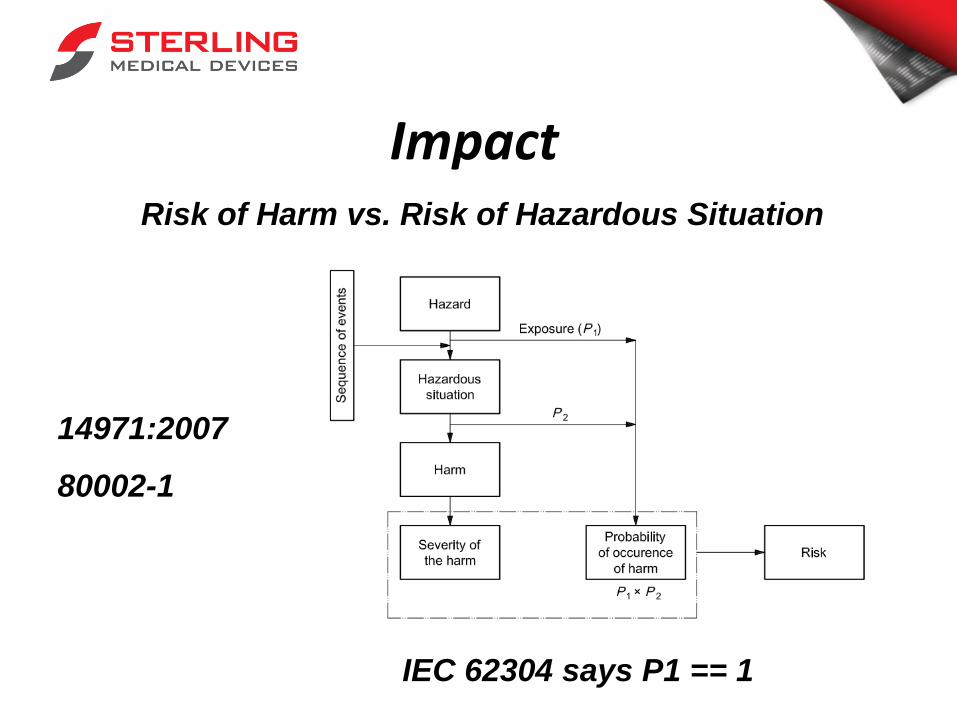
Impact Risk of Harm vs. Risk of Hazardous Situation
14971:2007
80002-1
IEC 62304 says P1 == 1

Recommendations
1. Develop Quality System per ISO 13485 and
QSR, Augment per IEC 62304
2. Reference ISO 90003 For Software Aspects of
the Quality System
3. Review Quality System and Trace to QSR and
13485
4. Develop Risk Plan per 14971/80002
a) Must Use 14971:2007 or later due to P1/P2
Distinction

Recommendations 5. Develop Software Portion of the Project Plan per IEC
62304, PMA Guidance, OTS Guidance, and Cyber
Security Guidance
• Be Sure To Integrate Risk Activities Into the
Development Plan!
6. Develop Risk/Hazard Analyses per 14971/80002
• Refer to IEC 80002-1 or 60601-1, ch14 for detailed
considerations
7. Develop Deliverable Templates and Work Product
Content per 62304, PMA, Validation, and OTS Guidance

What we do:
o System development and test
Software and Electronics Experts
Any Phase
o Risk planning and hazard identification
o DHF Remediation
o Project Rescue
o Quality System Consulting
300+ Projects with 100 Clients
ISO 13485
FM 543438
Registered
IEC 62304 Compliant
Your Partner in Medical Device Development
There when you need us!

A word about… Medical Device Data Systems (MDDS)
New rule: 21 CFR 880.6310 (not just guidance)
The Good:
Reclassification from III to I
The Bad:
Makes it clear that more devices and organizations fall
under Class I regulations
(e.g. hospitals developing/modifying their own systems
which meet the definition)

Medical Device Data Systems (MDDS)
Rule: Sec. 880.6310 Medical device data system.
(a)Identification. (1) A medical device data system (MDDS) is a
device that is intended to provide one or more of the
following uses, without controlling or altering the functions
or parameters of any connected medical devices:
(i) The electronic transfer of medical device data;
(ii) The electronic storage of medical device data;
(iii) The electronic conversion of medical device data from one
format to another format in accordance with a preset
specification; or
(iv) The electronic display of medical device data.

Medical Device Data Systems (MDDS)
Rule: Sec. 880.6310 Medical device data system.
(a)Identification. [continued]
(2) An MDDS may include software, electronic or electrical
hardware such as a physical communications medium
(including wireless hardware), modems, interfaces, and a
communications protocol. This identification does not
include devices intended to be used in connection with
active patient monitoring.

Medical Device Data Systems (MDDS)
Rule: Sec. 880.6310 Medical device data system.
(b)Classification. Class I (general controls). The device is
exempt from the premarket notification procedures in subpart
E of part 807 of this chapter, subject to the limitations in 880.9.
[76 FR 8649, Feb. 15, 2011]

References Standards: • ANSI/AAMI/IEC 62304:2006, Medical Device Software – Software Life Cycle
Processes
• EN/ISO 14971:2009, Medical Devices – Application of Risk Management to Medical Devices
• IEC/EN 60601-1-4, Medical electrical equipment — Part 1-4: General requirements for safety — Collateral standard: Programmable electrical medical systems (absorbed into 60601-1, ch 14 in latest version)
• EN/ISO 13485:2003, Medical devices - Quality Management Systems – Requirements for Regulatory Purposes

References FDA Guidance: • Guidance for Industry and FDA Staff -- Guidance for the Content of Premarket
Submissions for Software Contained in Medical Device, May 11, 2005
http://www.fda.gov/cdrh/ode/guidance/337.html
• General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 11, 2002
http://www.fda.gov/cdrh/comp/guidance/938.html
• Guidance for Industry, FDA Reviewers and Compliance on Off-The-Shelf Software Use in Medical Device, September 9, 1999
http://www.fda.gov/cdrh/ode/guidance/585.html

Daniel Sterling
President
Sterling Medical Devices 201-227-7569 x111
www.sterlingmedicaldevices.com
Mobile Medical Device
Compliance


















