
Mitochondrial Disorders
52
Mitochondrial Disorders Chenjie Xia (PGY-IV) Montreal Neurological Institute Wednesday, Jan. 25 th , 2012
description
Mitochondrial Disorders. Chenjie Xia (PGY-IV) Montreal Neurological Institute Wednesday, Jan. 25 th , 2012. Outline. Basic mitochondrial molecular biology Clinical implications of mitochondrial genetics Clinical approach to mitochondrial disorders General features - PowerPoint PPT Presentation
Transcript of Mitochondrial Disorders

Mitochondrial Disorders
Chenjie Xia (PGY-IV)
Montreal Neurological Institute
Wednesday, Jan. 25th, 2012

Outline
• Basic mitochondrial molecular biology
• Clinical implications of mitochondrial genetics
• Clinical approach to mitochondrial disorders– General features– Visual loss, ophthalmoplegia, peripheral
neuropathy, ataxia

Basic mitochondrial molecular biology

Mitochondrion Basics
• 4 compartments: – outer mb– inner mb (folds into
cristae)– intermembrane
space– matrix
Larsson and Oldfors, Acta Physiol Scand 2001.

Role of mitochondria = energy productionBerardo et al. Curr Neurol Neurosci Report, 2010

Respiratory Chain
http://www.photobiology.info/Hamblin.html

Mitochondrion Basics
• Mitochondrial respiratory chain:– Most “lucrative” step for ATP production– 5 enzymes complexes (90 protein subunits) – complexes I, II, III, IV creates proton gradient
(pump protons out of matrix) – complex V uses H gradient to generate ATP– Mobile electron carriers: CoQ, cyt c

Overview of clinical implications of mitochondrial genetics
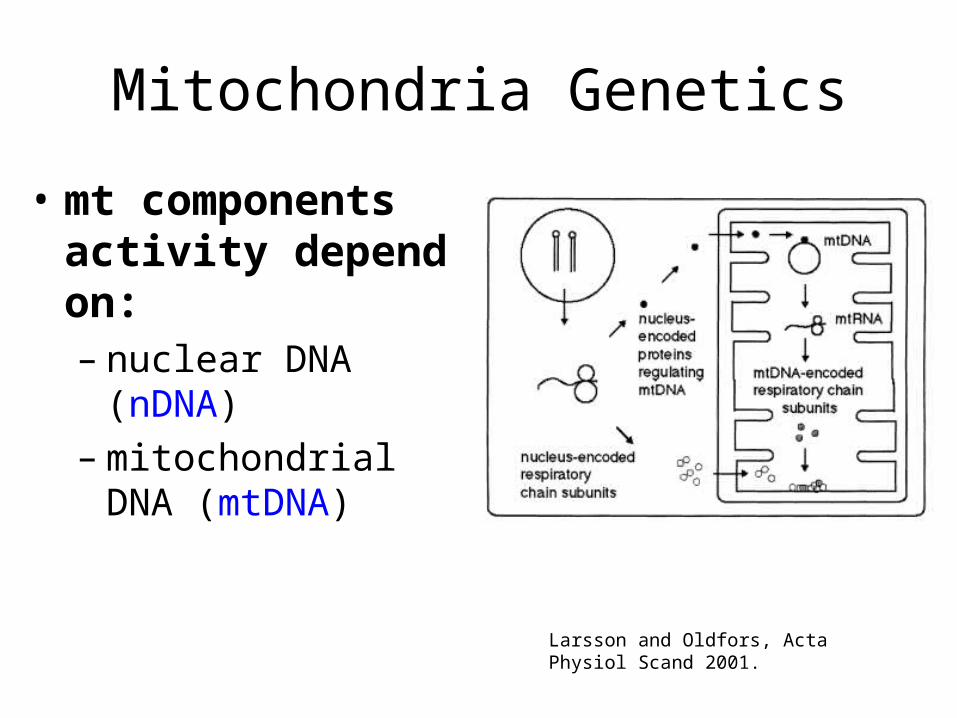
Mitochondria Genetics
• mt components activity depend on: – nuclear DNA (nDNA) – mitochondrial DNA
(mtDNA)
Larsson and Oldfors, Acta Physiol Scand 2001.

Mitochondrial Genetics
• mt genome encodes only 37 genes – 13/90 proteins of the RC– 2 rRNAs, 22 tRNAs
• Most mt proteins encoded by nDNA – e.g. complex II, CPTII, PDC– nDNA exerts +++ control on mt DNA & proteins– Concept of intergenomic communication
• Replication of mtDNA depend on factors encoded by nDNA

Mitochondrial Genetics
• nDNA mutations– Usually manifest in childhood– More severe and diffuse
• mtDNA mutations– Usually manifest in adulthood– More indolent and mosaic
• These principles hold less well given recent discoveries showing increasing clinical and genetic heterogeneity of mt disorders

Mitochondrial Genetics
• Concept of heteroplasmy– Mitochondrion contain mix of mutant and wild-type mtDNA– Proportion of mutant mtDNA differs in different tissues or even
cells of same tissue
• Concept of threshold effect– Sx develop only when mutant mtDNA reaches certain threshold
(usually high, >90%)– Threshold depends on energy metabolism of tissue
• Concept of replicative segregation– Mutant mtDNA are “selected out” with repeated mitoses, but
accumulate in tissues not undergoing mitoses (e.g. neurons, muscles)

Mitochondria Genetics
• Mitochondrial disorders can be sporadic or inherited
• If inherited, mainly maternally:– “Bottle-neck” effect in oogenesis – rare case report of paternally inherited
• Often no clear genotype-phenotype correlation

Mitochondrial Genetics
• THM = phenotypic expression of mt disorders depend on many factors:– Nuclear versus mitochondrial mutation– Pathogenicity of mutation itself– Heteroplasmy– Threshold effect– Mitotic activity of tissue– Energy demand of tissue– Age– Etc…

Clinical Approach to Mitochondrial Disorders

A few words on mitochondrial disorders in general

General features of MID
• Classic S/Sx:– Neurological:
• stroke, seizure, dev. delay, dementia, visual impairment, EOMs, deafness, neuropathy, myopathy
– Other: • DM, hepatopathy, cardiomyopathy, cardiac
conduction defects, short stature

Clinical Manifestations - Systemic
Nardin and Johns. Muscle and Nerve, 2001.

General features of MID
• Key points:– +++ multisystemic– +++ overlap b/w different syndromes– same mutation can cause different phenotypes
• proportion of 3243tRNA mutation determines CPEO vs MELAS vs Leigh’s
– same phenotype can result from different mutations
• MELAS can result from 3243tRNA, 3271tRNA, 11084 ND4

Classification of MIDs
• 1. Affected structure or pathway w/i mito.– RC subunits, tRNA, mt transport machinery, mt
maintenance, etc
• 2. Mono- vs multi-systemic
• 3. Syndromic vs non-syndromic– +++ genetic & phenotypic overlap b/w the two– Syndromic better known for acronyms and for
understanding of mito medicine– But non-syndromic more common, probably less
recognized in clinical practice (atypical & less spectacular presentations)

Presenting Phenotypes
• Visual Loss
• Ptosis / opthalmoplegia
• Neuropathy
• Ataxia
• (Myopathy)
• (Seizures)
• (Stroke)

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Presenting Phenotypes
• Visual Loss
• Ptosis / opthalmoplegia
• Neuropathy
• Ataxia
• (Myopathy)
• (Seizures)
• (Stroke)
Visual Loss – LHON• Leber’s Hereditary Optic Neuropathy
– Degeneration of retinal ganglion cells– Most common disease caused by mtDNA mutation
• Clinical presentation– Bilateral sequential acute or subacute visual failure– Central vision lost before peripheral, blue-yellow
perception lost early on (red-green more preserved)– Disc swelling and hyperemia followed by atrophy– Predominantly in young men– Little or no recovery (altho visual impairment seldom
complete)

Visual Loss = LHON
• When to think of LHON for visual loss:– Young men– no vascular comorbidities (less likely
ischemic)– Painless (less likely optic neuritis, either viral
or demyelinating)– No toxic or deficiency state (B12, thiamine,
tobacco-alcohol amblyopia, sildenafil)

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Visual Loss - RP
• Retinitis Pigmentosa– All retinal layers affected – Predominance in males – 1st Sx = nyctalopia (impairment of twilight vision)– Usu both eyes affected simultaneously– Perimacular zones affected first partial to complete
ring scotoma– Pigmentary changes spare fovea eventually pt
perceives world as if seeing through tubes

Visual Loss - RP
• DDx– Bardet-Biedl syndrome, Laurence-Moon
syndrome, Freidreich’s ataxia, Refsum, Cockayne syndrome, Bassen-Kornzweig disease
– Kearn-Sayre syndrome

Presenting Phenotypes
• Visual Loss
• Ptosis / opthalmoplegia
• Neuropathy
• Ataxia
• (Myopathy)
• (Seizures)
• (Stroke)

Ophthalmoplegia – KSS
• Kearns-Sayre syndrome– Obligatory triad: onset before 20, pigmentary
retinopathy, progressive external opthalmoplegia
– Other features: cardiac conduction abnormalities, can also have high CSF protein, cerebellar ataxia, seizures, sensorineural deafness, pyramidal signs

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Ophthalmoplegia – CPEO
• Chronic progressive external ophthalmoplegia• Clinical manifestations
– Ptosis, can be asym.; ophthalmoplegia, more symm. (rare diplopia, transient if occurs)
– Long durat’n of Sx before presentation (mean 26 years)– majority presents for ptosis (1/2 have less than 10% of
ocular motility fxn!!!)– Sx may worsen in the evening– Often no FMHx

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Ophthalmoplegia – CPEO
• DDx– KSS (ECG, age of onset, severity)– MG (anti-AchR, response to Mestinon)– OPMD (muscle biopsy)

Presenting Phenotypes
• Visual Loss
• Ptosis / opthalmoplegia
• Neuropathy
• Ataxia
• (Myopathy)
• (Seizures)
• (Stroke)

Classification of MIDs causing PNP
Finsterer, Journal of Neurological Sciences, 2011

PNP in MIDs – Leigh syndrome
• Clinical manifestations– Dev. delay, seizures– Ophthalmoparesis, nystamus– cerebellar ataxia, chorea, dystonia– Spasticity, muscle weakness– Brainstem involvement: respiratory insufficiency,
dysphagia, recurrent vomiting, abnormal thermoregulation
– Non-neurological: short stature, cardiomyopathy, anemia, RF, vomiting, diarrhea

Ataxia in MIDs – Leigh syndrome
• Other features– Most frequent childhood MID– Wide variety of abnormalities (from severe to absence
of neurological problems)– Wide genetic heterogeneity
• Features of peripheral neuropathy (collateral)– Sensori-motor, demyelinating– Can be confused with GBS (due to severe
demyelination)

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Saneto et al. Mitochondrion, 2008
Boy with LS: 7 mos
Boy with LS: 19 mos

PNP in MIDs – MNGIE
• Mitochondrial neuro-gastrointestinal encephalopathy– Severe gastrointestinal dysmotility (nausea,
postprandial emesis, early satiety, dysphagia, reflux, abdo pain, diarrhea, cachexia)
– Others: confusion, PEO, deafness, dysarthria, short stature
• Features of peripheral neuropathy (collateral)– Sensori-motor, with distal weakness, predominantly
affects lower limbs (may be confused with CIDP)– Mixed axonal and demyelinating on NCS

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Presenting Phenotypes
• Visual Loss
• Ptosis / opthalmoplegia
• Neuropathy
• Ataxia
• (Myopathy)
• (Seizures)
• (Stroke)

Finsterer, CJNS, 2009

PNP in MIDs – NARP
• Neurogenic weakness with ataxia and retinitis pigmentosa
• Clinical manifestations– Proximal muscle weakness due to PNP– Ataxia due to cerebellar atrophy– Visual impairment (optic atrophy, salt&pepper
retinopathy, bull’s eye maculopathy, or RP)– Others: short stature, opthalmoplegia, learning
difficulties, dementia, seizures, cardiac arrhythmias

Genetic Defects of MIDs
Finsterer, CJNS, 2009

Ataxia in MID – AHS
• Alpers-Huttenlocher Syndrome– Severe hepatocerebral syndrome
• Clinical manifestations– Starts in first years of life, early death– Neurological: intractable seizures, dev. delay,
psychomotor regression, stroke-like episodes, hypotonia, cortical blindness, ataxia
– Other: hepatic failure (avoid valproic acid!!), fasting hypoglycemia

Genetic Defects of MIDs
Finsterer, CJNS, 2009
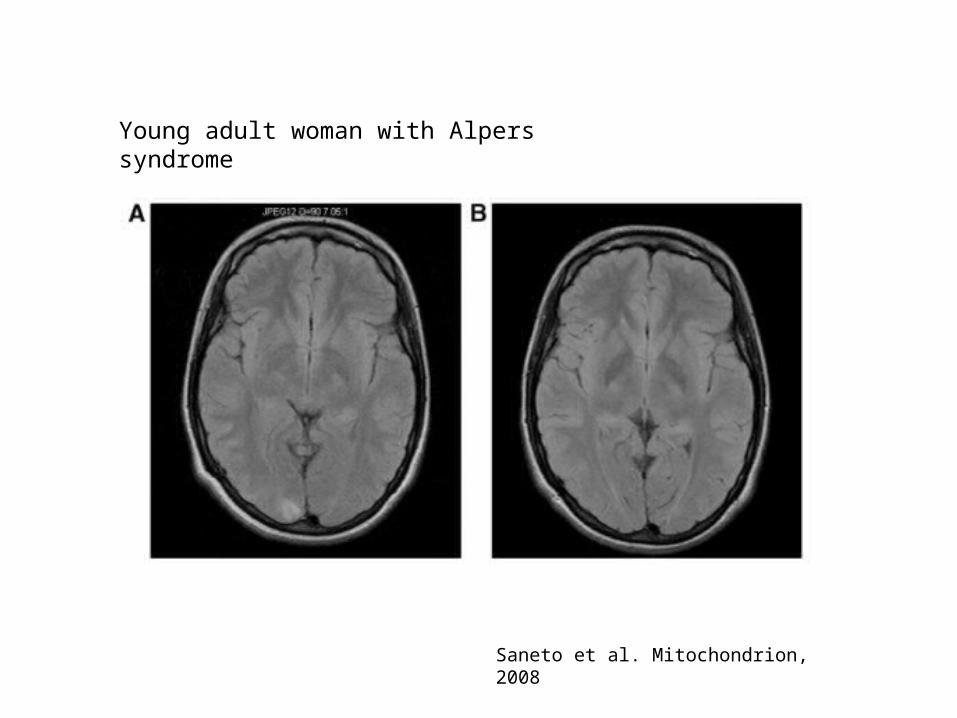
Saneto et al. Mitochondrion, 2008
Young adult woman with Alpers syndrome

Take Home Messages
• Main role of mitochondria = energy production; mitochondrial disorders predominantly affect high metabolism (energy dependent) tissues
• Both mtDNA and nDNA abnormalities are implicated in mitochondrial disorders
• Absence of FMHx by no means preclude Dx of mt disorder• Syndromic mt disorders are better known, but non-
syndromic mt disorders are more common• There is no clear genotype-phenotype correlation in
mitochondrial disorders• Mitochondrial disorders are often multisystemic• There is +++ overlap in phenotype b/w different mt disorders

References
• Caballero et al. Chronic progressive exertnal ophthalmoplegia, The Neurologist, 2007, 33-36.
• Finsterer, Mitochondrial ataxias, CJNS, 2009, 36, 543-553. • Finsterer, Inherited mitochondrial neuropathies, Journal of
Neurological Sciences, 304, 2011, 9-16• N.-G. Larsson and A. Oldfors. Mitochondrial myopathies,
Acta Physiol Scand 2001, 171, 385-393.• Rachel Nardin and Donald Johns. Mitochondrial dysfunction
and neuromuscular disease. Muscle and Nerve, 2001, 24: 170-191.
• Saneto et al. Neuroimaging of mitochondrial disease, Mitochondrion, 2008, 396-413.
• Schapira, Mitochondrial disease, Lancet, 2006, 70-82

Finsterer, Journal of Neurological Sciences, 2011
Classification of MID


















