
Michael Newton - University of...
87
Transcript of Michael Newton - University of...


Michael NewtonUW Madison
Why don’t we agree? Studying influenza with RNA interference
Buehler-Martin LectureUniversity of Minnesota Statistics

From a project on influenza biology with:
Qiuling HeLin HaoMark CravenPaul Ahlquist
Thanks Christina Kendziorski...

What human genes does influenza virus co-opt during its life cycle?

a bit about flu


RNA interference is a process within living cells that moderates the activity of their genes.
Experimental Method: RNAi
Fire and Mello, 2006, Nobel Prize

a bit about RNAi

Kim and Rossi Nature Reviews Genetics 8, 173–184 (March 2007) | doi:10.1038/nrg2006

Or, more simply, ...
RNA interference =

Or, more simply, ...
genome-wide...
RNA interference =


cell
target gene
expressionlevel

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
add siRNA
target gene
+

cell
expressionlevel
target gene

cell
expressionlevel
phenotype of interest changes
target gene

explain phenotype

Issues

gene
expressionlevel
Involvement: gene may not affect phenotype

involved gene
expressionlevel
Efficiency: knockdown may not be complete

gene
expressionlevel
Accessibility: something blocks the phenotype
a. no expression in these particular cells

g1
expressionlevel
Accessibility: something blocks the phenotype
b. Redundency/masking
g2

gene
expressionlevel
Accessibility: something blocks the phenotype
c.cytotoxicity

expressionlevel
Off target effects
target g1
off target g2
not involved
involved

Measurement error

Measurement error
non-involved gene
expressionlevel
false positive

Measurement error

Measurement error
expressionlevel
involved gene
false negative

one siRNA
+
knock down
no knock down

one siRNA
+
on target
off target
on target off targetX Xknock down
no knock down

one siRNA
+
on target
off target
on target off targetX Xknock down
no knock down
error
error
error
error
X
X
measurement

15
Meta analysis of four recent studies
2010
2010
2009
2008
A549DE
A549US
U2OS
Human
DL-1 Drosophila

Data
results (gene lists) from 4 two-stage RNAi studies

Data
results (gene lists) from 4 two-stage RNAi studies
1. detection2. confirmation

Data
results (gene lists) from 4 two-stage RNAi studies
1. detection2. confirmation
DL-1 U2OS A549DE A549US
Detection Screen 1 1 0 0
Confirmation Screen 1 0 0 0
Pattern Code 11 10 00 00
e.g., one genestudy
33
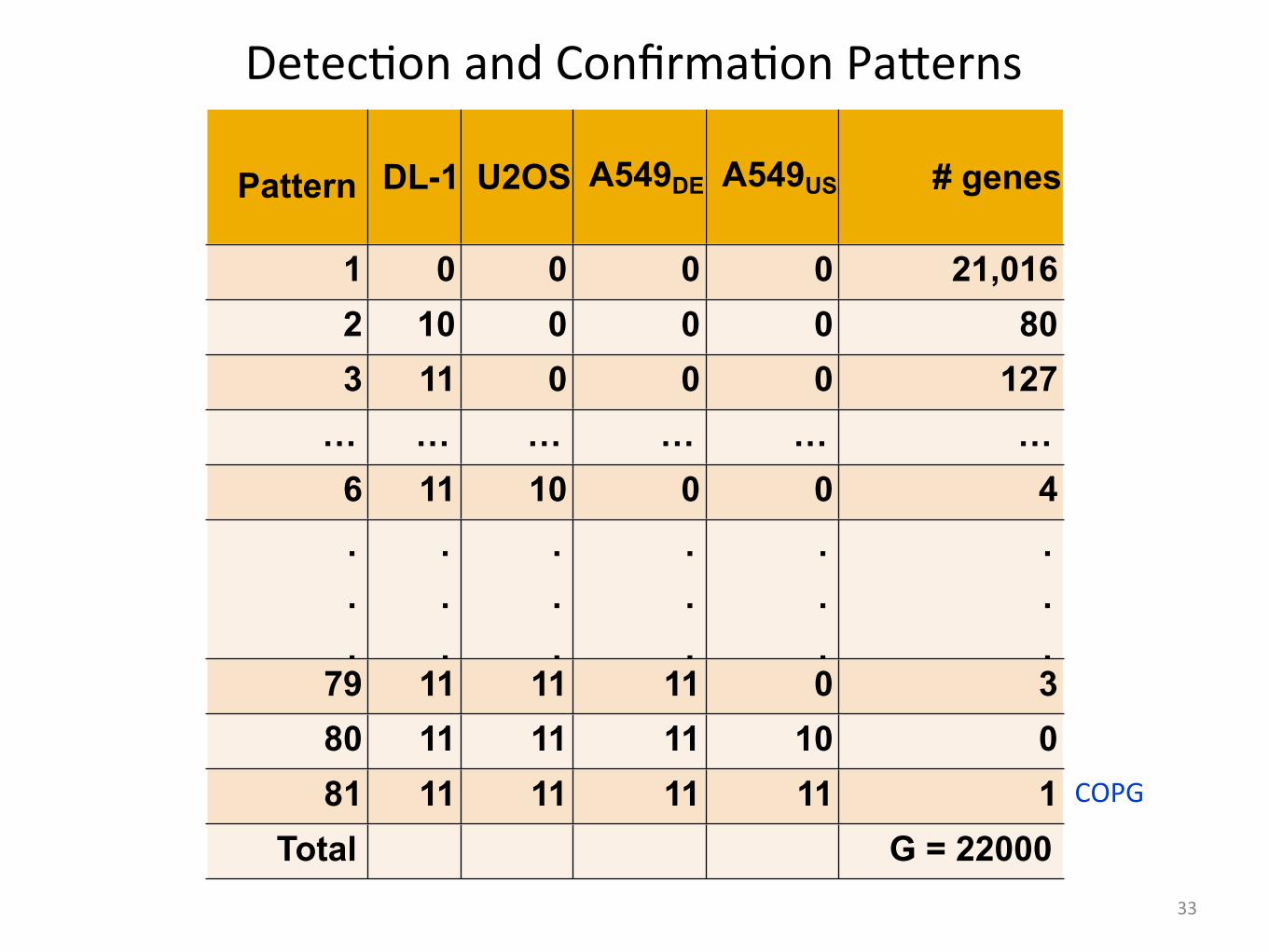
Pattern DL-1 U2OS A549DE A549US # genes
1 0 0 0 0 21,0162 10 0 0 0 803 11 0 0 0 127
… … … … … …6 11 10 0 0 4...
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.79 11 11 11 0 380 11 11 11 10 081 11 11 11 11 1
Total G = 22000
Detec9on and Confirma9on Pa>erns
COPG

Confirmed by 1 Study
Confirmed by 2 Studies
Confirmed by 3 Studies
Confirmed by 4 Studies
17
Agreement among studies is low.
Among the 614 genes confirmed by at least one study:

37
Is the limited overlap due more to false posi9ve or false nega9ve factors?

38
6.7% average pairwise overlap of confirmed gene lists is significantly higher than expected by chance

L =Q
⇡ Pn⇡⇡
P⇡ = Prob (gene shows detection/confirmation pattern ⇡ )
n⇡ = #(gene shows detection/confirmation pattern ⇡ )
39
Modeling approach
LikelihoodDL-1 U2OS A549DE A549US
1 0 0 0 0 21,016
2 10 0 0 0 80
3 11 0 0 0 127
… … … … … …
6 11 10 0 0 4
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.79 11 11 11 0 3
80 11 11 11 10 0
81 11 11 11 11 1
Total G = 22000
P⇡n⇡⇡

40
Cg,s = 1 [ gene g confirmed in study s ]
Dg,s = 1 [ gene g detected in study s ]
Ag,s = 1 [ g accessible in s ]
Ig = 1 [ g involved in flu ]
Data and latent variables
Tg,s = #{involved, accessible o↵ targets, study s, target g }

expression
target gene
Detection screen
pool of 4 distinct siRNA’s per target gene
one phenotype call Dg,s
+

Confirmation screen
Cg,s = 1
"X
k
Cg,s,k � 2
#
expression
target gene
expression
target gene
expression
target gene
expression
target gene
Cg,s,1 Cg,s,2 Cg,s,3 Cg,s,4
++++

43
���������������������������������������������
���
�
����
���
����
��
����
�����
��
���� ��
����
�����
��
���� �

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel
U1

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel
U1

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel
U1U2

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel
U1U2

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel Y
j
Uj

on target knock down model
U1, U2, U3, U4 ⇠iid Uniform(0, 1)
target gene
expressionlevel Y
j
Uj
Y
j
Uj < �
knock down effect if
threshold parameter
Prob: 1�G4 [log(1/!)]

off target knock down model
t = #{involved, accessible o↵ targets}
U1, U2, . . . , Ut ⇠i.i.d. Uniform(0, 1)
e.g. t = 3
U1 < !
U2 < !
U3 < !
knock down effect if any Uj < !
1� (1� !)tProb:

0.0 0.2 0.4 0.6 0.8 1.00.0
0.2
0.4
0.6
0.8
1.0
ω
Prob
4 hits on target4 hits off target
1�G4 [log(1/!)]
1� (1� !)4

detection screen:
knock down
no knock down
measurement
+
1� (1� !)t
(1� !)t{G4 [log(1/�)]}ai
1� {G4 [log(1/�)]}ai
�
1� �
↵
1� ↵
Dg,s | Ag,s = a, Ig = i, Tg,s = t

confirmation screen
Cg,s = 1
"X
k
Cg,s,k � 2
#
expression
target gene
expression
target gene
expression
target gene
expression
target gene
Cg,s,1 Cg,s,2 Cg,s,3 Cg,s,4
++++

confirmation screen:
knock down
no knock down
measurement
+
�
1� �
↵
1� ↵
1� !⇣1� !
4
⌘t
1�⇣1� !
4
⌘t
Cg,s,k | Ag,s = a, Ig = i, Tg,s = t
1� (1� !)ai

On the number of off targets per siRNA
• very limited data• libraries overlap among 4 studies

one target gene
K siRNA’s available to all studies (on average)
siRNA’s
involved off targets
Tg

one target gene
study s uses 4 siRNA’s
siRNA’s
involved, accessible off targets
Tg,s
x x

Tg = #{involved o↵-targets, target g }
Tg,s|Tg = t ⇠ Binomial
✓t,4�sK
◆detection
confirmationTg,s,k|Tg,s = t ⇠ Binomial
✓t,1
4
◆
Tg ⇠ Poisson(K✓⌫)

54
In these terms, the log-likelihood is
L = log Prob(data) =⇧
�
N� logP�, (1)
where pattern probabilities {P�} are defined by a smaller number of parameters through a stochasticmodel of genome-wide RNAi. The model is hierarchical, and is specified using latent random e�ects:
Ig = 1[g is involved in influenza virus replication]
Ag,s = 1[g is accessible in study s ]
Tg = number of involved o�-targets for gene g, relative to a pool of siRNAs
that might be used to target gene g
Tg,s = size of the accessible subset of Tg in study s
Vg,s,k = size of the accessible subset of Tg,s in assay k of secondary screen in study s .
One could alternatively classify the {Ig} as a high-dimensional parameter, but in doing Bayesianinference we would immediately cover it with a prior, so we treat it as a vector of latent factors inthe notation.
A number system-level parameters are used to specify the probability structure of latent vari-ables and observed data; they describe the basic system in terms of rates governing the latentvariables as well as quantities a�ecting false-positive and false-negative detections and confirma-tions:
⌅ = proportion of genome involved in influenza virus replication
� = false positive measurement error
⇥s = false negative measurement error, study s
⇤s = rate at which genes are accessible, study s
� = knockdown e⌅ciency per siRNA
⌃ = average number of o�-targets per siRNA .
The log-likelihood L in (1) is a function of these 12 parameters, which we collect in a vector⌥ = (⌅,�,⇥, ⇤,�, ⌃), where ⇥ = {⇥s}4s=1, ⇤ = {⇤s}4s=1. Thus L = L(⌥). The stochastic model itselfis:
Ig ⇤ Bernoulli(⌅) (2)
Ag,s ⇤ Bernoulli(⇤s)
Tg ⇤ Negative Binomial(⇧,K⌅⌃
K⌅⌃ + ⇧)
Tg,s |[Tg = t] ⇤ Binomial(t,4⇤sK
)
Vg,s,k |[Tg,s = u] ⇤ Binomial(u,1
4)
Dg,s |[Ig = i, Ag,s = a, Tg,s = t] ⇤ Bernoulli⌃1� ⇥s + (�+ ⇥s � 1) [G4,1(� log�)]ai (1� �)t
⌥
Cg,s,k |[Ig = i, Ag,s = a, Vg,s,k = v] ⇤ Bernoulli�1� ⇥s + (�+ ⇥s � 1)(1� �)ai+v
⇥
P (Cg,s = 1|Ig = i, Ag,s = a, Tg,s = u) = P
⇤4⇧
k=1
Cg,s,k ⇥ 2|Ig = i, Ag,s = a, Tg,s = u
⌅.
2
Parameters

55
In these terms, the log-likelihood is
L = log Prob(data) =⇤
�
N� logP�, (1)
where pattern probabilities {P�} are defined by a smaller number of parameters through a stochasticmodel of genome-wide RNAi. The model is hierarchical, and is specified using latent random e�ects:
Ig = 1[g is involved in influenza virus replication]
Ag,s = 1[g is accessible in study s ]
Tg = number of involved o�-targets for gene g, relative to a pool of siRNAs
that might be used to target gene g
Tg,s = size of the accessible subset of Tg in study s
Vg,s,k = size of the accessible subset of Tg,s in assay k of secondary screen in study s .
One could alternatively classify the {Ig} as a high-dimensional parameter, but in doing Bayesianinference we would immediately cover it with a prior, so we treat it as a vector of latent factors inthe notation.
A number system-level parameters are used to specify the probability structure of latent vari-ables and observed data; they describe the basic system in terms of rates governing the latentvariables as well as quantities a�ecting false-positive and false-negative detections and confirma-tions:
⌅ = proportion of genome involved in influenza virus replication
� = false positive measurement error
⇥s = false negative measurement error, study s
⇤s = rate at which genes are accessible, study s
� = knockdown e⌅ciency per siRNA
⌃ = average number of o�-targets per siRNA .
The log-likelihood L in (1) is a function of these 12 parameters, which we collect in a vector⌥ = (⌅,�,⇥, ⇤,�, ⌃), where ⇥ = {⇥s}4s=1, ⇤ = {⇤s}4s=1. Thus L = L(⌥). The stochastic model itselfis:
Ig ⇥ Bernoulli(⌅)
Ag,s ⇥ Bernoulli(⇤s)
Tg ⇥ Negative Binomial (mean = K⌅⌃, shape = ⇧)
Tg,s |[Tg = t] ⇥ Binomial (t, 4⇤s/K)
Dg,s |[Ig = i, Ag,s = a, Tg,s = t] ⇥ Bernoulli⌅1� ⇥s + (�+ ⇥s � 1) [G4,1(� log�)]ai (1� �)t
⇧
Cg,s,k |[Ig = i, Ag,s = a, Tg,s = t] ⇥ Bernoulli�1� ⇥s + (�+ ⇥s � 1)(1� �)ai(1� �/4)t
⇥
The shape parameter ⇧ = 0.11 is estimated from a predicted distribution of number of o�-targetgenes per dsRNA in a previous study that evaluates the o�-target e�ects of dsRNAs (Kullkarni etal.). G4,1(.) is the c.d.f. of a gamma distribution with shape parameter 4 and scale parameter 1.Another system-level parameter we fix a priori and do not estimate from the data is
K = average number of siRNAs that target a gene.
2
Poisson(K✓⌫)

P⇡ =X
i,a
P⇡(i, a)
56
studies are heterogeneous, both because they may entail di�erent sets of accessible genes, but alsobecause these accessibility rates (�s) are study specific. (We had considered a single parameter �,but saw substantial improvements when we allow the extra flexibility.) From study to study thedata are not independent, owing to gene specific factors Ig and Tg, which get marginalized in ourlikelihood computation.
The 81 multi-study pattern probabilities {P�} (and thus the log-likelihood L(⌃)) in (1) areobtained as a function of the 8 system-level parameters ⌃ by summing out the discrete-valuedlatent variables. Considering among-gene independence, we focus on a single gene, and sum outvalues of the involvement indicator Ig, the four accessibility indicators Ag,s, and the o�-targetcounts Tg and the four Tg,s. (We model Tg,s’s as subsets of a common Tg to reflect the possibilitythat di�erent studies share siRNAs.) All but the target counts are binary sums; more complicatedis the elimination of the o�-target counts. To investigate this calculation, write the vector a = {as}and the conditional probability of data pattern ⇧ as,
P�(i, a) = P (⇧| Ig = i, {Ag,s}4s=1 = a⇥.
Each multi-study pattern probability P� is computed as a summation of these P�(i, a) over the 25
values of its arguments. The trickier computation is the evaluation of each P�(i, a), which requiresmarginalization of the o�-target counts.
To marginalize the o�-target counts, first recognize that each pattern ⇧ is an intersection offour study-specific patterns ⇧ =
⇤s ⇧s. For example ⇧ = 3111 indicates that the gene is confirmed
and detected in the first study and neither detected nor confirmed in any of the remaining threestudies. The modeling assumptions give
P�(i, a) =�⌅
t=0
P (Tg = t) P�⇧|Ig = i, {Ag,s}4s=1 = a, Tg = t
⇥
=�⌅
t=0
Pois(t)4⇧
s=1
P (⇧s|Ig = i, Ag,s = as, Tg = t)
=�⌅
t=0
Pois(t)4⇧
s=1
t⌅
u=0
Bs(t, u) P (⇧s|Ig = i, Ag,s = as, Tg,s = u)
=�⌅
t=0
Pois(t)4⇧
s=1
t⌅
u=0
Bs(t, u) qs,i,as,u (3)
where Pois(t) = P (Tg = t) = exp{�4⇥⇤}(4⇥⇤)t/t! by the Poisson assumption, Bs(t, u) is theBinomial mass function at u with t trials and success probability �s, and where each contributionqs,i,as,u = P (⇧s|Ig = i, Ag,s = as, Tg,s = u) is computed from the stochastic model (2). Comingback to pattern ⇧ = 3111 for example, the four sub-pattern probabilities are:
q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
A key to simplifying the computation further is to recognize that with respect to the count variableu, each qs,i,as,u is a polynomial in ⌅ = (1 � ⌥)1/4, of degree at most 8u. By careful book-keeping,
3
studies are heterogeneous, both because they may entail di�erent sets of accessible genes, but alsobecause these accessibility rates (�s) are study specific. (We had considered a single parameter �,but saw substantial improvements when we allow the extra flexibility.) From study to study thedata are not independent, owing to gene specific factors Ig and Tg, which get marginalized in ourlikelihood computation.
The 81 multi-study pattern probabilities {P�} (and thus the log-likelihood L(⌃)) in (1) areobtained as a function of the 8 system-level parameters ⌃ by summing out the discrete-valuedlatent variables. Considering among-gene independence, we focus on a single gene, and sum outvalues of the involvement indicator Ig, the four accessibility indicators Ag,s, and the o�-targetcounts Tg and the four Tg,s. (We model Tg,s’s as subsets of a common Tg to reflect the possibilitythat di�erent studies share siRNAs.) All but the target counts are binary sums; more complicatedis the elimination of the o�-target counts. To investigate this calculation, write the vector a = {as}and the conditional probability of data pattern ⇧ as,
P�(i, a) = P (⇧| Ig = i, {Ag,s}4s=1 = a⇥.
Each multi-study pattern probability P� is computed as a summation of these P�(i, a) over the 25
values of its arguments. The trickier computation is the evaluation of each P�(i, a), which requiresmarginalization of the o�-target counts.
To marginalize the o�-target counts, first recognize that each pattern ⇧ is an intersection offour study-specific patterns ⇧ =
⇤s ⇧s. For example ⇧ = 3111 indicates that the gene is confirmed
and detected in the first study and neither detected nor confirmed in any of the remaining threestudies. The modeling assumptions give
P�(i, a) =�⌅
t=0
P (Tg = t) P�⇧|Ig = i, {Ag,s}4s=1 = a, Tg = t
⇥
=�⌅
t=0
Pois(t)4⇧
s=1
P (⇧s|Ig = i, Ag,s = as, Tg = t)
=�⌅
t=0
Pois(t)4⇧
s=1
t⌅
u=0
Bs(t, u) P (⇧s|Ig = i, Ag,s = as, Tg,s = u)
=�⌅
t=0
Pois(t)4⇧
s=1
t⌅
u=0
Bs(t, u) qs,i,as,u (3)
where Pois(t) = P (Tg = t) = exp{�4⇥⇤}(4⇥⇤)t/t! by the Poisson assumption, Bs(t, u) is theBinomial mass function at u with t trials and success probability �s, and where each contributionqs,i,as,u = P (⇧s|Ig = i, Ag,s = as, Tg,s = u) is computed from the stochastic model (2). Comingback to pattern ⇧ = 3111 for example, the four sub-pattern probabilities are:
q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
A key to simplifying the computation further is to recognize that with respect to the count variableu, each qs,i,as,u is a polynomial in ⌅ = (1 � ⌥)1/4, of degree at most 8u. By careful book-keeping,
3
Calcula9ng pa>ern probabili9es:
Ideally if all 4 studies use the same siRNA library, we would expect K = 4 i.e. there are exactly 4siRNAs targeting every gene as designed; if each study uses a di�erent library, then K = 16 if all li-braries are specific and there is no chance of genes being targeted by siRNAs other than the designedones. K controls the correlation in numbers of o�-targets between studies, i.e. cor(Tg,s,Tg,s0) � 0,as K � ⇥. In our case, we fix K = 12 since the 4 studies of interest use 3 libraries.
A full specification of conditional independence assumptions is in Figure 2. The various mod-eling elements have been introduced to address known features of genome-wide siRNA screening.For example, every additional siRNA applied to an involved gene increases the chance that theharboring cells exhibit a phenotype. The higher the rate of involved genes, the higher the rateof an o�-target phenotype. There is heterogeneity among genes, owing to whether or not theyare involved, and owing to varying amounts of o�-targets associated with their targeting pools ofsiRNAs, but there is among-gene independence in terms of siRNA detection/confirmation. Thestudies are heterogeneous, because they may entail di�erent sets of accessible genes and these ac-cessibility rates (⇥s) are study specific, and also there may be di�erent false negative measurementerrors involved in each study due to individual experimental environment. (We had considered asingle parameter ⇥ and �, but saw substantial improvements when we allow the extra flexibility.)From study to study the data are not independent, owing to gene specific factors Ig and Tg, whichget marginalized in our likelihood computation.
The 81 multi-study pattern probabilities {P�} (and thus the log-likelihood L(⌅)) in (1) areobtained as a function of the 12 system-level parameters ⌅ by summing out the discrete-valuedlatent variables. Considering among-gene independence, we focus on a single gene, and sum outvalues of the involvement indicator Ig, the four accessibility indicators Ag,s, and the o�-targetcounts Tg, the four Tg,s, and the {Vg,s,k}4k=1 for each study. (We model Tg,s’s as subsets of acommon Tg to reflect the possibility that di�erent studies share siRNAs.) All but the target countsare binary sums; more complicated is the elimination of the o�-target counts. To investigate thiscalculation, write the vector a = {as} and the conditional probability of data pattern ⇤ as,
P�(i, a) = P (⇤| Ig = i, {Ag,s}4s=1 = a⇥.
Each multi-study pattern probability P� is computed as a summation of these P�(i, a) over the 25
values of its arguments. The trickier computation is the evaluation of each P�(i, a), which requiresmarginalization of the o�-target counts.
To marginalize the o�-target counts, first recognize that each pattern ⇤ is an intersection offour study-specific patterns ⇤ =
⇤s ⇤s. For example ⇤ = 3111 indicates that the gene is confirmed
and detected in the first study and neither detected nor confirmed in any of the remaining threestudies. The modeling assumptions give
P�(i, a) =�⌅
t=0
P (Tg = t) P�⇤|Ig = i, {Ag,s}4s=1 = a, Tg = t
⇥
=�⌅
t=0
NB(t)4⇧
s=1
P (⇤s|Ig = i, Ag,s = as, Tg = t)
=�⌅
t=0
NB(t)4⇧
s=1
t⌅
u=0
Bs(t, u) P (⇤s|Ig = i, Ag,s = as, Tg,s = u)
=�⌅
t=0
NB(t)4⇧
s=1
t⌅
u=0
Bs(t, u) Qs,i,as,u (2)
3
Po(t)
Po(t)
Po(t)

⇠1 = 1� !
⇠2 = 1� !
4
57
where NB(t) = P (Tg = t) is the point mass of a Negative Binomial distribution with shape pa-rameter ⇤ and mean K⇥⌅, Bs(t, u) is the Binomial mass function at u with t trials and successprobability 4�s
K , and where each contribution Qs,i,as,u = P (⌃s|Ig = i, Ag,s = as, Tg,s = u) is com-puted from the stochastic model (??). Coming back to pattern ⌃ = 3111 for example, the foursub-pattern probabilities are:
Q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
Q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
Q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
Q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
where
P (Cg,s = 1|Ig = i, Ag,s = as, Tg,s = u) = P
⇤4⇧
k=1
Cg,s,k ⇥ 2|Ig = i, Ag,s = as, Tg,s = u
⌅
A key to simplifying the computation further is to recognize that with respect to the count variableu, each P (Dg,s|Ig, Ag,s, Tg,s) is a polynomial of ⇧1 = 1 � �, and each P (Cg,s|Ig, Ag,s, Tg,s) is apolynomial of ⇧2 = 1 � ⇥
4 . Thus, each Qs,i,as,u is a bivariate polynomial in ⇧1 and ⇧2, of degree atmost u and 4u respectively. By careful book-keeping, we identify coe�cients {bs,p,q} (dependingon system parameters ⌥ and the pattern ⌃) such that
Qs,i,as,u =1⇧
p=0
4⇧
q=0
bs,p,q (⇧p1 ⇧q2)
u
Thus the inner factor of (2)
t⇧
u=0
Bs(t, u) Qs,i,as,u =t⇧
u=0
Bs(t, u)8⇧
j=0
bs,j ⇧uj
=1⇧
p=0
4⇧
q=0
bs,p,q
t⇧
u=0
(⇧p1 ⇧q2)
uBs(t, u)
=1⇧
p=0
4⇧
q=0
bs,p,q
�1� 4�s
K+
4�sK
(⇧p1 ⇧q2)
u⇥t
=1⇧
p=0
4⇧
q=0
bs,p,qes,p,qt,
with the second-last line obtained from the moment generating function of a Binomial variable, andwith es,p,q = 1� 4�s
K + 4�sK (⇧p1 ⇧
q2)
u. Incorporating this back into (2), we obtain for the conditional
4
where NB(t) = P (Tg = t) is the point mass of a Negative Binomial distribution with shape pa-rameter ⇤ and mean K⇥⌅, Bs(t, u) is the Binomial mass function at u with t trials and successprobability 4�s
K , and where each contribution Qs,i,as,u = P (⌃s|Ig = i, Ag,s = as, Tg,s = u) is com-puted from the stochastic model (??). Coming back to pattern ⌃ = 3111 for example, the foursub-pattern probabilities are:
Q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
Q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
Q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
Q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
where
P (Cg,s = 1|Ig = i, Ag,s = as, Tg,s = u) = P
⇤4⇧
k=1
Cg,s,k ⇥ 2|Ig = i, Ag,s = as, Tg,s = u
⌅
A key to simplifying the computation further is to recognize that with respect to the count variableu, each P (Dg,s|Ig, Ag,s, Tg,s) is a polynomial of ⇧1 = 1 � �, and each P (Cg,s|Ig, Ag,s, Tg,s) is apolynomial of ⇧2 = 1 � ⇥
4 . Thus, each Qs,i,as,u is a bivariate polynomial in ⇧1 and ⇧2, of degree atmost u and 4u respectively. By careful book-keeping, we identify coe�cients {bs,p,q} (dependingon system parameters ⌥ and the pattern ⌃) such that
Qs,i,as,u =1⇧
p=0
4⇧
q=0
bs,p,q (⇧p1 ⇧q2)
u
Thus the inner factor of (2)
t⇧
u=0
Bs(t, u) Qs,i,as,u =t⇧
u=0
Bs(t, u)8⇧
j=0
bs,j ⇧uj
=1⇧
p=0
4⇧
q=0
bs,p,q
t⇧
u=0
(⇧p1 ⇧q2)
uBs(t, u)
=1⇧
p=0
4⇧
q=0
bs,p,q
�1� 4�s
K+
4�sK
(⇧p1 ⇧q2)
u⇥t
=1⇧
p=0
4⇧
q=0
bs,p,qes,p,qt,
with the second-last line obtained from the moment generating function of a Binomial variable, andwith es,p,q = 1� 4�s
K + 4�sK (⇧p1 ⇧
q2)
u. Incorporating this back into (2), we obtain for the conditional
4
Lemma:

58
where NB(t) = P (Tg = t) is the point mass of a Negative Binomial distribution with shape pa-rameter ⇤ and mean K⇥⌅, Bs(t, u) is the Binomial mass function at u with t trials and successprobability 4�s
K , and where each contribution Qs,i,as,u = P (⌃s|Ig = i, Ag,s = as, Tg,s = u) is com-puted from the stochastic model (??). Coming back to pattern ⌃ = 3111 for example, the foursub-pattern probabilities are:
Q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
Q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
Q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
Q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
where
P (Cg,s = 1|Ig = i, Ag,s = as, Tg,s = u) = P
⇤4⇧
k=1
Cg,s,k ⇥ 2|Ig = i, Ag,s = as, Tg,s = u
⌅
A key to simplifying the computation further is to recognize that with respect to the count variableu, each P (Dg,s|Ig, Ag,s, Tg,s) is a polynomial of ⇧1 = 1 � �, and each P (Cg,s|Ig, Ag,s, Tg,s) is apolynomial of ⇧2 = 1 � ⇥
4 . Thus, each Qs,i,as,u is a bivariate polynomial in ⇧1 and ⇧2, of degree atmost u and 4u respectively. By careful book-keeping, we identify coe�cients {bs,p,q} (dependingon system parameters ⌥ and the pattern ⌃) such that
Qs,i,as,u =1⇧
p=0
4⇧
q=0
bs,p,q (⇧p1 ⇧q2)
u
Thus the inner factor of (2)
t⇧
u=0
Bs(t, u) Qs,i,as,u =t⇧
u=0
Bs(t, u)8⇧
j=0
bs,j ⇧uj
=1⇧
p=0
4⇧
q=0
bs,p,q
t⇧
u=0
(⇧p1 ⇧q2)
uBs(t, u)
=1⇧
p=0
4⇧
q=0
bs,p,q
�1� 4�s
K+
4�sK
(⇧p1 ⇧q2)
u⇥t
=1⇧
p=0
4⇧
q=0
bs,p,qes,p,qt,
with the second-last line obtained from the moment generating function of a Binomial variable, andwith es,p,q = 1� 4�s
K + 4�sK (⇧p1 ⇧
q2)
u. Incorporating this back into (2), we obtain for the conditional
4
where NB(t) = P (Tg = t) is the point mass of a Negative Binomial distribution with shape pa-rameter ⇤ and mean K⇥⌅, Bs(t, u) is the Binomial mass function at u with t trials and successprobability 4�s
K , and where each contribution Qs,i,as,u = P (⌃s|Ig = i, Ag,s = as, Tg,s = u) is com-puted from the stochastic model (??). Coming back to pattern ⌃ = 3111 for example, the foursub-pattern probabilities are:
Q1,i,a1,u = P (Dg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u) P (Cg,1 = 1|Ig = i, Ag,1 = a1, Tg,1 = u)
Q2,i,a2,u = P (Dg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u) P (Cg,2 = 0|Ig = i, Ag,2 = a2, Tg,2 = u)
Q3,i,a3,u = P (Dg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u) P (Cg,3 = 0|Ig = i, Ag,3 = a3, Tg,3 = u)
Q4,i,a4,u = P (Dg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) P (Cg,4 = 0|Ig = i, Ag,4 = a4, Tg,4 = u) .
where
P (Cg,s = 1|Ig = i, Ag,s = as, Tg,s = u) = P
⇤4⇧
k=1
Cg,s,k ⇥ 2|Ig = i, Ag,s = as, Tg,s = u
⌅
A key to simplifying the computation further is to recognize that with respect to the count variableu, each P (Dg,s|Ig, Ag,s, Tg,s) is a polynomial of ⇧1 = 1 � �, and each P (Cg,s|Ig, Ag,s, Tg,s) is apolynomial of ⇧2 = 1 � ⇥
4 . Thus, each Qs,i,as,u is a bivariate polynomial in ⇧1 and ⇧2, of degree atmost u and 4u respectively. By careful book-keeping, we identify coe�cients {bs,p,q} (dependingon system parameters ⌥ and the pattern ⌃) such that
Qs,i,as,u =1⇧
p=0
4⇧
q=0
bs,p,q (⇧p1 ⇧q2)
u
Thus the inner factor of (2)
t⇧
u=0
Bs(t, u) Qs,i,as,u =1⇧
p=0
4⇧
q=0
bs,p,q
t⇧
u=0
(⇧p1 ⇧q2)
uBs(t, u)
=1⇧
p=0
4⇧
q=0
bs,p,q
�1� 4�s
K+
4�sK
(⇧p1 ⇧q2)
u⇥t
=1⇧
p=0
4⇧
q=0
bs,p,qes,p,qt,
with the second-last line obtained from the moment generating function of a Binomial variable, andwith es,p,q = 1� 4�s
K + 4�sK (⇧p1 ⇧
q2)
u. Incorporating this back into (2), we obtain for the conditional
4

59
probability of a pattern given accessibility and involvement:
P�(i, a) =�⌦
t=0
NB(t)4↵
s=1
1⌦
p=0
4⌦
q=0
bs,p,qes,p,qt
=�⌦
t=0
NB(t)1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅⇤4↵
s=1
es,ps,qs
⌅t
=1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅ �⌦
t=0
NB(t)
⇤4↵
s=1
es,ps,qs
⌅t
=1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅⇧
⌥ ⇥
⇥+K�⇤�1� 4
s=1 es,ps,qs
⇥
⌃
� .
where the last line comes from the moment generating function of a Negative Binomial distribution.Finally, the pattern probability P� is obtained by summing over the 25 states of i and a, as indicatedpreviously. This provides a route to computing all 81 multi-study pattern probabilities required forlikelihood evaluation.
5
Po(t)
Po(t)
Po(t)
exp
(�K✓⌫
1�
Y
s
es,ps,qs
!)

60
• 12 parameters• numerical (nlminb in R) (point es9ma9on)• extensive code tes9ng• MCMC (Bayes under flat prior) (induced parameters and predic9on)
Model fitting

61
0001
0002
0010
0011
0012
0020
0021
0022
0100
0101
0102
0110
0111
0112
0120
0121
0122
0200
0201
0202
0210
0211
0212
0220
0221
0222
1000
1001
1002
1010
1011
1012
1020
1021
1022
1100
1101
1102
1110
1111
1112
1120
1121
1122
1200
1201
1202
1210
1211
1212
1220
1221
1222
2000
2001
2002
2010
2011
2012
2020
2021
2022
2100
2101
2102
2110
2111
2112
2120
2121
2122
2200
2201
2202
2210
2211
2212
2220
2221
2222
0
50
100
150 data
0001
0002
0010
0011
0012
0020
0021
0022
0100
0101
0102
0110
0111
0112
0120
0121
0122
0200
0201
0202
0210
0211
0212
0220
0221
0222
1000
1001
1002
1010
1011
1012
1020
1021
1022
1100
1101
1102
1110
1111
1112
1120
1121
1122
1200
1201
1202
1210
1211
1212
1220
1221
1222
2000
2001
2002
2010
2011
2012
2020
2021
2022
2100
2101
2102
2110
2111
2112
2120
2121
2122
2200
2201
2202
2210
2211
2212
2220
2221
2222
0.000
0.002
0.004
0.006model fit
0001
0002
0010
0011
0012
0020
0021
0022
0100
0101
0102
0110
0111
0112
0120
0121
0122
0200
0201
0202
0210
0211
0212
0220
0221
0222
1000
1001
1002
1010
1011
1012
1020
1021
1022
1100
1101
1102
1110
1111
1112
1120
1121
1122
1200
1201
1202
1210
1211
1212
1220
1221
1222
2000
2001
2002
2010
2011
2012
2020
2021
2022
2100
2101
2102
2110
2111
2112
2120
2121
2122
2200
2201
2202
2210
2211
2212
2220
2221
2222
0.000.010.020.030.040.050.06 involved
0001
0002
0010
0011
0012
0020
0021
0022
0100
0101
0102
0110
0111
0112
0120
0121
0122
0200
0201
0202
0210
0211
0212
0220
0221
0222
1000
1001
1002
1010
1011
1012
1020
1021
1022
1100
1101
1102
1110
1111
1112
1120
1121
1122
1200
1201
1202
1210
1211
1212
1220
1221
1222
2000
2001
2002
2010
2011
2012
2020
2021
2022
2100
2101
2102
2110
2111
2112
2120
2121
2122
2200
2201
2202
2210
2211
2212
2220
2221
2222
0.00000.00050.00100.00150.00200.00250.0030 non−involved

62
MCMC looks good

63
●
●●
●
●
●
● ●
●●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●
● ●
●
●●
●
● ●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
● ●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
200 220 240 260 280 300 320 340
100
150
200
250
100 simulated data# of Primary Hits
# of
Con
firm
atio
n H
its
Comfirmed vs Detected
●
●
●
●
DL−1
U2OS
A549_DE
A549_US
● DL−1U2OSA549_DEA549_US
Posterior predictive checks look good

64
Table S3-2: Proportions of dsRNAs with predicted numbers of o�-targets in the DRSC collectionfrom Kulkarni et.al. (2006).
Number of o�-targets 0 1 2� 10 11� 50 51� 100 101+Proportion of dsRNAs (%) 61.21 18.96 13.11 3.04 0.91 2.77
Table S3-3: Estimated parameters and 95% credible intervals from Negative Binomial model atk = 0.110 and Poisson model.
Model Negative Binomial PoissonParameter Point Estimate 95% C.I. Point Estimate 95% C.I.
⇤̂ 0.118 (0.089, 0.150) 0.120 (0.092 0.152)⇥̂ : DL� 1 0.065 (0.047, 0.084) 0.065 (0.049 0.083)⇥̂ : U2OS 0.058 (0.042, 0.076) 0.058 (0.044 0.074)
⇥̂ : A549DE 0.076 (0.054, 0.096) 0.076 (0.057 0.097)⇥̂ : A549US 0.091 (0.068, 0.115) 0.091 (0.069 0.114)
�̂ 0.004 (0.003, 0.004) 0.003 (0.003 0.004)⌅̂ 0.016 (0.000, 0.145) 0.011 (0.000 0.056)⇧̂ 0.685 (0.602, 0.955) 0.676 (0.602 0.859)
Table S3-4: Estimated parameters by four ways of leaving out one study.
Leave out ⇤̂ ⇥̂ : DL� 1 ⇥̂ : U2OS ⇥̂ : A549DE ⇥̂ : A549US �̂ ⌅̂ ⇧̂DL-1 0.093 - 0.070 0.091 0.112 0.004 0.017 0.741U2OS 0.113 0.071 - 0.082 0.065 0.004 0.020 0.649
A549DE 0.181 0.040 0.034 - 0.056 0.004 0.021 0.855A549US 0.098 0.079 0.110 0.092 - 0.003 0.018 0.693
Table S3-5: Predicted number of extra genes confirmed by a 4th study based on modeling the otherthree studies.
Leave Out Predicted Additional 95% Prediction Interval Observed AdditionalDL-1 133 (76, 207) 136U2OS 128 (75, 199) 114A549US 144 (89, 212) 188A549DE 156 (80, 284) 131
8
Cross validation

65
MLE MEAN a b MLE+✓ 0.128 0.128 0.102 0.160 0.223↵ 0.003 0.003 0.002 0.004 0.000�1 0.083 0.164 0.010 0.337 0.298�2 0.340 0.400 0.266 0.500 0.404�3 0.312 0.375 0.240 0.474 0.336�4 0.038 0.125 0.007 0.277 0.208�1 0.063 0.072 0.049 0.101 0.069�2 0.094 0.107 0.073 0.147 0.086�3 0.113 0.127 0.088 0.169 0.008�4 0.084 0.095 0.068 0.129 0.076! 0.809 0.902 0.751 0.996 0.678⌫ 0.000 0.006 0.000 0.020 0.000
Point estimates

P (Cg,s = 1|Ig = 0)
P (Cg,s = 0|Ig = 1)
P (Ig = 1|Cg,s = 0)
P (Ig = 0|Cg,s = 1)
66
Density of False Discovery Rate (FDR) and False Non−Discovery Rate (FNDR)
Error rate
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40
FDRFNDR
Density of False Positive Rate (FP) and False Negative Rate (FN)
Error rate
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
FPFN

67
Predicted number of confirmed genes
Number of studies (log scale)
Cum
ulat
ive n
umbe
r of c
onfir
med
gen
es (s
quar
e ro
ot s
cale
)
0
154
277
426
614
1000
1500
2000
2500
3000
3500
10 20 30 40 50
1. D
L−1
2. U
2OS
3. A
549D
E
4. A
549U
S
2306
3342
●
●
●
●
95% confidence band of predicted number of confirmed genes50% confidence band of predicted number of confirmed genes95% confidence interval of number of involved genes
+1 study
+10 studies
+20 studies
+40 studies

Kulkarni et al. 2006, Nat. Meth.
- computational predictions- overestimate
What about low estimated off target rate ??

69
probability of a pattern given accessibility and involvement:
P�(i, a) =�⌦
t=0
NB(t)4↵
s=1
1⌦
p=0
4⌦
q=0
bs,p,qes,p,qt
=�⌦
t=0
NB(t)1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅⇤4↵
s=1
es,ps,qs
⌅t
=1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅ �⌦
t=0
NB(t)
⇤4↵
s=1
es,ps,qs
⌅t
=1⌦
p1=0
1⌦
p2=0
1⌦
p3=0
1⌦
p4=0
4⌦
q1=0
4⌦
q2=0
4⌦
q3=0
4⌦
q4=0
⇤4↵
s=1
bs,ps,qs
⌅⇧
⌥ ⇥
⇥+K�⇤�1� 4
s=1 es,ps,qs
⇥
⌃
� .
where the last line comes from the moment generating function of a Negative Binomial distribution.Finally, the pattern probability P� is obtained by summing over the 25 states of i and a, as indicatedpreviously. This provides a route to computing all 81 multi-study pattern probabilities required forlikelihood evaluation.
5
Po(t)
Po(t)
Po(t)
exp
(�K✓⌫
1�
Y
s
es,ps,qs
!)
very hard to extend to Nega9ve Binomial

• large K <==> independent studies• simpler likelihood• separate implementa9on with Nega9ve Binomial gives essen9ally the same fits
70
Sensitivity analysis

71
0.05 0.10 0.20 0.50 1.00 2.00 5.00
−7600−7400−7200−7000−6800−6600−6400
νpr
ofile
log
likel
ihoo
d
max_[other parameters] log Prob[ data given off−target rate ]
0.05 0.10 0.20 0.50 1.00 2.00 5.00
0.001
0.0050.010
0.0500.100
0.5001.000
ν
dete
ctio
n/co
nfirm
atio
n ra
te
Prob[ detected ]Prob[ confirmed if detected ]four study average
0.05 0.10 0.20 0.50 1.00 2.00 5.000.00
0.05
0.10
0.15
0.20
0.25
0.30
ν
erro
r rat
e
off target rate per siRNA
FDR = Prob[ not involved if confirmed ]FNDR = Prob[ involved if not confirmed ]
Profile analysis

1. gene-‐level agreement among studies2. func9onal category analysis3. protein interac9on analysis
72
Summary



















