Methods Genome Part Anu Rev Gen 09
24
Methods for Genomic Partitioning Emily H. Turner, Sarah B. Ng, Deborah A. Nickerson, and Jay Shendure Department of Genome Sciences, University of Washington, Seattle, Washington 98195-5065; email: [email protected], [email protected], [email protected], [email protected] Annu. Rev. Genomics Hum. Genet. 2009. 10:263–84 The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org This article’s doi: 10.1146/annurev-genom-082908-150112 Copyright c 2009 by Annual Reviews. All rights reserved 1527-8204/09/0922-0263$20.00 Key Words sequencing, genomic enrichment, multiplex analysis, exon capture, hybrid capture, resequencing Abstract The emergence of massively parallel DNA sequencing platforms has made resequencing an affordable approach to study genetic variation. However, the cost of whole genome resequencing remains too high to apply to large numbers of human samples. Genomic partitioning meth- ods allow enrichment for regions of interest at a scale that is matched to the throughput of the new sequencing platforms. We review general cat- egories of methods for genomic partitioning including multiplex PCR, capture-by-circularization, and capture-by-hybridization. Parameters that are relevant to the performance of any given method include multi- plexity, specificity, uniformity, input requirements, scalability, and cost. The successful development of genomic partitioning strategies will be key to taking full advantage of massively parallel sequencing, at least until resequencing of complete mammalian genomes becomes widely affordable. 263 Annu. Rev. Genom. Human Genet. 2009.10:263-284. Downloaded from arjournals.annualreviews.org by Universidad Nacional Autonoma de Mexico on 02/13/10. For personal use only.
description
Methods Genome Part Anu Rev Gen 09
Transcript of Methods Genome Part Anu Rev Gen 09

ANRV386-GG10-13 ARI 6 August 2009 6:45
Methods for GenomicPartitioningEmily H. Turner, Sarah B. Ng,Deborah A. Nickerson, and Jay ShendureDepartment of Genome Sciences, University of Washington, Seattle,Washington 98195-5065; email: [email protected], [email protected],[email protected], [email protected]
Annu. Rev. Genomics Hum. Genet. 2009.10:263–84
The Annual Review of Genomics and Human Geneticsis online at genom.annualreviews.org
This article’s doi:10.1146/annurev-genom-082908-150112
Copyright c© 2009 by Annual Reviews.All rights reserved
1527-8204/09/0922-0263$20.00
Key Words
sequencing, genomic enrichment, multiplex analysis, exon capture,hybrid capture, resequencing
AbstractThe emergence of massively parallel DNA sequencing platforms hasmade resequencing an affordable approach to study genetic variation.However, the cost of whole genome resequencing remains too high toapply to large numbers of human samples. Genomic partitioning meth-ods allow enrichment for regions of interest at a scale that is matched tothe throughput of the new sequencing platforms. We review general cat-egories of methods for genomic partitioning including multiplex PCR,capture-by-circularization, and capture-by-hybridization. Parametersthat are relevant to the performance of any given method include multi-plexity, specificity, uniformity, input requirements, scalability, and cost.The successful development of genomic partitioning strategies will bekey to taking full advantage of massively parallel sequencing, at leastuntil resequencing of complete mammalian genomes becomes widelyaffordable.
263
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
GWAS: genome-wideassociation studies
PCR: polymerasechain reaction
SNP: singlenucleotidepolymorphism
INTRODUCTION
The identification of genomic sequence varia-tion underlying specific phenotypes lies at thecore of human and model organism genetics.The most comprehensive and straightforwardapproach for genotype evaluation would be thecomplete sequencing of each genome of in-terest. To date, high costs make this largelyimpractical; instead, alternative approaches aretaken to focus on variation within particular ge-nomic intervals. In human genetics, for exam-ple, family-based linkage analyses of Mendelianphenotypes have been an enormously successfulparadigm. Combined with sequence analysis,these studies have defined the molecular ba-sis of over 2000 clinical disorders (47). Thegenetic architectures of complex phenotypes,such as quantitative traits and common dis-eases, have proven more elusive. Databasesof common sequence variants underlie therecent wave of successful genome-wide associa-tion studies (GWAS) (34). However, the follow-up to each of these GWAS studies frequentlyinvolves extensive regional sequencing in a pop-ulation of phenotyped individuals to search forthe causative variants (31). Furthermore, it is in-creasingly recognized that the heritable contri-bution to phenotypic variation in complex traitsmay arise from a combination of common, low-penetrance alleles and rare, high-penetrance al-leles (62, 64). Whereas the initial approach tocommon variants involves GWAS, the identifi-cation of rare variants underlying complex traitshas relied on the full sequencing of one or morecandidate genes in populations of phenotypedindividuals (13). The identification of sequencevariation is also central to cancer genetics, asthe recurrent observation of functional, non-synonymous somatic mutations in tumors is fre-quently the means by which a candidate gene isimplicated in oncogenesis (22).
Here, we define resequencing as the iden-tification of sequence variation in individualsof a species for which a canonical referencegenome is available. Conventional resequenc-ing pipelines rely on PCR amplification of eachregion of interest, followed by bidirectional
Sanger sequencing. Sequences of interest mightbe sets of exons, full genes, or larger intervals.As PCR amplification from a diploid genomeyields a mixture of products derived from eachhaploid equivalent, heterozygous variants areidentified in Sanger sequencing traces as mixedpeaks (61), whereas insertion-deletions requirea more complex analysis (8). Regions that aretoo large to be amplified and sequenced as a sin-gle PCR product can be “tiled” with multiplepairs of PCR primers, yielding a set of overlap-ping amplicons that collectively cover the tar-get. Although targeted resequencing pipelineshave grown increasingly sophisticated, theircosts are directly tied to those of the under-lying technologies, namely PCR amplificationand Sanger sequencing. Their expense remainsa major bottleneck for many studies.
An alternative approach that was appliedextensively for the discovery and genotypingof common variants in the human populationrelies on sequence determination by hybridiza-tion to a resequencing microarray with featuresdesigned to detect variation (20, 25, 30, 53). Fullregions or selected positions are interrogatedby amplifying and pooling long-range PCRproducts as input material for array-based rese-quencing. However, this approach has not beenwidely applied for targeted resequencing withinindividual studies. An exception involves thehigh-density SNP arrays used in GWAS stud-ies that do not necessarily require a complex-ity reduction prior to array hybridization (24).However, these are generally focused on posi-tions of common variation that are amenableto genotyping and for which an assay has beendesigned and validated.
A major development in genomics recentlyhas been the successful proof-of-concept,commercialization, and widespread adoptionof several alternative approaches to DNA se-quencing (6, 28, 46, 59). A common threadamong this crop of “second-generation” or“next-generation” sequencing platforms is thatall rely on the concept of massively parallel,iterative sequencing of a dense array of DNAfeatures (45, 57, 58). By the end of 2008, wellover 500 such instruments from several vendors
264 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
were in use (compared to less than 10 in 2005).These platforms vary widely in terms of per-formance and cost, but several generalizationscan be made. The primary motivator for theiradoption is a major reduction in cost-per-base,to several orders of magnitude below that ofhigh-throughput Sanger sequencing pipelines.Currently, the key drawbacks include shorterread-lengths and lower raw accuracies, as com-pared to conventional Sanger sequencing. Anadditional consideration is the amount of se-quencing analyzed in a single run; the mini-mal unit of sequencing is on the order of 108
bases, as opposed to 103 bases with Sangersequencing.
For genomic resequencing, the utility ofshort, low-accuracy reads is greatly enhancedby the availability of an assembled referencegenome for a given species. Individual readsonly need to contain sufficient informationto be uniquely mappable to that referencegenome in order to be useful. As the canonicalgenome sequences of most major model organ-isms and our own species are complete, signif-icant interest has emerged for applying thesenew sequencing technologies for resequenc-ing. These platforms have effectively rendered“whole genome genetics” feasible, first for bac-terial organisms (33, 46, 59), and more recentlyfor model organisms with larger genomes suchas Caenorhabditis elegans (29, 56). The genomeof an individual human, James Watson, wasresequenced using the 454 platform at a costof ∼$1 million (67), and three more humangenomes were resequenced using the Illu-mina/Solexa platform (6, 38, 66) at a reagentcost of ∼$250,000 per genome (6). By com-parison, J. Craig Venter’s genome (37) was se-quenced with conventional Sanger technologyfor >$10 million. The cost of complete humangenome resequencing remains high but is drop-ping rapidly as technical advances with the var-ious platforms are made. It remains an openquestion, however, how far we are from theso-called $1000 genome (58), and whether thisgoal will be achieved by extensions of second-generation platforms or other approaches suchas real-time (43) or nanopore sequencing (10).
Genomicpartitioning:methods to enrich asequence library forspecific regions of agenome
Complete genome resequencing is not al-ways necessary, however, as investigators are of-ten interested in identifying germline variantsor somatic mutations in a particular sub-set of the genome. Meaningful inferencesof genotype-phenotype associations necessar-ily require the analysis of multiple individuals.For at least the next few years, it is probable(though far from certain) that the routine re-sequencing of complete human genomes willcontinue to be prohibitively expensive in thecontext of studies requiring even modest samplesizes. Studies are therefore likely to initiallyfocus on the sequencing of specific subsets ofthe human genome across multiple individ-uals. Examples of genomic subsets that maybe highly relevant in the context of a specificstudy include: (a) positions of common varia-tion, primarily consisting of millions of scat-tered SNPs; (b) a specific megabase-scale re-gion of the genome that has been implicatedin a particular disease through family-basedlinkage or GWAS analysis; (c) specific candi-date genes belonging to a disease pathway; and(d ) the full complement of protein-codingDNA sequences (∼1% of the human genome).Although “fixed content” genotyping arrays areappropriate for the first example (7), the othersare best approached with cost-effective DNAresequencing. However, these subsets generallytotal to megabases, raising the critical questionof how they can be efficiently isolated from non-target sequences. Although performing PCRreactions remains an option, its scale (102 to104 bases per reaction unit) is poorly matchedto the granularity of next-generation sequenc-ing platforms (108 bases per reaction unit). Toaddress this need, significant effort has beendedicated over the past several years to thedevelopment of general genomic partitioningmethods for the selection and amplification ofcomplex subsets of a mammalian-scale genome(23). It is anticipated that the success of suchmethods will depend on their synergy withnew sequencing technologies, and will facili-tate their application to linkage studies, associ-ation studies, medical resequencing, cancer re-sequencing, and other areas.
www.annualreviews.org • Genomic Enrichment 265
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
Table 1 Summary of genomic partitioning strategies
SummarySource ofspecificity Multiplex Scalability References
Multiplex PCR andrelated methods
Amplification with multipleprimer pairs followed byenrichment steps
Hybridization +enzymatic
102–103 Moderate (21, 48, 65)
Capture bycircularization
Circularization directly on/fromgenomic DNA followed byenrichment steps
Hybridization +enzymatic
104–105 Excellent (6a, 15, 16, 35, 55)
Solution-basedhybrid selection
Shotgun library hybridized tobiotinylated probes in solution
Hybridization 104–105 Excellent (4, 23a, 50)
Array-based hybridselection
Shotgun library hybridized toprogrammable microarray
Hybridization 105–106 Moderate (2, 32, 51)
Multiplex PCR: PCRusing more than onepair of primers in thesame reaction;multiple reactionscarried out in a singlevolume
Capture specificity:the fraction ofmolecules in anenriched library thatcorrespond to targets
Uniformity: therelative coverage oftargets after genomicpartitioning
Allelic bias: unequalcapture of two allelesof a heterozygousvariant
Scalability: the easeof expansion to studiesinvolving largenumbers of samples
Here we review the variety of methods thathave been developed in recent years for car-rying out genomic partitioning at scales thatmove well beyond uniplex PCR (Table 1). Webegin by describing key performance metricsto consider in evaluating and comparing thesestrategies. The methods themselves have beendivided into three categories. First, we considermethods based around PCR amplification, in-cluding pooling of uniplex PCR products andseveral innovative approaches to multiplex PCRthat circumvent its usual limitations. Second,we describe two strategies that rely on targetcircularization as their primary means of multi-plex capture. Finally, we discuss both aqueous-phase and solid-phase methods for capture-by-hybridization.
PERFORMANCE METRICS
Each method described here aims to parti-tion or enrich a genomic DNA sample for se-quences derived from all target regions of in-terest, with the downstream goal of identifyingheterozygous or homozygous variants in thosetargets relative to a reference genome sequence.The fold-enrichment, calculated as the ratio ofabundances of the target sequences postenrich-ment vs pre-enrichment, provides a summarymeasure of performance. Its theoretical maxi-mum is the ratio of the size of the genome tothe aggregate size of the targets. In addition, atleast eight relevant performance metrics should
be considered—capture specificity, uniformity,completeness, allelic bias, multiplexity, inputrequirements, scalability, and cost. In looking atpostenrichment sequencing data, capture speci-ficity (related to degree of enrichment) is mea-sured as the fraction of sequence reads that mapto targeted regions. Uniformity refers to therelative abundance of individual targets afterenrichment, and along with capture specificityis a critical metric in determining the amountof sequencing required to adequately cover thetargets. Completeness (related to uniformity)can be defined as the fraction of targets (or tar-get bases) detectably captured by a given strat-egy. Allelic bias is present if there is a randomor systematic nonuniformity in the relative ef-ficiency with which the two alleles of a het-erozygous target are captured and amplified.A lack of allelic bias is critical, as preferen-tial capture of one allele over the other willsignificantly impair heterozygote calling. Theremaining metrics—multiplexity, scalability,input requirements, and cost—collectively de-scribe the general practicality of a method. Wecan define multiplexity as the total number ofdiscontiguous subsequences that can be simul-taneously targeted (which is relevant, for exam-ple, when targeting large numbers of exons). In-put requirements are the amount and quality ofgenomic DNA required to carry out a given tar-geting method. Scalability refers to the fact thatcertain methods may be more amenable thanothers to high-throughput sample processing.
266 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.
ANRV386-GG10-13 ARI 6 August 2009 6:45
Finally, provided that two methods perform ad-equately well with respect to other performancemetrics or when large sample sizes are beingconsidered, cost becomes the critical arbiter.As discussed below, current implementationsof genomic partitioning methods suffer froma significant deficit in at least of one of theseparameters, underscoring the need for furthertechnological development in this area.
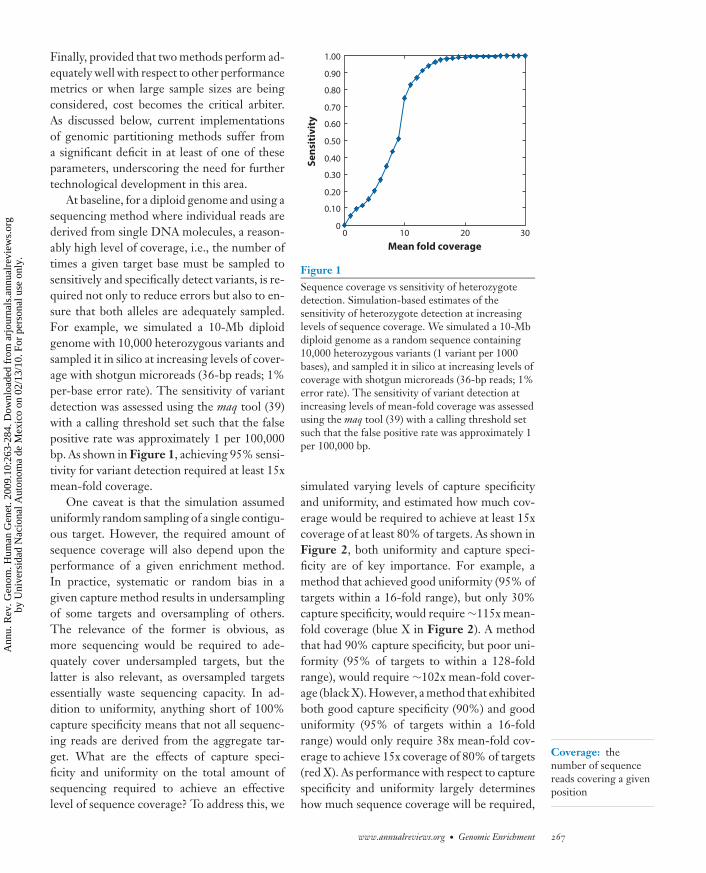
At baseline, for a diploid genome and using asequencing method where individual reads arederived from single DNA molecules, a reason-ably high level of coverage, i.e., the number oftimes a given target base must be sampled tosensitively and specifically detect variants, is re-quired not only to reduce errors but also to en-sure that both alleles are adequately sampled.For example, we simulated a 10-Mb diploidgenome with 10,000 heterozygous variants andsampled it in silico at increasing levels of cover-age with shotgun microreads (36-bp reads; 1%per-base error rate). The sensitivity of variantdetection was assessed using the maq tool (39)with a calling threshold set such that the falsepositive rate was approximately 1 per 100,000bp. As shown in Figure 1, achieving 95% sensi-tivity for variant detection required at least 15xmean-fold coverage.
One caveat is that the simulation assumeduniformly random sampling of a single contigu-ous target. However, the required amount ofsequence coverage will also depend upon theperformance of a given enrichment method.In practice, systematic or random bias in agiven capture method results in undersamplingof some targets and oversampling of others.The relevance of the former is obvious, asmore sequencing would be required to ade-quately cover undersampled targets, but thelatter is also relevant, as oversampled targetsessentially waste sequencing capacity. In ad-dition to uniformity, anything short of 100%capture specificity means that not all sequenc-ing reads are derived from the aggregate tar-get. What are the effects of capture speci-ficity and uniformity on the total amount ofsequencing required to achieve an effectivelevel of sequence coverage? To address this, we
0
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00
0 10 20 30
Se
nsi
tiv
ity
Mean fold coverage
Figure 1Sequence coverage vs sensitivity of heterozygotedetection. Simulation-based estimates of thesensitivity of heterozygote detection at increasinglevels of sequence coverage. We simulated a 10-Mbdiploid genome as a random sequence containing10,000 heterozygous variants (1 variant per 1000bases), and sampled it in silico at increasing levels ofcoverage with shotgun microreads (36-bp reads; 1%error rate). The sensitivity of variant detection atincreasing levels of mean-fold coverage was assessedusing the maq tool (39) with a calling threshold setsuch that the false positive rate was approximately 1per 100,000 bp.
Coverage: thenumber of sequencereads covering a givenposition
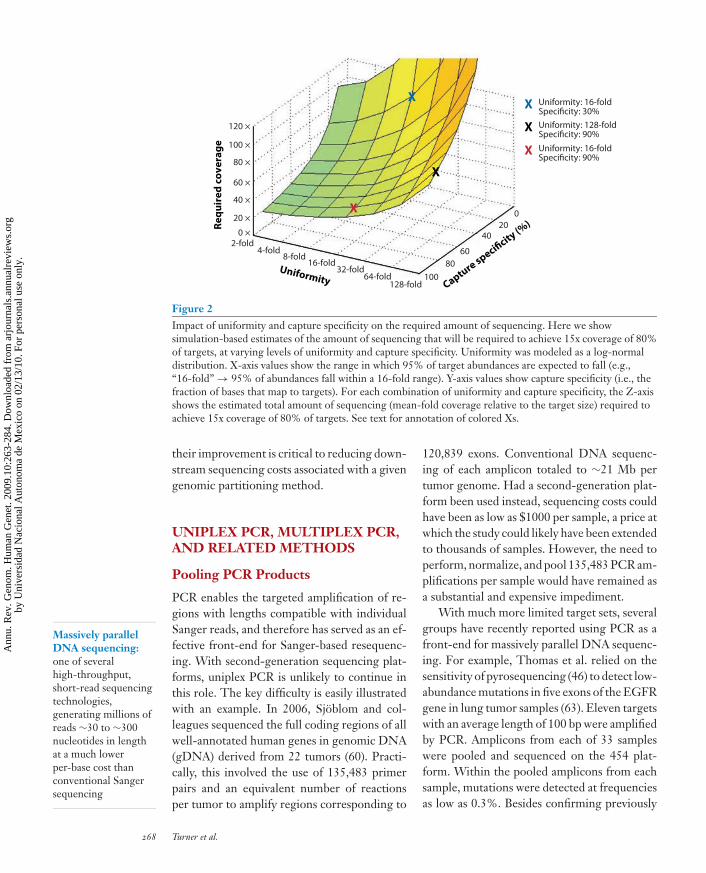
simulated varying levels of capture specificityand uniformity, and estimated how much cov-erage would be required to achieve at least 15xcoverage of at least 80% of targets. As shown inFigure 2, both uniformity and capture speci-ficity are of key importance. For example, amethod that achieved good uniformity (95% oftargets within a 16-fold range), but only 30%capture specificity, would require ∼115x mean-fold coverage (blue X in Figure 2). A methodthat had 90% capture specificity, but poor uni-formity (95% of targets to within a 128-foldrange), would require ∼102x mean-fold cover-age (black X). However, a method that exhibitedboth good capture specificity (90%) and gooduniformity (95% of targets within a 16-foldrange) would only require 38x mean-fold cov-erage to achieve 15x coverage of 80% of targets(red X). As performance with respect to capturespecificity and uniformity largely determineshow much sequence coverage will be required,
www.annualreviews.org • Genomic Enrichment 267
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.
ANRV386-GG10-13 ARI 6 August 2009 6:45
X
X
X
X
X
X
UniformityCaptu
re specificity
(%)R
eq
uir
ed
co
ve
rag
e
120 ×
80 ×
100 ×
40 ×
60 ×
0 ×
20 ×
2-fold
8-fold
32-fold
128-fold100
60
2040
80
0
4-fold
16-fold
64-fold
Uniformity: 16-foldSpecificity: 30%
Uniformity: 128-foldSpecificity: 90%
Uniformity: 16-foldSpecificity: 90%
Figure 2Impact of uniformity and capture specificity on the required amount of sequencing. Here we showsimulation-based estimates of the amount of sequencing that will be required to achieve 15x coverage of 80%of targets, at varying levels of uniformity and capture specificity. Uniformity was modeled as a log-normaldistribution. X-axis values show the range in which 95% of target abundances are expected to fall (e.g.,“16-fold” → 95% of abundances fall within a 16-fold range). Y-axis values show capture specificity (i.e., thefraction of bases that map to targets). For each combination of uniformity and capture specificity, the Z-axisshows the estimated total amount of sequencing (mean-fold coverage relative to the target size) required toachieve 15x coverage of 80% of targets. See text for annotation of colored Xs.
Massively parallelDNA sequencing:one of severalhigh-throughput,short-read sequencingtechnologies,generating millions ofreads ∼30 to ∼300nucleotides in lengthat a much lowerper-base cost thanconventional Sangersequencing
their improvement is critical to reducing down-stream sequencing costs associated with a givengenomic partitioning method.
UNIPLEX PCR, MULTIPLEX PCR,AND RELATED METHODS
Pooling PCR Products
PCR enables the targeted amplification of re-gions with lengths compatible with individualSanger reads, and therefore has served as an ef-fective front-end for Sanger-based resequenc-ing. With second-generation sequencing plat-forms, uniplex PCR is unlikely to continue inthis role. The key difficulty is easily illustratedwith an example. In 2006, Sjoblom and col-leagues sequenced the full coding regions of allwell-annotated human genes in genomic DNA(gDNA) derived from 22 tumors (60). Practi-cally, this involved the use of 135,483 primerpairs and an equivalent number of reactionsper tumor to amplify regions corresponding to
120,839 exons. Conventional DNA sequenc-ing of each amplicon totaled to ∼21 Mb pertumor genome. Had a second-generation plat-form been used instead, sequencing costs couldhave been as low as $1000 per sample, a price atwhich the study could likely have been extendedto thousands of samples. However, the need toperform, normalize, and pool 135,483 PCR am-plifications per sample would have remained asa substantial and expensive impediment.
With much more limited target sets, severalgroups have recently reported using PCR as afront-end for massively parallel DNA sequenc-ing. For example, Thomas et al. relied on thesensitivity of pyrosequencing (46) to detect low-abundance mutations in five exons of the EGFRgene in lung tumor samples (63). Eleven targetswith an average length of 100 bp were amplifiedby PCR. Amplicons from each of 33 sampleswere pooled and sequenced on the 454 plat-form. Within the pooled amplicons from eachsample, mutations were detected at frequenciesas low as 0.3%. Besides confirming previously
268 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
known SNPs, deep pyrosequencing detectedindels previously missed by Sanger sequencing.This report demonstrated the strength of mas-sively parallel sequencing over conventional re-sequencing for detecting low-abundance mu-tations in tumor samples, where normal tissuemay also be present and only a low fractionof tumor cells may carry a clinically significantmutation. This follows from sequencing largenumbers of single molecules individually ratherthan sequencing a mixture of molecules once.
To resequence a 136-kb region of human8q24 (implicated by GWAS in breast, colon,and prostate cancer), Yeager et al. generated andpooled PCR amplicons, each 2.0 to 5.5 kb inlength, tiling over the full region of interest with>150-bp overlaps (69). Primer pairs were care-fully designed and tested against several thermalcycling routines to ensure successful amplifica-tion of each locus. Pooling of amplicons fromeach individual at an equimolar ratio was fol-lowed by sequencing on the 454 platform. Meancoverage over the target region was 50x, withreasonably good uniformity. At positions withat least 20x coverage, genotypes were called byclassification based on the proportion of ob-servations. Overall, across 79 individuals, 442novel variants (i.e., not in dbSNP) were called.The global genotyping completion rate atpolymorphic positions was 93.5%. ComparingSNPs called by these data to SNPs at positionsof common variation called by array genotyp-ing, overall concordance per locus was 99.45%.
A study by Craig et al. (14) extended thepooling approach to incorporate sample-identifying barcodes, such that sequencinglibraries derived from multiple individualscould be mixed and sequenced simultaneously.Two libraries were prepared for each individ-ual, the first derived from 10 × 5 kb amplicons(covering a 50-kb region), and the second from14 × 5 kb amplicons (covering a 70-kb region).The 5-kb regions were individually amplifiedfrom each of 46 HapMap DNA samples andpooled by sample for library construction.Adaptors containing unique barcode sequenceswere ligated to fragmented PCR products,providing a sample-specific identifier for each
dbSNP: a publicdatabase of knownpolymorphismsmaintained at NCBI
HapMap: haplotypemap of the humangenome
library molecule. The 6-base barcodes were de-signed to tolerate at least one sequencing errorwhile maintaining correct index identification.The barcoded libraries derived from all individ-uals were then pooled and coamplified with aset of universal primers as part of the final stepsof library construction. Single-end sequencingwas performed on an Illumina Genome Ana-lyzer (5), with individual reads including boththe 6-bp barcode and 35 bp of sample-derivedsequence. Uniformity across the targeted re-gion for a given individual was quite good, witha 1.5- to 2.0-fold difference between ampliconswith the most and fewest reads. However, therepresentation of each barcode was more vari-able, with an 11-fold difference between themost and least abundant index (fivefold afteroptimization). The authors took a Bayesianapproach to polymorphism discovery, with aSanger sequencing–defined set of polymor-phisms from the ENCODE project availableas a set of true positives. Varying thresholdsdemonstrated a tradeoff between false positiveand false negative rates. At the most lenientcalling threshold, the false positive rate was88% and the false negative rate was 9%. At themost stringent threshold, the false positive ratewas 11% and the false negative rate was 91%.
Multiplex PCR and Related Methods
Although compatible with massively parallelsequencing, uniplex PCR with pooling ofproducts does not represent a viable long-termsolution for genomic partitioning. A morepromising category of methods includesmultiplex PCR and its derivatives. Firstreported in 1988 (12), multiplex PCR hasbeen widely applied in molecular diagnos-tics, e.g., for pathogen identification and inforensic studies. However, it is difficult toperform on more than a few dozen targetsper reaction (18), and even modest levels ofmultiplexing require significant optimiza-tion. The main challenges with multiplexPCR include the formation of primer-dimers, nonuniform amplification of targets,and high rates of mispriming events (17).
www.annualreviews.org • Genomic Enrichment 269
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
Multitemplate PCR:PCR using a singleprimer pair to amplifya population ofmolecules sharingcommon adaptorsequences. Used inmany methodsreviewed here
Introducing universal sequences to the 5′ endsof each primer pair and performing a two-stagePCR (i.e., with initial cycles directed at amplifi-cation from specific loci, followed by switchingto the 5′ universal sequences for continuedamplification via multitemplate PCR) substan-tially reduces artifacts (11, 40). Several groupshave recently reported on novel methods thatattempt to circumvent limitations on multiplexPCR and improve its relevance in the contextof genomic partitioning applications. Four ofthese approaches are summarized here.
As the formation of primer-dimers is a ma-jor issue for multiplex PCR, the immobiliza-tion of primers to a solid substrate can limitprimer-dimer formation by physically isolatingeach primer pair (1, 54). With the MegaPlexPCR method (48), for example, an initial in-solution multiplex PCR is carried out as a rel-atively nonspecific enrichment for targets ofinterest. This is followed by several cycles ofsolid-phase PCR, in which pairs of microbead-immobilized chimeric primers are used. Specif-ically, the immobilized chimeric primers aredesigned to append universal sequences to the 5′
ends of amplicons. Subsequent solution-phasemultitemplate PCR is driven by a single primerpair corresponding to the appended universalsequences. As a proof of concept, coamplifica-tion of 50 or 75 targets was carried out by thismethod and evaluated by both microarray and454 pyrosequencing. Most targets were recov-ered within a 100-fold range and less than 10%of products were observed to be primer-dimersartifacts.
A second approach in this area was re-cently developed by RainDance Technologies,and involves the use of emulsion PCR (68)and microfluidics to compartmentalize individ-ual PCR primer-pairs within the context of asingle reaction (K. Brown, personal communi-cation). Specifically, primer pairs correspond-ing to each target are separately emulsified intowater-in-oil droplets, and then the emulsionsare pooled to make a primer droplet library. Themixture of emulsions contains many primerpairs, but only a single primer pair withinany given droplet. A microfluidics platform is
applied to fuse each primer-containing dropletwith droplets from an emulsion containing ge-nomic DNA, dNTPs, and polymerase. Theemulsion is then thermocycled as a single PCRreaction, although individual primer pairs areeffectively compartmentalized from one an-other. The key advantage of the approach isthat this allows PCR reactions involving dif-ferent primer pairs to saturate within individ-ual compartments without directly competingwith one another, leading to an expectation ofhigh uniformity. In proof-of-concept experi-ments aimed at 384-plex amplification and withreadout on an Ilumina Genome Analyzer, it wasdemonstrated that 383 of 384 targets were de-tectably amplified. Capture specificity was ob-served at 50%–80%, and sequence coverageover the targeted exons did not appear to beoverly dependent on primer Tm, amplicon GCcontent, or amplicon length. Overall unifor-mity was high, with 89% of targeted ampli-cons within fivefold relative coverage, and 98%within tenfold relative coverage.
Varley & Mitra described a multiplex ampli-fication strategy termed “Nested Patch PCR”,illustrated in Figure 3 (65). The method re-lies on two rounds of target-specific enrich-ment. First, primer pairs are designed againsteach target, and the mixture of primers usedfor ten cycles of multiplex PCR. The primerscontain uracil bases in place of thymine, suchthat postamplification exposure to uracil DNAglycosylase, endonuclease VIII, and exonucle-ase I effectively removes the primer regionsfrom amplicons. For the second round of se-lection, Nested Patch adaptors are used. Theseconsist of a double-stranded universal segmentand a single-stranded overhang that is targetspecific. Hybridization and ligation of NestedPatch adaptors to primer-depleted ampliconsis followed by multitemplate PCR amplifica-tion with primers corresponding to the univer-sal sequences. Because this ligation is depen-dent on sequences immediately internal to theoriginal primers used in the limited multiplexPCR, the Nested Patch adaptors confer addi-tional specificity. The authors also incorporatedsample-specific barcodes into the Nested Patch
270 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
adaptors such that products from multiple cap-ture reactions could be pooled and sequencedtogether. Proof-of-concept experiments wereperformed by coamplifying 94 exons per reac-tion and sequencing amplification products onthe 454 platform. Of the total number of tar-gets, 96% were detectably amplified, with 75%falling within a 50-fold abundance range, and90% of all reads mapped to one of the 94 tar-gets. Seven of seven predicted polymorphismswere verified by conventional resequencing.
With the Gene-Collector method, a limitedmultiplex PCR is followed by a circularizationstep to confer additional specificity. In therecent report describing this method (21), 170unique primer pairs were designed to targetthe full coding sequences of 10 human cancergenes; amplicon lengths ranged from 160 to800 bp. Eight cycles of 170-plex PCR wereperformed. Then, as illustrated in Figure 4,170 “collector” oligos were introduced, eachof which could template the ligase-drivenself-circularization of PCR products flankedby one of the 170 intended primer pairings.After exonuclease digestion of uncircularizedmaterial (e.g., primer-dimers or nonspecificproducts, which would not be expected to cir-cularize efficiently), randomly primed rollingcircle amplification was carried out, and itsproducts converted to a shotgun sequencinglibrary. An estimated 58% of recovered prod-ucts were from expected targets. Additionally,enriched products were highly uniform inabundance, with 96% of all targets estimatedto be within fourfold of the average abundanceby quantitative PCR.
CAPTURE BY CIRCULARIZATION
With the Gene-Collector method, circu-larization is an effective means of boostingspecificity, but is secondary to an initial mul-tiplex PCR enrichment. In this section, wereview two promising methods with which theprimary capture event involves the multiplexcircularization of targets (or copies of targets)via DNA ligase (15, 55). In both approaches,the circularization step serves two purposes:
a
b
c
d
Left PCR primer Target region
Right PCR primerUDGEndo VIII
Exo I
Universal PCRprimer 1
Universal PCRprimer 2
Universal PCRprimer 2
Exonucleaseresistance3 carbon spacer
Left nested patch Right nested patch
AmpligaseExo I & IIIPCR
Universal PCRprimer 1
U
U
U U U
Figure 3Nested Patch PCR. (a and b) Primer pairs were designed for each target andused to perform an initial enrichment via limited cycles of multiplex PCR.Because the thymines were replaced with uracils in the primers but not thePCR reaction, treatment with UDG and Endo VIII, followed by end-repair,removed the primers while leaving the amplified target sequences intact.(c) Nested Patch primers were designed where each has a single-strandedinternal sequence specific to the target amplicon, and an external double-stranded universal adaptor. The ligation of the Nested Patch primers confersadditional specificity dependent on the newly exposed flanking sequences of theamplified targets. As the Nested Patch primers are exonuclease resistant at their5′ end, further specificity was achieved by exonuclease digestion.(d ) Multitemplate PCR with primers directed at the universal component ofthe Nested Patch primers allowed further amplification. Image adapted fromVarley & Mitra (65) with author’s permission.
(a) linking each target to a universal sequencethat can subsequently be used to prime PCRamplification of all targets with a single pair ofprimers; (b) protecting captured targets fromthe activity of exonucleases, which are used toreduce the concentration of background prod-ucts that might otherwise interfere with thepostcapture multitemplate PCR amplification.The key advantage of these approaches is that,like hybridization-based capture, they maybe compatible with a much higher degree of
www.annualreviews.org • Genomic Enrichment 271
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
a
b
c
i
ii
Collector probe
PCR product
Figure 4Gene-Collector method. (a) 170 primer pairs (colored bars) were designedagainst coding sequences in 10 genes, and used to perform multiplex PCR witha limited number of cycles as an initial enrichment. (b) Next, 170 “collector”probes, each containing segments complementary to a specific primer pair,were used to template circularization of intended products by a DNA ligase.Nonspecific products involving incorrect primer pairings and primer-dimerswere not efficiently circularized, permitting removal by exonuclease digestion.(c) Randomly primed rolling circle amplification yielded concatamers that wereconverted to a shotgun sequencing library. Image adapted from Fredrikssonet al. (21) with author’s permission.
RF: restrictionfragment
multiplexing than methods that rely on mul-tiplex PCR for initial enrichment. They alsoexhibit much greater capture specificity thanhybridization-based capture (discussed below),and scale well as they are aqueous-phasereactions.
Selective Circularization
This method, developed by Dahl and col-leagues (15, 16), achieves capture via the ligase-driven circularization of targeted restrictionfragments from genomic DNA, with a twistthat one end of a given restriction fragment
can be effectively trimmed to a desired posi-tion by endonucleolytic cleavage of an invasiveflap structure (Figure 5). The capture reactionincludes the following components: (a) a mix-ture of “selector” oligonucleotides (one per tar-get), each consisting of a common sequenceflanked by target-specific sequences; (b) a “vec-tor” oligonucleotide, the reverse complementof the common sequence internal to each selec-tor oligonucleotide; (c) genomic DNA restric-tion digested with one or several enzymes (or amix of genomic DNA that has been subjectedto different digests); and (d ) Taq DNA ligaseand Taq DNA polymerase. The boundaries ofselected targets depends in part on the expectedpattern(s) of restriction digestion of the refer-ence genome. For a subset of targets, the selec-tor oligo is designed to capture one strand ofan expected restriction fragment (RF) in its en-tirety, via hybridization of the RF to the 5′ and3′ single-stranded, target-specific overhangs ofa given selector-vector hybrid molecule. Thisis followed by ligation at both ends via Taqligase to yield a circular molecule that is es-sentially the full RF and the common vectoroligo. For a second subset of targets, one over-hang of each selector is again designed to cor-respond to the 3′ end of targeted RFs, but theother overhang is designed to hybridize to aregion internal to the 5′ end of targeted RFs.After hybridization of RFs to selector-vectorhybrids, the endonucleolytic activity of Taqpolymerase recognizes and cleaves the result-ing branched structure (44). Ligation at bothends again yields a circular molecule, here witha partial RF and the common vector oligo. Afterthe capture reaction, exonucleases are used toremove uncircularized material (e.g., incorrectligations that may not necessarily have resultedin circularization). A single multitemplate PCRwith a universal primer pair directed at the com-mon vector sequence is then used to amplify allcircularized targets in parallel. In a recent re-port using this method in the context of cancerresequencing, 177 exons from 10 genes weretargeted via 425 selectors, with individual tar-gets ranging in size from 138 bp to 238 bp andtotaling to ∼49 kb of genomic sequence (16).
272 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
Five restriction enzymes were required to ade-quately target the desired sequences, with someoverlap between targets of different selectorsin this panel. Products of capture from six ge-nomic DNA samples were sequenced by 454pyrosequencing. The length of captured ampli-cons was approximately matched to the read-length of this sequencing platform, such thatadditional steps to construct a shotgun sequenc-ing library were not required. A high specificityis expected [conferred by the hybridization ofthe RF and selector, the activity of Taq poly-merase (flap cleavage), Taq ligase (at both sidesof the selector), and the exonucleases] and wasobserved as ∼90% of all reads mapped to tar-gets. As with other enzymatic methods, nonuni-formity was significant, with the large majorityof reads falling within a 100-fold range. How-ever, the nonuniformity is highly systematic andreproducible across samples. It therefore can besubstantially mitigated through empirical ad-justment of the concentrations of individual se-lectors, which allows the majority of ampliconsto fall within a tenfold range (H. Ji, personalcommunication).
Gapped Molecular Inversion Probes
A padlock probe, or molecular inversion probe(MIP), is a single-stranded oligonucleotide con-sisting of two target-complementary arms sep-arated by a linker sequence (36, 49). The target-complementary arms are designed such that the5′ and 3′ ends of the padlock probe are immedi-ately adjacent when hybridized to the expectedtarget sequence. If the 5′ end is phosphorylated,DNA ligase will join the two ends, resulting ina circularized padlock probe that is catenatedto the target. Following circularization, exonu-clease digestion reduces the concentration ofuncircularized probe species by several ordersof magnitude. PCR or rolling circle amplifica-tion (RCA) with primer(s) directed against thelinker sequence can be used to amplify circu-larized probes in a multitemplate PCR (26).For detection of the presence/absence of a tar-get sequence, the reaction is sensitive in thatonly a single hybridization event is required,
i
ii
a
b
General primer-pair motif
Vector oligonucleotideselector probe
Target specific ends
Digested DNA Selectors
Exonucleasetreatment
Multiplex PCR with auniversal primer-pair
Selector
ssDNA
Ligation
Cleavage Ligation
Figure 5Capture by selective circularization. (a) Selector probes are generated with ashared internal sequence, flanked by target-specific ends. The vector oligosequence is complimentary to the shared internal sequence. (b) Single-strandedDNA targets, obtained by restriction digestion and denaturation of genomicDNA, are circularized with the selectors. With some designs (i ), the selectorprobe hybridizes to sequences at the 5′ and 3′ ends of targeted restrictionfragments and acts as a template for ligase-driven circularization of the targetto the vector oligo. For other designs (ii ), the selector probe hybridizes to the3′ end of the target and an internal sequence, such that there is a 5′ flap.Invasive cleavage by Taq polymerase removes the flap and circularization iscompleted with ligase. Following circularization, linear DNA is removed byexonuclease, and multitemplate PCR is carried out via a single primer pairdirected at the common vector sequence. Image adapted from Dahl et al. (15)with authors’ permission.
MIP: molecularinversion probe
RCA: rolling circleamplification
but is also highly specific, as the ends of theprobe must be brought into immediate prox-imity with no mismatches near the join site inorder to be efficiently ligated. The rapid kinet-ics of the intramolecular padlock reaction favor
www.annualreviews.org • Genomic Enrichment 273
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
target-probe hybridization over probe-probeinteractions; as a result, the padlock probe re-action can be highly multiplexed. The initialmethod was adopted for genotyping, eitherwith allele-specific padlock probes (3, 19, 26), orby the introduction of a single base-pair gap be-tween the targets of the arms, such that a single-base gap-fill (with polymerase) was required inaddition to ligation (26, 41). Extending the lat-ter concept, Hardenbol et al. demonstrated thatover 10,000 SNPs could be genotyped in par-allel via a padlock probe scheme requiring sin-gle base gap-fills at interrogated positions andfour-color readout on microarrays (27).
To adapt this approach for genomic par-titioning, Shendure and colleagues explored
a Anneal b Gap fill polymerization
c Gap fill ligation d Exonuclease selection
e Probe release f Amplification
Genome
Genome
Probe
Probe
C G G A G A T G G C C C AG C C T C T C C G G G T
C G G A G A T G G C C C AG C C T C T A C C G G G T
C G G A G A T G G C C C AG C C T C T A C C G G G T
Figure 6Gapped molecular inversion probes. (a) Probes are designed with a target-specific sequence at the ends, and an internal sequence that is commonto all MIPs. Probes hybridize to single-stranded genomic DNA, leaving agap over the target region. The gap can range from a single nucleotide for SNPgenotyping, as in References 26, 27, to several hundred nucleotides for exoncapture, as in References 35, 55. (b) Polymerase is used to fill in nucleotides overthe gap. (c) Ligase completes circularization of the molecular inversion probe.(d ) Exonuclease treatment removes linear DNA. (e) In some versions of thisprotocol, the probe is linearized. ( f ) Multitemplate PCR of the probes is carriedout with universal primers directed at the common backbone. Image adaptedwith permission from MacMillan Publishers Ltd: Nature Biotechnology 2003 (26).
MIP designs where the targeting arms of eachMIP flanked full exons (Figure 6), rather thansingle nucleotide variants (55). In the experi-ment described in Porreca et al. (55), a set of55,000 MIPs was designed to target 55,000exons as well as two base-pairs adjacent to thesplice junctions, with an aggregate target-sizeof 6.7 Mb. To mitigate the high cost ofcolumn-based oligonucleotide synthesis at thedesired scale, the 55,000 required MIPs wereobtained as a complex mixture of 100-mersby synthesis on and release from the surfaceof an Agilent microarray. After amplificationvia 15-bp universal sequences at each end, the100-mers were converted to 70-mer MIPsthrough a series of restriction digestions. Each70-mer MIP consisted of unique 20-bp target-ing sequences flanking a common 30-bp linker.The individual targets ranged in length from60 to 191 bp. With the amplification step, weestimate that the yield of one programmablearray is sufficient to support at least 1000independent capture reactions. Followinghybridization to genomic DNA, gap-fillingand circularization, and exonuclease treatment,capture products were rolling circle amplified,converted into shotgun sequencing library, andsequenced on the Illumina Genome Analyzer.Analysis of the resulting data demonstratedthat: (a) specificity was high, as ∼98% of readsthat could be confidently mapped to a locationoverlapping with one of the 55,000 targets;(b) completeness and specificity were poor,as only ∼10,000 of the 55,000 targets weredetectably captured, and the abundance withwhich individual targets were observed rangedover several logs; and (c) genotyping accuracywas high at homozygous positions, but lowat heterozygous positions, likely secondary tostochastic effects with poor capture efficiency.
We have subsequently observed that simpleoptimizations markedly improve the perfor-mance of this strategy (64a). These include:(a) increasing hybridization and gap-fill time;(b) increasing MIP and ligase concentration;(c) replacement of rolling circle amplificationsteps with multitemplate PCR with primersdirected at the linker; and (d ) direct sequencing
274 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.
ANRV386-GG10-13 ARI 6 August 2009 6:45
a bPost-optimization
Fold sequencing coverageFold sequencing coverage
Re
fere
nce
all
ele
fre
qu
en
cy
Re
fere
nce
all
ele
fre
qu
en
cy
Preoptimization1.0
0.8
0.6
0.4
0.2
0
1.0
0.8
0.6
0.4
0.2
0101 102 103 101 102 103
Homozygous,reference allele
Heterozygous
Homozygous,nonreference allele
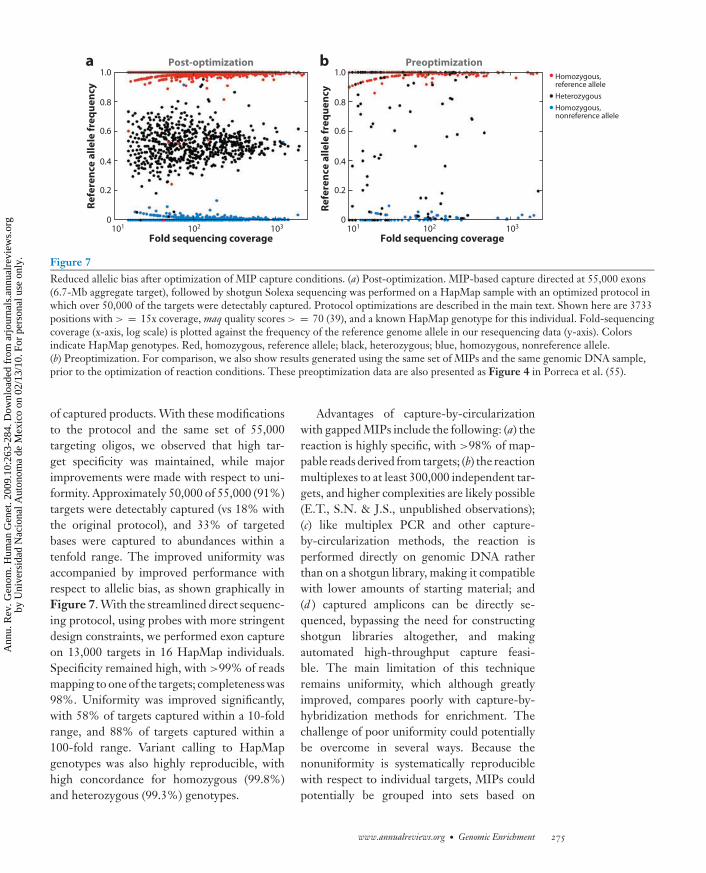
Figure 7Reduced allelic bias after optimization of MIP capture conditions. (a) Post-optimization. MIP-based capture directed at 55,000 exons(6.7-Mb aggregate target), followed by shotgun Solexa sequencing was performed on a HapMap sample with an optimized protocol inwhich over 50,000 of the targets were detectably captured. Protocol optimizations are described in the main text. Shown here are 3733positions with > = 15x coverage, maq quality scores > = 70 (39), and a known HapMap genotype for this individual. Fold-sequencingcoverage (x-axis, log scale) is plotted against the frequency of the reference genome allele in our resequencing data (y-axis). Colorsindicate HapMap genotypes. Red, homozygous, reference allele; black, heterozygous; blue, homozygous, nonreference allele.(b) Preoptimization. For comparison, we also show results generated using the same set of MIPs and the same genomic DNA sample,prior to the optimization of reaction conditions. These preoptimization data are also presented as Figure 4 in Porreca et al. (55).
of captured products. With these modificationsto the protocol and the same set of 55,000targeting oligos, we observed that high tar-get specificity was maintained, while majorimprovements were made with respect to uni-formity. Approximately 50,000 of 55,000 (91%)targets were detectably captured (vs 18% withthe original protocol), and 33% of targetedbases were captured to abundances within atenfold range. The improved uniformity wasaccompanied by improved performance withrespect to allelic bias, as shown graphically inFigure 7. With the streamlined direct sequenc-ing protocol, using probes with more stringentdesign constraints, we performed exon captureon 13,000 targets in 16 HapMap individuals.Specificity remained high, with >99% of readsmapping to one of the targets; completeness was98%. Uniformity was improved significantly,with 58% of targets captured within a 10-foldrange, and 88% of targets captured within a100-fold range. Variant calling to HapMapgenotypes was also highly reproducible, withhigh concordance for homozygous (99.8%)and heterozygous (99.3%) genotypes.
Advantages of capture-by-circularizationwith gapped MIPs include the following: (a) thereaction is highly specific, with >98% of map-pable reads derived from targets; (b) the reactionmultiplexes to at least 300,000 independent tar-gets, and higher complexities are likely possible(E.T., S.N. & J.S., unpublished observations);(c) like multiplex PCR and other capture-by-circularization methods, the reaction isperformed directly on genomic DNA ratherthan on a shotgun library, making it compatiblewith lower amounts of starting material; and(d ) captured amplicons can be directly se-quenced, bypassing the need for constructingshotgun libraries altogether, and makingautomated high-throughput capture feasi-ble. The main limitation of this techniqueremains uniformity, which although greatlyimproved, compares poorly with capture-by-hybridization methods for enrichment. Thechallenge of poor uniformity could potentiallybe overcome in several ways. Because thenonuniformity is systematically reproduciblewith respect to individual targets, MIPs couldpotentially be grouped into sets based on
www.annualreviews.org • Genomic Enrichment 275
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
similar capture efficiencies, or adjusted tonormalizing concentrations in the same reac-tion. Also, Krishnakumar et al. (35) recentlydescribed a modified probe generation proce-dure that lengthened the linker backbone, anddemonstrated that these long backbone probesyielded much greater capture uniformity whentested on a limited set of targets, and werecapable of capturing targets up to 500 bp long.
CAPTURE-BY-HYBRIDIZATION
Another approach to genomic partitioning is torely on the hybridization of shotgun genomicDNA libraries to a complex mixture of captureprobes, which may potentially be in solution ortethered to a solid substrate such as a microbeador a glass surface. Advantages of hybridization-based capture include the possibility of muchgreater degrees of multiplexing without signifi-cant interference, and potentially greater toler-ance for polymorphisms that overlap with thecapture probes themselves compared to pro-tocols based on extension or ligation by anenzyme. An unsurprising disadvantage is thathybridization-based capture tends to be muchless specific than enzymatic capture methods,due to cross-hybridization of similar sequences,for example. However, as described below, thisis offset by the fact that hybridization-basedmethods have generally resulted in significantlybetter uniformity than enzymatic methods. Inthis section, we review strategies that have beendescribed for both solution-based and array-based capture-by-hybridization.
Solution-Phase Hybrid Selection
Bashiardes and colleagues described a modifiedversion of genomic DNA-based cDNA selec-tion protocols (42, 52) directed at performinghybridization-based targeted capture of shot-gun library fragments corresponding to BAC-sized genomic regions (4). The method takesadvantage of widely available BAC clone li-braries as a source material for the captur-ing agent. In brief, an adaptor-flanked shot-gun library is generated from genomic DNA of
interest, and the library is hybridized in solu-tion to biotinylated DNA that is derived froma BAC corresponding to the region of inter-est. Streptavidin beads are used to pull downtarget-probe hybrids, followed by washing toremove nonspecifically bound molecules. Cap-tured targets are then eluted and amplified withprimers directed at the common adaptors priorto sequencing. In the described protocol, anadaptor-flanked shotgun library was generatedfrom pooled DNA from 14 individuals via re-striction digestion followed by linker ligation.Two rounds of in-solution selection were per-formed against the biotinylated BAC (∼70 heach), with PCR amplification of the capturedmaterial after each round of selection. Thetarget region (a ∼150-kb region correspond-ing to the BAC) was enriched 1000-fold af-ter the first round and 10,000-fold (total) af-ter the second round. On Sanger sequencingof 2119 clones generated from recovered mate-rial after two rounds of selection, 52% mappedunambiguously to the 150-kb target sequence.Nonrepetitive regions of the target were cov-ered ∼threefold, and both established and novelvariants were successfully detected. Within thepool, 69 previously known variants and 100 pu-tative novel variants were discovered, as well asseveral small indels.
In solution hybridization-based capture hasalso been successfully applied to the enrich-ment of target DNA sequences from ancientsources (50). The challenge in this contextis that contamination of ancient remains bymicrobial DNA prevents acquisition of a puresample for shotgun sequencing. To enrichfor specific Neanderthal sequences fromcontaminated sources, Noonan et al. evaluateddirect hybrid selection from metagenomicancient DNA libraries using capture probesderived from modern human DNA (Figure 8).Specifically, biotinylated capture probes weregenerated by PCR amplification from humangenomic DNA of regions correspondingto targets. These were hybridized againstPCR amplicons derived from metagenomicancient DNA libraries followed by pulldown ofheteroduplexes via streptavidin-coated beads.
276 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
Shotgun libraryor PCR amplifiedmetagenomiclibrary inserts
Biotinylatedprobes
Hybridizein solution
Capture probeson strepdavidin-coated beads
Wash, elutecaptured DNAs
Amplify by PCR withcommon primers
Sequenceproducts
Figure 8In solution hybrid selection. Target DNA is prepared as an in vitro shotgun library, with common adaptors flanking genomic DNAfragments. The library is hybridized in solution to a set of biotinylated probes. After hybridization, biotinylated probes are capturedwith streptavidin beads. Beads are washed to remove any nonspecific, unbound library molecules. Multitemplate PCR with primersdirected at the common adaptors is used to amplify eluted target molecules before high-throughput sequencing. Adapted from Noonanet al. (50). Images reprinted with permission from AASS.
Parallel capture of 29 of 35 human targetswas demonstrated, with the caveat that thesesequences were already known to be presentin the Neanderthal library via sequencing ofthe library prior to enrichment. Remarkably,the authors also demonstrated capture of 5 of96 targets using human capture probes corre-sponding to highly conserved regions againsta Pleistocene cave bear metagenomic library.In this case, the targets were not known to bepresent prior to enrichment and sequencing.
Andreas Gnirke, Chad Nusbaum, and col-leagues at the Broad Institute have recentlydeveloped a method for in solution hybridselection that relies on long RNA moleculesas capture probes (23a). First, as with thegapped MIP approach described above, a li-brary of DNA capture probe precursors is gen-erated by synthesis and release from a pro-grammable microarray. Each of these includes170 bp of target-specific sequence and com-mon flanking sequence. A T7 promoter is ap-pended, such that in vitro transcription candrive synthesis of large amounts of biotiny-lated RNA capture probes (aka baits) in thesame orientation. The mixture of RNA baitsis hybridized at high concentration in so-lution against a shotgun genomic DNA li-brary. After hybridization, streptavidin-coatedbeads are used to pull down RNA-DNA hy-brids, followed by amplification from univer-sal primers and sequencing. Key strengths
of this approach include the following:(a) because the RNA baits are single-strandedand present in only one orientation, a high con-centration and molar excess can drive the kinet-ics of hybridization; (b) relatively low amountsof input genomic DNA (0.5–3 μg) are suf-ficient; (c) the reaction is solution-based andtherefore more scalable, i.e., automatable, thansolid-phase array-based hybridization methods;(d ) allelic bias may be reduced with long cap-ture probes relative to short probes; (e) the RNAbaits can be prepared in large batches that canbe quality controlled for use in production-scale settings; ( f ) the approach can be appliedto many short, discontiguous targets or longcontiguous regions; and (g) high specificity hasbeen demonstrated with 85%–90% of posten-richment sequences overlapping with targets.Their initial experiments made use of as manyas 22,000 targeting oligos per reaction, thoughhigher levels of multiplexing will likely be feasi-ble. This approach has been licensed to Agilentfor development as a commercial product.
Array-Based Hybrid Selection
In 2007, several research groups reported ongenomic partitioning methods that made useof programmable oligonucleotide arrays (2, 32,51, 55). These arrays contain customizable setsof long, single-stranded probes (60 to 200 bp),and are available from vendors such as
www.annualreviews.org • Genomic Enrichment 277
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.
ANRV386-GG10-13 ARI 6 August 2009 6:45
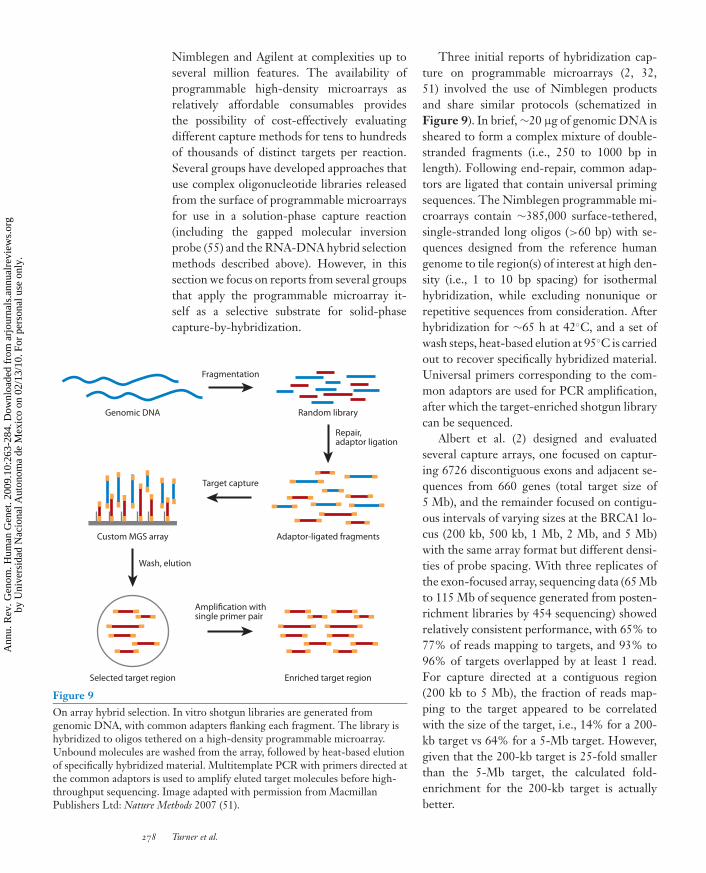
Nimblegen and Agilent at complexities up toseveral million features. The availability ofprogrammable high-density microarrays asrelatively affordable consumables providesthe possibility of cost-effectively evaluatingdifferent capture methods for tens to hundredsof thousands of distinct targets per reaction.Several groups have developed approaches thatuse complex oligonucleotide libraries releasedfrom the surface of programmable microarraysfor use in a solution-phase capture reaction(including the gapped molecular inversionprobe (55) and the RNA-DNA hybrid selectionmethods described above). However, in thissection we focus on reports from several groupsthat apply the programmable microarray it-self as a selective substrate for solid-phasecapture-by-hybridization.
Fragmentation
Genomic DNA Random library
Repair,adaptor ligation
Target capture
Adaptor-ligated fragmentsCustom MGS array
Wash, elution
Selected target region
Amplification withsingle primer pair
Enriched target region
Figure 9On array hybrid selection. In vitro shotgun libraries are generated fromgenomic DNA, with common adapters flanking each fragment. The library ishybridized to oligos tethered on a high-density programmable microarray.Unbound molecules are washed from the array, followed by heat-based elutionof specifically hybridized material. Multitemplate PCR with primers directed atthe common adaptors is used to amplify eluted target molecules before high-throughput sequencing. Image adapted with permission from MacmillanPublishers Ltd: Nature Methods 2007 (51).
Three initial reports of hybridization cap-ture on programmable microarrays (2, 32,51) involved the use of Nimblegen productsand share similar protocols (schematized inFigure 9). In brief, ∼20 μg of genomic DNA issheared to form a complex mixture of double-stranded fragments (i.e., 250 to 1000 bp inlength). Following end-repair, common adap-tors are ligated that contain universal primingsequences. The Nimblegen programmable mi-croarrays contain ∼385,000 surface-tethered,single-stranded long oligos (>60 bp) with se-quences designed from the reference humangenome to tile region(s) of interest at high den-sity (i.e., 1 to 10 bp spacing) for isothermalhybridization, while excluding nonunique orrepetitive sequences from consideration. Afterhybridization for ∼65 h at 42◦C, and a set ofwash steps, heat-based elution at 95◦C is carriedout to recover specifically hybridized material.Universal primers corresponding to the com-mon adaptors are used for PCR amplification,after which the target-enriched shotgun librarycan be sequenced.
Albert et al. (2) designed and evaluatedseveral capture arrays, one focused on captur-ing 6726 discontiguous exons and adjacent se-quences from 660 genes (total target size of5 Mb), and the remainder focused on contigu-ous intervals of varying sizes at the BRCA1 lo-cus (200 kb, 500 kb, 1 Mb, 2 Mb, and 5 Mb)with the same array format but different densi-ties of probe spacing. With three replicates ofthe exon-focused array, sequencing data (65 Mbto 115 Mb of sequence generated from posten-richment libraries by 454 sequencing) showedrelatively consistent performance, with 65% to77% of reads mapping to targets, and 93% to96% of targets overlapped by at least 1 read.For capture directed at a contiguous region(200 kb to 5 Mb), the fraction of reads map-ping to the target appeared to be correlatedwith the size of the target, i.e., 14% for a 200-kb target vs 64% for a 5-Mb target. However,given that the 200-kb target is 25-fold smallerthan the 5-Mb target, the calculated fold-enrichment for the 200-kb target is actuallybetter.
278 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
Okou et al. (51) targeted sequences near theFMR1 locus—a 50-kb region in one array de-sign, and 304 kb of unique sequence with an-other array design. Capture and recovery werecarried out similarly to Albert et al. (2), butthe resequencing was performed with an in-dependent microarray (70), rather than witha massively parallel sequencing platform. En-richment was instead measured by qPCR at∼1000-fold. The array-based resequencing re-sults were quite good, with a call-rate of 99.1%over 20 replicates, and an accuracy of 99.81%at segregating, genotyped sites in HapMapsamples.
Hodges et al. (32) targeted the full setof well-annotated human protein-coding se-quences and adjacent splice sites using a set ofseven Nimblegen 385K microarrays with 60- to90-bp probes. Each of these arrays targeted 6 to8 Mb of coding sequence, with a tiling densityof roughly one probe per 20 bp. Approximately20 μg of genomic DNA was used as input foreach of the seven array-based capture reac-tions. Eluted material was sequenced at highthroughput with either the 454 pyrosequencingplatform or the Illumina Genome Analyzer. Ininitial experiments, shotgun genomic librarieswere targeted with relatively large fragmentsizes (500 to 600 bp). Although successful, inthat 55% to 85% of reads mapped to targets,a substantial fraction of these were adjacent totargets rather than within a target. Additionalexperiments were performed that used shotgunlibraries sheared to a smaller size range (100 to200 bp). Although this resulted in a substantialreduction in capture specificity (to ∼29%), amuch greater fraction of sequenced bases werenow within target, rather than adjacent to tar-get. Given the high input material require-ments, the group primarily used genomic DNAthat had been subjected to whole genome am-plification (WGA) prior to hybridization, buta non-WGA sample was also evaluated againstone of the same array designs and performedsimilarly.
Based on the above reports as well as our ownexperiences, we can identify key advantages
WGA: whole genomeamplification
and disadvantages of hybridization on pro-grammable microarrays as a strategy for ge-nomic partitioning.
Some of these examples relate to the use ofhybridization as the capture method, and oth-ers to the use of arrays. A first advantage isthat there is greater flexibility (than the BAC-based approach, for example), in that eithera long contiguous region or many short, dis-contiguous regions can be targeted with thesame level of effort. Moreover, the aggregatesize of the target(s) of a programmable arraycan range from hundreds of kilobases to tensof megabases with the same protocol, simplyby varying the probe spacing. Another advan-tage is that as high-density programmable mi-croarrays are available for approximately $500to $1000, there is a relatively low cost to eval-uating any given array design, or to optimiz-ing an array by iterating through new designs.Finally, hybridization-based methods includingon-array capture have generally been observedto perform much better with respect to unifor-mity of target capture, a critical parameter indetermining the overall amount of sequencingthat will be required for variant discovery acrossthe full target.
A potential disadvantage is that from apractical perspective, array-based genomic par-titioning may be more difficult to scale tohundreds or thousands of samples than anaqueous-phase reaction. This potential diffi-culty is somewhat mitigated by the substantialthroughputs that have been achieved for arrayprocessing for high-throughput SNP genotyp-ing in the context of GWAS studies. A relateddisadvantage is that although there is a relativelylow cost to optimize an array design, the costof the arrays is still quite substantial when oneis considering scaling to many samples. More-over, the array cost does not necessarily scalelinearly with the size of the aggregate target.In contrast, in-solution capture reactions mightmake use of relatively small amounts of target-ing oligos, or libraries of targeting oligos canpotentially be amplified, reducing per-samplecosts. Potential workarounds for this concern
www.annualreviews.org • Genomic Enrichment 279
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
include multiplexing more than one sample onthe same array via barcoded libraries (S.N., E.T.& J.S., unpublished observations), or reusingarrays (51), though this carries a nontrivial riskfor contaminating new samples with old sam-ples on the array. A third disadvantage is thatmethods relying on hybridization alone gener-ally have lower capture specificity compared toother approaches, which increases the overallsequencing requirements and offsets the advan-tage of greater target uniformity. The originsof nonspecific capture have not been examinedin detail in this context, but possible sourcesinclude straightforward cross-hybridization aswell as “daisy-chaining” between adaptor se-quences that are present in both orientationsduring array hybridization (i.e., one librarymolecule hybridizes specifically to the array,then another library molecule hybridizes to itvia complementarity between the adaptor se-quences, etc.). The extent of nonspecific cap-ture may be correlated with the compositionand size of the aggregate target. For example,targeting of smaller genomic subsets may resultin significantly lower capture specificity thanlarger genomic subsets. In Albert et al., for ex-ample, 14% of postenrichment reads mappedto a 200-kb target, while 64% of reads mappedto a 5-Mb target. Target sizes between 200 kband 5 Mb exhibited intermediate capture speci-ficities [Table S3 in (2)]. Other aspects oflibrary preparation can significantly affect per-formance of array-based enrichment. For ex-ample, in Hodges et al. (32), longer genomicfragments were associated with increased cap-ture specificity, but this increase came at a costof more bases that were target-adjacent ratherthan target-within sequences. The relevance ofthis depends on whether one is targeting long,contiguous regions or short, discontiguous ex-ons. Allelic bias is a theoretical concern if multi-ple variants directly overlap a probe and reducehybridization efficiency; this has generally notbeen observed. One related point is that largedeletions overlapping the probes may be diffi-cult to detect when sequencing rather than ar-ray hybridization (51) is used as the readout.
SUMMARY AND FUTUREDIRECTIONS
Massively parallel DNA sequencing provides acost-effective means of identifying genetic vari-ation. However, for at least the time being, mostinvestigators interested in taking advantage ofthese technologies for human resequencing willuse a genomic partitioning method as a “front-end” to focus their finite resources. In the pastseveral years, much progress has been madein the development of diverse approaches thatmeet this need. We cannot offer any clear-cut answer on which of these strategies is thebest, in part because they are continuing toevolve and improve. There are tradeoffs inher-ent to the selection of any given method forgenomic partitioning. For example, capture-by-circularization methods are highly specificbut relatively less uniform, whereas capture-by-hybridization methods have shown higheruniformity but generally lower specificity. Im-provements in performance with respect tothese and other metrics (e.g., cost, scalability)and less tangible aspects (e.g., flexibility, ac-cess) may be important determinants of the ex-tent to which individual strategies are adopted.One’s choice of method may also depend onthe scale of a given study, both with respectto the size of the aggregate target and thenumber of samples to be analyzed. For exam-ple, multiplex PCR and derivative methods,combined with sequence-based barcodes, maybe most useful in the context of studies fo-cused on a small number of targets in a largenumber of individuals. Array-based capture-by-hybridization may be most appropriate for alarge aggregate target with limited numbers ofsamples. For large aggregate targets and manysamples, solution-based methods that multiplexand scale well (e.g., capture-by-circularizationor solution-based hybrid selection) may be thebest choice. Cost is difficult to estimate foreach method and may also depend on the scaleof application. For example, oligo libraries re-leased from programmable microarrays are ex-pensive and available from only a small numberof vendors, but are very cost-effective in this
280 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
context when amortized over a large number ofsamples.
A closing point is that these methods maywell be rendered obsolete before they achievewidespread use.
With second-generation sequencing plat-forms, costs are dropping quickly while readlengths and accuracies continue to improve.The practical implementation of real-time (43)or nanopore sequencing (10) methods may beimminent. If, for example, the all-inclusive costsof whole genome human resequencing were todrop to less than $1000, the demand for genomepartitioning methods might be expected to
significantly wane as increasing numbers ofinvestigators can afford whole genome rese-quencing of all samples of interest. Nonethe-less, these methods may continue to be usefulif investigators choose to continue to sequencegenomic subsets in larger numbers of sam-ples (i.e., 100 complete genomes for $100,000vs 1% of 10,000 genomes for the same cost).Another example where genomic partitioningmight continue to be useful is tumor resequenc-ing, where 1000x coverage of all coding se-quences (1% of the human genome) in a het-erogeneous sample might be more informativethan 10x coverage of the whole genome.
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting the objectivity of thisreview.
ACKNOWLEDGMENTS
The authors acknowledge support, in part, by grants from the U.S. National Institutes of Health(NIH) National Heart Lung and Blood Institute (RO1 HL094976 to D.A.N. and J.S.) and theNIH National Human Genome Research Institute (NHGRI) (R21 HG004749 to J.S.). E.H.T. issupported by a training fellowship from the NIH NHGRI (T32 HG00035). S.B.N. is supportedby the Agency for Science, Technology and Research, Singapore.
LITERATURE CITED
1. Adessi C, Matton G, Ayala G, Turcatti G, Mermod JJ, et al. 2000. Solid phase DNA amplification:characterisation of primer attachment and amplification mechanisms. Nucleic Acids Res. 28:E87
2. Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, et al. 2007. Direct selection of humangenomic loci by microarray hybridization. Nat. Methods 4:903–5
3. Antson DO, Isaksson A, Landegren U, Nilsson M. 2000. PCR-generated padlock probes detect singlenucleotide variation in genomic DNA. Nucleic Acids Res. 28:E58
4. Bashiardes S, Veile R, Helms C, Mardis ER, Bowcock AM, Lovett M. 2005. Direct genomic selection.Nat. Methods 2:63–69
5. Bentley DR. 2006. Whole-genome resequencing. Curr. Opin. Genet. Dev. 16:545–526. Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, et al. 2008. Accurate whole human
genome sequencing using reversible terminator chemistry. Nature 456:53–597. Bhangale TR, Rieder MJ, Nickerson DA. 2008. Estimating coverage and power for genetic association
studies using near-complete variation data. Nat. Genet. 40:841–438. Bhangale TR, Stephens M, Nickerson DA. 2006. Automating resequencing-based detection of insertion-
deletion polymorphisms. Nat. Genet. 38:1457–629. Bitinaite J, Rubino M, Varma KH, Schildkraut I, Vaisvila R, Vaiskunaite R. 2007. USER friendly DNA
engineering and cloning method by uracil excision. Nucleic Acids Res. 35:1992–200210. Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, et al. 2008. The potential and challenges of
nanopore sequencing. Nat. Biotechnol. 26:1146–53
www.annualreviews.org • Genomic Enrichment 281
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
11. Brownie J, Shawcross S, Theaker J, Whitcombe D, Ferrie R, et al. 1997. The elimination of primer-dimeraccumulation in PCR. Nucleic Acids Res. 25:3235–41
12. Chamberlain JS, Gibbs RA, Rainer JE, Nguyen PN, Thomas C. 1988. Deletion screening of theDuchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 16:11141–56
13. Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. 2004. Multiple rare allelescontribute to low plasma levels of HDL cholesterol. Science 305:869–72
14. Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, et al. 2008. Identification of genetic variantsusing bar-coded multiplexed sequencing. Nat. Methods 5:887–93
15. Dahl F, Gullberg M, Stenberg J, Landegren U, Nilsson M. 2005. Multiplex amplification enabled byselective circularization of large sets of genomic DNA fragments. Nucleic Acids Res. 33:e71
16. Dahl F, Stenberg J, Fredriksson S, Welch K, Zhang M, et al. 2007. Multigene amplification and massivelyparallel sequencing for cancer mutation discovery. Proc. Natl. Acad. Sci. USA 104:9387–92
17. Edwards MC, Gibbs RA. 1994. Multiplex PCR: advantages, development, and applications. PCR MethodsAppl. 3:S65–75
18. Fan JB, Chee MS, Gunderson KL. 2006. Highly parallel genomic assays. Nat. Rev. Genet. 7:632–4419. Faruqi AF, Hosono S, Driscoll MD, Dean FB, Alsmadi O, et al. 2001. High-throughput genotyping of
single nucleotide polymorphisms with rolling circle amplification. BMC Genomics 2:420. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. 2007. A second generation human
haplotype map of over 3.1 million SNPs. Nature 449:851–6121. Fredriksson S, Baner J, Dahl F, Chu A, Ji H, et al. 2007. Multiplex amplification of all coding sequences
within 10 cancer genes by Gene-Collector. Nucleic Acids Res. 35:e4722. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. 2004. A census of human cancer genes.
Nat. Rev. Cancer 4:177–8323. Garber K. 2008. Fixing the front end. Nat. Biotechnol. 26:1101–4
23a. Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, et al. 2009. Solution hybrid selection withultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27:182–89
24. Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. 2005. A genome-wide scalable SNPgenotyping assay using microarray technology. Nat. Genet. 37:549–54
25. Hacia JG, Brody LC, Chee MS, Fodor SP, Collins FS. 1996. Detection of heterozygous mutationsin BRCA1 using high density oligonucleotide arrays and two-colour fluorescence analysis. Nat. Genet.14:441–47
26. Hardenbol P, Baner J, Jain M, Nilsson M, Namsaraev EA, et al. 2003. Multiplexed genotyping withsequence-tagged molecular inversion probes. Nat. Biotechnol. 21:673–78
27. Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, et al. 2005. Highly multiplexed molecularinversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res.15:269–75
28. Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, et al. 2008. Single-molecule DNA sequencing ofa viral genome. Science 320:106–9
29. Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, et al. 2008. Whole-genome sequencing andvariant discovery in C. elegans. Nat. Methods 5:183–88
30. Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, et al. 2005. Whole-genome patterns of commonDNA variation in three human populations. Science 307:1072–79
31. Hirschhorn JN, Daly MJ. 2005. Genome-wide association studies for common diseases and complextraits. Nat. Rev. Genet. 6:95–108
32. Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, et al. 2007. Genome-wide in situ exon capture forselective resequencing. Nat. Genet. 39:1522–27
33. Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, et al. 2008. High-throughput sequencingprovides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40:987–93
34. IHC. 2005. A haplotype map of the human genome. Nature 437:1299–32035. Krishnakumar S, Zheng J, Wilhelmy J, Faham M, Mindrinos M, Davis R. 2008. A comprehensive assay
for targeted multiplex amplification of human DNA sequences. Proc. Natl. Acad. Sci. USA 105:9296–301
282 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
36. Landegren U, Schallmeiner E, Nilsson M, Fredriksson S, Baner J, et al. 2004. Molecular tools for amolecular medicine: analyzing genes, transcripts and proteins using padlock and proximity probes. J.Mol. Recognit. 17:194–97
37. Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, et al. 2007. The diploid genome sequence of an individualhuman. PLoS Biol. 5:e254
38. Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, et al. 2008. DNA sequencing of a cytogeneticallynormal acute myeloid leukaemia genome. Nature 456:66–72
39. Li H, Ruan J, Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mappingquality scores. Genome Res. 18(11):1851–58
40. Lin Z, Cui X, Li H. 1996. Multiplex genotype determination at a large number of gene loci. Proc. Natl.Acad. Sci. USA 93:2582–87
41. Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. 1998. Mutation detection andsingle-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 19:225–32
42. Lovett M, Kere J, Hinton LM. 1991. Direct selection: a method for the isolation of cDNAs encoded bylarge genomic regions. Proc. Natl. Acad. Sci. USA 88:9628–32
43. Lundquist PM, Zhong CF, Zhao P, Tomaney AB, Peluso PS, et al. 2008. Parallel confocal detection ofsingle molecules in real time. Opt. Lett. 33:1026–28
44. Lyamichev V, Brow MA, Dahlberg JE. 1993. Structure-specific endonucleolytic cleavage of nucleic acidsby eubacterial DNA polymerases. Science 260:778–83
45. Mardis ER. 2008. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9:387–402
46. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. 2005. Genome sequencing in microfab-ricated high-density picolitre reactors. Nature 437:376–80
47. McKusick VA. 2007. Mendelian inheritance in man and its online version, OMIM. Am. J. Hum. Genet.80:588–604
48. Meuzelaar LS, Lancaster O, Pasche JP, Kopal G, Brookes AJ. 2007. MegaPlex PCR: a strategy formultiplex amplification. Nat. Methods 4:835–37
49. Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U. 1994. Padlockprobes: circularizing oligonucleotides for localized DNA detection. Science 265:2085–88
50. Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, et al. 2006. Sequencing and analysis of Nean-derthal genomic DNA. Science 314:1113–18
51. Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME. 2007. Microarray-based genomicselection for high-throughput resequencing. Nat. Methods 4:907–9
52. Parimoo S, Patanjali SR, Shukla H, Chaplin DD, Weissman SM. 1991. cDNA selection: efficient PCRapproach for the selection of cDNAs encoded in large chromosomal DNA fragments. Proc. Natl. Acad.Sci. USA 88:9623–27
53. Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, et al. 2001. Blocks of limited haplotype diversityrevealed by high-resolution scanning of human chromosome 21. Science 294:1719–23
54. Pemov A, Modi H, Chandler DP, Bavykin S. 2005. DNA analysis with multiplex microarray-enhancedPCR. Nucleic Acids Res. 33:e11
55. Porreca GJ, Zhang K, Li JB, Xie B, Austin D, et al. 2007. Multiplex amplification of large sets of humanexons. Nat. Methods 4:931–36
56. Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. 2008. Caenorhabditis elegans mutant allele identifi-cation by whole-genome sequencing. Nat. Methods 5:865–67
57. Shendure J, Ji H. 2008. Next-generation DNA sequencing. Nat. Biotechnol. 26:1135–4558. Shendure J, Mitra RD, Varma C, Church GM. 2004. Advanced sequencing technologies: methods and
goals. Nat. Rev. Genet. 5:335–4459. Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, et al. 2005. Accurate multiplex polony
sequencing of an evolved bacterial genome. Science 309:1728–3260. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. 2006. The consensus coding sequences of
human breast and colorectal cancers. Science 314:268–7461. Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. 2006. Automating sequence-based
detection and genotyping of SNPs from diploid samples. Nat. Genet. 38:375–81
www.annualreviews.org • Genomic Enrichment 283
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

ANRV386-GG10-13 ARI 6 August 2009 6:45
62. Stratton MR, Rahman N. 2008. The emerging landscape of breast cancer susceptibility. Nat. Genet.40:17–22
63. Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, et al. 2006. Sensitive mutation detection inheterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med. 12:852–55
64. Topol EJ, Frazer KA. 2007. The resequencing imperative. Nat. Genet. 39:439–4064a. Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. 2009. Massively parallel exon capture and
library-free resequencing across 16 genomes. Nat. Methods 6:315–1665. Varley KE, Mitra RD. 2008. Nested Patch PCR enables highly multiplexed mutation discovery in can-
didate genes. Genome Res. 18:1844–5066. Wang J, Wang W, Li R, Li Y, Tian G, et al. 2008. The diploid genome sequence of an Asian individual.
Nature 456:60–6567. Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, et al. 2008. The complete genome of an
individual by massively parallel DNA sequencing. Nature 452:872–7668. Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD. 2006. Amplification of
complex gene libraries by emulsion PCR. Nat. Methods 3:545–5069. Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, et al. 2008. Comprehensive resequence analysis of
a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum. Genet.124:161–70
70. Zwick ME, McAfee F, Cutler DJ, Read TD, Ravel J, et al. 2005. Microarray-based resequencing ofmultiple Bacillus anthracis isolates. Genome Biol. 6:R10
284 Turner et al.
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

AR386-GG10-FM ARI 6 August 2009 7:15
Annual Review ofGenomics andHuman Genetics
Volume 10, 2009
Contents
Chromosomes in Leukemia and Beyond: From Irrelevantto Central PlayersJanet D. Rowley � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 1
Unraveling a Multifactorial Late-Onset Disease: From GeneticSusceptibility to Disease Mechanisms for Age-Related MacularDegenerationAnand Swaroop, Emily Y. Chew, Catherine Bowes Rickman, and Goncalo R. Abecasis � � �19
Syndromes of Telomere ShorteningMary Armanios � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �45
Chronic Pancreatitis: Genetics and PathogenesisJian-Min Chen and Claude Ferec � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �63
The Genetics of Crohn’s DiseaseJohan Van Limbergen, David C. Wilson, and Jack Satsangi � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �89
Genotyping Technologies for Genetic ResearchJiannis Ragoussis � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 117
Applications of New Sequencing Technologies for TranscriptomeAnalysisOlena Morozova, Martin Hirst, and Marco A. Marra � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 135
The Posttranslational Processing of Prelamin A and DiseaseBrandon S.J. Davies, Loren G. Fong, Shao H. Yang, Catherine Coffinier,
and Stephen G. Young � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 153
Genetic Testing in Israel: An OverviewGuy Rosner, Serena Rosner, and Avi Orr-Urtreger � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 175
Stewardship of Human Biospecimens, DNA, Genotype, and ClinicalData in the GWAS EraStephen J. O’Brien � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 193
Schistosoma Genetics: New Perspectives on Schistosome Biologyand Host-Parasite InteractionZe-Guang Han, Paul J. Brindley, Sheng-Yue Wang, and Zhu Chen � � � � � � � � � � � � � � � � � � � � 211
Evolution of Genomic Imprinting: Insights from Marsupialsand MonotremesMarilyn B. Renfree, Timothy A. Hore, Geoffrey Shaw,
Jennifer A. Marshall Graves, and Andrew J. Pask � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 241
v
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.

AR386-GG10-FM ARI 6 August 2009 7:15
Methods for Genomic PartitioningEmily H. Turner, Sarah B. Ng, Deborah A. Nickerson, and Jay Shendure � � � � � � � � � � � � � 263
Biased Gene Conversion and the Evolution of MammalianGenomic LandscapesLaurent Duret and Nicolas Galtier � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 285
Inherited Variation in Gene ExpressionDaniel A. Skelly, James Ronald, and Joshua M. Akey � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 313
Genomic Analyses of Sex Chromosome EvolutionMelissa A. Wilson and Kateryna D. Makova � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 333
Sequencing Primate Genomes: What Have We Learned?Tomas Marques-Bonet, Oliver A. Ryder, and Evan E. Eichler � � � � � � � � � � � � � � � � � � � � � � � � � � � 355
Genotype ImputationYun Li, Cristen Willer, Serena Sanna, and Goncalo Abecasis � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 387
Genetics of Athletic PerformanceElaine A. Ostrander, Heather J. Huson, and Gary K. Ostrander � � � � � � � � � � � � � � � � � � � � � � � � 407
Genetic Screening for Low-Penetrance Variantsin Protein-Coding DiseasesJill Waalen and Ernest Beutler � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 431
Copy Number Variation in Human Health, Disease, and EvolutionFeng Zhang, Wenli Gu, Matthew E. Hurles, and James R. Lupski � � � � � � � � � � � � � � � � � � � � � 451
The Toxicogenomic Multiverse: Convergent Recruitment of ProteinsInto Animal VenomsBryan G. Fry, Kim Roelants, Donald E. Champagne, Holger Scheib,
Joel D.A. Tyndall, Glenn F. King, Timo J. Nevalainen, Janette A. Norman,Richard J. Lewis, Raymond S. Norton, Camila Renjifo,and Ricardo C. Rodríguez de la Vega � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 483
Genetics, Medicine, and the Plain PeopleKevin A. Strauss and Erik G. Puffenberger � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 513
Indexes
Cumulative Index of Contributing Authors, Volumes 1–10 � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 537
Cumulative Index of Chapter Titles, Volumes 1–10 � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 541
Errata
An online log of corrections to Annual Review of Genomics and Human Genetics articlesmay be found at http://genom.annualreviews.org/
vi Contents
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
9.10
:263
-284
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
idad
Nac
iona
l Aut
onom
a de
Mex
ico
on 0
2/13
/10.
For
per
sona
l use
onl
y.