
Metabolic Diseases of the Liver
35
Metabolic Diseases of the Liver Capungcol, Marc Steven D. Mengullo, Ramon Kristoffer A. Noel, Lauro Gabriel G. Remandaban, David III C.
-
Upload
valcrist-balder -
Category
Documents
-
view
224 -
download
2
description
Metabolic diseases of liver several examples
Transcript of Metabolic Diseases of the Liver
Metabolic Diseases of the Liver
Capungcol, Marc Steven D.Mengullo, Ramon Kristoffer A.
Noel, Lauro Gabriel G.Remandaban, David III C.
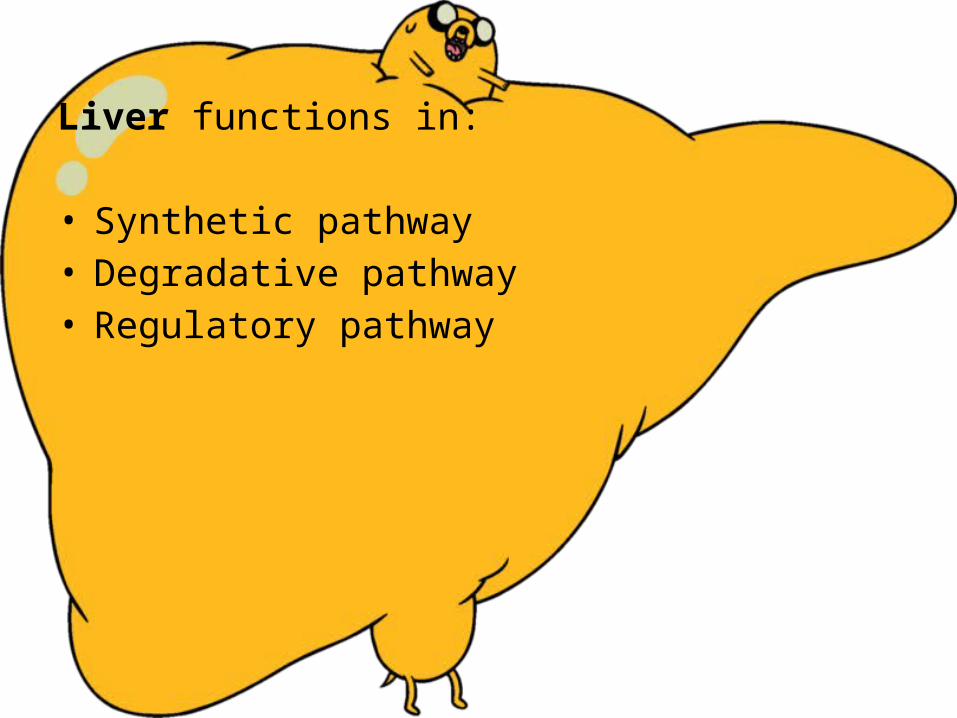
Liver functions in: • Synthetic pathway • Degradative pathway• Regulatory pathway
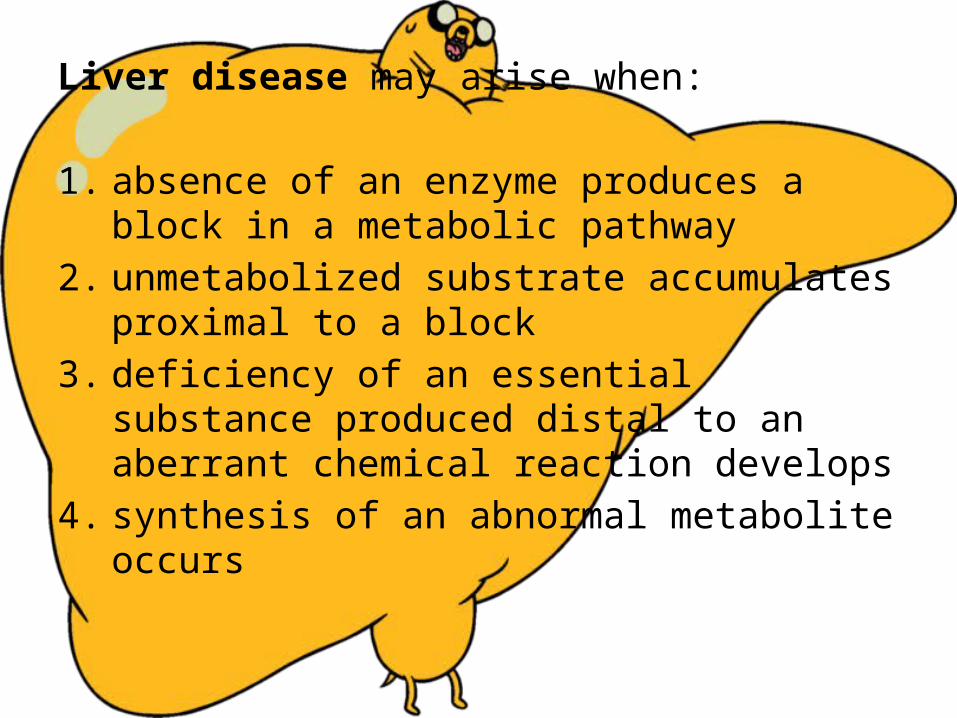
Liver disease may arise when: 1. absence of an enzyme produces a block in a
metabolic pathway2. unmetabolized substrate accumulates proximal to a
block3. deficiency of an essential substance produced distal
to an aberrant chemical reaction develops4. synthesis of an abnormal metabolite occurs
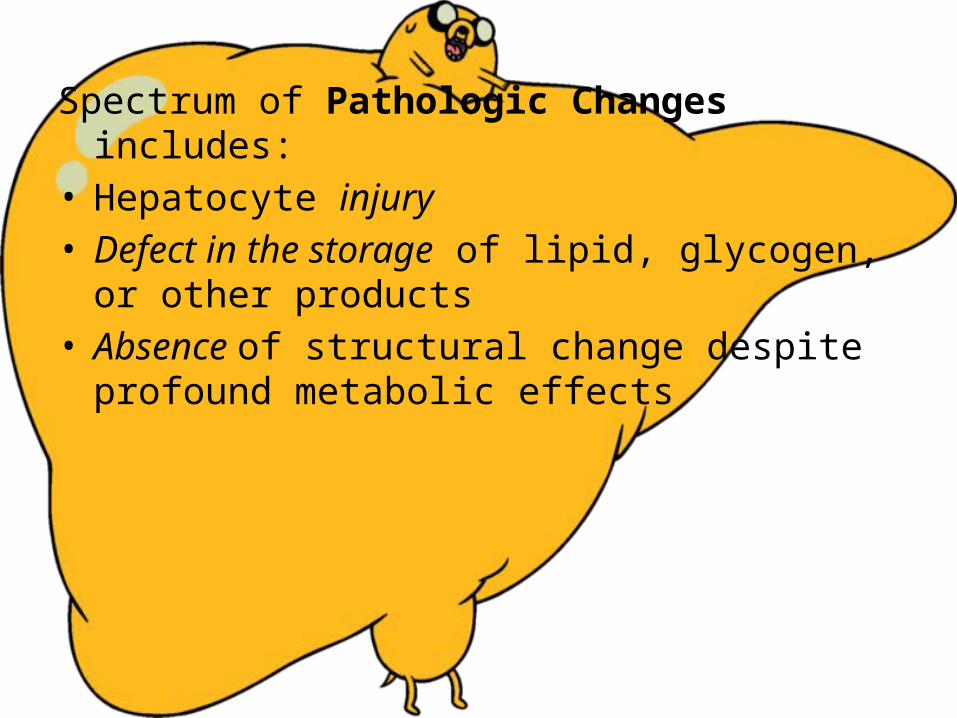
Spectrum of Pathologic Changes includes:• Hepatocyte injury• Defect in the storage of lipid, glycogen, or other
products• Absence of structural change despite profound
metabolic effects

Clinical Manifestations That Suggest the Possibility of Metabolic Disease
Recurrent vomiting Failure to thrive Short stature and dysmorphic features Jaundice, hepatomegaly, splenomegaly Hypoglycemia, organic acidemia, lactic acidemia,
hyperammonemia Bleeding (coagulopathy) Cardiac dysfunction/failure, unusual odors, rickets,
cataracts
INHERITED DEFICIENT CONJUGATION OF BILIRUBIN (FAMILIAL NONHEMOLYTIC UNCONJUGATED HYPERBILIRUBINEMIA)
Crigler-Najjar Syndrome (Type I Glucuronyl Transferase Deficiency)
• autosomal recessive• producing congenital nonobstructive,
nonhemolytic, unconjugated hyperbilirubinemia • glucuronyl transferase activity is deficient
Clinical Manifestations Severe unconjugated hyperbilirubinemia Stools are pale yellow An almost universal complication of this disorder ---
Kernicterus
Diagnosis• measure Hepatic Glucuronyl Transferase activity in a
liver specimen obtained by a closed biopsy
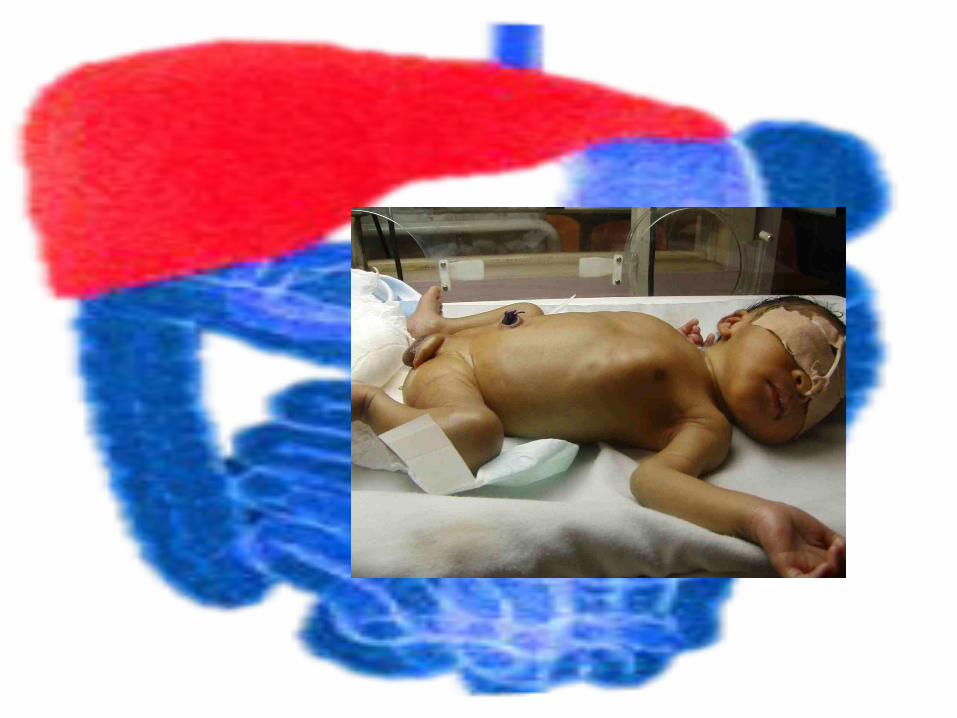
Treatment • Serum bilirubin kept below 20 mg/dL for at least the
first 2-4 wk of life • Repeated exchange transfusions and phototherapy• Phenobarbital therapy should be considered to
determine responsiveness and differentiation between type I and II
• Orthotopic hepatic transplantation cures the disease and has been successful in a small number of patients

II. Crigler-Najjar Syndrome (Type II Glucuronyl Transferase Deficiency)
• autosomal recessive disease • caused by homozygous missense mutation in
glucuronyl transferase isoform I resulting in only partial enzymatic activity
• may present in a manner similar to type I syndrome

Clinical Manifestations Unconjugated hyperbilirubinemia occurs during the first 3 days of life Stool color is normal Onset of kernicterus is unusual Infants are without clinical signs or symptoms of disease Diagnosis• respond readily to 5 mg/kg/24 hr of oral phenobarbital with a
decrease in serum bilirubin concentration to 2-3 mg/dL within 7-10 days
Treatment • continued administration of phenobarbital at 5 mg/kg/24 hr

INHERITED CONJUGATED HYPERBILIRUBINEMIA • Autosomal recessive • Characterized by mild jaundice• The transfer of bilirubin and other organic anions
from liver to bile is defective• Chronic mild conjugated hyperbilirubinemia is
usually detected during adolescence or early adulthood

I. Dubin-Johnson Syndrome • Autosomal recessive inherited defect in hepatocyte
secretion of bilirubin glucuronide • Bile acid excretion is normal, and serum bile acid
levels are normal• Liver cells contain black pigment similar to melanin
II. Rotor Syndrome • Have an additional deficiency in organic anion
uptake.• Urinary coproporphyrin excretion is normal in
quantity • Liver cells contain no black pigment
WILSON DISEASE • Hepatolenticular degeneration• Autosomal recessive disorder characterized by
degenerative changes in the brain, liver disease, and Kayser-Fleischer rings in the cornea

Pathogenesis • Abnormal gene for Wilson disease on chromosome
13
(The gene encodes amino acid structural motifs consistent with a role in copper transport.Fetal and neonatal liver normally contains relatively high concentrations of sulfur-rich copper-binding protein (metallothionein) and of copper; serum ceruloplasmin and copper levels are relatively low. The mechanisms responsible for copper homeostasis in older children reach maturity by 2 yr of age)
• Defective mobilization of copper from lysosomes in liver cells for excretion into bile
• accumulation of copper in the liver• Copper enters the circulation in a non-
ceruloplasmin-bound form• damage other organs, particularly the brain and
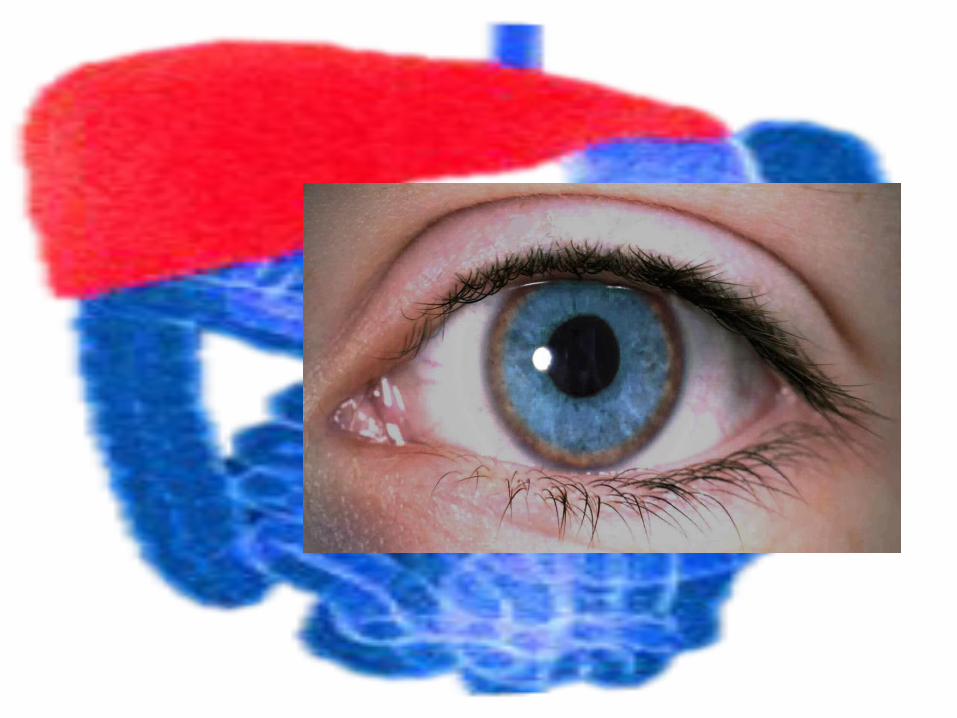
kidneys, and accumulates in the cornea, visible as Kayser-Fleischer rings
Clinical Manifestations Younger patient ---hepatic involvement
asymptomatic hepatomegaly subacute or chronic hepatitis, fulminant hepatic failureCryptogenic cirrhosis portal hypertensionascites edema, variceal bleeding, or other effects of hepatic
dysfunction
>20 yr of age -- neurologic symptoms predominatemay develop insidiously or precipitously intention tremor dysarthria dystonia deterioration in school performancebehavioral changes
Diagnosis
• Best screening test is to measure the serum ceruloplasmin level
• Serum copper level may be elevated in early Wilson disease, and urinary copper excretion (usually <40 μg/ day) is increased
• Liver biopsy
Treatment • Administration of copper-chelating agents
Penicillamine 1 g/day in two doses before meals for adults 0.5-0.75 g/day for patients younger than 10 yr
• Restrict copper intake to less than 1 mg/day (liver, shellfish, nuts, and chocolate)• Zinc,as adjuvant therapy or as maintenance
therapy owing to its unique ability to impair the gastrointestinal absorption of copper
INDIAN CHILDHOOD CIRRHOSIS (ICC) • Occurs predominantly in rural areas of India• Linked to excess dietary ingestion of copper, through use of
contaminated utensils• Increased hepatic copper content up to > 700 ug/g Clinical Manifestation Jaundice Pruritus Lethargy Splenomegaly
Histological Picture • Characterized by hepatocyte necrosis, Mallory
bodies, intralobular fibrosis and inflammation Treatment • Penicillamine- early in the course of disease• Avoidance of using contaminated feeding
utensils
NEONATAL IRON STORAGE DISEASE (NISD)/ NEONATAL HEMOCHROMATOSIS
• Rare form of fulminant liver disease that presents in
the 1st few days of life• High rate of recurrence in families (80%)• May be sustained alloimmune disease, target of the
natural alloimmune response is a fetal liver autism• Coagulopathy is refractory to therapy of Vit. K
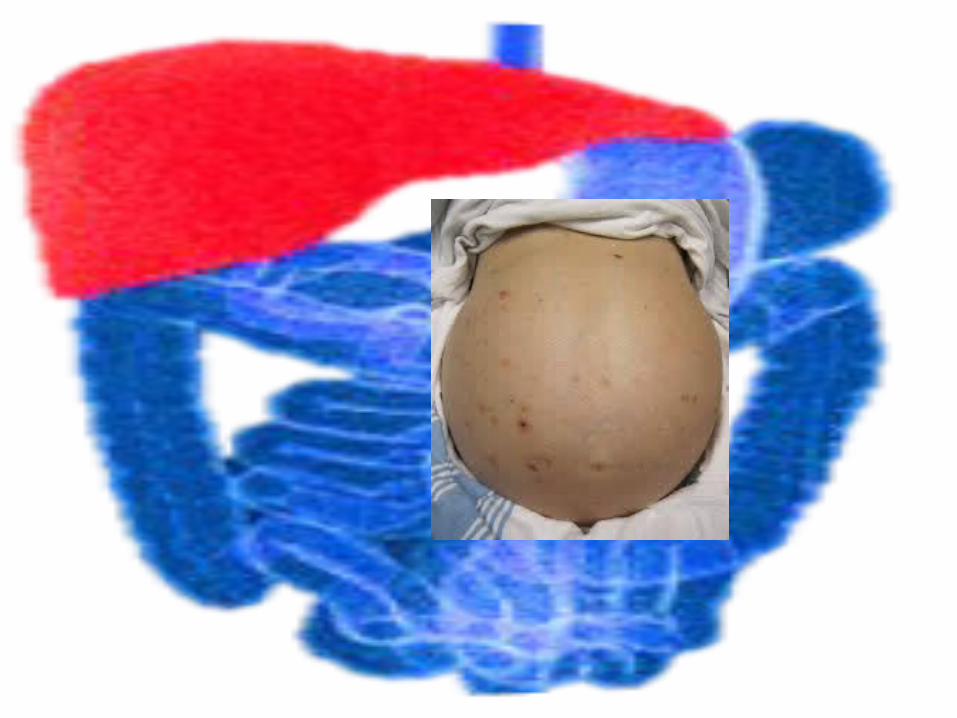
Clinical Manifestation Hepatomegaly Hypoglycemia Hypoprothrombinemia Hypoalbuminemia Hyperferritenima Hyperbilirubinemia
Diagnosis • Documentation of extrahepatic stenosis (biopsy
material at buccal mucosa glands)- confirmatory• MRI Treatment • IVIG’s for recurrence of diseases (once a week) from
8th wk AOG IVI’s until delivery• Deforoxamine- agents with antioxidant therapy• Liver Taransparent
MISCELLANEOUS METABOLLIC DISEASE OF THE LIVER

ALPHA-1 ANTITRYPSIN DEFICIENCY • Deficiency of major serum protease inhibitor alpha-
1 antitrypsin• Normal phenotype PiMM (Protease inhibitor with
the most common allele- “M”) is replaced with Z allele (PIZZ) which predisposes to clinical deficiency
• Serum alpha-1 antitrypsin levels <2 mg/ml (equal to 10-20% of normal)
• In patients with PIZZ, rate of a1-antitrypsin peptide folds is decreased which allows the formation of polymers retain in the ER
Diagnosis• Liver Biopsy Clinical Manifestation 1st week of life Jaundice Alcoholic stools Hepatomegaly Older children Asymptomatic Hepatomegaly Manifestations of chronic liver disease or Cirrhosis- portal hypertension Treatment• Liver Transplantation

Thank you for your listening…


















