
Megakaryopoiesis and Thrombopoiesis
90
Megakaryopoiesis and Thrombopoiesis Williams Hematology: Chapter 113 Ranjita Pallavi Fellow 1 Department of Hematology and Oncology
-
Upload
ranjita-pallavi -
Category
Healthcare
-
view
374 -
download
2
Transcript of Megakaryopoiesis and Thrombopoiesis

Megakaryopoiesis and Thrombopoiesis
Williams Hematology: Chapter 113
Ranjita Pallavi
Fellow 1
Department of Hematology and Oncology

Kinetics of Thrombopoiesis
The circulatory life span of a platelet is approximately 10 days in humans with
normal platelet counts, but somewhat shorter in patients with moderate (7 days)
to severe (5 days) thrombocytopenia, as a higher proportion of the total-body
platelet mass is consumed in the day-to-day function of maintaining vascular
integrity.
Based on a “normal” level of 200,000 platelets/μL, a blood volume of 5 L, and a half-life of 10 days, 1 × 1011 platelets per day are produced.
If 1 megakaryocyte produces approximately 1000 platelets, approximately 1 ×
108 megakaryocytes are generated in the marrow each day.

Kinetics of Thrombopoiesis
The transit time from megakaryocyte progenitor cell to release of platelets into
the circulation ranges from 4 to 7 days.
For example, following platelet apheresis, the platelet count falls, recovers
substantially by day 4, and completely recovers by day 7.
In most physiologic and pathologic states, the platelet count is inversely related
to plasma thrombopoietin levels.

Kinetics of Thrombopoiesis
Liver failure is associated with moderate thrombocytopenia as a result of
splenomegaly and thrombopoietin deficiency. Within the first week following
orthotopic liver transplantation, the platelet count rises substantially, with kinetics
matching those of thrombopoietin infusion.

Cellular Physiology of
Thrombopoiesis
Platelets form by fragmentation of megakaryocyte membrane extensions
termed proplatelets, in a process that consumes nearly the entire cytoplasmic
complement of membranes, organelles, granules, and soluble macromolecules.
Each megakaryocyte is estimated to give rise to 1000 to 3000 platelets before
the residual nuclear material is engulfed and eliminated by marrow
macrophages.
The continuum of megakaryocyte development is arbitrarily divided into four
stages. The major criteria differentiating these stages are the quality and quantity
of the cytoplasm and the size, lobulation, and chromatin pattern of the nucleus.

Cellular Physiology of
Thrombopoiesis

Megakaryoblast
Stage I megakaryocytes, also termed megakaryoblasts, account for
approximately 20 percent of all cells destined to form platelets.
These cells in human marrow are 8 to 24 μm in spherical diameter, contain a
relatively large, minimally indented nucleus with loosely organized chromatin and multiple nucleoli, and scant basophilic cytoplasm containing a small Golgi
complex, a few mitochondria and α granules, and abundant free ribosomes.

Date of download: 12/21/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Electron micrograph of a normal human megakaryoblast stained for platelet peroxidase. The small cell (<9 μm) exhibits dense
platelet peroxidase in the perinuclear space and endoplasmic reticulum (arrows) (magnification ×12,150). (Inset) Enlargement of
the Golgi zone. The Golgi saccules and vesicles are devoid of platelet peroxidase (open arrows), whereas the endoplasmic
reticulum contains platelet peroxidase activity (closed arrow) (magnification ×25,000).
(Courtesy of Dr. J. Breton-Gorius.)
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Megakaryoblast: Surface Adhesion
Molecule Expression
Integrin αIIbβ3 is an integral transmembrane protein of two subunits, but only the α subunit is megakaryocyte-lineage specific.
Absence of integrin αIIbβ3 leads to Glanzmann thrombasthenia resulting from failure of the defective platelets to engage fibrinogen and other adhesive
ligands during hemostasis.
Megakaryocytes and platelets contain in their cytoplasmic membranes about
twice the amount of integrin αIIbβ3 as is present on the cell surface.

Megakaryoblast: Surface Adhesion
Molecule Expression
The granule compartment serves as a mobilizable pool that is exteriorized upon platelet activation.
During the early and mid-stages of megakaryocyte development,
the granule content of integrin rises.
Moreover, as developing megakaryocytes do not synthesize but
contain fibrinogen in their α-granules and cells from patients with
Glanzmann thrombasthenia do not, integrin αIIbβ3 clearly begins to
function, at least at the level of fibrinogen binding and uptake, long before platelet formation.

Megakaryoblast: Surface Adhesion
Molecule Expression
The glycoprotein (GP) Ib-IX complex is expressed only slightly after the
appearance of integrin αIIbβ3.
Although endothelial cells reportedly express GPIb, its levels are very low;
otherwise, GPIb is a second megakaryocyte-specific protein.
Glycoprotein V also is expressed in complex with GPIb and GPIX, in a ratio of
1:2:2.

Megakaryoblast: Surface Adhesion
Molecule Expression
However, the genetic elimination of GPV has little effect on platelet adhesion, and unlike GPIb and GPIX, no mutations of GPV are
associated with Bernard-Soulier disease.
Therefore, GPV does not appear to be required for the GPIb-V-IX
complex to function as a von Willebrand factor receptor.
Rather, GPV is a target of thrombin, potentially playing a role in
platelet activation.

Megakaryoblast: Demarcation
Membranes
Another feature of the megakaryoblast is the initial development of demarcation
membranes, which begin as invaginations of the plasma membrane and
ultimately develop into a highly branched interconnected system of channels
that course through the cytoplasm.
The demarcation membrane system is in open communication with the
extracellular space, based on studies using electron dense tracers.
Biochemical analysis indicates the composition of these membranes is very
similar to the plasma membrane at each stage of megakaryocyte development.

Megakaryoblast: Demarcation
Membranes
Over the 72 hours required for stage III/IV cells to develop from megakaryoblasts,
the demarcation membrane system grows substantially.
The purpose of the demarcation membrane system has been disputed for
several decades. As the term implies, many believed the demarcation membrane system acts to compartmentalize the megakaryocyte cytoplasm into
“platelet territories,” which ultimately fragment into mature platelets along the
cleavage planes so formed.
In contrast, the current belief is that these membranes provide the material
necessary for development of proplatelet processes, structures that form in stage
IV megakaryocytes and give rise upon fragmentation to mature platelets.

Megakaryoblast: Endomitosis
One of the most characteristic features of megakaryocyte development is
endomitosis, a unique form of mitosis in which the DNA is repeatedly replicated in
the absence of nuclear or cytoplasmic division.
The resultant cells are highly polyploid. Endomitosis begins in megakaryoblastsfollowing the many standard cell divisions required to expand the number of
megakaryocytic precursor cells and is completed by the end of stage II
megakaryocyte development.
During the endomitotic phase, each cycle of DNA synthesis produces an exact
doubling of all the chromosomes, resulting in cells containing DNA content from 8
to 128 times the normal chromosomal complement in a single, highly lobated
nucleus.

Megakaryoblast: Endomitosis
Endomitosis is not simply the absence of mitosis but rather consists of recurrent
cycles of aborted mitoses.
Cell-cycle kinetics in endomitotic cells also are unusual, characterized by a short
G1 phase, a relatively normal DNA synthesis phase, a short G2 phase, and a very
short endomitosis phase.
During the endomitosis phase, megakaryocytic chromosomes condense, the
nuclear membrane breaks down, and multiple (at advanced stages) mitotic
spindles form upon which the replicated chromosomes assemble.
However, following initial chromosomal separation, individual chromosomes fail
to complete their normal migration to opposite poles of the cell, the spindle
dissociates, the nuclear membrane reforms around the entire chromosomal complement, and the cell again enters G1 phase.

Date of download: 12/21/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Origin and development of megakaryocytes. The pluripotential stem cell produces a progenitor committed to megakaryocyte
differentiation (colony-forming unit–megakaryocyte [CFU-MK]), which can undergo mitosis. Eventually the CFU-MK stops mitosis
and enters endomitosis. During endomitosis, neither cytoplasm nor nucleus divides, but DNA replication proceeds and gives rise to
immature polyploid progenitors, which then enlarge and mature into morphologically identifiable, mature megakaryocytes that shed
platelets. This figure does not necessarily imply that endomitosis and platelet formation are sequential but they can occur
simultaneously. Meg-CFC, megakaryocyte colony-forming cells.
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Megakaryoblast: Regulation of Gene
Expression
The promoters for integrin αIIb, GPIb, GPVI, GPIX, and platelet factor-4 are active
at the megakaryoblast stage of development.
Consensus sequences for both GATA-1 and members of the Ets family of
transcription factors (e.g., Fli-1) are present in the 5′ flanking regions of these
genes, deletion of which reduces or eliminates reporter gene expression, at least
in mature hematopoietic cells.
MafB also enhances GATA-1 and Ets activity during megakaryoblast
differentiation, induced by activation of ERK1/2, one of the primary downstream
events of thrombopoietin stimulation.

Megakaryoblast: Cytokine Dependency
The cytokines, hormones, and chemokines that affect the survival and
proliferation of megakaryoblasts include thrombopoietin, interleukin (IL)-3, stem
cell factor (also termed mast cell growth factor, steel factor, and c-kit ligand),
and stromal cell-derived factor (SDF)-1.
Thrombopoietin is the most critical, as genetic elimination of the TPO gene in
mice leads to circulating platelet levels approximately 10 percent of normal.
Homozygous or complex heterozygous mutation of the gene encoding the
thrombopoietin receptor cMpl leads to congenital amegakaryocytic
thrombocytopenia, in which platelet levels are approximately 10 percent of
normal because of a near absence of megakaryocytic progenitors and megakaryoblasts.

Megakaryoblast: Cytokine Dependency
Genetic reduction in expression of stem cell factor or its receptor c-kit leads to a
50 percent reduction in circulating platelet levels.
The cytokine acts in synergy with thrombopoietin to enhance megakaryocyte
production in semisolid and suspension culture systems.
Evidence that IL-3 contributes to normal or accelerated megakaryopoiesis in
vivo is weak. Genetic elimination of the IL-3 gene fails to affect platelet counts,
even when combined with thrombopoietin receptor deficiency, but the cytokine
can induce growth of marrow progenitors into colonies containing immature
megakaryocytes in vitro in the absence of thrombopoietin.
The chemokine SDF-1 appears to play a role in megakaryocyte proliferation. In
vitro, SDF-1 acts in synergy with thrombopoietin to support the survival and proliferation of megakaryocyte progenitors.

Megakaryoblast: Signal Transduction
The survival and proliferation of megakaryoblasts depends on at least two
thrombopoietin-induced signaling pathways: PI3K and mitogen-activated
protein kinase (MAPK).
In the presence of chemical inhibitors of PI3K, the favorable effects of
thrombopoietin on megakaryocyte progenitor survival and proliferation are
eliminated, although constitutively activating this pathway is not sufficient for
thrombopoietin-induced growth.
MAPK is another important signaling pathway stimulated by thrombopoietin.
Using purified marrow megakaryocytic progenitors and model cell lines, several groups showed that inhibition of MAPK blocks megakaryoblast
maturation because of its effect of activating Ets transcription factors.

Stage II Megakaryocytes
Stage II megakaryocytes contain a lobulated nucleus and more abundant, but
less intensely basophilic, cytoplasm.
Ultrastructurally, the cytoplasm contains more abundant α granules and organelles.
The demarcation membrane system begins to expand at this stage of
development.
Stage II megakaryocytes measure up to 30 μm in diameter, constitute
approximately 25 percent of marrow megakaryocytes, and are the stage of
development during which endomitosis is most prominent, generating cells
displaying ploidy values of 8N to 64N.

Stage II Megakaryocyte: Cytoplasmic
Development
Early in megakaryocyte development, the cytoplasm acquires a rich network of
microfilaments and microtubules.
Toward stages III and IV, the proteins accumulate in the cell periphery, creating
an organelle poor peripheral zone.
Biochemically, the megakaryocyte cytoskeleton is composed of actin, α-actinin,
filamin, non-muscle myosin (including the product of the MYH9 gene), mutated
in several giant platelet thrombocytopenic syndromes, β1-tubulin, talin, and
several other actin-binding proteins.
Like platelets, megakaryocytes can respond to external stimuli by changing
shape, transporting organelles around the cytoplasm, and secreting granules.
These functions are dependent on the microfilament and microtubule systems of
the cell. In addition, microtubules play a vital role during the later stages of platelet formation.

Stage II Megakaryocyte: Platelet
Granule Formation
Although more prominent in later stages of differentiation, platelet-specific α
granules first begin to form adjacent to the Golgi apparatus as 300- to 500-nm
round or oval organelles in stage II megakaryocytes.
Three distinct compartments are recognized in α granules: (1) a central, electron-dense nucleoid, containing fibrinogen, platelet factor-4, β-
thromboglobulin, transforming growth factor (TGF)-β1, vitronectin, and tissue
plasminogen activator-like plasminogen activator; (2) a peripheral zone,
containing tubules and von Willebrand factor (arranged much like that seen in
endothelial cell Weibel-Palade bodies); and (3) the granule membrane,
containing many of the critical platelet receptors for cell rolling (P-selectin), firm
adhesion (GPIb-V-IX), and aggregation (integrin αIIbβ3).

Stage II Megakaryocyte: Platelet
Granule Formation
Proteins present in α granules arise from de novo megakaryocyte synthesis (e.g.,
GPIb-V-IX, GPIV, integrin αIIbβ3, von Willebrand factor, P-selectin, β-
thromboglobulin, platelet-derived growth factor), nonspecific pinocytosis of
environmental proteins (albumin and immunoglobulin G), or cell surface
membrane receptor-mediated uptake from the environment (e.g., fibrinogen, fibronectin, factor V).
Insights into platelet granule formation have come from a molecular
understanding of Hermansky-Pudlak Syndrome (HPS).
In this disorder, characterized by oculocutaneous albinism and a qualitative
platelet bleeding disorder, a complex of at least eight proteins form in various
granule associated complexes such as the biogenesis of lysosome-related organelles complexes, which affect δ granule formation.
These complexes are thought to be involved in cargo transport of a number of subcellular granules, such as lysosomes, melanosomes, and platelet δ granules.

Stage III/IV Megakaryocytes
Continued cytoplasmic maturation characterizes stage III/IV megakaryocyte
development.
Cells are extremely large (40–60 μm in diameter) and display a low nuclear-to-
cytoplasmic ratio.
Cytoplasmic basophilia disappears as cells progress from stage III to IV.
The demarcation membrane system gradually replaces the endoplasmic
reticulum and Golgi apparatus during the final stages of maturation.
The nucleus usually is eccentrically placed.

Stage III/IV Megakaryocytes
The nucleus remains highly lobulated but single at all stages of megakaryocyte
development.
In occasional marrow sections, neutrophils or other marrow cells are seen
transiting through the cytoplasm of the mature megakaryocyte, a process
termed emperipolesis, and is of no pathologic significance.

Date of download: 12/21/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Megakaryocyte morphology. A. Normal human marrow biopsy. Two megakaryocytes are evident. In one case the section is through
the cell at the level of the nuclei (horizontal arrow), and in the other it is through the cytoplasm above or below the nucleus (vertical
arrow). B. Normal human marrow aspirate. Mature (stage III) megakaryocyte with a multilobated nucleus and abundant cytoplasm.
C. Normal human marrow aspirate. Mature megakaryocyte with a neutrophil embedded in the cytoplasm. Many ultrastructural
studies have confirmed that this appearance represents marrow cells entering the canalicular system of megakaryocyte cytoplasm
through its opening to the exterior of the cell (emperipolesis).
(Used with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Date of download: 12/21/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Megakaryocyte morphology. A. Normal human marrow biopsy. Two megakaryocytes are evident. In one case the section is through
the cell at the level of the nuclei (horizontal arrow), and in the other it is through the cytoplasm above or below the nucleus (vertical
arrow). B. Normal human marrow aspirate. Mature (stage III) megakaryocyte with a multilobated nucleus and abundant cytoplasm.
C. Normal human marrow aspirate. Mature megakaryocyte with a neutrophil embedded in the cytoplasm. Many ultrastructural
studies have confirmed that this appearance represents marrow cells entering the canalicular system of megakaryocyte cytoplasm
through its opening to the exterior of the cell (emperipolesis).
(Used with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Date of download: 12/21/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Megakaryocyte morphology. A. Normal human marrow biopsy. Two megakaryocytes are evident. In one case the section is through
the cell at the level of the nuclei (horizontal arrow), and in the other it is through the cytoplasm above or below the nucleus (vertical
arrow). B. Normal human marrow aspirate. Mature (stage III) megakaryocyte with a multilobated nucleus and abundant cytoplasm.
C. Normal human marrow aspirate. Mature megakaryocyte with a neutrophil embedded in the cytoplasm. Many ultrastructural
studies have confirmed that this appearance represents marrow cells entering the canalicular system of megakaryocyte cytoplasm
through its opening to the exterior of the cell (emperipolesis).
(Used with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Stage III/IV Megakaryocytes:
Proplatelet formation
Careful microscopic studies have localized marrow megakaryocytes to the
abluminal surface of sinusoidal endothelial cells.
In specially prepared specimens, the megakaryocytes can be seen issuing long,
slender cytoplasmic processes between endothelial cells and into the sinusoidal lumen, structures termed proplatelet processes.
The processes have been reproduced in vitro and in vivo.
The processes consist of a β-tubulin cytoskeleton and highway, transporting
organelles and platelet constituents from the megakaryocyte to the terminal
projection, the nascent platelet.

Date of download: 12/23/2014 Copyright © 2014 McGraw-Hill Education. All rights reserved.
Megakaryocyte proplatelet processes in the marrow sinusoid. Scanning electron micrograph showing the luminal view of the
confluence of two marrow sinusoids with two proplatelet processes protruding through the lining endothelial cells. One of the
processes has intermittent constrictions (arrows), indicating potential sites for platelet formation. Other cells depicted include
lymphocytes and erythrocytes (magnification ×3000).
(From Becker RP, De Bruyn P,54 with permission.)
Legend:
From: Chapter 113. Megakaryopoiesis and Thrombopoiesis
Williams Hematology, 8e, 2010

Stage III/IV Megakaryocytes: Membrane
Composition
Most of the specific characteristics of platelet membranes are present at stages
III and IV of megakaryocyte development.
Megakaryocyte membrane lipid composition progressively changes through
development, achieving approximately four times the content of phospholipids and cholesterol as found in immature cells.
Megakaryocytes contain about the same amounts of membrane neutral and
phospholipid as platelets, but contain relatively more phosphatidylinositol and
less phosphatidylserine and arachidonic acid.

Platelet Formation
Platelet formation involves massive reorganization of megakaryocyte
cytoskeletal components, including actin and tubulin, during a highly active,
motile process in which the termini of the process branch and issue platelets.
The size of the individual platelets formed is of interest.
Unfortunately, little is known about this aspect of platelet formation except that
tubulin is proposed to act as a measuring device for the proper site to pinch off platelets from proplatelet processes.
The mechanism of platelet formation clearly must be affected in some way by
the transcription factor GATA-1, the glycoprotein Ib-IX complex, the Wiskott-
Aldrich syndrome protein, and platelet myosin, as defects in each of these genes
leads to unusually large or small platelets.
Finally, localized cytoplasmic membrane proteolysis, a sublethal form of
apoptosis, likely plays a role in initiating the final stages of platelet formation

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
The term thrombopoietin was first coined in 1958 to describe the primary
regulator of platelet production.
A major impetus to the discovery of thrombopoietin in 1986 was the identification of the myeloproliferative leukemia virus (MPLV), which induces a vast expansion
of hematopoietic cells.
The responsible viral oncogene was characterized in 1990, and its cellular
homologue c-Mpl was cloned in 1992.
The gene for thrombopoietin encodes a 36-kDa polypeptide, which also is
predicted to be extensively posttranslationally modified, resulting in an approximately 50- to 70-kDa protein.

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
Thrombopoietin bears striking homology to erythropoietin, the primary regulator
of erythropoiesis, within the amino-terminal half of the predicted polypeptide.
The two proteins are more closely related than any other two cytokines within the
hematopoietic cytokine family, sharing 20 percent identical amino acids, an
additional 25 percent conservative substitutions, and identical positions of three
of the fourcysteine residues.
Unlike any of the other cytokines in the family, thrombopoietin contains a 181-
residue carboxyl-terminal extension, which bears homology to no known
proteins.
Two functions have been assigned to this region: it prolongs the circulatory half-
life of the hormone, and it aids in its secretion from the cells that normally synthesize the hormone.

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
Incubation of marrow cells with thrombopoietin stimulates megakaryocyte
survival and proliferation, alone and in combination with other cytokines.
In vivo, thrombopoietin stimulates platelet production in a log-linear manner to
levels 10-fold higher than baseline without affecting the blood red or white cell
counts.
In addition, because of its affect on hematopoietic stem cells, the number of erythroid and myeloid progenitors and mixed myeloid progenitors in marrow and
spleen also are increased, an effect that is particularly impressive when
the hormone is administered following myelosuppressive therapy.
This effect likely results from the synergy between thrombopoietin and the other
hematopoietic cytokines circulating at high levels in this condition.
MPL gene is located on chromosome 1p34.

c-MPL Receptor

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
Based on genetic studies, thrombopoietin clearly is the primary regulator of thrombopoiesis.
Elimination of either the c-Mpl or Tpo gene leads to profound thrombocytopenia in mice as a result of a greatly reduced number of megakaryocyte progenitors, mature megakaryocytes, and the reduced polyploidy of the remaining megakaryocytes.
A similar result occurs in humans. Patients with congenital amegakaryocyticthrombocytopenia (CAMT) display numerous homozygous or mixed heterozygous nonsense or severe missense mutations of the thrombopoietinreceptor c-Mpl.
The effect of thrombopoietin on hematopoietic stem cells is particularly revealed by consideration of children with CAMT.
Within 5 years of birth, nearly every patient with CAMT develops aplastic anemia as a result of stem cell exhaustion.

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
The thrombopoietin gene displays an unusual 5′ flanking structure.
Unlike the majority of genes that initiate translation of the encoded polypeptide with the first ATG codon present in the messenger ribonucleic acid (mRNA), thrombopoietin translation initiates at the eighth ATG codon located within the third exon of a full-length transcript.
However, since the eighth ATG of thrombopoietin mRNA is embedded in the short, open reading frame of the seventh ATG, its translation is particularly inefficient because of the mechanism of ribosomal initiation.
As such, little thrombopoietin protein is produced for any given amount of mRNA. Although this molecular arrangement has no known physiologic consequences, it forms the basis for an unusual form of disease, a disorder of translation efficiency.
Four cases of autosomal dominant familial thrombocytosis have been linked to mutations in the region surrounding the initiation codon.

Extrinsic Regulation of Megakaryocyte
Production: Thrombopoietin
It has been seen that plasma hormone concentrations vary inversely with platelet counts, rising to maximal levels within 24 hours of onset of profound thrombocytopenia.
Two nonmutually exclusive models have been advanced to explain these findings.
In the first model, thrombopoietin production is constitutive, but its consumption, and hence the level remaining in the blood to affect megakaryopoiesis, is determined by the mass of c-Mpl receptors present on platelets and megakaryocytes accessible to the plasma.
In this way, states of thrombocytosis result in increased thrombopoietinconsumption (by the expanded platelet mass of c-Mpl receptors), reducing megakaryopoiesis.
Conversely, thrombocytopenia reduces blood thrombopoietindestruction, resulting in elevated blood levels of the hormone that drive megakaryopoiesis and platelet recovery.

Extrinsic Regulation of
Megakaryocyte Production: Thrombopoietin A second model suggests thrombopoietin expression is a regulated
event.
Very low platelet levels can induce thrombopoietin-specific mRNA production.
Several studies show that thrombopoietin mRNA levels are modulated in response to moderate to severe thrombocytopenia, at least in the marrow.
The signal(s) responsible for this form of thrombopoietin regulation is being uncovered, but is, at least in part, mediated by transcriptional enhancement.
CD40 ligand, platelet-derived growth factor, fibroblast growth factor, TGF-β, platelet factor-4, and thrombospondin modulate thrombopoietinproduction from marrow stromal cells.

Thrombopoietin Mimetics:
Romiplostim
It has been approved for use in refractory ITP.
In immune thrombocytopenia, immune dysregulation leads to the production of
autoantibodies or immune complexes that accelerate peripheral platelet
destruction by binding to platelets, causing platelet phagocytosis, along with T-cell and possibly complement-mediated lysis.
The production of new platelets is also suppressed; antiplatelet antibodies have
been shown to bind to megakaryocytes in the bone marrow, causing both a
decrease in the number of megakaryocytes and the inhibition of
megakaryocyte maturation.

Thrombopoietin Mimetics:
Romiplostim
Plasma levels of endogenous thrombopoietin are typically high in
patients who have thrombocytopenia associated with bone
marrow failure syndromes. However, thrombopoietin levels are usually normal or only slightly increased in patients with immune
thrombocytopenia, for reasons that remain unclear.
The fact that thrombopoietin levels in immune thrombocytopenia
are lower than anticipated led to the concept of treating the
disorder by means of exogenous stimulation of thrombopoietin
receptors.

Thrombopoietin Mimetics:
Romiplostim
Initial trials involved the use of recombinant forms of thrombopoietin. However, clinical trials were stopped when thrombocytopenia —resulting from the development of autoantibodies against endogenous thrombopoietin — developed in healthy volunteers receiving these agents.
Thus, development began on a second generation of thrombopoietinmimetics or agonists that are structurally dissimilar to thrombopoietinand thus do not lead to the formation of autoantibodies.
Romiplostim (Nplate, Amgen) is a synthetic fusion protein that has four peptides consisting of 14 amino acid residues connected to an IgG Fc fragment, producing a “peptibody”.
Romiplostim binds to the thrombopoietin receptor and activates intracellular signaling pathways (the JAK–STAT and MAP kinase pathways), which stimulates platelet production.

Thrombopoietin Mimetics:
Romiplostim
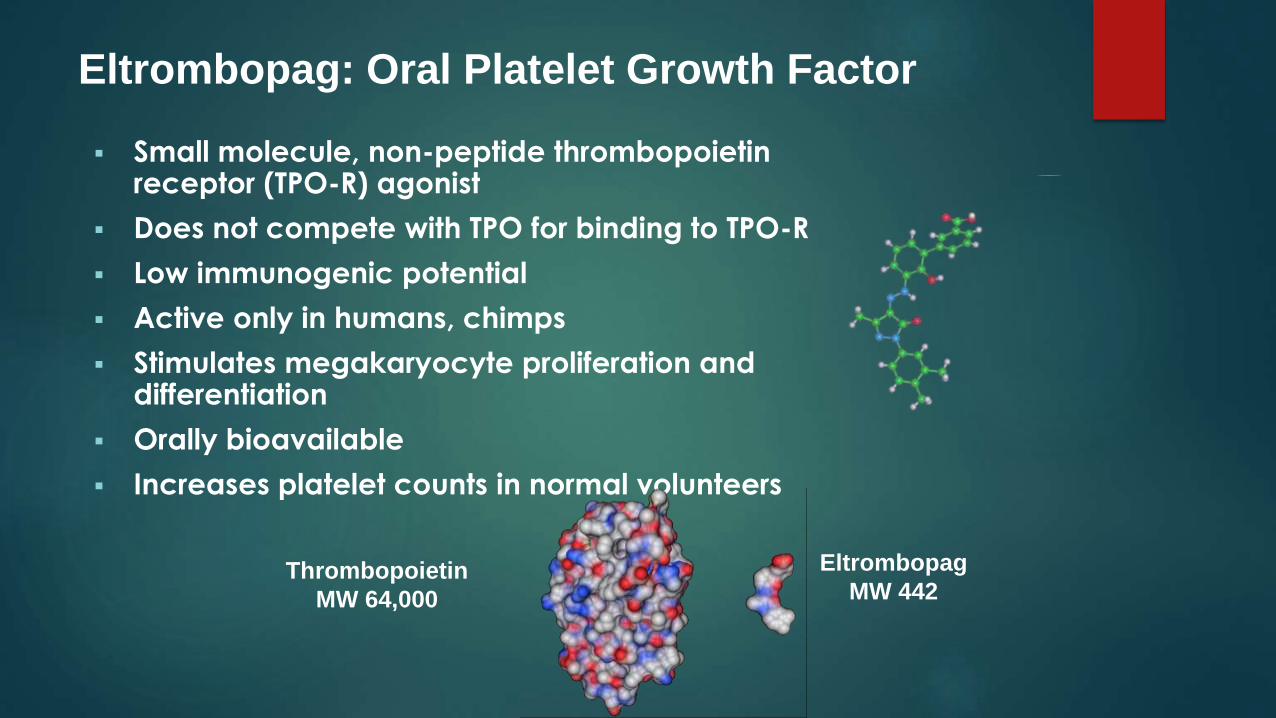
Eltrombopag (Promacta, GlaxoSmithKline) is a small nonpeptide
molecule that binds to the transmembrane region of the
thrombopoietin receptor, which activates the same intracellular pathways that are activated by romiplostim.
In initial pharmacodynamic studies, a single intravenous or
subcutaneous dose of romiplostim resulted in an increase in the
platelet count after 5 to 8 days in a dose-dependent fashion.
The peak platelet count was reached between days 12 and 16, and
by day 28 the platelet count had fallen to baseline.
Eltrombopag has shown a similar pattern of response in platelet
count.


AMG 531
• Unique platform “peptibody”• Made in E. coli • Molecular weight = 60,000 D• 4 Mpl binding sites
Bussel JB et al. N Engl J Med. 2006;355:1672.
• No sequence homology with TPO• Cleared endothelial FcRn
Recycled• Cleared RES
Fc Carrier Domain
TPO Agonist
PeptidesFc Carrier Domain
TPO Agonist
Peptides

The romiplostim trials included two 24-week placebo-controlled phase 3 studies,
one enrolling 63 splenectomized patients and the other enrolling 62 non splenectomized patients.
The primary efficacy end point was a durable response, which was defined as a
platelet count of at least 50,000 per cubic millimeter for at least 6 of the final 8
weeks.

Thrombopoietin Mimetics:
Romiplostim
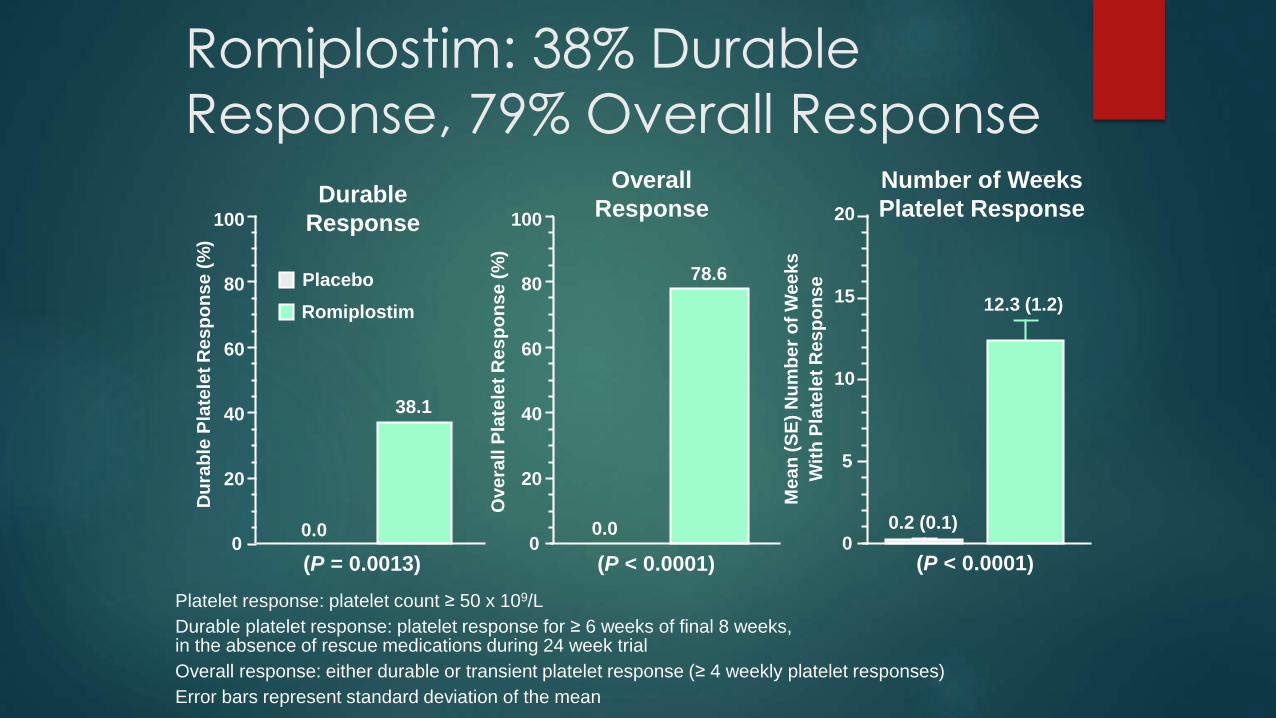
This end point was achieved in 38% of splenectomized patients and in 61% of nonsplenectomized patients in the romiplostim groups, as compared with no splenectomized patients and 5% of nonsplenectomized patients in the placebo groups (P<0.001 for both comparisons).
Patients receiving romiplostim were more likely to reduce or discontinue concurrent medications (primarily glucocorticoids) and to require less rescue medication than patients in the placebo group.
In an open-label extension study involving 292 patients, 94.5% achieved a platelet count of at least 50,000 per cubic millimeter during the study, and more than 50% had a platelet count of at least 50,000 per cubic millimeter during at least 90% of all visits at a median of 78 weeks.
Romiplostim: 38% Durable
Response, 79% Overall Response
Durable
Response
Overall
Response
Number of Weeks
Platelet Response
Platelet response: platelet count ≥ 50 x 109/L
Durable platelet response: platelet response for ≥ 6 weeks of final 8 weeks,in the absence of rescue medications during 24 week trial
Overall response: either durable or transient platelet response (≥ 4 weekly platelet responses)
Error bars represent standard deviation of the mean
0.0
38.1
(P = 0.0013)
0.0
78.6
0
20
40
60
80
100
(P < 0.0001)
Du
rab
le P
late
let
Re
sp
on
se
(%
)
Ove
rall
Pla
tele
t R
es
po
ns
e (
%)
Me
an
(S
E)
Nu
mb
er
of
We
ek
s
Wit
h P
late
let
Re
sp
on
se
0.2 (0.1)
12.3 (1.2)
0
5
10
15
20
(P < 0.0001)0
20
40
60
80
100
Placebo
Romiplostim

Lancet 2008;371:395

Romiplostim (AMG 531):
Summary
In splenectomized patients:
38% durable response, 79% overall response
Increased and maintained platelet counts over24 weeks
Significantly decreased the use of rescue medications
All romiplostim patients discontinued or reduced concurrent ITP therapy (corticosteroids, azathioprine, danazol)
Romiplostim appeared to be well tolerated

Romiplostim: Summary of Long-term
Dosing
Efficacy Data Summary
The majority of patients achieved long-term platelet
counts > 50 x 109/L and double the baseline value
Mean platelet count maintained between 50 and 250 x 109/L over
2 years
Use of concomitant and rescue medications was
substantially reduced over time
No trend in this study for adverse events to increase
in frequency with longer drug exposure
One patient had neutralizing antibodies to AMG
531; negative on retesting.
Small molecule, non-peptide thrombopoietinreceptor (TPO-R) agonist
Does not compete with TPO for binding to TPO-R
Low immunogenic potential
Active only in humans, chimps
Stimulates megakaryocyte proliferation and differentiation
Orally bioavailable
Increases platelet counts in normal volunteers
Thrombopoietin
MW 64,000
Eltrombopag
MW 442
Eltrombopag: Oral Platelet Growth Factor

Eltrombopag was approved by the Food and Drug Administration (FDA) on the basis of the results of two 6-week, placebo-controlled clinical trials and the initial results of an open-label extension study.
The primary efficacy measurement in both randomized studies was the proportion of patients achieving a platelet count of at least 50,000 per cubic millimeter on day 43.
In a dose-adjustment study involving 118 patients, this platelet count was achieved more often in the groups who received daily oral eltrombopag (at a dose of either 50 mg or 75 mg) than in those receiving placebo.

Primary Endpoint: Percentage of
Patients With Platelets ≥50,000/µL at
Day 43 Visit†
0
20
40
60
80
100
Resp
on
ders
(%
)
Placebo§ Eltrombopag
P <0.001‡
OR = 9.61 (3.31, 27.86)
†Last observation carried forward.‡Indicates significance at 5% (2-sided) level of significance.§1 patient received IVIg on Day 1.
Logistic regression analysis adjusted for randomization stratification variables.

Conclusions
The EXTEND data suggest that oral eltrombopag was well tolerated and safe
Eltrombopag up to 75 mg/day increased and sustained platelet counts >50,000/μL in the majority of patients
Eltrombopag reduced the incidence and severity of bleeding

Thrombopoietin Mimetics:
Eltrombopag
In a subsequent phase 3 placebo-controlled trial involving 114
patients who were treated with 50 mg of eltrombopag per day, a
platelet count of at least 50,000 per cubic millimeter was achieved
by day 43 in 59% of patients in the eltrombopag group, as
compared with 16% of those in the placebo group (P<0.001).
In an open-label extension study involving 299 patients who
completed a previous eltrombopag study, 87% of patients achieved
a platelet count of at least 50,000 per cubic millimeter during
treatment.

Thrombopoietin Mimetics
Both romiplostim and eltrombopag have been found to reduce
bleeding complications.
In the romiplostim extension study, the rate of moderate-to-severe bleeding decreased from 23% to 12% within the first 24 weeks and to
6% during weeks 24 through 48.
In the eltrombopag extension study, the incidence of any bleeding
symptoms declined from 56% at baseline to 16% by week 52 and to
20% by week 104.

Thrombopoietin Mimetics
Romiplostim and eltrombopag are approved by the FDA for
patients with chronic immune thrombocytopenia who have an
insufficient response to glucocorticoids, intravenous immune
globulin, or splenectomy.
Clinically, these agents are typically used in patients who have
persistent or chronic immune thrombocytopenia and ongoing
bleeding, with or without previous splenectomy and one or more
courses of rituximab.
Since thrombopoietin-receptor agonists cross the placenta, their
safety in pregnancy has not been shown, so their use in such cases is
not recommended.

Thrombopoietin Mimetics
The recommended initial dose of romiplostim is 1 μg per kilogram of body weight, administered subcutaneously once weekly, with subsequent dose adjustment on the basis of the platelet count.
The mean therapeutic dose is 3 to 4 μg per kilogram, with a maximum dose of 10 μg per kilogram.
Romiplostim is available in 250-μg and 500-μg vials as a lyophilized powder.
Only providers who are enrolled in a regulated prescriber program called NEXUS (Network of Experts Understanding and Supporting Nplateand Patients) may prescribe romiplostim. The NEXUS program requires providers to enroll all patients receiving romiplostim in a registry and to enter baseline data for patients as well as periodic safety information. Details regarding the NEXUS program are available at www.nplatenexus.com. These restrictions do not apply to the use of romiplostim outside the United States. In most other countries, the drug can be self-administered by the patient at home.

Thrombopoietin Mimetics
The recommended initial dose of eltrombopag for most patients is 50 mg daily given orally, with subsequent dose adjustment on the basis of the platelet count (to a maximum of 75 mg daily or a minimum of 25 mg daily).
Patients with hepatic dysfunction and patients of Asian ethnic background (in whom plasma concentrations of the drug are higher than in white patients) should initiate treatment at a dose of 25 mg once daily.
Eltrombopag should be taken 1 to 2 hours after a meal because of interactions with food.
It should not be taken within 4 hours after taking antacids, dairy products, or supplements that contain polyvalent cations, such as calcium, magnesium, and aluminum.
Like romiplostim, eltrombopag was approved by the FDA under restricted terms. In the United States, the drug can be prescribed only by participants in a regulated prescriber program called Promacta Cares. Details regarding the program are available at www.promactacares.com.

Thrombopoietin Mimetics
When either romiplostim or eltrombopag is given, the platelet count
should be measured weekly until a stable count (>50,000 per cubic
millimeter for at least 4 weeks without dose adjustment) has been achieved and monthly thereafter.
Treatment should be withheld temporarily when the platelet count is
200,000 to 400,000 per cubic millimeter; it is not a goal of therapy to
achieve and maintain a normal platelet count.

Thrombopoietin Mimetics
Eltrombopag therapy can cause hepatic injury. Thus, patients
receiving eltrombopag should have levels of serum aspartate
aminotransferase, alanine aminotransferase, and bilirubin checked every 2 weeks during the dose-adjustment phase of therapy and
monthly after the establishment of a stable dose.

Thrombopoietin Mimetics: Adverse
Effects
The most common adverse effects of thrombopoietin-receptor
agonists in clinical trials included headache, nausea, vomiting,
fatigue, diarrhea, arthralgia, and nasopharyngitis.
Worsened thrombocytopenia after the discontinuation of the
thrombopoietin-receptor agonist occurs in 8 to 10% of patients, with
an increased risk of bleeding during the first 4 weeks.
Tapering of the agent or reinitiation of other treatment for immune
thrombocytopenia is recommended if severe thrombocytopenia
supervenes. The platelet count typically recovers to pretreatment
levels after several weeks.

Thrombopoietin Mimetics
In prospective studies, patients receiving eltrombopag had hepatobiliary laboratory abnormalities; 11% of patients receiving eltrombopag and 7% receiving placebo had aminotransferase values at least 3 times the upper limit of the normal range and alkaline phosphatase or total bilirubin values at least 1.6 times the upper limit of the normal range.
These abnormalities may resolve despite continued therapy. However, the package insert specifies that eltrombopag therapy should be discontinued if alanine aminotransferase levels increase to 3 times the upper limit of the normal range or higher and are progressive, are persistent for 4 weeks or more, are accompanied by an increase in the direct bilirubin level, or are accompanied by clinical symptoms of liver injury or evidence of hepatic decompensation.
No similar effects have been seen with romiplostim.

Thrombopoietin Mimetics
In a study of extended romiplostim treatment involving 291 patients,
25 venous or arterial thromboembolic events occurred in 17
patients.
In an eltrombopag extension study involving 299 patients, 16 patients had 21 thromboembolic events.
The frequency of thromboembolic events did not increase with the
duration of treatment in either study.
Most thromboembolic events that have been associated with the
use of thrombopoietin-receptor agonists have been observed in
patients with at least one additional risk factor for thrombosis.

Thrombopoietin Mimetics
The American Society of Hematology Guidelines:
The use of thrombopoietin-receptor agonists is recommended for
adult patients at risk for bleeding who have a relapse after
splenectomy or who have a contraindication to splenectomy and do not have a response to at least one other therapy.
It was also suggested that these agents could be considered for
adult patients at risk for bleeding who have not had a response to
one line of therapy and have not undergone splenectomy.

c-mpl mutations are the cause of
congenital amegakaryocytic
thrombocytopenia (CAMT)
CAMT is a rare autosomal recessive bone marrow failure syndrome that presents with severe thrombocytopenia which can evolve into
aplastic anemia and leukemia.
The disorder is expressed in infancy with or without physical
anomalies.
It is often recognized on day 1 of life or at least within the first month.
It is often initially confused with fetal and neonatal alloimmunethrombocytopenia, but the neonate fails to improve and responds
only to platelet transfusion.

CAMT
A recent classification was proposed in 2005.
Type I—early onset of severe pancytopenia, decreased bone
marrow activity and very low platelet counts. In this group, there is complete loss of functional c-Mpl. Median platelet count is usually
21 × 109/L or below.
Type II—milder form with transient increases of platelet counts up to
nearly normal values during the first year of life and an onset of
bone marrow failure at age 3 to 6 years or later. In this group, there
are partially functional receptors for the c-Mpl gene. Median
platelet count is usually 35 × 109/L to 132 × 109/L.
Type III—there is ineffective megakaryopoeisis with no defects in the
c-Mpl gene.

CAMT
Differential diagnosis for severe CAMT includes thrombocytopenia
with absent radii (TAR) and Wiskott-Aldrich syndrome (WAS).
The primary treatment for CAMT is bone marrow transplantation.
Newer modalities are on the way, such as TPO-mimetics for binding
towards partially functioning c-Mpl receptors and gene therapy.
Prognosis of CAMT patients is poor, because all develop in
childhood a tri-linear marrow aplasia that is always fatal when
untreated. Thirty percent of patients with CAMT die due to bleeding
complications and 20% -due to HSCT if it has been done.

GATA1 Mutation leading to X linked
Thrombocytopenia
X-linked thrombocytopenia is a well-known clinical condition, found
most often in the context of the Wiskott-Aldrich syndrome (WAS) and
consisting of thrombocytopenia, defective humoral and cellular immunity, and eczema.
Mutations in the WASP gene lead either to the full-blown WAS
picture or to isolated X-linked thrombocytopenia.
The platelets in this syndrome are typically small sized. However,
hereditary macrothrombocytopenia with or without associated
thrombopathy has been identified in a variety of syndromes, such as
the May-Hegglin anomaly, Bernard-Soulier syndrome, Fechtnersyndrome, or Epstein syndrome.

GATA1 Mutation leading to X linked
Thrombocytopenia
Very recently, the May-Hegglin anomaly and Fechtner syndrome
have been linked to mutations in the nonmuscle myosin heavy
chain 9 gene on chromosome 22.
GATA1 is the founding member of the GATA-binding family of
transcription factors and has been shown to be an essential protein
for normal erythropoiesis and megakaryocyte differentiation.
The human gene encoding GATA1 has been mapped to Xp11.23.
Shivdasani et al developed a lineage-selective knockout mouse of
GATA1, leading to megakaryocyte-specific loss of GATA1 expression
and established the critical role of this transcription factor for megakaryocyte growth and platelet development.

GATA1 Mutation leading to X linked
Thrombocytopenia
Very recently Nichols et al described for the first time a mutation in
the GATA1 gene in a family with X-linked dyserythropoietic anemia
and macrothrombocytopenia.
This missense mutation (V205M) leads to a reduced interaction of
the N-terminal zinc finger of GATA1 with its essential cofactor FOG1
(for Friend of GATA1).

MYH9-Related Platelet Disorders
Myosin heavy chain 9 (MYH9)-related platelet disorders belong to the group of
inherited thrombocytopenias.
The MYH9 gene encodes the nonmuscle myosin heavy chain IIA (NMMHC-IIA), a cytoskeletal contractile protein.
Several mutations in the MYH9 gene lead to premature release of platelets from the bone marrow, macrothrombocytopenia,andcytoplasmic inclusion bodies within leukocytes.
Four overlapping syndromes, known as May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian platelet syndrome, describe different clinical manifestations of MYH9 gene mutations.

MYH9-Related Platelet Disorders
Macrothrombocytopenia is present in all affected individuals,
whereas only some develop additional clinical manifestations such
as renal failure, hearing loss, and presenile cataracts.
The bleeding tendency is usually moderate, with menorrhagia and easy bruising being most frequent.
The biggest risk for the individual is inappropriate treatment due to
misdiagnosis of chronic autoimmune thrombocytopenia.
To date, 31 mutations of the MYH9 gene leading to macro-
thrombocytopenia have been identified, of which the upstream
mutations up to amino acid 1400 are more likely associated with
syndromic manifestations than the downstream mutations.

MYH9-Related Platelet Disorders
In 1909, May described a family in which several members had
enlarged platelets but minor if any bleeding symptoms. In 1945,
Hegglin found Do¨hle body like inclusions within the leukocytes of
affected individuals with a dominantly-inherited giant platelet disorder. This led to the term May-Hegglin anomaly (MHA) to
describe the triad of thrombocytopenia, giant platelets, and
leukocyte inclusion bodies.
These inclusion bodies are spindle shaped and appear bright blue in
standard blood films.

MYH9-Related Platelet Disorders
In 1972, Epstein et al described Epstein syndrome (EPS) as the first
macro-thrombocytopenic syndrome characterized by giant
platelets, associated with deafness and nephritis; however, in
contrast with MHA leukocyte inclusion bodies were absent.
In 1985, Peterson et al characterized another dominantly inherited
macro-thrombocytopenic syndrome, characterized by interstitial
nephritis, cataract, deafness (i.e., a syndrome complex resembling
Alport syndrome), and leukocyte inclusions. These inclusion bodies
were much smaller than those observed in the MHA and typically
round rather than spindle shaped. These authors called the disorder
Fechtner syndrome (FS).

MYH9-Related Platelet Disorders
In 1990, Greinacher et al described a milder variant macrothrombocytopenia, Sebastian platelet syndrome (SPS), with small inclusion bodies in leukocytes.Individuals were recognized to have developed cataracts at a young age (45 to 50 years), and all affected family members that were >50 years of age had developed high-tone hearing impairment.
In 1999, the inheritance of these giant platelet disorders was linked to a 5.5-Mb region on the short arm of chromosome 22q.2,15
The syndromes MHA, EPS, FS, and SPS are now recognized to be related disorders, caused by mutations in the MYH9 gene, with phenotypic differences related to the presence or absence of the additional features of cataracts, nephritis, and sensorineural hearing loss.

MYH9-Related Platelet Disorders
An MYH9-related macrothrombocytopenia should be suspected in
individuals that have large platelets, a high MPV, a broad platelet
histogram, and a peak preceding the leukocyte histogram.

MYH9-Related Platelet Disorders

MYH9-Related Platelet Disorders
Platelet aggregometry and platelet function studies using the PFA-
100 do not show major defects in MYH9 disorders.
Because of the altered composition of the platelet cytoskeleton, the shape change in the aggregation curve is typically absent.

MYH9-Related Platelet Disorders

Hypothesis on the Mechanism of
Bleeding
with MYH9-Related Disorders The most
important reason for bleeding is the reduced clot stability due to impaired clot retraction by platelets with a disturbed cytoskeleton. This is aggravated in case of iron-deficiency anemia.

Bernard-Soulier syndrome (BSS)
Bernard-Soulier syndrome (BSS) is an autosomal recessive giant platelet disorder that is usually caused by reduced or absent expression of the glycoprotein (GP) Ib-IX-V receptor complex.
Platelets in BSS are as large as in the MYH9 disorders and appear similar to MYH9 platelets by light microscopy and electron microscopy ultrastructural analysis.
Aggregation studies can help to distinguish a MYH9 disorder from the rare, dominant Bolzano type of BSS, in which the GP IbIX-V complex is expressed but functionally impaired.
In BSS, there is typically absent agglutination with ristocetin, despite aggregation responses to other agonists.
The diagnosis of BSS is typically confirmed by quantitative analysis of GP Ib-IX complex expression on the platelets or genetic testing.

Paris-Trousseau
syndrome/Jacobson syndrome
This is a disorder with large platelets that contain giant a-granules.
This syndrome is distinguished from MYH9 disorders by its features of
mental retardation, facial and cardiac abnormalities, and its
genetic cause, which is a heterozygous deletion of one part of
chromosome 11q23.
X-linked macrothrombocytopenia
X-linked macrothrombocytopenia can be caused by a mutation in
the GATA-1 gene Xp11–12, a transcription factor important for
megakaryopoiesis and erythropoiesis.
The disorder is associated with splenomegaly and red cell
abnormalities (reticulocytosis and anemia) and thrombocytopenia
with platelet function defects (decreased agglutination with
ristocetin, weak aggregation with collagen).

Gray platelet syndrome (GPS)
It is an autosomal recessive bleeding disorder associated with giant
platelets containing empty a-granules and moderate
thrombocytopenia—the appearance of pale and gray platelets in the blood smear is an important diagnostic clue to distinguish this
condition from MYH9 disorders. The genetic defect is unknown.

Thank You


















