
MASTOCITOSIS - Cátedra de Dermatología...
53
39 FACULTAD DE CIENCIAS MÉDICAS UNIVERSIDAD NACIONAL DE ROSARIO CURSO UNIVERSITARIO SUPERIOR EN DERMATOLOGÍA TÍTULO: MASTOCITOSIS Rosso, Pablo Joaquín Mat 15.911 Tercer año en curso. Hospital Provincial Rosario
Transcript of MASTOCITOSIS - Cátedra de Dermatología...

39
FACULTAD DE CIENCIAS MÉDICAS
UNIVERSIDAD NACIONAL DE ROSARIO
CURSO UNIVERSITARIO SUPERIOR EN DERMATOLOGÍA
TÍTULO: MASTOCITOSIS
Rosso, Pablo Joaquín
Mat 15.911
Tercer año en curso.
Hospital Provincial Rosario
TUTOR: Molteni, Ana Gabriela

39
ÍNDICE GENERAL
MASTOCITOSIS…………………………………………………………………… 3
OBJETIVOS………………………………………………………………………… 3
INTRODUCCIÓN…………………………………………...……………………… 4
CLASIFICACIÓN DE MASTOCITOSIS(WHO)…….…..………………………. 7
SUBCLASIFICACIÓN DE MASTOCITOSIS CUTÁNEA……………………... 7
EPIDEMIOLOGÍA…………………………………………………………………… 8
MANIFESTACIONES CLÍNICAS…..……………………………………………… 9
C-KIT................................................................................................................15
EVALUACIÓN DIAGNÓSTICA……………………………………………………19
DIAGNÓSTICO Y ESTUDIO DE MASTOCITOSIS……………………………. 20
CRITERIOS DE GRAVEDAD………………………..…………………………… 27
TRATAMIENTO……………………………………………………………………. 28
TRATAMIENTO DE LAS MASTOCITOSIS PEDIÁTRICAS………………..… 28
TRATAMIENTO DE MASTOCITOSIS DE ADULTOS………………………… 30
MANIFESTACIONES GASTROINTESTINALES……………………………… 30
MANIFESTACIONES ÓSEAS……………………………………………………. 31
PROTOCOLOS ESPECÍFICOS ………………………………………………… 32
MASTOCITOSIS Y ANAFILAXIA POR PICADURA DE HIMENÓPTEROS………………………………………………………………… 34
DISCUSIÓN-CONCLUSIÓN…………………………………………………...… 35
BIBLIOGRAFÍA……………………………………………………………………. 37

39
MASTOCITOSIS
En 1869, Nettership y Tay fueron los primeros que escribieron sobre las típicas lesiones mastocíticas como una forma rara de urticaria: la llamaron urticaria mastocítica. Inmediatamente después del descubrimiento del mastocito, Paul Elrich en 1879; quien descubrió que estas lesiones contenían acúmulos de estas células, sin embargo no fue hasta 1949 que Ellis describió variantes sistémicas de esta enfermedad con compromiso de múltiples órganos (Allergol et Inmunophathol 2008,36-3_154-63).
OBJETIVOS
Explicar porqué una patología con base genética en células mastocitarias, tiene expresión fenotípica variada tanto desde el punto de vista cutáneo como sistémico a diferentes edades.
Plantear un protocolo de estudio, dado que tiene múltiples órganos de choque, es idóneo para el dermatólogo que dirija una evaluación clínica exhaustiva en cada caso.
Actualizar una terapéutica eficaz y posibles combinaciones ante casos refractarios, teniendo siempre presente que en la fisiopatogenia de la misma existe la liberación de mediadores químicos pre y neo-formados, responsables de la sintomatología.

39
INTRODUCCIÓN
El término de mastocitosis denota un grupo heterogéneo de desórdenes caracterizados por un anormal desarrollo, crecimiento y acumulación de mastocitos en uno o más tejidos. (1)
La Mastocitosis se manifiesta clínicamente en sitios donde las células mastocitarias se encuentran normalmente presentes. El órgano más frecuentemente involucrado es la Piel, aunque el Aparato Gastrointestinal, Médula Ósea, Tejido óseo, Bazo, Hígado y Ganglios linfáticos pueden estar comprometidos. (2)
El mastocito es una célula que participa en gran cantidad de procesos biológicos, desde la inflamación alérgica hasta la inmunidad innata contra bacterias y helmintos. Cumple función fisiológica en la cicatrización de heridas, remodelación de los tejidos y en la angiogénesis.
Se encuentran distribuidos en todo el organismo y una gran proporción, en las superficies mucosas, donde están expuestos a estímulos extraños, como “pastores inmunológicos” en sitos de contacto con el exterior: piel, mucosa del aparato respiratorio y digestivo. En los tejidos inflamados aumentan en cantidad. Los Mastocitos se localizan principalmente alrededor de los vasos sanguíneos, nervios, linfáticos y en la unión dermoepidérmica con una densidad entre 7.000 y 20.000 células por milímetro cúbico. (3)
Los mastocitos presentan gran afinidad por receptores de IgE, FcξRI, los cuales al ser activados promueven la degranulación. La activación y degranulación mastocitaria también puede ocurrir a partir de otros estímulos: opiodes, componentes de la cascada del Complemento, neuropéptidos (péptido intestinal vasoactivo, péptido relacionado al gen de la calcitonina y sustancia P), superóxido aniónico, contraste radiológico, lipoproteínas de baja densidad, factores liberadores de histamina, quemoquinas, bacterias patógenas, parásitos, enterotoxina B, cambios en la osmolaridad, etc. Ha sido demostrado que la Interleuquina 1(IL-1), catecolaminas e interacciones celulares (mastocito-fibroblasto) pueden activar al mastocito y expresar sus citoquinas. Otro estudio revela que los principales factores desencadenantes en niños son los alimentos, seguidos por venenos y fármacos. Bussman et al. revelan que la manipulación mecánica induce la degranulación mastocitaria con desencadenamiento de síntomas sistémicos y elevación transitoria de la triptasa sérica. (4)
Los mastocitos pueden ser identificados por su gran cantidad de gránulos, presentan metacromasia de color violeta cuando se tiñen de colorante básico azul de toluidina. La tinción metacromática de los gránulos se debe a la

39
presencia de proteoglucanos, heparina y condroitinsulfato. La diferencia entre mastocitos depende del contenido de sus gránulos, siendo identificados por inmuno-histoquímica y microscopía electrónica. Están los que sólo contienen triptasa (Mastocito reactivo), predominando en mucosa intestinal y parénquima pulmonar; y los que contienen triptasa, quimasa, y carboxipeptidasa A (Mastocito tisular constitutivo), alojándose en piel y submucosa de muchos tejidos. (3)
Estas células se originan a partir de las células madres hematopoyéticas pluripotenciales (Stem Cells) de la médula ósea, y maduran y se diferencian en tejidos específicos. Las células madres mononucleares CD 34+, c-kit+, CD 14 presentes en la médula ósea llegan a los tejidos finales a través de la circulación periférica y se diferencian y maduran de acuerdo con las influencias del SCF (Factor de crecimiento celular también llamado ligando del c-KIT) (3) (4)
Para su desarrollo en los tejidos dependen de su receptor c-kit con actividad tirosinquinasa (CD117) y su ligando Factor de células madres (SCF) producido por células de la estroma o fibroblastos. Otras citocinas derivadas del linfocito T como la IL 4, la IL5, la IL6 y la IL 9, funcionan en forma sinérgica con el SCF para multiplicar la cantidad de mastocitos cutáneos.
Hay estudios que demuestran que mutaciones en el c-kit están asociadas al desarrollo de Mastocitosis. El c-kit tiene muchos efectos biológicos sobre los mastocitos, incluidos diferenciación, crecimiento, localización, inducción e hiperplasia y producción de sus mediadores. (4) (12)
Así mismo otros factores son responsables de la regulación de la apoptosis del mastocito, incluidos: Factor de necrosis tumoral alfa (TNF-α), Ig E, receptores Toll-like y proteínas bcl-2.
La disminución de la transmisión de la señal a través de c-kit produce una deficiencia de mastocitos, mientras una excesiva activación de este receptor produce una proliferación aberrante. (3)
Generalmente hay una población de Mastocitos constitutivos, primordialmente en el tejido conectivo perivascular y se desarrolla una subpoblación celular reactiva, diferente desde el punto de vista inmunohistoquímico, que se disemina en las superficies mucosas en respuesta a factores de crecimiento derivados de los linfocitos T.
Durante su diferenciación adquieren propiedades morfológicas y fenotípicas con 4 diferentes estados de maduración: los no granulados, las células metacromáticas, el promastocito/mastocito atípico tipo II y el mastocito maduro. (1)

39
Los mediadores mastocitarios almacenados en sus gránulos secretorios se originan a partir de los componentes fosfolipídicos de la membrana o pueden ser inducidos por medio de la expresión de genes de novo. De los mediadores prefabricados (preformados), se encuentra la histamina, amina biógena producida tanto por los mastocitos como por los basófilos y plaquetas, liberada por exocitosis en respuesta a estímulos inmunológicos como no inmunológicos y ejerce sus funciones a través de su receptor H1 para aumentar la permeabilidad vascular, la contracción del musculo liso bronquial e intestinal, y la producción de moco nasal; y, a través del H2 para aumentar la permeabilidad vascular, la secreción de ácido gástrico y la producción de moco en la vía aérea. Las triptasas se almacenan junto con heparina. La triptasa B se almacena en los gránulos secretorios y es eliminada por medio de exocitosis. Los niveles séricos de ésta aumentan dentro de los primeros 15 minutos del comienzo de los episodios de anafilaxia y son máximos entre 1 y 2 horas después. Los mastocitos secretan triptasa alfa en forma constitutiva, y en los pacientes con mastocitosis se observa un incremento de los niveles de triptasa alfa y beta. La quimasa es una endopeptidasa que se une con heparina para formar un complejo macro molecular. Esta tiene la función de transformar la Angiotensina I en Angiotensina II. (3)
Los mediadores lipídicos lo hacen de novo a partir de los fosfolípidos de la membrana en el momento de la activación celular; tomando como sustrato el ácido araquidónico para formar prostaglandinas y leucotrienos. La fosfolipasa A2 es la que libera el ácido araquidónico que sirve de sustrato para formar los eicosanoides y factor activador plaquetario (PAF). Las enzimas ciclooxigenasa 1 y ciclooxogenasa 2 metabolizan el ácido araquidónico para formar PGG2 y PGH2 y por las PG sintetasa terminales convierte la PGH2 en PGD2. La primera enzima de la vía de los leucotrienos es la de la lipooxigenasa, la que transforma al ácido araquidónico en leucotrieno A4, éste a su vez es convertido en leucotrieno C4 por la leucotrieno C4 sintetasa. Estos son transformados en metabolitos activos: LTD4 y LTE4. (3)

39
CLASIFICACIÓN DE MASTOCITOSIS DE WHO
MASTOCITOSIS CUTÁNEA
MACULO-PAPULAR
DIFUSA O BULLOSA
MASTOCITOMA
MASTOCITOSIS SITÉMICA INDOLENTE
LATENTE
LOCALIZADA EN MÉDULA ÓSEA
MASTOCITOSIS SISTÉMICA ASOCIADA CON ENFERMEDAD HEMÁTICA CLONAL QUE NO PERTENECE AL LINAJE DE LOS MASTOCITOS
MASTOCITOSIS SISTÉMICA AGRESIVA CON LINFADENOPATÍA Y EOSINOFILIA
LEUCEMIA MASTOCÍTICA
SARCOMA MASTOCÍTICO
MASTOCITOMA EXTRACUTÁNEO
SUBCLASIFICACIÓN DE MASTOCITOSIS CUTÁNEA (MC)
URTICARIA PIGMENTOSA – UP (MC MÁCULO-PAPULAR)
URTICARIA PIGMENTOSA TÍPICA
FORMA EN PLACA
FORMA NODULAR
TELANGIECTASIA MACULAR ERUPTIVA PERSTANS (TMEP)
MASTOCITOSIS CUTÁNEA DIFUSA
MASTOCITOMA SOLITARIO
FUENTE ADAPTADA: JAFFE ES ET AL (World Health Classification of Tumors).

39
La clasificación de World Health Organization (WHO 2001 = OMS 2001) está basada en distintos criterios, en las formas cutáneas tienen como pilar fundamental la clínica (urticaria pigmentosa, el signo de Darier, mastocitoma) y hallazgos histológicos, junto con la ausencia de criterios que podrían permitir el diagnóstico de enfermedad sistémica. Esta clasificación agrega dos subvariantes en el año 2008: la mastocitosis sistémica oculta, y la mastocitosis confinada a la médula ósea. Un tercer grupo que no se identifica con estos se los llamó otras mastocitosis sistémicas indolente. Cabe agregar que esta clasificación no reconoce a la telangiectasia macularis eruptiva perstans dentro de las formas cutáneas. (5)
EPIDEMIOLOGÍA
La prevalencia estimada es de un caso entre 1.000 a 8.000 pacientes. En los niños es de aproximadamente 5,4 casos cada 1.000 pacientes. La mastocitosis se presenta en ambos sexos por igual y se han descriptos en diferentes grupos étnicos, sin embargo se han reportado más en caucásicos.

39
MANIFESTACIONES CLÍNICAS
Existen tres tipos de manifestaciones clínicas; las que se producen como consecuencia de una liberación masiva de mediadores mastocitarios, las secundarias a la liberación crónica de los mismos y las debidas a la infiltración celular de mastocitos en los tejidos.
Sistémicos
Inestabilidad vascular
Incremento de la permeabilidad vascular
Fibrosis
Eosinofilia
Infiltración linfocitaria
Anticoagulación local
Hiperplasia mastocitaria
Caquexia
Histamina,LTC4, LTE4,PGD2,PAF, endotelina
Histamina, LTC4, LTD4, PAF
TGF beta
IL5
IL 16, linfotaxina
Heparina
IL 3, IL 6, SCF
TNF alfa, IL 6
Piel
Prurito
Urticaria
Histamina
Histamina, LTC4, PAF
Tracto grastrointestinal
Hipersecreción gástrica
Dolor abdominal
Diarrea
Histamina
Histamina, LTC4, LTD4, PAF
Histamina
Pulmón
Broncoconstricción
Secresión de moco
Edema pulmonar
Histamina, PGD2, LTC4, PAF, endotelina
Histamina, proteasas, PGD2, LTC4
Histamina, LTC4, PAF

39
Esqueleto
Remodelado óseo
Osteoporosis
SNC
Cefaleas, cambios en el humor y cognitivos
Triptasa, quimiotripsina, IL 6
Heparina, proteasas
Histamina, PGD2
La Mastocitosis está constituída por máculas pigmentarias y dermografismo en el 50% de los casos, tumores con aspecto de histiocitoma, pápulas, nódulos, ampollas sub o intraepidérmicas, más comunes en el lactante y más excepcional en el adulto, que pueden aparecer sobre piel sana, infiltraciones dérmicas difusas con tono marfil viejo, y de consistencia pastosa, lisa o granulosa; y eritrodermia. (6)
Cuanto más joven y menor es el número de lesiones clínicas, mayor es la probabilidad de remisión espontánea. El comienzo en edad adulta aumenta los riesgos de compromiso sistémico y persistencia prolongada de la enfermedad. (1)
La patología es usualmente esporádica y la forma familiar es una condición muy rara. Aproximadamente en el 65% la enfermedad se presenta durante la niñez; donde el 55% de estas manifestaciones lo hacen dentro de los 2 años de vida; y se considera que los que presentan manifestaciones clínicas después de la pubertad (35%), son de comienzo adulto. La mayoría de los casos pediátricos son asintomáticos o mínimamente sintomáticos con resolución espontánea antes o durante la adolescencia en el 50% de los casos. Alrededor del 90% de éstos pacientes tienen escasos síntomas cutáneos, mientras el 10% tienen compromiso sistémico. (1)
La Urticaria pigmentosa (UP) es la manifestación cutánea más frecuente, se la observa en el 90 % de las formas inactivas y en la mitad de los casos de mastocitosis sistémica con enfermedad hemática clonal asociada, que no pertenece al linaje de los mastocitos o mastocitosis sistémica agresiva. Es más común en los niños y su comienzo es antes de los 6 meses de vida en la mitad de los casos. La incidencia es de 1 por 1.000 a 1 por 8.000 nacimientos. Se presenta como una erupción generalizada de lesiones eritematosas, amarronadas, redondeadas, en forma de máculas, pápulas o placas. El

39
número de lesiones varía con cada paciente entre 10 y 1.000, y la extensión que involucra no es predictora de enfermedad sistémica. (3)(7)(8)
Todas las regiones del cuerpo pueden estar comprometidas incluyendo piel y mucosas; ubicándose generalmente en tronco y respetando palmas, plantas y cuero cabelludo. La lesión puede ser redonda u oval, variar entre un milímetro a algunos centímetros de tamaño. Prurito, dermografismo, y signo de Darier (caracterizado por la formación de ronchas urticarianas posterior a la fricción de la lesión), pueden acompañar a las lesiones. Las ampollas pueden ocurrir en respuesta exagerada al signo de Darier. Esto se debe a la liberación de quimasas, quienes actúan en la unión dermo-epidérmica. La urticaria pigmentosa se estabiliza a los 3 años de vida, donde deja de producir las mismas. En la dermatoscopía muestra lesiones pigmentarias. Se describen tres subvariantes de la misma basadas en los aspectos clínicos: Forma en Placa, Forma Nodular y con Telangiectasias (Telangiectasia Macular Eruptiva Pertans). (7) (17)
La telangiectasia macular eruptiva perstans (TMEP) es de rara presentación, observada en el 1% de los pacientes con mastocitosis, principalmente en adultos. Se manifiesta por máculas pigmentadas marrones reticuladas surcadas con telangiectasias, de 2 a 4 mm de diámetro irregularmente definidas, comprometiendo principalmente el tronco. Algunos autores no la consideran como variante clínica por su número pequeño de grandes lesiones (≥ 1cm) con mínimo número de mastocitos. Es común la asociación con enfermedad sistémica o con varias anormalidades hematológicas, como mielodisplasias, desórdenes mieloproliferativos, leucemia mieloide aguda y trastornos linfoproliferativos. Pueden acompañarse de episodios de flushing, palpitaciones, síncopes, cefaleas y molestias gastrointestinales. (3) (7) (8)
La mastocitosis cutánea difusa es extremadamente rara y usualmente se presenta como una eritrodermia con compromiso de casi la totalidad de la superficie corporal de los niños. Se observa antes de los 3 años de edad y presenta un edema difuso con engrosamiento y consistencia pastosa de la piel que se acentúa en los pliegues cutáneos. En las variantes más severas las ampollas producidas por infiltración mastocitaria, deben ser diferenciadas de enfermedades congénitas bullosas. Tiene una resolución espontánea antes de los 5 años de vida. Se acompañan de síntomas sistémicos como hipotensión, shock, flushing, diarrea, sangrado gastrointestinal, cefalea, síncope, disfonía y palpitaciones. (7) (8)
El mastocitoma se manifiesta antes del año de vida y en la mayoría de los casos lo hace antes de los 3 meses. Se encuentra un ligero predominio en los

39
varones y representa un 10 a un 15% de las mastocitosis cutánea de los niños. Es una lesión generalmente única, macular, indurada, castaño rojizo, o como pápula, placa o tumor; con un diámetro de 4 centímetros alojada principalmente en tronco al que le siguen en orden de frecuencia extremidades, cabeza y cuello. Esta variedad muestra la más alta concentración de mastocitos llegando a unas 150 veces la población normal de la piel. Los síntomas asociados incluyen ampollas, prurito, flushing, signo de Darier y se han reportado asociaciones con Dermatitis Atópica, Dermografismo y Asma. (7) (8)
En los pacientes con comienzo en la edad adulta, generalmente hay evidencia de enfermedad sistémica. Puede seguir un comportamiento benigno e indolente, como un desorden clonal y persistente (en la médula ósea), o puede estar asociado a trastornos hematológicos. Su forma indolente representa las dos terceras partes de todos los casos sistémicos y muestra un curso clínico prolongado con una sobrevida de 20 años o más. La forma sistémica asociada a enfermedad hematológica clonal de linaje no mastocítico combina 2 histologías diferentes. El mastocitario en sí mismo y no mastocitario (Síndrome Mielodisplásico, Síndrome Mieloproliferativo, Leucemia mieloide aguda, Linfoma no Hodgkin). La enfermedad hematológica asociada más frecuente, en el 90% de los casos, es la del linaje mieloide. La forma agresiva se caracteriza por una infiltración progresiva de órganos con función comprometida como citopenias severas, malabsorción, fracturas óseas, eosinofilia periférica, y hepatopatía; pero usualmente sin lesiones cutáneas. La leucemia tiene infiltración de varios órganos por células neoplásicas inmaduras. Las manifestaciones localizadas para el mastocitoma, son de muy buen pronóstico pero el sarcoma es localmente destructivo y de peor evolución. (9) (10)
Los trastornos gastrointestinales se deben a la hipersecreción gástrica secundaria a un aumento del nivel plasmático de la histamina con gastritis y enfermedad ulcerosa péptica presentando pirosis, diarrea, dolor abdominal y hasta la instalación de un síndrome de malabsorción.
La infiltración tisular masiva puede dar lugar a signos y síntomas secundarios a la existencia de hepatomegalia y esplenomegalia, como dolor, distención abdominal y alteraciones en la circulación portal con ascitis. (10)
Las manifestaciones de enfermedad hepática en la mastocitosis sistémica indolente son infrecuentes, aunque pueden hallarse las pruebas de función hepática algo por encima de los valores normales.
El compromiso esplénico se puede observar en el 50% de los casos, donde la esplenomegalia está presente en las variantes de pronóstico menos favorable.

39
En la médula ósea los acúmulos o agregados multifocales de mastocitos triptasa positivas, se hallan entremezclados con eosinófilos, linfocitos T y B. La presencia de anemia, leucopenia, trombocitopenia y eosinofilia, sugieren cualquiera de las manifestaciones sistémicas.
La osteoporosis es cercana al 17%, estos pacientes tienen un riesgo de sufrir fracturas patológicas, como aplastamientos vertebrales, fundamentalmente en los casos de larga evolución no tratados en forma adecuada. Por el contrario la esclerosis ósea difusa y la esclerosis parcheada, aunque son más frecuentes en las mastocitosis agresivas, pueden darse también en formas indolentes, llegando a afectar un 5 % de los casos. (10)
Con respecto a las subvariantes de la forma indolente propuesto por WHO, el 14% presentan mastocitosis sistémica oculta y el 23% se encuentra confinada a la médula ósea sin repercusión cutánea, en contraste con la anterior que la tenía en el 72%. (5)
Los cuadros anafilácticos o el colapso vascular con riesgo vital se dan en un porcentaje variable de adultos, hasta un 22% de los pacientes adultos y un 6% de las formas pediátricas. En muchas ocasiones no se descubre un desencadenante claro, mientras que en otras situaciones existe el antecedente de un fármaco, como la aspirina y otros antiinflamatorios no esteroideos, inductores usados en la anestesia general, relajantes musculares, morfina y todos sus derivados, medios de contraste yodados de alto peso molecular y las picaduras de los insectos.
La mastocitosis puede afectar a los niños y adultos y dependiendo de la edad de comienzo pueden diferir las manifestaciones clínicas y pronosticas. En la niñez, generalmente es de buen pronóstico y la presentación típica es en los menores de dos años de edad. En ellos la regresión es espontánea, y raramente está asociada con desórdenes hematológicos, con afección de otros órganos que no sea la piel. En la mayoría de los pacientes resuelven completamente antes de la pubertad. La patología de comienzo adulto suele ser severa, es comúnmente considerada incurable; puede asociarse con compromiso sistémico de múltiples órganos. Puede estar asociado también con enfemedades clonales hematológicas de linaje celular no mastocítico, como mielodisplasia y desórdenes mieloproliferativos. (13)
Subtipos de mastocitosis, en relación a edad promedio y características clínicas de comienzo:

39
Subtipo Edad Características
Mastocitoma 0–6 meses 1–5 lesiones con hiperpigmentación marrón rojizas/ nodulo/nodulares
Urticaria pigmentosa
3–9 meses; si el comienzo es después de la edad de 10 años, el promedio es de 26.5 años
Multiples máculas y places marrones rojizas, hiperpigmentadas
Mastocitosis Cutánea Difusa
En general antes de los 3 años, frecuente al nacimiento
Engrosamiento cutáneo con liquenificación, papulas, y formación de bullas al menor trauma
TMEP Casi exclusivamente en adultos
Maculas hiperpigmentadas con telangiectasias
Mastocitosis Sistémica
En adultos, raro en niños Infiltrados de células mastocíticas en piel y órganos internos
Leukemia de Células Mastocíticas
Edad adulta Células mastocíticas en sangre periférica.
Dermatology 2007.

39
C-KIT
El complejo formado por SCF-KIT es requerido para una normal hematopoyesis, melanogénesis, gametogénesis y regulación del flujo gástrico además del crecimiento de células cebadas. Por lo que una deficiencia de esta unión se encuentra ligada a una carencia de células mastocitarias, anemia macrocítica, pérdida de pigmentación del cabello, esterilidad y un reducido número de células marcapaso gastrointestinales. La pérdida de función de c-kit está relacionado con el piebaldismo, patología autosómica dominante, un desorden caracterizado por parches blancos de piel y pelo, en contraste la ganancia de mutaciones de c-kit están asociadas a tumores de células estromales gastrointestinales y otros cánceres. (12)
La mastocitosis de comienzo adulto tiene un pronóstico más reservado, en alguno de los casos está asociado a enfermedad hemática clonal que no pertenece al linaje de los mastocitos como mielodisplasia y desórdenes mieloproliferativos. También puede presentarse como una forma de leucemia con una alta mortalidad.
Está claramente demostrado que la enfermedad está emparentada con una proliferación clonal de células, secundariamente a la disfunción del proto-oncogen c- kit; localizado en el cromosoma 4q12. El c-Kit está expresado en células progenitoras hematopoyéticas, melanocitos, células germinales, además de células intersticiales. El gen de c- kit codifica un receptor de transmembrana tipo 3, tirosin- kinasa, para su ligando factor de células madres (SCF). Este receptor posee un dominio extracelular (compuesto por 5 regiones Ig.) que se une a su ligando, un dominio de transmembrana, un dominio intracelular yuxtamembranoso, y un dominio citoplasmático proteína tirosin- kinasa que se une al ATP y regiones de fosfotransferasa. Las 2 isoformas de kit difieren en la ausencia o presencia de 4 aminoácidos (GNNK) en el dominio extracelular.

39
Una variedad de mutaciones han sido encontradas en c- kit. Sin embargo, no está clara la heterogeneidad de las mutaciones que comprometen la mastocitosis de adultos, que pueden contar con una gran variedad de presentaciones clínicas. Debido a que la presencia de ciertas mutaciones son relevantes para el pronóstico y sensibilidad a la terapéutica, es importante demostrar estas mutaciones.
De todas maneras la posible relación entre las variantes en las mutaciones y las presentaciones clínicas aún está en investigación. La heterogeneidad en Mastocitosis Sistémica en cuanto a su manifestación clínico-patológica y pronóstico sugiere que podría haber otras alteraciones genéticas además de la mutación KIT (ej: Activación constitutiva del ras-proteína relacionada con m-ras). Ésto podría contribuir a explicar los variables patrones inmunofenotípicos y las interacciones de los Mastocitos con su ambiente, similar a lo demostrado en otras enfermedades hematológicas malignas. (11) (2) (1)
En la mayoría de los pacientes con mastocitosis de comienzo adulto se han reportado que tienen la mutación D816V (sustitución de valina por aspartamo en el codón 816 del exón 17). La V560G es también encontrada, pero con menos frecuencia. La activación constitutiva de Kit involucra otros desórdenes proliferativos, como los tumores estromales gastrointestinales (GISTs). (11)

39
La frecuencia y rol de las mutaciones c-KIT en Mastocitosis de la infancia y si la Mastocitosis es una enfermedad clonal o reactiva; siguen siendo tema de gran debate científico.
La mayoría de los estudios del rol de las mutaciones de c-KIT en la mastocitosis de la infancia han sido focalizados en el codón 816 e incluyen relativamente pocos pacientes.
En el último trabajo del Journal ol Investigative Dermatology de Abril de 2010 se estudiaron 50 niños entre 0 y 16 años con formas esporádicas y familiares de Mastocitosis, siendo el trabajo con mayor estadística de casos publicados. El 86% de los pacientes presentaron mutaciones en el c-KIT. Esto demuestra claramente la idea de que al igual que en el adulto, la Mastocitosis de la Infancia es una enfermedad clonal y está asociada a la activación de mutaciones de c-KIT. Estos descubrimientos enfatizan la importancia de la activación de KIT tanto en Mastocitosis pediátricas como del adulto. Ahora bien, cómo es posible que siendo la variante pediátrica una enfermedad clonal, resuelva espontáneamente? Ésta es la principal pregunta cuya respuesta deberá ser encontrada.
No hubo correlación entre los fenotipos (compromiso cutáneo) y los genotipos de los pacientes. Además tampoco hubo clara correlación entre genotipo y formas familiares o esporádicas.
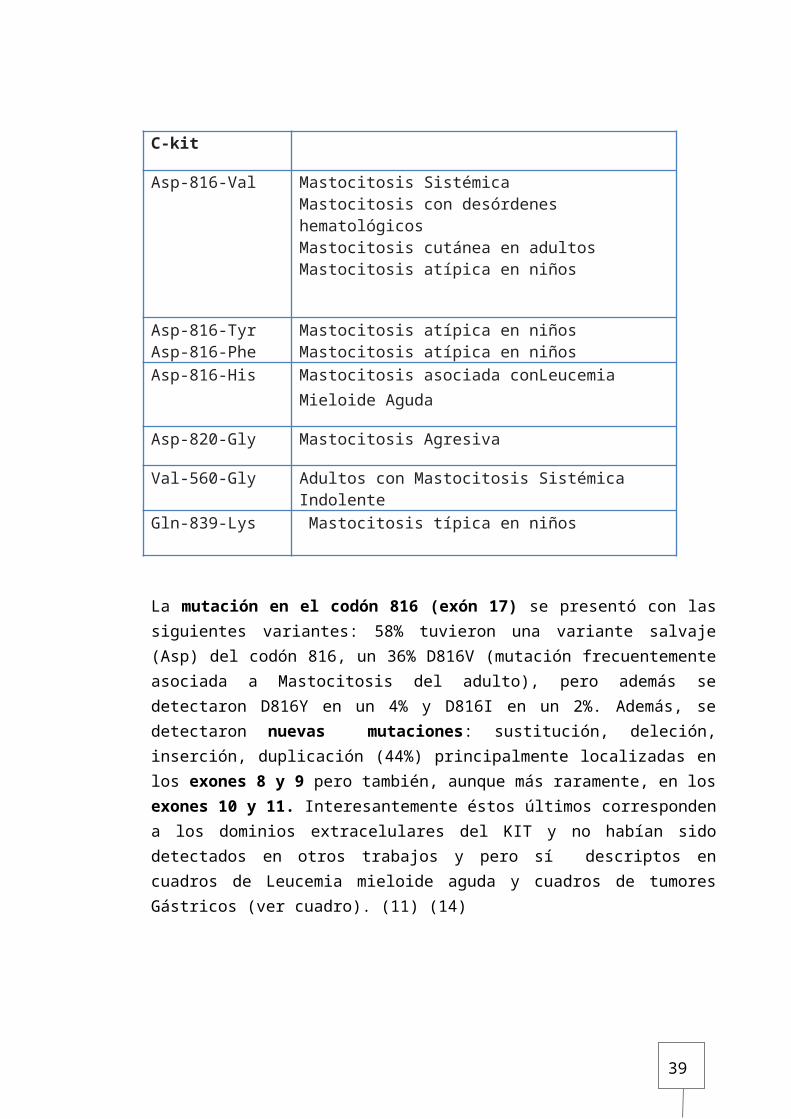
Mutaciones en c-kit en pacientes con mastocitosisDermatology 2007 (2)
mutación de C-kit Demonstrado en:
Asp-816-Val Mastocitosis Sistémica Mastocitosis con desórdenes hematológicosMastocitosis cutánea en adultosMastocitosis atípica en niños
Asp-816-Tyr Asp-816-Phe
Mastocitosis atípica en niñosMastocitosis atípica en niños
Asp-816-His Mastocitosis asociada conLeucemia Mieloide Aguda
Asp-820-Gly Mastocitosis Agresiva
Val-560-Gly Adultos con Mastocitosis Sistémica Indolente
Gln-839-Lys Mastocitosis típica en niños

39
La mutación en el codón 816 (exón 17) se presentó con las siguientes variantes: 58% tuvieron una variante salvaje (Asp) del codón 816, un 36% D816V (mutación frecuentemente asociada a Mastocitosis del adulto), pero además se detectaron D816Y en un 4% y D816I en un 2%. Además, se detectaron nuevas mutaciones: sustitución, deleción, inserción, duplicación (44%) principalmente localizadas en los exones 8 y 9 pero también, aunque más raramente, en los exones 10 y 11. Interesantemente éstos últimos corresponden a los dominios extracelulares del KIT y no habían sido detectados en otros trabajos y pero sí descriptos en cuadros de Leucemia mieloide aguda y cuadros de tumores Gástricos (ver cuadro). (11) (14)
Summary of c-kit mutations found in the patients with chilhood mastocytosis. (J. Invest. Dermatol. 2010.)
Es importante destacar que todas las mutaciones detectadas tanto en los dominios extracelulares como intracelulares (yuxtamembrana) son sensibles al tratamiento con Imatinib con excepción de la mutación D816V que representa una resistencia espontánea al tratamiento.(11)
Los mastocitos que tienen el D816V u otra activación de mutación del gen receptor de factor de crecimiento stem cell se traducen en una morfología atípica, transformación funcional, e inmunofenotipo aberrante.

39
En la forma sistémica típicamente muestra aberrancia sobre CD25 y CD2 que son utilizados como criterios menores en Mastocitosis Sistémica, a los que se agregan CD63, CD58; CD69, CD33, CD11c, CD35, CD59 y CD88. En contraste de la expresión de KIT (CD117), el CD 71 receptor de transferrina, y el CD29 B1 integrina, son reguladores negativos de la expresión.
En el último Journal Allergy Clin Inmunol. de Marzo 2010, se estudiaron 123 casos de pacientes con Mastocitosis Sistémica e interesantemente arrojó que la forma sistémica abarca una hematopoyesis clonal con compromiso multilinaje, asociada a un bloqueo temprano de la maduración de los mastocitos en las variantes sistémicas más agresivas. Ésto se demuestra en el compromiso de médula ósea y ganglios linfáticos y menor compromiso de piel en pacientes con Mastocitosis sistémica agresiva y Leucemia mastocitaria en contraste con Mastocitosis sistémica indolente. Más aún, estudios genéticos/moleculares son necesarios para dilucidar si el bloqueo en la maduración podría ser una coexistencia adicional de cambios genéticos en células progenitoras hematopoyéticas D816V (+).
EVALUACIÓN DIAGNÓSTICA
Inicialmente se debería realizar una biopsia de piel, con el análisis de la mutación c-kit D816V y el monitoreo de la triptasa sérica con seguimiento periódico del mismo. A estos estudios se puede agregar, análisis de sangre periférica que incluyen hemograma con recuento celular, enzimología hepática, parámetros de coagulación, bioquímica, y fosfatasa alcalina. La enfermedad en pacientes pediátricos está usualmente restringida a la piel, por lo que el examen de médula ósea no se requiere a menos que tenga una presentación tardía de las lesiones (mayores de 5 años), organomegalia, anormalidades en sangre periférica y una elevación persistente de triptasa sérica (mayor de 20 ug/l).
En los casos de adultos, aún sin síntomas sistémicos, la biopsia de médula ósea se debería hacer, ya que el pronóstico y compromiso vital no es el mismo al de los niños. De presentar síntomas sistémicos, se requiere ampliar la metodología diagnóstica con evaluación gastrointestinal, con ecografía abdominal, tomografía computada, seriada gastroduodenal, tránsito de intestino delgado y endoscopía. Se completa además con scan óseo radiográfico y orina de 24 horas para determinar mediadores. (1)
Los criterios diagnósticos planteados por WHO para la Mastocitosis Sistémica son los siguientes:

39
Mayor:
1. Presencia múltiples infiltrados densos de mastocitos (≥15 mastocitos) en la biopsia de médula ósea o en otro tejido extracutáneo, confirmado por inmunohistoquímica u otra coloración específica.
Menores:
1. Detección de un 25% de mastocitos elongados o con morfología atípica en la biopsia de médula ósea o en otro tejido extracutáneo; o la presencia de más del 25% de mastocitos inmaduros o atípicos en el aspirado de médula ósea.
2. Presencia de mutación activante del c-kit (codón 816) en los mastocitos de la médula ósea u otro tejido.
3. Expresión de antígenos CD2 y/o CD2 por citometría de flujo coexpresados con KIT(+)
4. Triptasa sérica total > 20 ug/l (excepto en cuadros con asociación clonal hematológica en los cuales este parámetro se ha perdido)
1 criterio mayor + 1 menor o 3 criterios menores = Diagnóstico de Mastocitosis Sistémica.
DIAGNÓSTICO Y ESTUDIOS EN MASTOCITOSIS
Evaluación general
Hemograma completo y VES
Enzimas hepáticas y Fosfatasa alcalina
Estudios de coagulación
Triptasa sérica
Examen de piel: macroscopia y microscopía (biopsia)
Aspirado de Médula Ósea y biopsia:
Coloraciones recomendadas: Giemsa, Azul de Toluidina, Antitriptasa, (cloro acetato estera, anti CD25 y CD117- no específico para mastocitos)

39
Patrón histológico y patrón de infiltración
Número y morfología celular
Investigación de mutaciones del c-kit ( codón 816 u otros)
Niveles séricos de Kit soluble o CD25
Rx de Tórax y Centellograma óseo
Biopsia ganglionar
EEG/evaluación neuropsiquiátrica (1)
En pacientes adultos con afectación cutánea sin organomegalia ni otros signos de actividad:
Biopsia cutánea compatible
Estudio de MO
Morfología y % de mastocitos normales, no agregados de mastocitos, IF (inmunofenotipo) normal, no mutación de C kit
Morfología normal, % mastocitos aumentado, agregados mastocitos, IF normal, no mutación de C kit
Morfología de mastocitos anormales, IF aberrante más agregados de mastocitos. Mutación C kit
MASTOCITOSIS CUTÁNEA PURA
MASTOSITOSIS SISTÉMICA BIEN DIFERENCIADA
MASTOSITOSIS SISTÉMICA INDOLENTE NO ASOCIADA A OTRA HEMOPATÍA CLONAL

39
Pacientes sin lesión cutánea y con cuadros recurrente de anafilaxia y colapso vascular:
(An. Sist. Sanit. Navar 2008, Vol. 31 Nº1 Enero- Abril).
La expresión fenotípica la mutación del c-kit se halló en el 90% de los casos en la urticaria pigmentosa, D816V se encontró en el 45% de los pacientes y el 48 % tenía cualquier otra de las mutaciones como D816Y e D816I. El 42% las manifestaba en los exones 8, 9 y 11. En la mastocitosis cutánea difusa se encontraron mutaciones en el 100% de los casos pero solo un 25% fueron
Triptasa elevada, fuera de la crisis, comprobada al menos en 2 determinaciones sucesivas con 3 meses de separación
Estudio completo de alergia
Desencadenante detectado
Desencadenante no detectado
Evitar el o /los alérgenos o anticuerpos responsables
Repetir triptasa cada 2 meses
Estudio de médula ósea
Criterios de mastocitosis
Mastocitosis sitémica sin lesión cutánea asociada a anafilaxia recurrente o colapso vascular
No criterios de mastocitosis
Si persiste elevada
39
D816V y el 50% estaban codificadas en los exones 8, 9 y 11. El mastocitoma tiene mutación en el 100% de los casos, pero ninguna de ellas es laD816V, y todas ellas se hallaron en los exones 8, 9 y 11. (12)
En las formas sistémicas, donde se realiza la citometría de flujo a partir de biopsia de médula ósea, se observa un incremento considerable de células aberrantes en las formas agresivas, leucémica y asociada con enfermedad clonal de linaje no mastocitario, mucho mayor que las formas indolentes. Cualquiera de las manifestaciones sistémicas muestran similares patrones de expresión CD 117, HLA I y CD34. Se identifica expresión aberrante de CD25, CD2, CD123, en más del 70% de los casos. Se agregan además CD22, HLA DR, CD64, CD16, HLA DQ, los cuales están anormalmente incrementados en la mayoría de las formas sistémicas, en los infiltrados de médula ósea. Las células mastocitarias de la médula ósea mostraron anormalmente baja expresión de triptasa total citoplasmática con aumento de triptasa madura citoplasmática y triptasa sérica. (15)
Los diferentes perfiles inmunofenotípicos de biopsia de médula ósea, dejan claramente tres patrones definidos. El primero de ellos es la mastocitosis sistémica indolente con la expresión de CD25 y en la mayoría de los casos de CD 2, pero también CD 16 y CD45 y alta reactividad para CD69, CD203c, CD32, CD64, CD123, y HLA DR con un incremento de los niveles de triptasa sérica y disminución de la triptasa total citoplasmática. La mastocitosis sistémica bien diferenciada también expresa CD25 y CD2 pero con mucha menos frecuencia; anormalmente exhibe un leve incremento de CD16, CD22, triptasa total citoplasmática, asociado con un bajo nivel de triptasa sérica. Los de peor pronóstico son las formas agresivas y leucémica que muestran aberrancia en el 100% de los casos para el CD25 pero en menos de 10% para el CD2; además, la expresión de CD63 y CD69, fue significativamente incrementada en pacientes con mastocitosis sistémica agresiva, pero no lo fue para aquellos que presentaban formas leucémicas. Estas últimas son las variantes de peor pronóstico, que muestran los niveles más altos de triptasa sérica en asociación con una disminución de la triptasa total citoplasmática. Las formas intermedias o aquellas asociadas a linaje de células no mastocíticas adquieren perfiles inmunofenotípicos de acuerdo a si se aproximan a formas indolentes o agresivas. (15)
La Red Española de Mastocitosis (REMA) incluye dos formas clínicas de mastocitosis sistémicas no consideradas en la de la WHO: la Mastocitosis Sistémica bien diferenciada, caracterizada por su inicio pediátrico, la presencia de lesión cutánea generalmente difusa, un aumento marcado del porcentaje de mastocitos de la médula ósea, cuya morfología e inmunofenotipos son normales, y la ausencia de mutaciones somáticas

39
activadoras del C-kit. La otra variante clínica considerada es la Mastocitosis Sistémica sin lesión cutánea asociada a anafilaxia o colapso vascular que, además de la presencia de mastocitos en la médula ósea con morfología normal, inmunofenotipo aberrante y frecuente mutación activadora del C-kit, se caracteriza por la persistencia de valores elevados de triptasa sérica fuera de las crisis agudas.
La dermatoscopía es un método no invasivo, que no sólo facilita el diagnóstico, sino que también ayuda a diferenciar distintas formas de mastocitosis cutánea y de otras enfermedades de la piel. La variedad más frecuente es la urticaria pigmentosa, donde la histopatología muestra una hiperpigmentación basal y denso infiltrado intersticial de células cebadas en la dermis papilar, correlacionándose con líneas reticulares marrones a la dermatoscopía. La Telangiectasia Macular Eruptiva Perstans tiene una infiltración de células en la dermis superficial y telangiectasia de vasos dérmicos, por lo que se puede observar líneas reticuladas marrones sobre una base eritematosa y una delicada red de vasos sanguíneos. (17)
Hay una correlación positiva entre SCORMA Index y los niveles de triptasa sérica indicando el valor del SCORMA Index en la evaluación de la mastocitosis con compromiso cutáneo. En el año 2001 se desarrollo un esquema de puntuación para la evaluación de la extensión clínica de los pacientes con mastocitosis. Este sistema es conocido como SCORMA (SCORing MAstocitosis). Este consiste en tres partes y está basado en los principios de SCORAD, Scoring Atopic Dermatitis Index. En SCORMA, la extensión de la piel comprometida es usada en primer lugar (A). El área marcada representa el porcentaje de piel expuesta. Si la lesión es un mastocitoma solitario, el compromiso es del 1%, y bien, si es difuso, con la totalidad del tegumento afectado, corresponde al 100%. La intensidad de la manifestación cutánea está representada en la parte B, en donde una lesión típica, en forma y color representa la mayoría de las lesiones examinadas. Se prefiere aquella lesión que no es afectada por la luz solar por lo que generalmente se elige una de la espalda. Esta es juzgada en pigmentación/eritema, vesiculación, elevación y signo de Darier. Cada ítem tiene un puntaje de 0 a 3, donde 0 representa ausencia y 3 la respuesta más severa. 5 síntomas subjetivos son los que se ubican en la parte C (factores provocadores, flushing, diarrea, prurito y dolor óseo), el puntaje otorgado es a partir de una escala visual análoga, donde 0 representa la ausencia de síntomas y 10, los son persistentes. La fórmula A/5 +5B+2C/5 es usada para calcular el SCORMA Index, el valor se encuentra entre 5.2 y 100. (19)

39

39
Los niveles de triptasa sérica son normales en la mayoría de los casos no complicados de mastocitosis cutáneas, pero son más de 20 ug/l en las formas sistémicas. En adultos con manifestaciones sistémicas y compromiso cutáneo hay una correlación positiva entre la densidad de las lesiones en piel, duración de la enfermedad, síntomas constitucionales, organomegalia y altos niveles de triptasa. En cambio, en los niños esa relación no se observa. Además en pacientes con desórdenes hematológicos asociados como mielodisplasia, la regresión de las lesiones cutáneas puede acompañarse de una enfermedad en progresión. En contraste con esto, la regresión de mastocitosis cutánea en pacientes con curso indolente paralelo, decae la severidad de la enfermedad, sin embargo hallazgos anormales de la médula ósea pueden aún estar presentes.
Los niveles de triptasa sérica y en sangre de la médula ósea se observan significativamente elevados en las formas sistémicas de la enfermedad. La triptasa es almacenada en los gránulos de las células mastocíticas y sus niveles se encuentran elevados después de la activación de las células, como el proceso anafiláctico. El nivel total de la triptasa sérica (alfa y beta) se correlaciona con la acumulación de células mastocíticas cargadas en sus gránulos y esto es especialmente importante en el descubrimiento de mastocitosis oculta. El valor de la triptasa sérica mayor a 11,4 ug/l es considerado elevado y sugestivo de una enfermedad mastocitocítica. Cuando éste valor es mayor a 20 ug/l es fuertemente sugestivo de manifestación sistémica. Se han detectado niveles de triptasa sérica mucho mayor en pacientes con enfermedad sistémica que en aquellos con sólo repercusión cutánea. Las células mastocíticas presentan una acumulación focal que puede no ser reconocida en una muestra de biopsia. Se han detectado valores significativamente elevados de los niveles de triptasa en sangre de médula ósea de pacientes con compromiso extracutáneo, de médula ósea y síntomas sistémicos, cuyo valor inferior encontrado es de 141 ug/l. Entonces se asume que altos niveles de triptasa en sangre de médula ósea, pueden tener una sensibilidad mayor que la histología sola. (18)

39
CRITERIOS DE GRAVEDAD
Se han establecido criterios de gravedad, los cuales pasan por distintos estadíos hasta que tienen un desenlace ominoso con falla multiorgánica. Estos son:
Signos B:
Carga mastocitaria elevada--. Infiltración de médula ósea >30% y triptasa > 200 ug/ml
Dismielopoyesis:
Hipercelularidad de médula ósea o signos de mielodisplasia o alteraciones de los recuentos en sangre periférica sin progresión.
Hepato y/o esplenomegalia sin alteración funcional. Adenomegalias > a 2 cm en ECO o TAC.
Signos C:
Alteración de la función de los órganos.
Citopenias: leucocitos < a 1.000/ mm3; hb < 10gr/dl, plaquetas < 100.000/mm3.
Hepatomegalia palpable con ascitis, test de función hepática anormal o hipertensión portal.
Bazo palpable con hiperesplenismo.
Malabsorción intestinal con hipoalbuminemia y pérdida de peso.
Lesión ósea con osteólisis y/o osteoporosis severa con fracturas patológicas.
Por último el óbito se produce por fallo orgánico, pancitopenia severa y es característico el fallo hepático. (10)

39
TRATAMIENTO
El tratamiento debe establecerse sobre la base de 4 pilares fundamentales:
Una información cuidadosa sobre la enfermedad a los pacientes o a los padres en las formas pediátricas
Evitar todos aquellos agentes que pueden desencadenar una liberación de mediadores por el mastocito
Tratamiento de los síntomas asociados a la liberación aguda o mantenida de los mediadores mastocitarios
Tratamiento citorreductor en aquellos casos con infiltración orgánica extensa. (10)
Tratamiento de las mastocitosis pediátricas:
Los síntomas asociados a la liberación de los mediadores mastocitarios pueden ser muy heterogéneos, y sus causas muy diversas. En los mastocitomas se suelen evitar únicamente los factores desencadenantes. En la urticaria pigmentosa el tratamiento dependerá de los síntomas en cada caso, ya que estos pueden ser leves, moderados y raramente severos. En las formas difusas los síntomas son siempre graves. Tanto en la urticaria pigmentosa como la mastocitosis cutánea difusa, es habitual que los síntomas vayan mejorando a partir de uno o dos años después de la aparición de la enfermedad. (10)
En todas las formas cutáneas se deba mantener al paciente con medidas generales que eviten la liberación de mediadores mastocitarios. Los factores que estimulan esta liberación pueden ser físicos, emocionales y medicamentosos. Entre los primeros encontramos al calor, frío, presión, rozamiento de las lesiones cutáneas, endoscopías y manipulación de asas intestinales durante la cirugía abdominal. Para el baño se recomienda utilizar agua templada y no frotar con la toalla para secar la piel, terminar el secado con un secador de aire a temperatura templada. Entre los factores emocionales, el estrés, la ansiedad y la irritabilidad pueden actuar como desencadenantes. Es larga la lista de drogas y medicamentos, pero los más frecuentes son: aspirina y antiinflamatorios no esteroideos :ácido mefenámico, diclofenac, fenilbutazona, ibuprofeno, indometacina, ketoprofeno, ketorolac, metimazol, nabumetona, naproxeno, piroxicam, propifenazona. Otros: codeína, morfina y sus derivados, antitusígenos, alcohol., relajantes musculares e

39
inductores empleados en la anestesia general, anestésicos locales, contrastes empleados para estudios radiológicos diversos e interferón alfa. (16)
Los alimentos a evitar son: fresas, tomates, espinacas, embutidos. Frutos de mar, pescados ahumados, pescados de carne roja, salsa china a base de pescados. Quesos de pasta cocida (grouyere, gouda), clara de huevo en gran cantidad. Se agregan chocolates, vinos, alimentos muy condimentados, y abuso de cafeína o té. (20)
Otras causas menos frecuentes son los procesos infecciosos y síndromes febriles de cualquier etiología; dentición y vacunación en los niños. (16)
Con respecto al tratamiento tópico, se puede utilizar crema de cromoglicato disódico. Se aplica varias veces por día cuando exista prurito, vesículas y ampollas, excepto que la piel esté denudada en la cual se utiliza mupirocina u otro antibiótico tópico. En el caso de no disponer de cromoglicato disódico, los corticoides tópicos son una opción válida. Uno de los cuales se puede utilizar es el propionato de fluticasona 0.05% en crema diluido al 25% en oclusión húmeda, ya que de esta forma, disminuyen los efectos adversos de los mismos. Su utilidad clínica está demostrada en la disminución del SCORMA Index ; y en la disminución de entre un 10 y un 60% en la cantidad de mastocitos por mm3 , esto puede verse en la biopsia de piel después del tratamiento tópico a las seis semanas.(2) Otros de los corticoides estudiados, es el clobetasol en crema. Los corticoides demostraron que inducen apoptosis de las células mastocíticas por la disminución de la producción de FSC por parte de las células estromales. En comparación con el clobetasol, se encuentra en Miltefosine tópico (Miltex), que disminuye en menor medida el volumen de las ronchas y también tiene tendencia a disminuir la cantidad de células mastocitarias en la dermis apical, pero con mayores efectos adversos. Estos son eccema e irritación. (23)
El tratamiento sistémico se inicia con antihistamínicos H1 no sedantes más antihistamínicos H1 sedantes si fuera necesario en forma reglada o a demanda. Otros a administrar son los antihistamínicos H2. El cromoglicato disódico oral, si no ceden los síntomas o si el paciente presenta dolor abdominal con o sin diarrea, irritabilidad o trastornos del sueño, o descenso del nivel de colesterol, ferritina, vitamina B12, o folatos, no debidos a otra causa. La dosis del cromoglicato disódico es de 10 a 20 mg/kilo de peso/día.
Otros de los fármacos que se pueden utilizar son los antileucotrienos, el montelukast; en casos con síntomas persistentes.
Puva, se utiliza en casos excepcionales con cuadros repetidos y severos de formación de ampollas que no ceden con el tratamiento anterior.

39
La mastocitosis cutánea difusa constituye una verdadera emergencia médica. Es aconsejable el ingreso a una unidad de cuidados críticos y utilizar dosis plenas de: antihistamínicos H1 no sedantes más antihistamínicos H1 sedantes, antihistamínicos H2, cromoglicato disódico oral y antileucotrienos. Si persisten episodios severos de liberación y formación de ampollas la fotoquimioterapia está indicada. (21)
Tratamiento de las mastocitosis de adultos:
Se recomienda, al igual que los casos pediátricos, evitarse los desencadenantes antes mencionados. La crema de cromoglicato disódico se utilizará sólo en casos excepcionales con síntomas cutáneos localizados. El tratamiento farmacológico estará en relación con la frecuencia e intensidad de los síntomas de liberación y debe ser individualizado en cada caso. En pacientes asintomáticos se debe instaurar el tratamiento con cromoglicato disódico oral desde el diagnóstico. En los sintomáticos el tratamiento debe ir encaminado a controlar los síntomas de liberación de mediadores tales como el prurito, el enrojecimiento facial, y la sensación de calor, la cefalea y el malestar general, para los cuales los antihistamínicos antiH1 de primera generación (dexclorfeniramina,difenhidramina, hidroxicina) son más eficaces para el control de los síntomas que los de segunda generación (loratadina, fexofenadina, desloratadina, cetirizina, levocetirizina). Como regla general, si un paciente ha respondido de manera adecuada con un antihistamínico H1, éste no debería ser sustituido. Una buena combinación es la asociación de antihistamínicos H1 sedantes y no sedantes para un control adecuado de los síntomas; asimismo, la adición de bloqueadores de los receptores H2 de la histamina (ranitidina, cimetidina) puede proporcionar un alivio de los síntomas.
En casos con síntomas persistentes, se iniciará un tratamiento con AINES si el paciente los ha tolerado previamente. Si la respuesta no fuera positiva o si hay contraindicación para emplear los mismos, se utilizará doxepina de forma reglada.
Manifestaciones gastrointestinales
El tratamiento de elección para la hipersecreción gástrica o úlcera péptica son los antihistamínicos H2 asociados o no a los inhibidores de la bomba de protones. El cromoglicato disódico es el tratamiento más eficaz para los cólicos abdominales de repetición, con o sin diarrea. Las dosis para los adultos es de

39
200 mg por vía oral, cuatro veces en el día, media hora antes de las comidas principales y al acostarse. Si la respuesta no es adecuada se puede asociar un AINE siempre que el paciente lo haya tolerado previamente sin problemas, y se puede añadir un antagonista de los leucotrienos, como el montelukast. (10)
Para el tratamiento de la malabsorción debe emplearse el cromoglicato disódico con un seguimiento estricto. En casos persistentes se puede asociar dosis bajas de corticoesteroides. En estos casos es necesario mantener dosis mínima necesaria para el control. (10) (16)
Manifestaciones óseas
El objetivo básico es la prevención antes que la osteoporosis se desarrolle, por lo que debe realizarse un estricto control del metabolismo cálcico y un detallado estudio óseo desde el diagnóstico. La absorción de calcio debe regularse con el cromoglicato disódico y, si la osteoporosis está presente, se debe utilizar calcio, vitamina D, y los bifosfonatos por vía oral. Se ha descrito necrosis maxilar en pacientes tratados con pamidronato, zolendronato o alendronato, especialmente en casos tratados por vía intravenosa; por ello es aconsejable el uso de bifosfonatos por vía oral, realizar un estudio radiológico de los maxilares antes de iniciar el tratamiento y evitar las intervenciones dentales agresivas durante la terapia. En los pacientes con antecedentes de fracturas patológicas podrá estar indicado el tratamiento citorreductor con interferón a dosis bajas (1,5 a 3 millones de unidades tres días por semana). Este tratamiento debería llevarse a acabo en una unidad de cuidados intensivos y después de una valiosa valoración de las contraindicaciones del mismo (depresión, cardiopatía isquémica, hepatopatía grave), y de los efectos colaterales. (10)
A la fecha, no hay un tratamiento citorreductor estándar y que tampoco permita remisión y recuperación a largo plazo. Entre las drogas disponibles para las formas sistémicas agresivas se encuentran el mesilato de imatinib, interferón alfa y los análogos de las purinas. Sin embargo la duración de la respuesta del tratamiento es corta y puede producir numerosos efectos adversos. (22)
El origen de la mastocitosis sitémica es considerada una enfermedad de las células stem cells, con compromiso clonal multi linaje. World Health Organization (who) en el 2008 posicionó a las enfermedades de las células mastocíticas en la categoría de la neoplasias mieloproliferativas. Al mismo tiempo, Who distingue, la mutación de c- Kit de la mastocitosis sistémica, de la mutación del Receptor de Factor de Crecimiento derivado de Plaquetas (PDGFR) de neoplasias mieloides asociadas con mastocitosis de la médula ósea y eosinofilia. Es importante distinguir entre estos dos grupos de pacientes,

39
por sus diferencias clínicas, pronosticas y selección del tratamiento. La Mastocitosis sistémica con mutaciones de PDGFR es altamente sensible al tratamiento con imatinib, un inhibidor de ABL, y PDGFR kinasa, mientras que en la mutación KIT D816Ves usualmente resistente al imatinib; esto es porque la mutación KIT D816V induce un cambio conformacional en los sitios de fijación del ATP del receptor C-kIT. (22)
Otra droga a utilizar es el interferón alfa, que a los tres meses de tratamiento, la biopsia de médula ósea muestra una recuperación de las células hematopoyéticas, un descenso de las células triptasa positivas, y se acompaña de tendencia a la normalización de los niveles de triptasa sérica. (22)
El transplante de médula ósea puede ser considerado un tratamiento opcional para pacientes con categorías avanzadas de mastocitosis con pobre sobrevida, como una mastocitosis sistémica asociada con enfermedad hemática clonal que no pertenece al linaje de los mastocitos, mastocitosis sistémica agresiva con linfadenopatía y eosinofilia y, leucemia mastocítica. (22)
La Talidomida se la utiliza en varias enfermedades hematológicas. Esta tiene efectos antiangiogénicos, inmunomoduladores, y antiinflamatorios. También actúa directamente sobre las células tumorales, inhibiendo el crecimiento celular e induciendo la apoptosis. Se reduce la secreción de IL6, factor de crecimiento del endotelio vascular y la producción de factor de necrosis tumoral alfa y estimula a las células Natural Killer. La dosis se inicia con 100mg/d, hasta llegar a un máximo de 300 mg/d y es mantenida según evolución. La respuesta al tratamiento con Talidomida se demuestra tanto con la disminución de los síntomas sistémicos como con la reducción de los acúmulos celulares en las lesiones de piel, como en los que producen organomegalia. (24)
PROTOCOLOS ESPECÍFICOS QUE DEBEN SER USADOS EN LOS PACIENTES:
ANESTESIA GENERAL EN PACIENTES ADULTOS CON MASTOCITOSIS
Muchas de las drogas empleadas en la preanestesia, en la fase de inducción de la misma o en la post anestesia pueden ser causa de reacciones anafilácticas o anafilactoides; así como graves alteraciones de la coagulación sanguínea. Estos cuadros se producen por la liberación de mediadores químicos, tanto preformados como originados en el proceso de activación mastocitario.

39
El mecanismo por el cual se originan estos cuadros puede ser mediado por los receptores para inmunoglobulina E, los receptores Fc gamma o a través del sistema del complemento. Los mediadores liberados por cualquiera de los mecanismos, actúan sobre los órganos diana como el corazón, los vasos, piel, pulmón y otros, pueden dar lugar a trastornos hemodinámicos y metabólicos similares a los que se presentan en una reacción anafiláctica o a graves trastornos de la coagulación.
Las drogas a administrar son las siguientes:
Preparación General
Prednisona: 60mg, 13, 7 y 1hora antes de la anestesia
maleato de dexclorfeniramina: 5mg IV, 1 hora antes de la anestesia
Ranitidina: 2 ampollas de 50 mg, 1 hora antes de la anestesia
Montelukast 1 comprimido de 10 mg, 24 horas antes y otro 1 hora antes.
Pre medicación:
El valium u otros ansiolíticos pueden ser usados sin inconvenientes.
Inducción:
Etomidato. Hipnótico imidazólico.
Relajante muscular:
Vecuronio.
Mantenimiento:
Inhalatorios fluorados.
Hay que tener en cuenta, que ciertas drogas que se utilizan en procedimientos quirúrgicos pueden desencadenar liberación del contenido de los matocitos. Alguna de ellas con efectos analgésicos como el clorhidrato de morfina y sus derivados (fentanilo entre otros). Los coloides como el dextrán, utilizados para los casos de hipotensión, pueden provocar reacción anafiláctica. (16)
Además se recomienda no administrar drogas alfa adrenérgicas, no beta adrenérgicas, así también los antagonistas de receptores colinérgicos. Es dable la determinación de la triptasa total en suero, antes, durante y después de la cirugía. (16)

39
Las técnicas de anestesia regional deben considerarse como procedimientos de elección en éstos pacientes. Por ello, se debería sustituir a la anestesia general siempre que sea posible. (16)
MASTOCITOSIS Y ANAFILAXIA POR PICADURAS DE HIMENÓPTIROS
Evitar las picaduras de insectos y en particular los himenópteros (abejas y avispas) hace que disminuya la posibilidad de reacciones anafilácticas. Se ha detectado un aumento de la triptasa basal y se ha sugerido una clara correlación entre los niveles de triptasa y la gravedad de la reacción; por lo que ante una reacción anafiláctica debe realizarse toma de triptasa sérica. (10)
El porcentaje de Mastocitosis con historia de anafilaxia por picadura de himenópteros es mucho mayor entre los casos sin afectación cutánea (43%) frente a un 2% en los pacientes con lesión cutánea, por lo que los pacientes con historia de reacción sistémica tras picadura de insectos deben solicitarse una triptasa sérica basal. Una cifra de triptasa sérica basal normal no excluye el diagnóstico de Mastocitosis, sobre todo en los casos con anafilaxia grave no medida con inmunoglobulina E.
Con respecto a la administración de inmunoterapia a los pacientes con Mastocitosis y antecedentes de anafilaxia, se han descrito resultados dispares. Se han publicado casos en los que ha resultado efectiva, y se los recomienda de por vida; de la misma manera que se han descrito reacciones mortales tras una picadura a pesar de su administración y no recomienda la inmunoterapia, debido a la ausencia de eficiencia y falsa seguridad. La inmunoterapia está solo indicada en aquellos en los que se demuestre una sensibilización mediada por la inmunoglobulina E tras la reacción de pruebas cutáneas y determinación de inmunoglobulina E específica frente al correspondiente veneno. (10)
La presencia de sensibilización por IgE no siempre se sucede en la Mastocitosis, sugiriendo que existen otros mecanismos no mediados por IgE desencadenantes de la anafilaxia, por lo que no puede descartarse que el mecanismo sea múltiple y que, por lo tanto la inmunoterapia no confiera al paciente la protección esperada. (10)
En caso de anafilaxia se administrará inmediatamente ADRENALINA, en una dilución 1/1.000, 0.3 a 0.5ml por vía subcutánea en los adultos, y en los niños de acuerdo a su peso. En las reacciones locales, o en las reacciones sistémicas sin colapso vascular se emplearán antihistamínicos H1 (Maleato de Dexclorfeniramina 6 mg VO) y H2 (ranitidina 300mg por vía oral), junto con prednisona 0,5 a 1mg/kg de peso. El paciente debe ser trasladado con urgencia al hospital más próximo. En estos casos, es de buena práctica el seguimiento con triptasa sérica. (16)

39
DISCUSIÓN - CONCLUSIÓN
La Mastocitosis puede afectar tanto a niños como adultos, dependiendo de la edad de comienzo las manifestaciones clínicas y pronósticas pueden diferir. Las repercusiones en los niños, generalmente son de buen pronóstico y típicamente se presentan antes de los 2 años de edad. La Mastocitosis pediátrica tiende a la regresión espontánea y está raramente asociada a desórdenes hematológicos, con afección de otros órganos distintos a la piel. La mayoría de los pacientes mejoran sustancialmente o su enfermedad resuelve completamente antes del comienzo de la pubertad.
La enfermedad de comienzo adulto suele ser severa y es comúnmente considerada incurable, puede estar asociada con compromiso sistémico de múltiples órganos, e incluso manifestarse con trastornos hematológicos clonales que no pertenecen al linaje de los mastocitos como desórdenes mielodisplásicos o mieloproliferativos. También, puede presentarse como una Leucemia de células mastocíticas, con alta mortalidad. La forma de comienzo de edad adulta está asociada con mutaciones en c-kit, D816V, la mayoría ubicadas en el exón 17.
No está clara la heterogeneidad de las mutaciones y su relación con la expresión fenotípica. No está tampoco definido, si el ligando que activa al c-KIT, el cambio conformacional del c-KIT o la cascada de señales que este origina, serían el agente causal del acúmulo de células mastocitarias en los distintos órganos y tejidos. Es aún resorte de investigación el por qué de las manifestaciones predominantemente cutáneas en infantes y sistémicas en adultos con evolución desfavorable.
Las publicaciones acerca de la mutación del receptor c-kit del mastocito, muestran resultados dispares, desde receptores inactivos hasta activaciones independientes de sus ligandos. El avance del artículo publicado por Bodemer y col., estudia la secuencia del ADN, del cromosoma 4q12 y, encuentra las mutaciones más frecuentes, en el codón 816 (D816V), exón 17 en el 42% de los casos. La correlación con el estudio de la medula ósea de los pacientes se ve limitada, porque la mayoría de ellos tenía manifestaciones cutáneas, sin síntomas sistémicos, por lo que el comité de ética que supervisó el trabajo prohibió dicha práctica en infantes.
Se han encontrado otras ubicaciones que portan la mutación y, a algunas de ellas, se las relaciona como factor de mal pronóstico, como la D816Y que se presenta en la forma difusa de pacientes pediátricos. Otras mutaciones son el asp.820gly, asp.816his., asp.816val., las que se encuentran en formas graves de Mastocitosis Sistémicas. Entre ellas están las Mastocitosis Sistémicas

39
asociados a desórdenes hematológicos, zleucemia mieloide aguda y Mastocitosis agresiva.
La biopsia de médula ósea es elegida como criterio para los pacientes con manifestaciones clínicas sistémicas, porque la mayoría de las veces está comprometida, y no hay comparación disponible para la evaluación de los mastocitos patológicos en otros órganos que no sea la médula ósea. Ésta, tiene alta especificidad para la detección de células mastocíticas pero la sensibilidad es baja debido a la heterogeneidad en la distribución de la muestra. En los pacientes con diagnóstico de Mastocitosis Sistémicas, con manifestaciones cutáneas y extracutáneas, con niveles aumentados de triptasa sérica; se correlacionan en todos los casos, con aumento en los niveles de triptasa en médula ósea, y son considerablemente más bajos que en las formas cutáneas puras.
De esta forma podemos asumir que el compromiso de la médula ósea, con niveles de triptasa mayores a 100ug/l supone diagnóstico de Mastocitosis Sistémica.
En la Mastocitosis Sistémica se demuestran tres perfiles inmunofenotípicos con diferente maduración, que pueden ser utilizados como marcadores pronósticos, y los de peor evolución son los de la Leucemia mastocítica y la forma Sistémica Agresiva. El 100% de los casos de ésta última presenta como marcador aberrante al CD25, mientras que está en el 67% de los casos de las Leucemias. Con respecto al CD2, sólo el 10% de la forma Agresiva lo manifiesta, junto al 30% en la Leucemia. Se agrega además, únicamente en la forma Agresiva la expresión de CD63 y CD69.
Existe además correlación entre el SCORMA Index y la triptasa sérica, y su utilidad puede ser definida cuando en la evaluación inicial de un paciente se realiza la medición del SCORMA y de la triptasa sérica. Si bien es clara la relación con el aumento de ambos en forma consecutiva, hay casos en que uno no va acompañado del otro; pero sigue siendo clara la utilidad sobre todo en los pacientes pediátricos para no someterlos a instrumentaciones reiteradas cuando hay cambios evolutivos en el cuadro clínico.
Los tratamientos tópicos con clobetasol, en comparación con miltefosine, sigue siendo más efectivo el corticoide de alta potencia, ya que disminuye más el prurito, volumen de las ronchas y a nivel histopatológico, la concentración de células mastocíticas en la dermis papilar. Además, el miltefosine, produce más intolerancia, por inducir irritación y eccema.

39
BIBLIOGRAFÍA
1) I.SILVA, S. CARVALHO, P.L. PINTO, S. MACHADO AND ROSADO PINTO. MASTOCYTOSIS: A RARE CASE OF ANAPHYLAXIS IN PAEDIATRIC AGE AND LITERATURE REVIEW. ALLERGOL ET INMUNOPATHOL 2008; 36 (3):154-63.
2) VERZIJIL, R. HEIDE, A.P. ORANGE, R.H.N. SCHAIK. C KIT ASP-816-VAL MUTATION ANALYSIS IN PATIENTS WITH MASTOCYTOSIS. DERMATOLOGY 2007; 214:15-20.
3) DEAN METCALFE. SÍNDROME DE MASTOCITOSIS. FITZPATRICK EN MEDICINA GENERAL. EDICIÓN 6ª.TOMO II, PÁG.1809.
4) LUCIANA CIRILLO MALUF,CARLOS DOS SANTOS MACHADO FILHO, JEFFERSON AFREDO DE BARROS. MASTOCYTOSIS. AN BRAS DERMATOL. 20009; 84 (3):213-25.
5) PARDADANANI, A. TEFFERI. PROPOSAL FOR A REVISED CLASSIFICATION OF SISTEMIC MASTOCYTOSIS. BLOOD. ABRIL 1010. VOLÚMEN 115.Nº13.
6) MIGUEL MAZZINI. MASTOCITOSIS. DERMATOLOGÍA CLÍNICA. 2º EDICIÓN 1985. PAG. 764.
7) LAURA BRILEY AND CHARLES PHILLIPS. CUTANEOUS MASTOCYTOSIS: A REVIEW FOCUSING ON THE PEDIATRIC POPULATION. CLINICAL PEDIATRICS VOLME 47, NUMBER 8, OCTOBER 2008, 757-761.
8) VELANA BRUDAT, LIBORIJA LUGOVIÉ, MIRNA SITUM, MARIJA BULJAN, IVA BLAJIÉ AND JANA PUSIÉ. MOST COMMON CLINICAL PRESENTATIONS OF CUTANEOUS MASTOCYTOSIS. ACTA CLIN CROAT 2009, 48:59-64.
9) SANZ ANQUELA, JOSE MIGUEL; YEBENES GREGORIO; LAURA BUSTEROS MORAZA; JOSE RAMOS GUILLEN. MASTOCITOSIS SISTÉMICA DEL ADULTO. HOSPITAL UNIVERSITARIO PRINCIPE DE ASTURIAS. JUNIO 2006.
10) B. DE LA HOZ, D. GONZÁLEZ DE OLANO, I. ÁLVAREZ, L.SANCHEZ, R. NÚÑEZ, I. SÁNCHEZ, L. ESCRIBANO. GUIDELINES FOR THE

39
DIAGNOSIS, TREATMENT AND MANAGEMENT OF MASTOCYTOSIS. AN. SIST. SANIT. NAVAR. 2008; 31 (1):11-32.
11) BODEMER C., HERMINE O, PALMIERINI ET AL. PEDIATRIC MASTOCYTOSIS IS A CLONAL DISEASE ASSOCIATED WITH D816V AND OTHER ACTIVATING C-KIT MUTATIONS. J INVESTIGATIVE 2010.130: 804-15
12) NAOTOMO KAMBE, JACK LONGLEY, YOSHIKI MIYACHI AND KENJI KABASHIMA. KIT MASTERS MAST CELLS IN KIDS, TOO. JOURNAL OF INVESTIGATIVE DERMATOLOGY(2010) 130,648-50.
13) CYNTHIA PRICE, JEREMY GREEN, ROBERT KIRSNER. MASTOCYTOSIS IN CHILDREN IS ASSOCIATED WITH MUTATIONS IN C-KIT. JOURNAL OF INVESTIGATIVE DERMATOLOGY(2010) 130,639.
14) J. ROCHA, M. LUZ DUARTE, HM MARQUES, F.TORRES, P. TAVARES, A. SILVA, C. BRITO. ASSOCIATION OF ADULT MASTOCYTOSIS WITH M541L IN THE TRANSMEMBRANE DOMAIN OF KIT. J. EUR ACAD. DERMATOL VENEREOL 2010.
15) CRISTINA TEODOSIO, ANDRÉS GARCÍA MONTERO, MARÍA JARA ACEVEDO Y COL.MAST CELLS FROM DIFFERENT MOLECULAR AND PROGNOSTIC SUBTYPES OF SYSTEMIC MASTOCYTOSIS DISPLAY DISTINTC INMUNOPHENOTYPES.J ALLERGY CLIN INMUNOL, MARCH 2010, VOLUME 125, NUMBER 3.
16) LUIS ESCRIBANO, C. ULIBARRENA REDONDO, I. ÁLVAREZ TOWSE. FACTORES DESENCADENANTES Y PROTOCOLOS ESPECÍFICOS PARA SITUACIONES DE RIESGO.RED ESPAÑOLA DE MASTOCITOSIS. AGOSTO 2006.
17) BENGÜ NISA AKAY, HARALD KITTLER, HATICE SANLI, KAAN HARMANKAYA, RANA ANADOLU. DERMATOSCOPIC FINDINGS OF CUTANEOUS MASTOCYTOSIS. DERMATOLOGY 2009; 218:226-30.
18) JULIA PROELSS, JOERG WENZEL, YON KO, THOMAS BIEBER, RALF BAUER. TRYPTASA DETECTION IN BONE MARROW BLOOD: A NEW DIAGNOSTIC TOOL IN SYSTEMIC MASTOCYTOSIS .J AMERICAM ACADEMT OF DERMATOLOGY 2007(56)3-453-57.
19) R.HEIDE, K. VAN DOORN, P. G. MULDER, A. W. TOORENENBERGEN, A. BEISHUIZEN, H. DE GROOT, B. TANK AND A. P. ORANJE. SERUM TRYPTASE AND SCORMA (SCORING

39
MASTOCYTOSIS) INDEX AS DISEASE SEVERITY PARAMETERS IN CHILDHOOD AND ADULT CUTANEOUS MASTOCYTOSIS. CLINICAL AND EXPERIMENTAL DERMATOLOGY.2008, 34, 462-468.
20) MARTINE MORISSET, PATRICIA SERGEANT, SIMONA WIDMER, GISELE KANNY.TRATAMIENTO SINTOMÁTICO DE UNA MASTOCITOSIS SISTÉMICA.CICBAA 2006. WWW.CICBAA.ORG .
21) A.P.ORANJE. MASTOCYTOSIS IN CHILDREN: A PROTOCOL FOR MANEGEMENT. PEDIATRIC DERMATOLOGY VOL.25 Nº4 AUGUST 2008.
22) CHIKAMASA YOSHIDA, MAKOTO TAKEUCHI, JUNJIRO TSUCHIYAMA AND YOSHITO SADAHIRA. SUCSSESFUL TRAEMENT OF KIT D816V-POSITIVE, IMATINIB-RESISTANT SYSTEMIC MASTOCYTOSIS WITH INTERFERON –ALPHA. INTERNAL MEDINCINE (48) 1973-1978, 2009.
23) GANDHI DAMAJ, EMMANUELLE BERNIT, DAVID GHEZ, JEAN FRANCOISE CLAISSE, NICOLAS SCHEINITZ, JEAN ROBERT HARLÉ, DANIELLE CANIONI AND OLIVIER HERMINE. THALIDOMIDE IN ADVENCED MASTOCYTOSIS. BRITSH JOURNAL OF HAEMATOLOGY 2008, 141, PAG 249-253.
24) K HARTMANN, F. SIEBENHAAR, B. BELLONI, K. BROCKOW, B HARTMANN, F. RUËFF, N. CHOEPKE, P. STAUBACH, A. WEBER, AND M. MAURER.EFFECTS OF TOPICAL TREATMENT WITH THE RAFT-MODULATOR MILTEFOSINE AND CLOBETASOL IN CUTANEOUS MASTOYTOSIS: A RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED TRIAL. BRITISH JOURNAL OF DERMATOLOGY, 2010 (162), PP 185-190.
25) ROGIER HEIDE, FLORA B. DE WAARD- VAN DERSPEK, JAN C DEN HOLLANDER Y COL, EFFICACY OF 25% DILUTED FLUTICASONE PROPIONATE 0,05% CREAM AS WET-WRAP TREATMENT IN CUTANEUS MASTOCITOSIS. DERMATOLOGY 2007; 214:333-335.


















