
Mast Cell-Associated TNF Promotes Dendritic Cell · PDF fileMast Cell-Associated TNF Promotes...
12
of May 2, 2018. This information is current as Dendritic Cell Migration Mast Cell-Associated TNF Promotes Sedgwick, Mindy Tsai and Stephen J. Galli Hajime Suto, Susumu Nakae, Maki Kakurai, Jonathon D. http://www.jimmunol.org/content/176/7/4102 doi: 10.4049/jimmunol.176.7.4102 2006; 176:4102-4112; ; J Immunol References http://www.jimmunol.org/content/176/7/4102.full#ref-list-1 , 25 of which you can access for free at: cites 69 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2006 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on May 2, 2018 http://www.jimmunol.org/ Downloaded from by guest on May 2, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Mast Cell-Associated TNF Promotes Dendritic Cell · PDF fileMast Cell-Associated TNF Promotes...

of May 2, 2018.This information is current as
Dendritic Cell MigrationMast Cell-Associated TNF Promotes
Sedgwick, Mindy Tsai and Stephen J. GalliHajime Suto, Susumu Nakae, Maki Kakurai, Jonathon D.
http://www.jimmunol.org/content/176/7/4102doi: 10.4049/jimmunol.176.7.4102
2006; 176:4102-4112; ;J Immunol
Referenceshttp://www.jimmunol.org/content/176/7/4102.full#ref-list-1
, 25 of which you can access for free at: cites 69 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2006 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Mast Cell-Associated TNF Promotes Dendritic Cell Migration1
Hajime Suto,2*† Susumu Nakae,2* Maki Kakurai,* Jonathon D. Sedgwick,3‡ Mindy Tsai,* andStephen J. Galli4*
Mast cells represent a potential source of TNF, a mediator which can enhance dendritic cell (DC) migration. Although theimportance of mast cell-associated TNF in regulating DC migration in vivo is not clear, mast cells and mast cell-derived TNF cancontribute to the expression of certain models of contact hypersensitivity (CHS). We found that CHS to FITC was significantlyimpaired in mast cell-deficient KitW-sh/W-sh or TNF�/� mice. The reduced expression of CHS in KitW-sh/W-sh mice was fully repairedby local transfer of wild-type bone marrow-derived cultured mast cells (BMCMCs), but was only partially repaired by transferof TNF�/� BMCMCs. Thus, mast cells, and mast cell-derived TNF, were required for optimal expression of CHS to FITC. Wefound that the migration of FITC-bearing skin DCs into draining lymph nodes (LNs) 24 h after epicutaneous administration ofFITC in naive mice was significantly reduced in mast cell-deficient or TNF�/� mice, but levels of DC migration in these mutantmice increased to greater than wild-type levels by 48 h after FITC sensitization. Mast cell-deficient or TNF�/� mice also exhibitedsignificantly reduced migration of airway DCs to local LNs at 24 h after intranasal challenge with FITC-OVA. Migration ofFITC-bearing DCs to LNs draining the skin or airways 24 h after sensitization was repaired in KitW-sh/W-sh mice which had beenengrafted with wild-type but not TNF�/� BMCMCs. Our findings indicate that mast cell-associated TNF can contribute signif-icantly to the initial stages of FITC-induced migration of cutaneous or airway DCs. The Journal of Immunology, 2006, 176:4102–4112.
C ontact hypersensitivity (CHS)5 is a cutaneous allergic in-flammatory response mediated by contact allergen- (i.e.,hapten-) specific T cells (1). A critical initial step in sen-
sitization for expression of CHS is the migration of hapten-bearingLangerhans cells (LCs), immature dendritic cells (DCs) located inthe epidermis, and/or dermal DCs, into draining lymph nodes(LNs) (2, 3). After completing their maturation in the LNs, themature hapten-bearing DCs then present the Ags to naive T cells,leading to induction of contact allergen-specific memory T cells inthe sensitization phase of the response (1, 2). In the subsequentelicitation phase of CHS responses, re-exposure to the cognatehapten results in the recruitment of allergen-specific T cells andother, Ag-nonspecific leukocytes to the site of cutaneous haptenchallenge. These recruited leukocytes then orchestrate a local in-flammatory response (1).
Classically, CHS was regarded as a Th1/type 1 cytotoxic (Tc1)cell-mediated immune response (1). However, recent evidence in-
dicates that some haptens, including FITC, can induce CHS re-sponses that are dependent on the activity of Th2 cells and which,for optimal expression, also require TNF (4–6). The potentialroles of TNF in the sensitization phase of such responses includeenhancing LC or dermal DC migration (3, 7–9), as well as thepromotion of LC maturation (7). However, the important cellularsource(s) of TNF in CHS to FITC are not known.
Although mast cells represent a significant potential source ofTNF (10, 11), the extent to which mast cell-associated TNF mightcontribute to the DC migration associated with the sensitizationphase of CHS is not yet clear. For example, studies performed byseveral groups using genetically mast cell-deficient mice haveshown that, under certain (but not all) experimental conditions,mast cells can be required for optimal expression of CHS (12–14).In one such model, CHS to 2,4,6-trinitro-1-chlorobenzine (TNCB),mast cell-derived TNF was required for the optimal expression ofthe response at sites of hapten challenge (13). In another model,CHS to oxazolone (Ox) in ethanol, mast cell-deficient mice exhib-ited impaired migration of LCs from the epidermis during an 18-hperiod after initial exposure to the hapten at the time of sensitiza-tion (14). Finally, Jawdat et al. (15) reported that the IgE- andspecific Ag-dependent activation of skin mast cells can promotethe migration of LCs to local LNs. Taken together, such studiesraise the possibility that one mechanism by which mast cells, andmast cell-associated TNF, might contribute to the development ofCHS responses is by promoting LC/DC migration from the skin atsites of hapten exposure in the sensitization phase of the response.
In the present study, we found that both the optimal migration ofFITC-bearing DCs to local LNs during the first 24 h after theepicutaneous application of the hapten, and the optimal expressionof CHS to FITC, required mast cells and mast cell-associated TNF.Moreover, mast cells and mast cell-associated TNF also were re-quired for optimal induction of airway DC migration to thoracicLNs within 24 h of intranasal administration of FITC-OVA tomice. However, neither mast cells nor TNF were absolutely re-quired for DC migration in response to FITC, and equivalent levels
*Department of Pathology, Stanford University School of Medicine, Stanford, CA94305; †Atopy Research Center, Juntendo University, Tokyo, Japan; and ‡DNAXResearch, Palo Alto, CA 94304
Received for publication September 26, 2005. Accepted for publication January19, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by U.S. Public Health Service Grants (to S.J.G.) AI-23990,CA-72074, and HL-67674. DNAX Research is supported by Schering-Plough Re-search Institute.2 H.S. and S.N. contributed equally to this study.3 Current address: Lily Research Laboratories, Eli Lilly, Indianapolis, IN 46285.4 Address correspondence and reprint requests to Dr. Stephen J. Galli, Department ofPathology, Stanford University School of Medicine, L-235, 300 Pasteur Drive, Stan-ford, CA 94305-5324. E-mail address: [email protected] Abbreviations used in this paper: CHS, contact hypersensitivity; LC, Langerhanscell; DC, dendritic cell; LN, lymph node; Tc1, type 1 cytotoxic T cell; TNCB, 2,4,6-trinitro-1-chlorobenzine; Ox, oxazolone; PMN, polymorphonuclear leukocyte; BM-CMC, bone marrow-derived cultured mast cell; i.d., intradermally; i.n., intranasally;WT, wild type.
The Journal of Immunology
Copyright © 2006 by The American Association of Immunologists, Inc. 0022-1767/06/$02.00
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

of T cell sensitization, as reflected in T cell proliferation inducedin response to Ag challenge in vitro, were detected in either mastcell-deficient or TNF-deficient mice at 6 days after epicutaneoussensitization to FITC.
Materials and MethodsMice
Completely TNF-deficient mice (C57BL/6J-TNF�/� mice), as well as miceexpressing membrane TNF but lacking the ability to release a secreted TNFform (C57BL/6J-memTNF�/� mice), were generated from C57BL/6 em-bryonic stem cells (16, 17). FcR��/� mice on the C57BL/6 backgroundwere obtained from Taconic Farms. WBB6F1-Kit�/� and -KitW/W-v miceand C57BL/6J-TNFR1�/� and -TNFR2�/� mice were obtained from TheJackson Laboratory. Mast cell-deficient KitW-sh/W-sh mice on the C57BL/6background were generously provided by Dr. P. Besmer (Molecular Biol-ogy Program, Memorial Sloan-Kettering Cancer Center and Cornell Uni-versity Graduate School of Medical Sciences, New York, NY) (18); theKitW-sh/W-sh mice used herein were backcrossed in our laboratory for two tothree generations onto the C57BL/6J background. Female 6- to 10-wk-oldmice were used in all experiments. All mice were housed at the AnimalCare Facilities at Stanford University Medical Center (Stanford, CA) keptunder standard temperature, humidity, and timed lighting conditions, pro-vided mouse chow and water ad libitum, and sacrificed by CO2 inhalation;all experiments were performed in compliance with the “Guide for the Careand Use of Laboratory Animals” prepared by the Institute of LaboratoryAnimal Resources, National Research Council, and published by the Na-tional Academy Press (revised 1996) and the Stanford University Com-mittee on Animal Welfare.
FITC-induced CHS
To analyze both DC migration in response to a hapten and the subsequentdevelopment and expression of CHS to that hapten at the same anatomicalsite, we developed in preliminary experiments a protocol in which bothsensitization and challenge for CHS could be performed using the earpinna. Mice were sensitized with a total of 80 �l of 2% FITC isomer-I(FITC; Sigma-Aldrich) in a vehicle consisting of acetone-dibutylphthalate(1:1) administered to both ears (20 �l to each side of each ear). Five daysafter sensitization with FITC, mice were challenged with a total of 40 �lof vehicle alone to the right ear (20 �l to each side) and 0.5% FITC to theleft ear (20 �l to each side). Each mouse was housed in a separate cage toprevent contact with other mice after FITC challenge. Ear thickness wasmeasured before and at multiple intervals after FITC challenge with anengineer’s microcaliper (Ozaki). Some mice from each group were killedat 24 h after FITC or vehicle challenge for histological analysis to quantifynumbers of mast cells or polymorphonuclear leukocytes (PMNs) in tolu-idine blue-stained or H&E-stained, respectively, 4-�m sections of ear skin.Cells were quantified by light microscopy at �400, by observers not awareof the identity of the individual specimens. Mast cells and PMNs werecounted in the entire area of dermis present in a strip of skin extending6.6 � 0.3 mm in length from the base to the tip of the ear pinna (repre-senting 1.25 � 0.07 mm2 of dermis); mast cells also were counted sepa-rately in 10 randomly selected areas of the dermis (�0.625 mm2 in totalarea) in the central part of the ear pinna (representing the area in whichbone marrow-derived cultured mast cells (BMCMCs) had been injectedintradermally (i.d.) in the BMCMC-engrafted KitW-sh/W-sh mice). All dataare expressed as mast cells per mm2 of dermis.
Preparation of BMCMCs and local mast cell engraftment ofmast cell-deficient mice
BMCMCs were obtained by culturing bone marrow cells in WEHI-3-con-ditioned medium (containing IL-3) for 4–6 wk, at which time �98% of thecells were identified as mast cells by toluidine blue staining and by flowcytometry analysis. For mast cell reconstitution studies, BMCMCs wereinjected i.d. (1.3 � 106 cells in 40 �l/ear for studies of LC/DC migrationand CHS) or i.v. (1.0 � 107 cells for studies of lung DC migration) in 4-to 6-wk-old KitW/W-v mice or KitW-sh/W-sh mice. The mice were used for DCmigration experiments 6 wk (in the setting of CHS) or 8 wk (in airwaystudies) after transfer of BMCMCs.
LC/DC migration: LC/DC release from epidermis.
Naive mice were treated with 2% FITC (left ear) or vehicle alone (rightear) as described above. Twenty-four hours after FITC treatment, micewere killed and ear skin was harvested and incubated in PBS containing 20mM EDTA at 37°C for 2 h. Epidermal sheets then were collected on glass
slides and fixed in cold acetone for 20 min. Specimens were washed in PBSfor 20 min three times and incubated in PBS containing 3% BSA (blockingsolution) at room temperature for 1 h. After blocking, the specimens wereincubated with PE anti-mouse I-A/I-E (M5/114.15.2; eBioscience) at 4°Covernight, then washed with PBS containing 0.2% Tween 20 (Sigma-Al-drich) at room temperature for 1 h. The number of epidermal LCs werecounted by fluorescence microscopy in 10 randomly selected areas of epi-dermal sheets (�400).
Skin LC/DC migration to draining LNs
Naive mice were treated with 2% FITC (left ear) or vehicle alone (rightear) as described above. Twenty-four, 48, or 72 hours after FITC treatment,mice were killed for harvesting of submaxillary and brachial LNs on boththe left (FITC-treated) and right (vehicle alone) sides, and a single-cellsuspension was prepared as described (19). After incubation with anti-CD16/CD32 mAb (2.4G2; BD Pharmingen), cells were incubated with PEanti-mouse I-Ab (M5/114.15.2; eBioscience) and allophycocyanin anti-mouse CD11c (N418; eBioscience). The proportion (percentage) of FITC�
cells among 5000 7-aminoactinomycin D-negative, I-Ab�, CD11c� cellswas determined on a FACSCalibur (BD Biosciences) using CellQuest soft-ware (BD Biosciences).
Lung DC migration
Mice were treated with 20 �l � 5 (total 100 �l) of 10 mg/ml FITC-conjugated OVA (FITC-OVA) (20) intranasally (i.n.) (21). At 24 h afterFITC-OVA inhalation, submaxillary or thoracic LNs were collected andsingle-cell suspensions were prepared (19). Cells were incubated with bi-otin anti-33D1 and allophycocyanin-anti-mouse CD11c mAbs after FcRblocking and then incubated with PE-streptavidin (BD Pharmingen). Theproportion of FITC� cells among 5000 7-aminoactinomycin D-negative,CD11c�33D1� cells was determined by FACS as described above.
Depletion of skin LCs/DCs
Depletion of skin LCs/DCs were performed as described elsewhere (22).Briefly, 0.2 g of corticosteroid cream (0.05% clobetasol-17-propionate;Glaxo) per mouse (or vehicle as a control) was applied topically to bothsides each ear, daily for up to 5 days. After 1–5 days (see Fig. 3A) or 4 days(see Fig. 3B), epidermal sheets or draining LNs were harvested, respec-tively, for quantification of LC number after anti-I-Ab mAb staining in theepidermal sheets and for assessment of FITC�MHCII�CD11c� DCs inthe LNs.
FITC-specific LN cell proliferation
Mice were sensitized with 2.0% FITC on both left and right ears as de-scribed above. Six days after FITC treatment, submaxillary LNs were col-lected and pooled, single-cell suspensions were prepared, and the LN cellswere cultured in the presence or absence of 40 �g/ml FITC at 37°C for72 h. Proliferation was assessed by pulsing with 0.25 �Ci [3H]thymidine(Amersham Bioscience) for 6 h, harvesting the cells using a Harvester 96Mach IIIM (Tomtec), and measuring incorporated [3H]thymidine using theMicro � System (Amersham Bioscience).
Quantification of numbers of DCs resident in the lung by FACS
Single-cell suspensions of lungs were prepared as described elsewhere(23). Lung DCs were stained with FITC-anti-mouse CD11c (N418; eBio-science), PE-anti-mouse I-Ab (M5/114.15.2; eBioscience), and biotin-anti-mouse 33D1 (33D1; eBioscience) after FcR blocking. The cells were thenincubated with allophycocyanin-streptavidin (BD Pharmingen) and 7-ami-noactinomycin D-negative, I-Ab highCD11chigh33D1� cells were countedon a FACSCalibur (BD Biosciences) using CellQuest software (BDBiosciences).
Quantification of numbers of lung mast cells
Single-cell suspensions of lungs were prepared as described above andmast cells were quantified after 2 � 105 of these cells were cytospun ontoglass slides. The glass slides were air-dried overnight, fixed in methanol for5 min, and then incubated with blocking buffer (Protein Block Serum-Free,Ready-to-Use; DakoCytomation) at room temperature for 1 h. After re-moval of blocking buffer, the specimens were incubated with rhodamine-conjugated avidin (Vector Laboratories) in DakoCytomation Ab diluentwith background-reducing components (DakoCytomation) at room tem-perature for 4 h. The specimens were then washed five times in PBS atroom temperature for 30 min each time. Rhodamine-positive cells (i.e.,mast cells) were counted by fluorescence microscopy.
4103The Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Statistics
The Student t test (two-tailed), paired or unpaired, as appropriate, was usedfor statistical evaluation of the results; unless otherwise specified, all re-sults are presented as mean � or � SEM.
ResultsOptimal expression of CHS responses to FITC is both mast celland TNF dependent
Wild-type (WT) C57BL/6 mice developed robust CHS responsesto FITC, as assessed by tissue swelling at the site of hapten chal-lenge (Fig. 1A), and also exhibited infiltration of PMNs at these
sites (Fig. 1D). By contrast, the responses elicited by hapten chal-lenge in the congenic mast cell-deficient KitW-sh/W-sh mice devel-oped only slightly, albeit significantly, greater swelling than thecontrol reactions elicited by vehicle (Fig. 1A) and exhibited sig-nificantly reduced numbers of PMNs compared with the corre-sponding values in the WT mice (Fig. 1D). TNF�/� mice on theC57BL/6 background also exhibited weak CHS responses to FITC,with minimal levels of tissue swelling (Fig. 1A) and significantlylower than WT levels of PMN infiltration (Fig. 1D).
As shown in Fig. 1, B and C, the ear pinnae of KitW-sh/W-sh mice,which were the sites of sensitization and challenge for CHS, wereprofoundly mast cell-deficient (containing no detectable dermalmast cells), whereas levels of mast cells at this site in the TNF�/�
mice were statistically indistinguishable from those in the WT an-imals. In the central part of the ear pinnae, mast cell counts permm2 of dermis were nearly identical in WT and TNF�/� mice(Fig. 1B), whereas, over the entire length of the ear pinnae, num-bers of mast cells per mm2 were slightly lower in the TNF�/� micethan in the WT mice, a difference that did not achieve statisticalsignificance (Fig. 1C).
To determine whether the abnormalities in CHS in KitW-sh/W-sh
mice reflected the lack of mast cells in these animals, as opposedto other consequences of their c-kit mutations, we testedKitW-sh/W-sh mice that had been selectively engrafted with mastcells in the ear pinnae. We found that engraftment with WTBMCMCs fully restored CHS reactivity in KitW-sh/W-sh mice, asjudged by tissue swelling over the course of the reaction (Fig. 1A)or by PMN infiltration at 24 h after hapten challenge (Fig. 1D).However, KitW-sh/W-sh mice that had been engrafted with TNF�/�
BMCMCs exhibited only partial reconstitution of both the tissueswelling response associated with CHS to FITC (to �50% of theWT level) (Fig. 1A) and only minimal enhancement of the PMNinfiltration associated with the response compared with the levelsobserved in mast cell-deficient KitW-sh/W-sh mice (Fig. 1D).
Engraftment of KitW-sh/W-sh mice with TNF�/� BMCMCs re-sulted in numbers of mast cells in the central part of the ear dermis(the site injected with BMCMCs) that were statistically indistin-guishable from those in WT mice or in KitW-sh/W-sh mice that hadbeen engrafted with WT BMCMCs (Fig. 1B). As expected, whenmeasured over the entire length of the ear pinna, numbers of mastcells per mm2 of dermis were substantially lower in the WTBMCMC- or TNF�/� BMCMC-engrafted KitW-sh/W-sh mice thanin the WT or TNF�/� mice; mast cells per mm2 also were slightlylower in the KitW-sh/W-sh mice that had been engrafted with WT asopposed to TNF�/� BMCMCs, but this difference was not statis-tically significant (Fig. 1C).
Taken together, these findings show that, under the conditionstested in our experiments, mast cells, and mast cell-associatedTNF, are required for optimal expression of CHS to FITC. Theseresults are in accord with those of Biedermann et al. (13), whoshowed that mast cell-derived TNF contributes to optimal expres-sion of CHS reactions to TNCB.
Mast cells, and mast cell-associated TNF, are required foroptimal DC migration to LNs during the first 24 h of thesensitization phase of CHS to FITC
Skin LCs can firmly adhere to keratinocytes through interactionsof adhesion molecules such as E-cadherin (24). After epicutaneousexposure to haptens, such adhesion molecule expression is down-regulated and LCs can then migrate from the epidermis. We foundthat the numbers of LCs in epidermal sheets were significantlyreduced (by �30%) 24 h after epicutaneous application of FITC tothe ear pinnae of WT C57BL/6 mice, compared with the corre-sponding values after vehicle treatment (Fig. 2A). In accord with
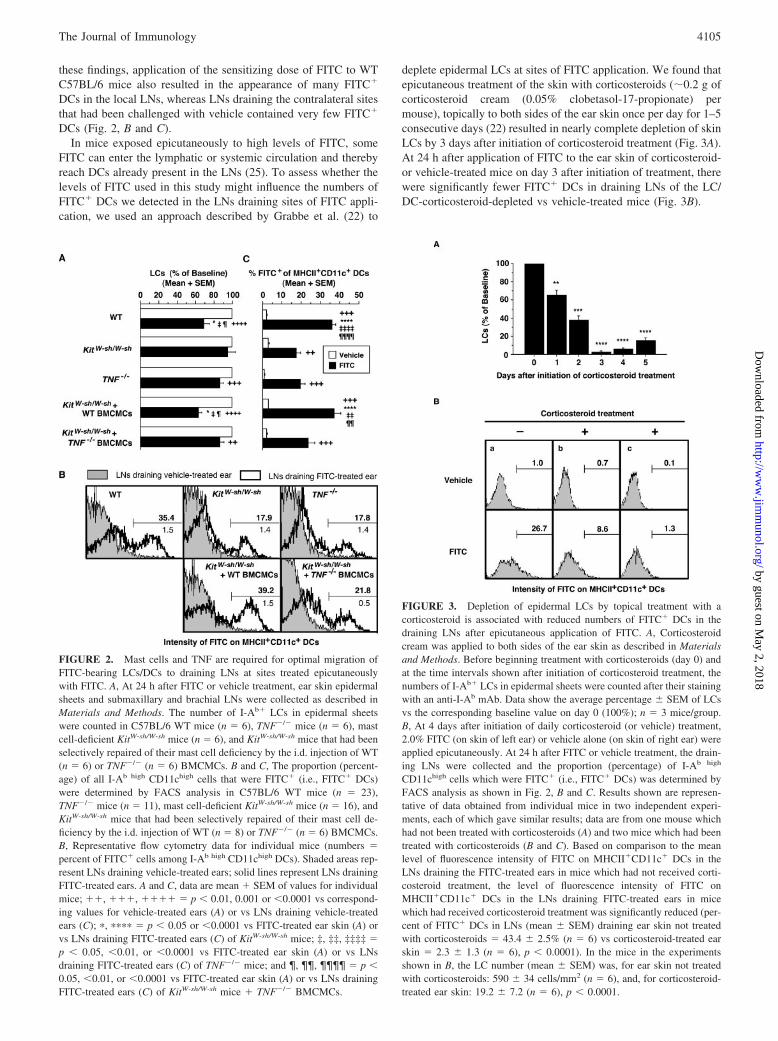
FIGURE 1. Mast cells and TNF are required for the optimal expressionof FITC-induced CHS responses. A, Tissue swelling responses to vehicleor FITC in FITC-sensitized C57BL/6 WT mice, TNF�/� mice, mast cell-deficient KitW-sh/W-sh mice, and KitW-sh/W-sh mice that had been selectivelyrepaired of their mast cell deficiency by the i.d. injection of WT or TNF�/�
BMCMCs. The first and second numbers in parentheses show total numberof mice in each group in all measurements up to 24 h and at all intervalsafter 24 h, respectively; the numeral after # is the total number of inde-pendent experiments performed that included mice in that group. Ear swell-ing responses are shown for vehicle-treated right ears and FITC-challengedleft ears. �, ��, ��� � p 0.05, 0.01, or 0.001 vs corresponding24 h values for vehicle-treated ears; ���, ���� � p 0.001 or 0.0001 vs24 h values for KitW-sh/W-sh mice; �, ��, ���� � p 0.05, 0.01, or0.0001 for the 24 h comparisons shown in parentheses; NS (p � 0.05).B–D, Four mice from each group shown in A were killed for assessment ofmast cell numbers, expressed as number per mm2 of dermis, in the centralpart of the ear pinna (B, Central Site) or for numbers of mast cells (C,Entire Ear) or PMNs (D) in a strip of FITC-challenged skin extending fromthe base to the tip of the ear pinna. �, ��, ���� � p 0.05, 0.01, or0.0001 vs corresponding values for WT mice; ††, ††† � p 0.01 or0.001 vs corresponding values for KitW-sh/W-sh � WT BMCMCs mice;‡‡ � p 0.01 vs corresponding values for TNF�/� or KitW-sh/W-sh �TNF�/� BMCMCs mice; NS (p � 0.05).
4104 MAST CELL TNF PROMOTES DC MIGRATION
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
these findings, application of the sensitizing dose of FITC to WTC57BL/6 mice also resulted in the appearance of many FITC�
DCs in the local LNs, whereas LNs draining the contralateral sitesthat had been challenged with vehicle contained very few FITC�
DCs (Fig. 2, B and C).In mice exposed epicutaneously to high levels of FITC, some
FITC can enter the lymphatic or systemic circulation and therebyreach DCs already present in the LNs (25). To assess whether thelevels of FITC used in this study might influence the numbers ofFITC� DCs we detected in the LNs draining sites of FITC appli-cation, we used an approach described by Grabbe et al. (22) to
deplete epidermal LCs at sites of FITC application. We found thatepicutaneous treatment of the skin with corticosteroids (�0.2 g ofcorticosteroid cream (0.05% clobetasol-17-propionate) permouse), topically to both sides of the ear skin once per day for 1–5consecutive days (22) resulted in nearly complete depletion of skinLCs by 3 days after initiation of corticosteroid treatment (Fig. 3A).At 24 h after application of FITC to the ear skin of corticosteroid-or vehicle-treated mice on day 3 after initiation of treatment, therewere significantly fewer FITC� DCs in draining LNs of the LC/DC-corticosteroid-depleted vs vehicle-treated mice (Fig. 3B).
FIGURE 2. Mast cells and TNF are required for optimal migration ofFITC-bearing LCs/DCs to draining LNs at sites treated epicutaneouslywith FITC. A, At 24 h after FITC or vehicle treatment, ear skin epidermalsheets and submaxillary and brachial LNs were collected as described inMaterials and Methods. The number of I-Ab� LCs in epidermal sheetswere counted in C57BL/6 WT mice (n � 6), TNF�/� mice (n � 6), mastcell-deficient KitW-sh/W-sh mice (n � 6), and KitW-sh/W-sh mice that had beenselectively repaired of their mast cell deficiency by the i.d. injection of WT(n � 6) or TNF�/� (n � 6) BMCMCs. B and C, The proportion (percent-age) of all I-Ab high CD11chigh cells that were FITC� (i.e., FITC� DCs)were determined by FACS analysis in C57BL/6 WT mice (n � 23),TNF�/� mice (n � 11), mast cell-deficient KitW-sh/W-sh mice (n � 16), andKitW-sh/W-sh mice that had been selectively repaired of their mast cell de-ficiency by the i.d. injection of WT (n � 8) or TNF�/� (n � 6) BMCMCs.B, Representative flow cytometry data for individual mice (numbers �percent of FITC� cells among I-Ab high CD11chigh DCs). Shaded areas rep-resent LNs draining vehicle-treated ears; solid lines represent LNs drainingFITC-treated ears. A and C, data are mean � SEM of values for individualmice; ��, ���, ���� � p 0.01, 0.001 or 0.0001 vs correspond-ing values for vehicle-treated ears (A) or vs LNs draining vehicle-treatedears (C); �, ���� � p 0.05 or 0.0001 vs FITC-treated ear skin (A) orvs LNs draining FITC-treated ears (C) of KitW-sh/W-sh mice; ‡, ‡‡, ‡‡‡‡ �p 0.05, 0.01, or 0.0001 vs FITC-treated ear skin (A) or vs LNsdraining FITC-treated ears (C) of TNF�/� mice; and ¶, ¶¶, ¶¶¶¶ � p 0.05, 0.01, or 0.0001 vs FITC-treated ear skin (A) or vs LNs drainingFITC-treated ears (C) of KitW-sh/W-sh mice � TNF�/� BMCMCs.
FIGURE 3. Depletion of epidermal LCs by topical treatment with acorticosteroid is associated with reduced numbers of FITC� DCs in thedraining LNs after epicutaneous application of FITC. A, Corticosteroidcream was applied to both sides of the ear skin as described in Materialsand Methods. Before beginning treatment with corticosteroids (day 0) andat the time intervals shown after initiation of corticosteroid treatment, thenumbers of I-Ab� LCs in epidermal sheets were counted after their stainingwith an anti-I-Ab mAb. Data show the average percentage � SEM of LCsvs the corresponding baseline value on day 0 (100%); n � 3 mice/group.B, At 4 days after initiation of daily corticosteroid (or vehicle) treatment,2.0% FITC (on skin of left ear) or vehicle alone (on skin of right ear) wereapplied epicutaneously. At 24 h after FITC or vehicle treatment, the drain-ing LNs were collected and the proportion (percentage) of I-Ab high
CD11chigh cells which were FITC� (i.e., FITC� DCs) was determined byFACS analysis as shown in Fig. 2, B and C. Results shown are represen-tative of data obtained from individual mice in two independent experi-ments, each of which gave similar results; data are from one mouse whichhad not been treated with corticosteroids (A) and two mice which had beentreated with corticosteroids (B and C). Based on comparison to the meanlevel of fluorescence intensity of FITC on MHCII�CD11c� DCs in theLNs draining the FITC-treated ears in mice which had not received corti-costeroid treatment, the level of fluorescence intensity of FITC onMHCII�CD11c� DCs in the LNs draining FITC-treated ears in micewhich had received corticosteroid treatment was significantly reduced (per-cent of FITC� DCs in LNs (mean � SEM) draining ear skin not treatedwith corticosteroids � 43.4 � 2.5% (n � 6) vs corticosteroid-treated earskin � 2.3 � 1.3 (n � 6), p 0.0001). In the mice in the experimentsshown in B, the LC number (mean � SEM) was, for ear skin not treatedwith corticosteroids: 590 � 34 cells/mm2 (n � 6), and, for corticosteroid-treated ear skin: 19.2 � 7.2 (n � 6), p 0.0001.
4105The Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The simplest interpretation of all of our findings is that most orall of the FITC� DCs, which we detected in draining LNs afterepicutaneous application of FITC to the ear pinnae, were thosewhich originally had been resident in the skin as LCs or dermalDCs and then migrated to the LNs, rather than DCs originallyresident in the LNs, which acquired FITC via the systemic distri-bution or local lymphatic drainage of the hapten.
The migration of LCs 24 h after application of FITC, as reflectedin the reduced numbers of LCs in epidermal sheets, was pro-foundly impaired in mast cell-deficient KitW-sh/W-sh mice and, to asomewhat lesser extent, in TNF�/� mice (Fig. 2A). Consistentwith these data, numbers of FITC� DCs in draining LNs 24 h afterhapten application also were markedly reduced in mast cell-defi-cient KitW-sh/W-sh or TNF�/� mice (by mean values, in comparisonto the corresponding levels in WT mice, of 49 or 54%, respec-tively, Fig. 2, B and C).
Local transfer of BMCMCs did not significantly influence thenumber of LCs in the epidermis at the site of mast cell engraftment(LC numbers per mm2 (mean � SEM) were, for KitW-sh/W-sh mice:533 � 14 (n � 5), for KitW-sh/W-sh mice � WT BMCMCs: 474 �31 (n � 3) and, for KitW-sh/W-sh mice � TNF�/� BMCMCs: 486 �25.3 (n � 3)). However, we found that local engraftment ofKitW-sh/W-sh mice with WT BMCMCs fully restored WT levels ofhapten-induced release of LCs from the epidermis and appearanceof FITC� DCs in draining LNs, as assessed 24 h after haptenapplication (Fig. 2). By contrast, KitW-sh/W-sh mice that had beenengrafted with TNF�/� BMCMCs exhibited significantly lowerlevels of both hapten-induced release of LCs from epidermis andnumbers of FITC� DCs in local LNs (Fig. 2). Indeed, numbers ofFITC� DC in draining LNs from KitW-sh/W-sh mice that had beenengrafted with TNF�/� BMCMCs were only slightly higher thanthose in KitW-sh/W-sh or TNF�/� mice, differences that did notachieve statistical significance (Fig. 2, B and C).
Two types of mast cell-engrafted genetically mast cell-deficientmice can now be used for studies of mast cell function in vivo:C57BL/6-KitW-sh/W-sh mice (26, 27) and WBB6F1-KitW/W-v mice(28–30). We found that both the migration of FITC� DCs to drain-ing LNs within 24 h of initial sensitization with FITC, and theexpression of CHS reactions to FITC in sensitized animals, werealso markedly reduced in WBB6F1-KitW/W-v mast cell-deficientmice in comparison to values in the congenic WT (i.e., WBB6F1-Kit�/�) mice; moreover, both of these abnormalities in WBB6F1-
KitW/W-v mice were repaired in animals that had undergone localselective repair of their cutaneous mast cell deficiency (Fig. 4).
TNF can express biological functions in either its membrane-associated or its secreted form (17) and via interactions with cellsbearing TNFR1 and/or TNFR2 (31). The findings shown in Fig. 5indicate that C57BL/6J-memTNF�/� mice, which are defective intheir ability to produce the secreted form of TNF, are able to sus-tain apparently normal levels of DC migration to LNs within thefirst 24 h or FITC application, and that, in this setting, TNF acts viaTNFR1 to a significantly greater extent than via TNFR2.
Taken together, our findings in the FITC-induced model of CHSshow that, under the conditions tested in our experiments, mastcells, and mast cell membrane-associated TNF, were required foroptimal migration of hapten-bearing cutaneous DCs to local drain-ing LNs within the first 24 h of initial application of the hapten, aswell as for optimal expression of the elicitation phase of the CHSresponse to this hapten. In contrast, detectable, albeit quite weak,
FIGURE 4. CHS responses to FITC, and FITC-in-duced DC migration, in WBB6F1 WT mice, mast cell-deficient KitW/W-v mice and WT BMCMC-engraftedKitW/W-v mice. A, Ear swelling responses for vehicle-treated right ears and FITC-challenged left ears. Thefirst and second numbers in parentheses show total num-ber of mice in each group used for all measurements upto 24 h and at all intervals after 24 h, respectively; thenumeral after # is the total number of independent ex-periments performed using mice in that group. ��,���� � p 0.01 or 0.0001 vs corresponding 24 hvalues for vehicle-treated ears; ���, ���� � p 0.001or 0.0001 for the 24 h comparisons shown in brackets;NS � not significant (p � 0.05). B, Migration of FITC�
DCs was assessed, and the data expressed, as in Fig. 2,B and C. �, �� � p 0.05 or 0.01 vs correspondingvalues from LNs draining the FITC-treated ears of KitW/
W-v mice). For all individual groups of mice, the valuesfor LNs draining the FITC-challenged vs correspondingvehicle-treated sites were significantly different at thep 0.001 level.
FIGURE 5. Membrane-associated TNF and TNFR1 are important foroptimal skin DC migration in response to the application of FITC. Themigration of FITC� DCs was assessed, and the data expressed, as in Fig. 2, Band C. The proportion of I-Ab high CD11chigh FITC� cells (i.e., % FITC� DCs)was determined by FACS analysis in C57BL/6 WT mice, TNF�/� mice,memTNF�/�, TNFR1�/�, and TNFR2�/� mice. The numbers in parenthesesshow total number of mice in each group; the numeral after # is the totalnumber of independent experiments performed using mice in that group. Dataare mean � SEM of values for individual mice. ��, p 0.01 vs correspondingvalues for LNs draining the FITC-treated ears of TNF�/� mice; ‡, p 0.05 vscorresponding values for LNs draining the FITC-treated ears of TNFR1�/�
mice. For all individual groups of mice, the values for LNs draining theFITC-challenged vs corresponding vehicle-treated sites were significantlydifferent at the p 0.001 level.
4106 MAST CELL TNF PROMOTES DC MIGRATION
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
CHS reactions to this hapten occurred in the virtual absence ofmast cells or in mice that completely lacked TNF (Fig. 1). Simi-larly, some migration of FITC� DCs from the skin to local drain-ing LNs was elicited within 24 h of FITC application in mast cell-or TNF-deficient mice, or in mice that contained only mast cellsthat were unable to produce TNF (Fig. 2).
Thus, while our results clearly show that mast cells and mastcell-associated TNF can significantly enhance both the FITC-in-duced migration of hapten-bearing DCs upon initial hapten expo-sure and the robust expression of CHS upon subsequent haptenchallenge of the sensitized mice, neither mast cells nor TNF areabsolutely required for either the FITC-induced DC migration(Figs. 2 and 4) or expression of CHS at sites of hapten challengein sensitized mice (Figs. 1 and 4). Nakae et al. (9) previouslyreported that DC migration from the skin to draining LNs wasimpaired in TNF�/� mice at 24 h after epicutaneous application ofFITC, but that the levels of DC migration in TNF�/� mice latercaught up with those observed in wild-type mice. We found thatTNF�/� or KitW-sh/W-sh mice, which showed significantly impairedDC migration at 24 h after FITC treatment, exhibited significantlyincreased levels of DCs in the draining LNs, compared with cor-responding levels in wild-type mice, at 48 h after FITC treatment(Fig. 6, A and B). At 72 h after FITC treatment, mice of all threegenotypes (WT, TNF�/�, or KitW-sh/W-sh) exhibited very similarlevels of FITC-bearing DC in the draining LNs (Fig. 6, A and B).
These results indicate that the initial impairment of DC migra-tion to local LNs observed in the absence of mast cells or TNFeventually resolves, presumably by the engagement of mast cell-or TNF-independent mechanisms. To assess whether the delay inDC migration observed in mast cell-deficient or TNF�/� mice re-sulted in significantly impaired expansion of populations of hap-ten-specific memory T cells, we harvested draining LNs 6 daysafter FITC sensitization and examined the proliferation induced insuch LN cells in the presence of FITC. We found essentially iden-tical levels of FITC-specific LN T cell proliferation in cells derivedfrom wild-type, KitW-sh/W-sh, or TNF�/� mice (Fig. 6C). Similarly,no differences among the responses of wild-type, KitW-sh/W-sh, orTNF�/� mice were detected when FITC-induced proliferation wasmeasured in draining LN T cells obtained from these mice at 5days after the onset of sensitization with FITC, in experiments inwhich various numbers of T cells were challenged with any ofmultiple different concentrations of FITC (data not shown).
Thus, under the conditions of FITC sensitization examined, adeficiency in mast cells or TNF delayed the migration of hapten-bearing DCs to the local LNs but this did not significantly influ-ence the expansion of hapten-specific memory T cells during thesensitization phase of FITC-induced CHS, at least as assessed 5 or6 days after the initial application of hapten.
The mechanism(s) by which mast cell-associated TNF can in-fluence the early stages of DC migration remain to be elucidated.
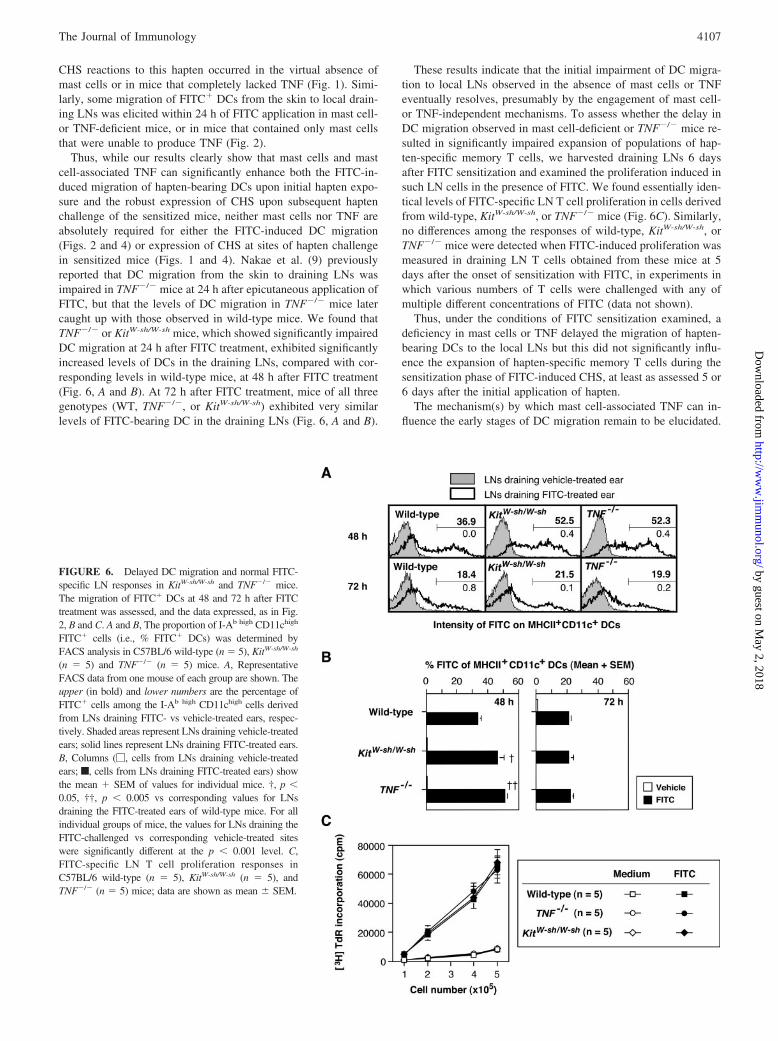
FIGURE 6. Delayed DC migration and normal FITC-specific LN responses in KitW-sh/W-sh and TNF�/� mice.The migration of FITC� DCs at 48 and 72 h after FITCtreatment was assessed, and the data expressed, as in Fig.2, B and C. A and B, The proportion of I-Ab high CD11chigh
FITC� cells (i.e., % FITC� DCs) was determined byFACS analysis in C57BL/6 wild-type (n � 5), KitW-sh/W-sh
(n � 5) and TNF�/� (n � 5) mice. A, RepresentativeFACS data from one mouse of each group are shown. Theupper (in bold) and lower numbers are the percentage ofFITC� cells among the I-Ab high CD11chigh cells derivedfrom LNs draining FITC- vs vehicle-treated ears, respec-tively. Shaded areas represent LNs draining vehicle-treatedears; solid lines represent LNs draining FITC-treated ears.B, Columns (�, cells from LNs draining vehicle-treatedears; f, cells from LNs draining FITC-treated ears) showthe mean � SEM of values for individual mice. †, p 0.05, ††, p 0.005 vs corresponding values for LNsdraining the FITC-treated ears of wild-type mice. For allindividual groups of mice, the values for LNs draining theFITC-challenged vs corresponding vehicle-treated siteswere significantly different at the p 0.001 level. C,FITC-specific LN T cell proliferation responses inC57BL/6 wild-type (n � 5), KitW-sh/W-sh (n � 5), andTNF�/� (n � 5) mice; data are shown as mean � SEM.
4107The Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
We considered the possibility that, in CHS to FITC, as in CHS toOx administered in ethanol (14), activation of mast cells via theFcR� chain can contribute to the ability of mast cells to enhancethe early stages of DC migration. We found that FcR� chain�/�
mice did exhibit a defect in their ability to express tissue swellingduring the elicitation phase of CHS to FITC (Fig. 7A), a result thatis in accord with our findings with FcR� chain�/� mice in CHS toOx (14). However, we detected no impairment of the ability ofFcR� chain�/� mice to exhibit FITC-induced migration of FITC�
DCs to draining LNs in the first 24 h of the sensitization phase ofthe response (Fig. 7B). Taken together, these results indicate thatthe requirements for FcR� chain (and, by implication, Ab) func-tion in various components of the sensitization and elicitationphases of CHS may vary, depending on the specific hapten testedand/or on other conditions of the experiment.
The same also appears to be true with respect to the roles ofmast cells in the expression of CHS responses. Studies using ge-netically mast cell-deficient KitW/W-v mice have shown that, withsome hapten/vehicle combinations, mast cells significantly con-tribute to the expression of various aspects of the responses (12–14). By contrast, with other hapten/vehicle combinations, no sig-nificant contributions of mast cells have been detected (32, 33).These findings suggest that the roles of mast cells in influencingdifferent aspects of CHS responses may vary significantly depend-ing on the hapten and/or conditions of sensitization and challengetested.
Mast cells, and mast cell-associated TNF, are required foroptimal DC migration to LNs during the first 24 h after i.n.administration of FITC-OVA
To examine possible effects of mast cells and mast cell-associatedTNF on DC migration in a different context, we administeredFITC-OVA to mice i.n. We used an Ab to 33D1 as a DC marker,rather than an Ab to MHC class II, to help to identify FITC�
airway DCs (34). We found that the percentage of FITC� DCs inthe thoracic LNs of FITC-OVA-treated WBB6F1-Kit�/� (WT)mice 24 h after intranasal application of FITC-OVA was signifi-cantly higher than the corresponding values for mast cell-deficientWBB6F1-KitW/W-v mice, and that the systemic engraftment ofKitW/W-v mice with mast cells by i.v. administration of WT BM-CMCs permitted these mice to express a response which was sta-tistically indistinguishable from that of the WT mice (Fig. 8).
We also examined the migration of FITC� airway DCs in re-sponse to i.n. administration of FITC-OVA in the same strains of
mice used in our studies of FITC-induced CHS (Fig. 9, A and B).In accord with our findings in the CHS model, the appearance ofFITC� DCs in the local (i.e., thoracic) LNs 24 h after airwaychallenge with hapten was significantly impaired, in comparison tovalues in WT mice, in mice deficient in mast cells, TNF or TNFR1,and in mast cell-deficient mice engrafted with TNF�/� BMCMCs,but not in mice defective in their ability to produce secreted TNF(i.e., memTNF�/� mice) or lacking TNFR2, or in mast cell-defi-cient mice engrafted with WT BMCMCs. In accord with findingsreported in our prior work (26, 35, 36), the numbers of mast cellsin the lungs of naive mast cell-deficient mice (i.e., mice not chal-lenged i.n. with FITC-OVA) which had received i.v. transfer ofBMCMCs were significantly higher than those in naive WT mice(Fig. 9C). Moreover, in naive FcR� chain�/� mice, the lungs con-tained a percentage of I-Ab�33D1�CD11c� DCs (among all lungcells) which were significantly greater than those in naive WTmice (Fig. 9D).
DiscussionWe found that, in naive mice, mast cells and mast cell-associatedTNF were required for optimal migration of hapten-bearing DCsfrom the skin or airways to local LNs within the first 24 h ofepicutaneous or airway administration of FITC or FITC-OVA, re-spectively. We also found that migration of FITC� DCs 24 h afterapplication of FITC or FITC-OVA was normal in mice which weredefective in their ability to secrete TNF, strongly indicating thatthe membrane-associated form of TNF fully or largely accountedfor the TNF-dependent effects on DC migration in these settings.
Mast cells and mast cell-associated TNF also were required foroptimal expression of the tissue swelling and neutrophil infiltrationat sites of FITC challenge in sensitized mice. However, in thismodel of CHS, significantly greater numbers of FITC� DCs weredetected in draining LNs 48 h after epicutaneous application ofFITC in mast cell- or TNF-deficient mice than in WT mice. Thesefindings indicate that, at the later stages of the sensitization pro-cess, the engagement of mast cell- and/or TNF-independent mech-anisms can contribute significantly to the migration of hapten-bearing DCs from the skin to local LNs. Moreover, the delay inmigration of FITC� DCs from the skin to local LNs in the absenceof mast cells or TNF was not associated with reduced levels ofproliferation upon Ag challenge of T cells obtained from drainingLNs 5 or 6 days after the onset of sensitization. Thus, the defect inthe expression of CHS at sites of FITC challenge in sensitized
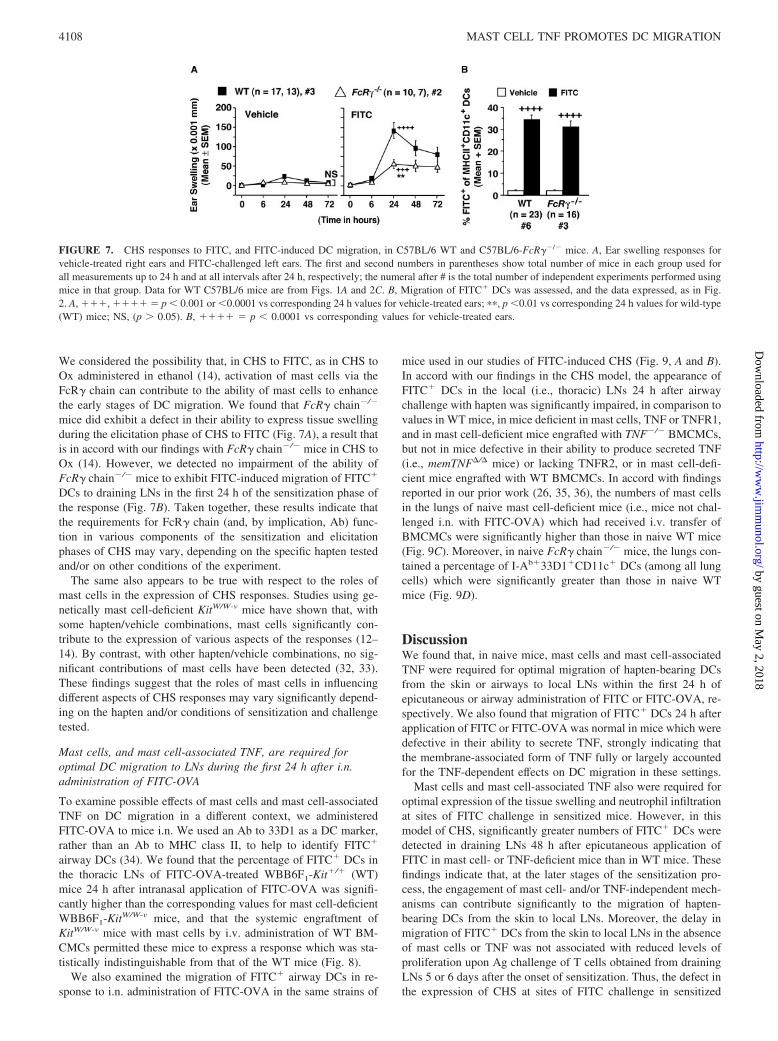
FIGURE 7. CHS responses to FITC, and FITC-induced DC migration, in C57BL/6 WT and C57BL/6-FcR��/� mice. A, Ear swelling responses forvehicle-treated right ears and FITC-challenged left ears. The first and second numbers in parentheses show total number of mice in each group used forall measurements up to 24 h and at all intervals after 24 h, respectively; the numeral after # is the total number of independent experiments performed usingmice in that group. Data for WT C57BL/6 mice are from Figs. 1A and 2C. B, Migration of FITC� DCs was assessed, and the data expressed, as in Fig.2. A, ���, ���� � p 0.001 or 0.0001 vs corresponding 24 h values for vehicle-treated ears; ��, p 0.01 vs corresponding 24 h values for wild-type(WT) mice; NS, (p � 0.05). B, ���� � p 0.0001 vs corresponding values for vehicle-treated ears.
4108 MAST CELL TNF PROMOTES DC MIGRATION
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

mice may have solely reflected defects in mast cell- and/or TNF-associated effector function at these sites.
The findings that mast cells, and mast cell-associated TNF, canmake important contributions to the migration of hapten-bearingDCs during the initial stages of the sensitization phase of acquired,T cell-dependent immune responses are in accord with prior ob-servations indicating that mast cells, and mast cell-derived TNF,can promote the migration of other hemopoietic cell types, includ-ing neutrophils, monocytes, and T cells, in the context of bothinnate and acquired immune responses (13, 37–39). Our observa-tions are also consistent with work with other haptens demonstrat-ing defects in both DC migration and the expression of CHS inTNF�/� mice (4, 9).
Although our experiments show that mast cell-associated TNFis necessary for optimal DC migration in response to FITC in thetwo model systems tested, we have not excluded the possibilitiesthat other mast cell-derived mediators also can contribute to theseresponses or have important roles in other settings associated withDC migration. Mast cells represent a rich source of a diverse arrayof biologically active mediators, including histamine and manycytokines and chemokines (40, 41), many of which have potentialeffects on DC migration, maturation, or function (7, 42–45). AndJawdat et al. (15) showed that activation of skin mast cells withIgE and specific Ag can enhance epidermal LC migration bymechanisms that are, at least in part, both histamine and H2Rdependent. Accordingly, it is possible that histamine and/or othermast cell-derived mediators, as well as TNF, may also have sig-nificant effects on DC migration during responses to FITC.
Just as multiple mast cell-derived mediators have the potentialto influence DC migration, maturation, or function during the sen-
sitization phase of acquired immune responses, multiple mecha-nisms probably can contribute to mast cell activation in such set-tings. Bryce et al. (14) showed that Ag nonspecific IgE-Fc�RIsignals, as well as mast cells, significantly promoted the egress ofepidermal LCs when mice were sensitized with 2% Ox in ethanol.Demeure et al. (46) reported that mosquito saliva can directly in-duce mast cell degranulation in vitro and in vivo, in an apparentlyIgE- and specific Ag-independent manner, and that such mast cellactivation can promote the migration of leukocytes, including skinDCs, to draining LNs. Although Demeure et al. (46) did not for-mally rule out a role for Ag-independent effects of IgE on mastcells in this model, their study showed that mosquito saliva candirectly or indirectly promote the mast cell-dependent enhance-ment of DC migration.
We found that the migration FITC� DCs to local LNs during thefirst 24 h after application of FITC or FITC-OVA was not signif-icantly diminished in mice which lacked expression of FcR�.Thus, in these skin or airway models, mast cells apparently exerttheir effects on DC migration independently of activation by eitherIgE/Fc�RI or IgG1/Fc�RIII signaling. Although we have no dataproving that FITC or FITC-OVA induced FcR�-independent mastcell activation in our experiments, and, if so, by what mecha-nism(s) this occurred, there are a number of possibilities which canbe investigated.
For example, the vehicle used for administration of FITC con-sists of acetone and the dialkyl phthalate, dibutylphthalate. A 1:1mixture of acetone and dibutylphthalate can induce the migrationof F4/80 Ag� and macrophage C-type lectin� macrophages fromthe mouse dermis by a mechanism which is at least in part IL-1�dependent (47). Perhaps the vehicle-induced local production ofcytokines (e.g., by keratinocytes, which can produce TNF andother proinflammatory mediators; Ref. 1), can contribute to mastcell activation independently of FcR�-dependent signals. Alterna-tively, in our airway model, FITC-OVA might first activate alve-olar macrophages to produce mediators which can then induceFcR�-independent mast cell activation. Finally, dialkyl phthalatesnot only can enhance the Ag- and IgE-dependent activation of ratbasophilic leukemia cells, but they also can directly increase con-centrations of cytosolic calcium ions in the cells and, at high con-centrations, weakly promote degranulation independently of IgEand specific Ag (48). However, these represent only a few of themany possible direct or indirect mechanisms of mast cell activa-tion which could contribute to our observation that mice lackingFcR� exhibited no apparent defects in their ability to express mastcell-dependent enhancement of DC migration in response to FITC.
Moreover, in attempting to understand how mast cell function iselicited in this setting, it is important to recognize that the form ofmast cell TNF which contributes to FITC-induced, mast cell-de-pendent enhancement of DC migration appears to be the mem-brane-associated form of the cytokine. Perhaps relatively subtlechanges in the expression of the membrane-associated form ofTNF by mast cells at sites of hapten challenge are sufficient topermit the elicitation of mast cell-dependent enhancement of DCmigration. Possibly, even constitutive levels of expression ofmembrane-associated TNF are sufficient to allow mast cells to pro-mote DC migration. In that case, initiation of DC migration byhapten may require a local signal other than an alteration of levelsof mast cell-associated TNF.
Even though FcR�-dependent signals were not essential for theenhancement of skin (or airway) DC migration, expression ofFcR� was required for the optimal local expression of CHS re-sponses upon subsequent FITC challenge of sensitized mice. Thismay have reflected an important role for FcR� in either the sen-sitization or elicitation phases of CHS to FITC. For example, FcR�
FIGURE 8. Mast cells are required for optimal DC migration from theairways to thoracic LNs in response to intranasal administration of FITC-OVA. A and B, At 24 h after intranasal administration of FITC-OVA,thoracic and submaxillary LNs were separately collected from WBB6F1-Kit�/� (WT) mice, KitW/W-v mice and KitW/W-v mice after i.v. transfer, 8 wkbefore the experiment, of 1.0 � 107 congenic WT BMCMCs. Then, theproportion (percent) of FITC� cells among the 33D1�CD11chigh DCs wasdetermined by FACS. A, Representative FACS data from one mouse ofeach group. The upper (in bold) and lower numbers are the percentage ofFITC� cells among the thoracic and submaxillary LNs, respectively.Shaded areas, submaxillary LNs; bold lines, thoracic LNs. B, Columns (�,submaxillary LNs; f, thoracic LNs) show the average � SEM proportion(percent) of FITC� cells among the 33D1�CD11chigh DCs in mice of eachgroup. The numbers in parentheses represent the total number of mice ineach group; the numeral after # is the total number of independent exper-iments performed using mice in that group. ����, p 0.0001 vs corre-sponding values for thoracic LNs from Kit�/� (WT) mice or from KitW/W-v
mice which had been engrafted with congenic �/� (WT) BMCMCs. Forall individual groups of mice, the values for thoracic vs the correspondingsubmaxillary LNs were significantly different at the p 0.0001 level.
4109The Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
(on DCs and/or other cell types) may be required for key steps inDC maturation or function, and/or for the optimal orchestration ofthe elicitation phase of the response.
The findings that mast cell-associated TNF can significantly en-hance DC migration from the skin or airways in mice, as well ascontribute to the effector phase of CHS responses, offer a newperspective on a large body of work implicating TNF, and specif-ically mast cell-derived TNF, in a wide variety of mouse models ofdisease, and, by extension, in the corresponding human disorders.Thus, in mice, mast cell-derived TNF can contribute to the devel-opment of the pathology associated with certain models of sepsis(37, 49–51), cutaneous granuloma formation (52), skin vasculitisand glomerulonephritis (53), and CHS (13). In such settings, it isgenerally thought that mast cell-derived TNF mediates “proinflam-matory” effects at sites of disease.
However, our data indicate that mast cell-associated TNF alsocan promote DC migration during the early stages of the sensiti-zation phase of acquired immune responses, and thereby may con-tribute to the development of such responses. The actual extent towhich mast cell-associated TNF can contribute to immune sensi-tization in various settings will have to be established on a case-by-case basis. For example, in the FITC-induced CHS model in-vestigated herein, we so far have not detected a defect in immunesensitization in the absence of either mast cells or TNF, at least ascan be assessed by quantifying Ag-induced proliferation of drain-ing LN T cells obtained 5 or 6 days after application of FITC.
Nevertheless, our data indicate that mast cell-associated TNFhas the potential to contribute to the development of acquired im-mune responses involved in host defense or immunological disor-ders. One might even speculate that some of the clinical benefit ofanti-TNF therapy in humans reflects suppression of either “effec-tor/proinflammatory” or “immunoregulatory” functions of mastcell-derived TNF. Anti-TNF therapy has been reported to be help-ful in rheumatoid arthritis (54), Crohn’s disease (55), and asthma(56), each of which has been associated with increased numbersand/or activation of mast cells at sites of disease (57–65). In suchdisorders, anti-TNF treatment may confer benefit in part by antag-onizing effector functions of TNF derived from mast cells, as wellas from the many other potential cellular sources of this cytokine.
Although the work reported herein indicates that anti-TNF treat-ment also has the potential to interfere with an “immunoregula-tory” role of mast cell-associated TNF, the literature does not re-veal a clear picture regarding the importance of TNF in thesensitization phase of CHS. On the one hand, anti-TNF Ab treat-ment in the sensitization phase of a model of Ox-induced CHS inmice almost completely suppressed DC migration to local LNs,and this was associated with attenuated inflammation in the elic-itation phase (66). Neutralization of TNF in the elicitation phasealso can suppress the development of CHS reactions to TNCB inmice (67). In contrast, normal levels of Ag-specific proliferationwere observed in LN T cells obtained from TNF�/� mice sensi-tized with TNCB, and mice that received TNCB-sensitized T cells
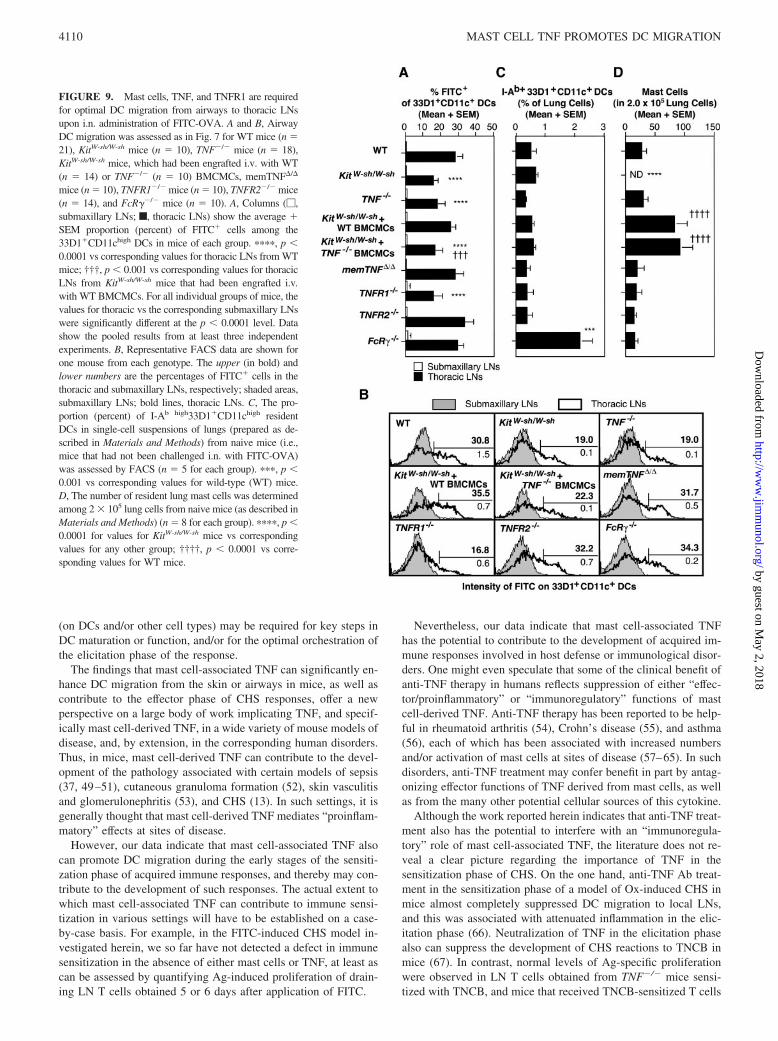
FIGURE 9. Mast cells, TNF, and TNFR1 are requiredfor optimal DC migration from airways to thoracic LNsupon i.n. administration of FITC-OVA. A and B, AirwayDC migration was assessed as in Fig. 7 for WT mice (n �21), KitW-sh/W-sh mice (n � 10), TNF�/� mice (n � 18),KitW-sh/W-sh mice, which had been engrafted i.v. with WT(n � 14) or TNF�/� (n � 10) BMCMCs, memTNF�/�
mice (n � 10), TNFR1�/� mice (n � 10), TNFR2�/� mice(n � 14), and FcR��/� mice (n � 10). A, Columns (�,submaxillary LNs; f, thoracic LNs) show the average �SEM proportion (percent) of FITC� cells among the33D1�CD11chigh DCs in mice of each group. ����, p 0.0001 vs corresponding values for thoracic LNs from WTmice; †††, p 0.001 vs corresponding values for thoracicLNs from KitW-sh/W-sh mice that had been engrafted i.v.with WT BMCMCs. For all individual groups of mice, thevalues for thoracic vs the corresponding submaxillary LNswere significantly different at the p 0.0001 level. Datashow the pooled results from at least three independentexperiments. B, Representative FACS data are shown forone mouse from each genotype. The upper (in bold) andlower numbers are the percentages of FITC� cells in thethoracic and submaxillary LNs, respectively; shaded areas,submaxillary LNs; bold lines, thoracic LNs. C, The pro-portion (percent) of I-Ab high33D1�CD11chigh residentDCs in single-cell suspensions of lungs (prepared as de-scribed in Materials and Methods) from naive mice (i.e.,mice that had not been challenged i.n. with FITC-OVA)was assessed by FACS (n � 5 for each group). ���, p 0.001 vs corresponding values for wild-type (WT) mice.D, The number of resident lung mast cells was determinedamong 2 � 105 lung cells from naive mice (as described inMaterials and Methods) (n � 8 for each group). ����, p 0.0001 for values for KitW-sh/W-sh mice vs correspondingvalues for any other group; ††††, p 0.0001 vs corre-sponding values for WT mice.
4110 MAST CELL TNF PROMOTES DC MIGRATION
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

from either WT or TNF�/� mice developed very similar CHSresponses upon subsequent hapten challenge (9).
We found that the levels of Ag-specific proliferation in T cellsisolated from draining LNs 5 or 6 days after FITC application werenearly identical in WT, mast cell-deficient, and TNF�/� mice (Fig.6C and data not shown). Thus, as in TNF�/� or IL-1�/� mice,which exhibited a transient defect in DC migration after epicuta-neous application of FITC (9, 19), the delayed migration of FITC�
DCs observed in KitW-sh/W-sh and TNF�/� mice apparently re-sulted in adequate expansion of hapten-specific T cells duringsensitization.
In summary, our findings indicate that mast cell-membrane-as-sociated TNF can enhance skin or airway DC migration during thesensitization phase of acquired immune responses in mice. It maybe very difficult to determine whether mast cell-associated TNFalso can contribute to the sensitization phase of acquired immuneresponses in humans. However, this possibility should be kept inmind, both when considering the consequences of anti-TNF treat-ment in human subjects and when investigating approaches to en-hance the development of protective immune responses. Oneshould also keep in mind that some mast cell-associated mediators,such as PGD2, can have effects that suppress the migration and/orfunction of skin LCs (68) and airway DCs (69). Thus, dependingon factors yet to be fully understood, it is possible that mast cellsand their products can mediate a complex spectrum of positive andnegative effects on the biology of DCs.
AcknowledgmentsWe thank Z.-S. Wang for histological analysis, Peter Besmer for providingKitW-sh/W-sh mice, and Hideoki Ogawa and Juntendo University for provid-ing support for H. Suto.
DisclosuresThe authors have no financial conflict of interest.
References1. Grabbe, S., and T. Schwarz. 1998. Immunoregulatory mechanisms involved in
elicitation of allergic contact hypersensitivity. Immunol. Today 19: 37–44.2. Steinman, R. M., M. Pack, and K. Inaba. 1997. Dendritic cells in the T-cell areas
of lymphoid organs. Immunol. Rev. 156: 25–37.3. Kissenpfennig, A., S. Henri, B. Dubois, C. Laplace-Builhe, P. Perrin, N. Romani,
C. H. Tripp, P. Douillard, L. Leserman, D. Kaiserlian, et al. 2005. Dynamics andfunction of Langerhans cells in vivo: dermal dendritic cells colonize lymph nodeareas distinct from slower migrating Langerhans cells. Immunity 22: 643–654.
4. Pasparakis, M., L. Alexopoulou, V. Episkopou, and G. Kollias. 1996. Immuneand inflammatory responses in TNF�-deficient mice: a critical requirement forTNF� in the formation of primary B cell follicles, follicular dendritic cell net-works and germinal centers, and in the maturation of the humoral immune re-sponse. J. Exp. Med. 184: 1397–1411.
5. Takeshita, K., T. Yamasaki, S. Akira, F. Gantner, and K. B. Bacon. 2004. Es-sential role of MHC II-independent CD4� T cells, IL-4 and STAT6 in contacthypersensitivity induced by fluorescein isothiocyanate in the mouse. Int. Immu-nol. 16: 685–695.
6. Yokozeki, H., M. Ghoreishi, S. Takagawa, K. Takayama, T. Satoh, I. Katayama,K. Takeda, S. Akira, and K. Nishioka. 2000. Signal transducer and activator oftranscription 6 is essential in the induction of contact hypersensitivity. J. Exp.Med. 191: 995–1004.
7. Cumberbatch, M., R. J. Dearman, C. E. Griffiths, and I. Kimber. 2000. Langer-hans cell migration. Clin. Exp. Dermatol. 25: 413–418.
8. Wang, B., L. Zhuang, H. Fujisawa, G. A. Shinder, C. Feliciani, G. M. Shivji,H. Suzuki, P. Amerio, P. Toto, and D. N. Sauder. 1999. Enhanced epidermalLangerhans cell migration in IL-10 knockout mice. J. Immunol. 162: 277–283.
9. Nakae, S., Y. Komiyama, S. Narumi, K. Sudo, R. Horai, Y. Tagawa,K. Sekikawa, K. Matsushima, M. Asano, and Y. Iwakura. 2003. IL-1-inducedtumor necrosis factor-� elicits inflammatory cell infiltration in the skin by in-ducing IFN-�-inducible protein 10 in the elicitation phase of the contact hyper-sensitivity response. Int. Immunol. 15: 251–260.
10. Gordon, J. R., and S. J. Galli. 1990. Mast cells as a source of both preformed andimmunologically inducible TNF-�/cachectin. Nature 346: 274–276.
11. Gordon, J. R., and S. J. Galli. 1991. Release of both preformed and newly syn-thesized tumor necrosis factor � (TNF-�)/cachectin by mouse mast cells stimu-lated via the Fc�RI: a mechanism for the sustained action of mast cell-derivedTNF-� during IgE-dependent biological responses. J. Exp. Med. 174: 103–107.
12. Askenase, P. W., H. Van Loveren, S. Kraeuter-Kops, Y. Ron, R. Meade,T. C. Theoharides, J. J. Nordlund, H. Scovern, M. D. Gerhson, and W. Ptak.
1983. Defective elicitation of delayed-type hypersensitivity in W/Wv and SI/SIdmast cell-deficient mice. J. Immunol. 131: 2687–2694.
13. Biedermann, T., M. Kneilling, R. Mailhammer, K. Maier, C. A. Sander,G. Kollias, S. L. Kunkel, L. Hultner, and M. Rocken. 2000. Mast cells controlneutrophil recruitment during T cell-mediated delayed-type hypersensitivity re-actions through tumor necrosis factor and macrophage inflammatory protein 2.J. Exp. Med. 192: 1441–1452.
14. Bryce, P. J., M. L. Miller, I. Miyajima, M. Tsai, S. J. Galli, and H. C. Oettgen.2004. Immune sensitization in the skin is enhanced by antigen-independent ef-fects of IgE. Immunity 20: 381–392.
15. Jawdat, D. M., E. J. Albert, G. Rowden, I. D. Haidl, and J. S. Marshall. 2004.IgE-mediated mast cell activation induces Langerhans cell migration in vivo.J. Immunol. 173: 5275–5282.
16. Korner, H., M. Cook, D. S. Riminton, F. A. Lemckert, R. M. Hoek,B. Ledermann, F. Kontgen, B. Fazekas de St. Groth, and J. D. Sedgwick. 1997.Distinct roles for lymphotoxin-� and tumor necrosis factor in organogenesis andspatial organization of lymphoid tissue. Eur. J. Immunol. 27: 2600–2609.
17. Ruuls, S. R., R. M. Hoek, V. N. Ngo, T. McNeil, L. A. Lucian, M. J. Janatpour,H. Korner, H. Scheerens, E. M. Hessel, J. G. Cyster, et al. 2001. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient tosecreted TNF in driving autoimmune inflammation. Immunity 15: 533–543.
18. Duttlinger, R., K. Manova, T. Y. Chu, C. Gyssler, A. D. Zelenetz,R. F. Bachvarova, and P. Besmer. 1993. W-sash affects positive and negativeelements controlling c-kit expression: ectopic c-kit expression at sites of kit-ligand expression affects melanogenesis. Development 118: 705–717.
19. Nakae, S., C. Naruse-Nakajima, K. Sudo, R. Horai, M. Asano, and Y. Iwakura.2001. IL-1�, but not IL-1�, is required for contact-allergen-specific T cell acti-vation during the sensitization phase in contact hypersensitivity. Int. Immunol.13: 1471–1478.
20. Lutz, M. B., C. U. Assmann, G. Girolomoni, and P. Ricciardi-Castagnoli. 1996.Different cytokines regulate antigen uptake and presentation of a precursor den-dritic cell line. Eur. J. Immunol. 26: 586–594.
21. Vermaelen, K. Y., I. Carro-Muino, B. N. Lambrecht, and R. A. Pauwels. 2001.Specific migratory dendritic cells rapidly transport antigen from the airways tothe thoracic lymph nodes. J. Exp. Med. 193: 51–60.
22. Grabbe, S., K. Steinbrink, M. Steinert, T. A. Luger, and T. Schwarz. 1995. Re-moval of the majority of epidermal Langerhans cells by topical or systemic ste-roid application enhances the effector phase of murine contact hypersensitivity.J. Immunol. 155: 4207–4217.
23. Chensue, S. W., N. W. Lukacs, T. Y. Yang, X. Shang, K. A. Frait, S. L. Kunkel,T. Kung, M. T. Wiekowski, J. A. Hedrick, D. N. Cook, et al. 2001. Aberrant invivo T helper type 2 cell response and impaired eosinophil recruitment in CCchemokine receptor 8 knockout mice. J. Exp. Med. 193: 573–584.
24. Tang, A., M. Amagai, L. G. Granger, J. R. Stanley, and M. C. Udey. 1993.Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cad-herin. Nature 361: 82–85.
25. Pior, J., T. Vogl, C. Sorg, and E. MacHer. 1999. Free hapten molecules aredispersed by way of the bloodstream during contact sensitization to fluoresceinisothiocyanate. J. Invest. Dermatol. 113: 888–893.
26. Grimbaldeston, M. A., C. C. Chen, A. M. Piliponsky, M. Tsai, S. Y. Tam, andS. J. Galli. 2005. Mast cell-deficient W-sash c-kit mutant KitW-sh/W-sh mice as amodel for investigating mast cell biology in vivo. Am. J. Pathol. 167: 835–848.
27. Wolters, P. J., J. Mallen-St Clair, C. C. Lewis, S. A. Villalta, P. Baluk, D. J. Erle,and G. H. Caughey. 2005. Tissue-selective mast cell reconstitution and differen-tial lung gene expression in mast cell-deficient KitW-sh/KitW-sh sash mice. Clin.Exp. Allergy 35: 82–88.
28. Galli, S. J., S. Nakae, and M. Tsai. 2005. Mast cells in the development ofadaptive immune responses. Nat. Immunol. 6: 135–142.
29. Galli, S. J., J. Kalesnikoff, M. A. Grimbaldeston, A. M. Piliponsky,C. M. Williams, and M. Tsai. 2005. Mast cells as “tunable” effector and immu-noregulatory cells: recent advances. Annu. Rev. Immunol. 23: 749–786.
30. Tsai, M., M. A. Grimbaldeston, M. Yu, S. Y. Tam, and S. J. Galli. 2005. Usingmast cell knock-in mice to analyze the roles of mast cells in allergic responses invivo. Chem. Immunol. Allergy 87: 179–197.
31. Bazzoni, F., and B. Beutler. 1996. The tumor necrosis factor ligand and receptorfamilies. N. Engl. J. Med. 334: 1717–1725.
32. Galli, S. J., and I. Hammel. 1984. Unequivocal delayed hypersensitivity in mastcell-deficient and beige mice. Science 226: 710–713.
33. Mekori, Y. A., and S. J. Galli. 1985. Undiminished immunologic tolerance tocontact sensitivity in mast cell-deficient W/Wv and Sl/Sld mice. J. Immunol. 135:879–885.
34. Nussenzweig, M. C., R. M. Steinman, M. D. Witmer, and B. Gutchinov. 1982. Amonoclonal antibody specific for mouse dendritic cells. Proc. Natl. Acad. Sci.USA 79: 161–165.
35. Martin, T. R., T. Takeishi, H. R. Katz, K. F. Austen, J. M. Drazen, and S. J. Galli.1993. Mast cell activation enhances airway responsiveness to methacholine in themouse. J. Clin. Invest. 91: 1176–1182.
36. Williams, C. M., and S. J. Galli. 2000. Mast cells can amplify airway reactivityand features of chronic inflammation in an asthma model in mice. J. Exp. Med.192: 455–462.
37. Malaviya, R., T. Ikeda, E. Ross, and S. N. Abraham. 1996. Mast cell modulationof neutrophil influx and bacterial clearance at sites of infection through TNF-�.Nature 381: 77–80.
38. McLachlan, J. B., J. P. Hart, S. V. Pizzo, C. P. Shelburne, H. F. Staats,M. D. Gunn, and S. N. Abraham. 2003. Mast cell-derived tumor necrosis factorinduces hypertrophy of draining lymph nodes during infection. Nat. Immunol. 4:1199–1205.
4111The Journal of Immunology
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

39. Wershil, B. K., Z. S. Wang, J. R. Gordon, and S. J. Galli. 1991. Recruitment ofneutrophils during IgE-dependent cutaneous late phase reactions in the mouse ismast cell-dependent: partial inhibition of the reaction with antiserum against tu-mor necrosis factor-�. J. Clin. Invest. 87: 446–453.
40. Kawakami, T., and S. J. Galli. 2002. Regulation of mast-cell and basophil func-tion and survival by IgE. Nat. Rev. Immunol. 2: 773–786.
41. Mekori, Y. A., and D. D. Metcalfe. 2000. Mast cells in innate immunity. Immu-nol. Rev. 173: 131–140.
42. Caron, G., Y. Delneste, E. Roelandts, C. Duez, N. Herbault, G. Magistrelli,J. Y. Bonnefoy, J. Pestel, and P. Jeannin. 2001. Histamine induces CD86 ex-pression and chemokine production by human immature dendritic cells. J. Im-munol. 166: 6000–6006.
43. Mazzoni, A., H. A. Young, J. H. Spitzer, A. Visintin, and D. M. Segal. 2001.Histamine regulates cytokine production in maturing dendritic cells, resulting inaltered T cell polarization. J. Clin. Invest. 108: 1865–1873.
44. Kabashima, K., D. Sakata, M. Nagamachi, Y. Miyachi, K. Inaba, andS. Narumiya. 2003. Prostaglandin E2-EP4 signaling initiates skin immune re-sponses by promoting migration and maturation of Langerhans cells. Nat. Med.9: 744–749.
45. Stoitzner, P., G. Ratzinger, F. Koch, K. Janke, T. Scholler, A. Kaser, H. Tilg,W. W. Cruikshank, P. Fritsch, and N. Romani. 2001. Interleukin-16 supports themigration of Langerhans cells, partly in a CD4-independent way. J. Invest. Der-matol. 116: 641–649.
46. Demeure, C. E., K. Brahimi, F. Hacini, F. Marchand, R. Peronet, M. Huerre,P. St-Mezard, J. F. Nicolas, P. Brey, G. Delespesse, and S. Mecheri. 2005.Anopheles mosquito bites activate cutaneous mast cells leading to a local inflam-matory response and lymph node hyperplasia. J. Immunol. 174: 3932–3940.
47. Chun, K. H., Y. Imai, N. Higashi, and T. Irimura. 2000. Migration of dermal cellsexpressing a macrophage C-type lectin during the sensitization phase of delayed-type hypersensitivity. J. Leukocyte Biol. 68: 471–478.
48. Nakamura, R., R. Teshima, and J. Sawada. 2002. Effect of dialkyl phthalates onthe degranulation and Ca2� response of RBL-2H3 mast cells. Immunol. Lett. 80:119–124.
49. Echtenacher, B., D. N. Mannel, and L. Hultner. 1996. Critical protective role ofmast cells in a model of acute septic peritonitis. Nature 381: 75–77.
50. Prodeus, A. P., X. Zhou, M. Maurer, S. J. Galli, and M. C. Carroll. 1997. Im-paired mast cell-dependent natural immunity in complement C3-deficient mice.Nature 390: 172–175.
51. Maurer, M., B. Echtenacher, L. Hultner, G. Kollias, D. N. Mannel, K. E. Langley,and S. J. Galli. 1998. The c-kit ligand, stem cell factor, can enhance innateimmunity through effects on mast cells. J. Exp. Med. 188: 2343–2348.
52. von Stebut, E., M. Metz, G. Milon, J. Knop, and M. Maurer. 2003. Early mac-rophage influx to sites of cutaneous granuloma formation is dependent on MIP-1�/� released from neutrophils recruited by mast cell-derived TNF�. Blood 101:210–215.
53. Watanabe, N., B. Akikusa, S. Y. Park, H. Ohno, L. Fossati, G. Vecchietti,J. E. Gessner, R. E. Schmidt, J. S. Verbeek, B. Ryffel, et al. 1999. Mast cellsinduce autoantibody-mediated vasculitis syndrome through tumor necrosis factorproduction upon triggering Fc� receptors. Blood 94: 3855–3863.
54. Feldmann, M., and R. N. Maini. 2003. Lasker clinical medical research award:TNF defined as a therapeutic target for rheumatoid arthritis and other autoim-mune diseases. Nat. Med. 9: 1245–1250.
55. Papadakis, K. A., and S. R. Targan. 2000. Role of cytokines in the pathogenesisof inflammatory bowel disease. Annu. Rev. Med. 51: 289–298.
56. Babu, K. S., D. E. Davies, and S. T. Holgate. 2004. Role of tumor necrosis factor� in asthma. Immunol. Allergy Clin. North Am. 24: 583–597, v-vi.
57. Bradding, P., J. A. Roberts, K. M. Britten, S. Montefort, R. Djukanovic,R. Mueller, C. H. Heusser, P. H. Howarth, and S. T. Holgate. 1994. Interleukin-4,-5, and -6 and tumor necrosis factor-� in normal and asthmatic airways: evidencefor the human mast cell as a source of these cytokines. Am. J. Respir. Cell Mol.Biol. 10: 471–480.
58. Beil, W. J., P. F. Weller, M. A. Peppercorn, S. J. Galli, and A. M. Dvorak. 1995.Ultrastructural immunogold localization of subcellular sites of TNF-� in colonicCrohn’s disease. J. Leukocyte Biol. 58: 284–298.
59. Koshino, T., Y. Arai, Y. Miyamoto, Y. Sano, M. Itami, S. Teshima, K. Hirai,T. Takaishi, K. Ito, and Y. Morita. 1996. Airway basophil and mast cell densityin patients with bronchial asthma: relationship to bronchial hyperresponsiveness.J. Asthma 33: 89–95.
60. Denburg, J. A. 1998. Basophils and mast cells in airway inflammation andasthma. Can. Respir. J. 5(Suppl. A): 41A–44A.
61. Lilja, I., C. Gustafson-Svard, L. Franzen, and R. Sjodahl. 2000. Tumor necrosisfactor-� in ileal mast cells in patients with Crohn’s disease. Digestion 61: 68–76.
62. Furusu, H., K. Murase, Y. Nishida, H. Isomoto, F. Takeshima, Y. Mizuta,B. R. Hewlett, R. H. Riddell, and S. Kohno. 2002. Accumulation of mast cellsand macrophages in focal active gastritis of patients with Crohn’s disease. Hepa-togastroenterology 49: 639–643.
63. Brightling, C. E., P. Bradding, F. A. Symon, S. T. Holgate, A. J. Wardlaw, andI. D. Pavord. 2002. Mast-cell infiltration of airway smooth muscle in asthma.N. Engl. J. Med. 346: 1699–1705.
64. Nigrovic, P. A., and D. M. Lee. 2005. Mast cells in inflammatory arthritis. Ar-thritis Res. Ther. 7: 1–11.
65. Juurikivi, A., C. Sandler, K. A. Lindstedt, P. T. Kovanen, T. Juutilainen,M. J. Leskinen, T. Maki, and K. K. Eklund. 2005. Inhibition of c-kit tyrosinekinase by imatinib mesylate induces apoptosis in mast cells in rheumatoid syno-via: a potential approach to the treatment of arthritis. Ann. Rheum. Dis. 64:1126–1131.
66. Cumberbatch, M., and I. Kimber. 1995. Tumour necrosis factor-� is required foraccumulation of dendritic cells in draining lymph nodes and for optimal contactsensitization. Immunology 84: 31–35.
67. Piguet, P. F., G. E. Grau, C. Hauser, and P. Vassalli. 1991. Tumor necrosis factoris a critical mediator in hapten induced irritant and contact hypersensitivity re-actions. J. Exp. Med. 173: 673–679.
68. Angeli, V., C. Faveeuw, O. Roye, J. Fontaine, E. Teisser, A. Capron,I. Wolowczuk, M. Capron, and F. Trottein. 2001. Role of the parasite-derivedprostaglandin D2 in the inhibition of epidermal Langerhans cell migration duringschistosomiasis infection. J. Exp. Med. 193: 1135–1147.
69. Hammad, H., H. J. de Heer, T. Soullie, H. C. Hoogsteden, F. Trottein, andB. N. Lambrecht. 2003. Prostaglandin D2 inhibits airway dendritic cell migrationand function in steady-state conditions by selective activation of the D prostanoidreceptor 1. J. Immunol. 171: 3936–3940.
4112 MAST CELL TNF PROMOTES DC MIGRATION
by guest on May 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















