
Non-coding RNA William Liu CS374: Algorithms in Biology November 23, 2004.
date post
21-Dec-2015Category
view
216download
0
Marina Sirota
CS374
October 19, 2004
PROTEIN MULTIPLE
SEQUENCE ALIGNMENT

OUTLINE
• Introduction
• Alignments
• Pairwise vs. Multiple
• DNA vs. Protein
• MUSCLE
• PROBCONS
• Conclusion

INTRODUCTION
1. Sequence Analysis – Look at DNA and protein
sequences, searching for clues about structure, function and control
2. Structure Analysis – Examine biological structures, to
learn more about structure, function and control
3. Functional Analysis – Understand how the sequences
and structures lead to the biological function

PAIRWISE SEQUENCE ALIGNMENT (REVIEW)
• The Problem: Given two sequences of letters, and a scoring scheme for evaluating matching letters, find the optimal pairing of letters from one sequence to letters of the other sequence
• Basic Idea: The score of the best possible alignment that ends at a given pair of positions (i, j) is the score of the best alignment previous to those two positions plus the score for aligning those two positions.
PAIRWISE SEQUENCE ALIGNMENT (REVIEW)

PAIRWISE vs. MULTIPLE
PAIRWISE
• Evaluated by addition of match or mismatch scores for aligned pairs and affine gap penalties for unaligned pairs
• O(L2) time and O(L) space via dynamic programming
MULTIPLE
• Lack of proper objective scoring functions to measure alignment quality
• High computational cost and no efficient algorithm that can be applied
L = sequence length

PROTEIN VS. DNA
• DNA (4 characters) • Protein (20 characters)• DNA – 50% similarity• Protein – 20% similarity
• DNA – fewer sequences to compare• Protein – many sequences to compare• DNA aligners need to be able to handle
long sequences, protein aligners do not

PROTEIN MULTIPLE SEQUENCE ALIGNMENT
• Note that areas that are considered very similar don’t necessarily contain the same amino acids

MOTIVATION• Find similarity between known and unknown
sequences• Protein sequence similarity implies
divergence from a common ancestor and functional similarity
PROBLEM• Given n sequences and a scoring scheme for
evaluating matching letters, find the optimal pairing of letters between the sequences
• Can be done using dynamic programming with time and space complexity O(Ln) which is not practical!!!
• Need new algorithms and approaches

APPLICATIONS
• Evolutionary research
• Isolation of most relevant regions
• Characterization of protein families

MORE APPLICATIONS
• 3Dimentional structure prediction
• Phylogenetic Studies

PAPERS
MUSCLE: a Multiple Sequence Alignment Method with Reduced Time and Space Complexity
by Robert C. Edgar
ProbCons: Probabilistic Consistency-based Multiple Sequence Alignment
by Chuong B. Do, Michael Brudno, and Serafim Batzoglou

MUSCLE – OVERVIEW
• Basic Idea: A progressive alignment is built, to which horizontal refinement is applied
• 3 stages of the algorithm
• At the completion of each, a multiple alignment is available and the algorithm can be terminated
• Significant improvement in accuracy and speed

MUSCLE – THE ALGORITHM
Stage 1: Draft Progressive – Builds a progressive alignment
• Similarity of each pair of sequences is computed using
• K-mer counting
• Constructing a global alignment and determining fractional identity of the sequences
• A tree is constructed and a root is identified
• A progressive alignment is built by following the branching order of the tree, yielding a multiple alignment

MUSCLE – PROGRESSIVE ALIGNMENT
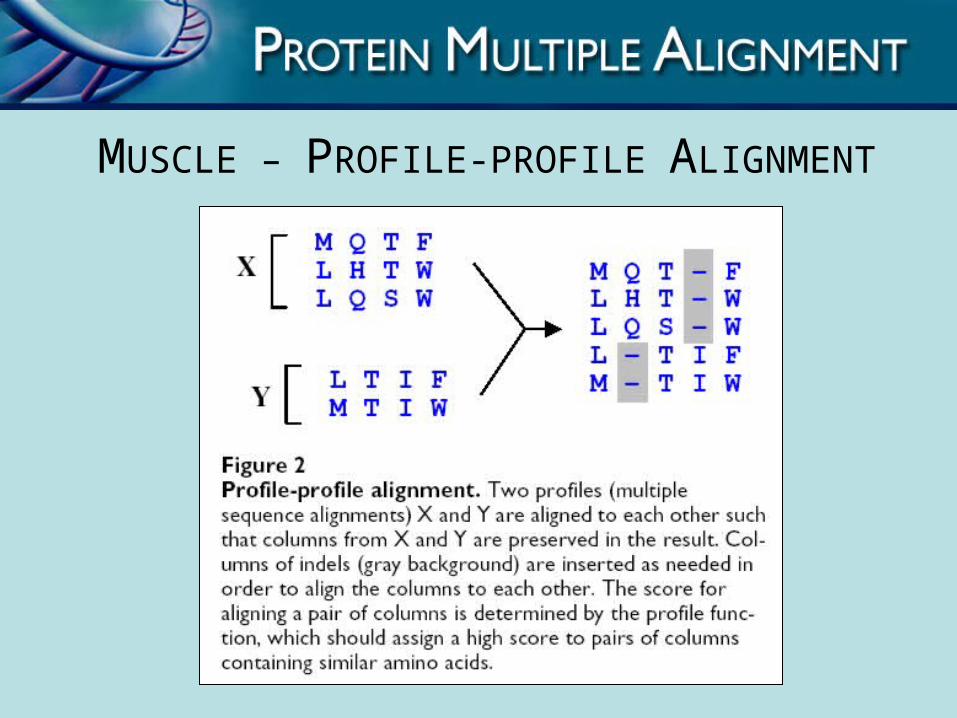
MUSCLE – PROFILE-PROFILE ALIGNMENT

MUSCLE – THE ALGORITHM
Stage 2: Improved Progressive – Improves the tree
• Similarity of each pair of sequences is computed using fractional identity from the mutual alignment
• A tree is constructed by applying a clustering method to the distance matrix
• The trees are compared; a set of nodes for which the branching order has changed is identified
• A new alignment is built, the existing one is retained if the order is unchanged

MUSCLE – TREE COMPARISON

MUSCLE – THE ALGORITHM
Stage 3: Refinement – Iterative Refinement is performed
• An edge is deleted from a tree, dividing the sequences into two disjoint subsets
• The profile (MA) of each subset is extracted
• The profiles are re-aligned to each other
• The score is computed, if the score has increased, the alignment is retained, otherwise it is discarded
• Algorithm terminates at convergence

MUSCLE – ITERATIVE REFINEMENT
S
T
U
X
Z
Delete this edge
Realign these resulting profiles to each other
S
T
U
X
Z

MUSCLE
Results:
• O(N2 + L2) Space and O(N4 + NL2) Time Complexity
• Improvements in selection of heuristics
• Close attention paid to implementation details
• Enables high-throughput applications to achieve good accuracy
• http://www.drive5.com/muscle

PROBCONS - OVERVIEW
• Alignment generation can be directly modeled as a first order Markov process involving state emissions and transitions
• Uses maximum expected accuracy alignment method
• Probabilistic consistency used as a scoring function
• Model parameters obtained using unsupervised maximum likelihood methods
• Incorporate multiple sequence information in scoring pairwise alignments

PROBCONS – HIDDEN MARKOV MODEL
• Deletion penalties on Match => Gap transitions
• Extension penalties on Gap => Gap transitions
• Match/Mismatch penalties on Match emissions

INSERT X INSERT Y
MATCH ABRACA-DABRAAB-ACARDI---
xxyy
xxiiyyjj
――yyjj
xxii
――
• Basic HMM for sequence alignment between two sequences
• M emits two letters, one from each sequence
• Ix emits a letter from x that aligns to a gap
• Iy emits a letter from y that aligns to a gap
PROBCONS – HIDDEN MARKOV MODEL

PROBCONS - MAXIMUM EXPECTED
ACCURACY
• LAZY TEACHER ANALOGY
• 10 students take a 10 question true/false quiz
• How do you make up the answer key?
1. Use the answers of the single best student (Viterbi Algorithm)
2. Use weighted majority rule (Maximum Expected Accuracy)

PROBCONS – MAXIMUM EXPECTED
ACCURACY
• Viterbi
• Picks a single alignment with the highest chance of being completely correct (analogous to Needleman-Wunch)
• Mathematically, finds the alignment a which maximizes
Ea*[1{a = a*}] (maximum probability alignment)
• Maximum Expected Accuracy
• Picks alignment with the highest expected number of correct predictions
• Mathematically, finds the alignment a which maximizes
Ea*[accuracy(a, a*)]

PROBCONS – COMPUTING MEA• Define accuracy (a, a*) = the expected
number of correctly aligned pairs of letters divided by the length of the shorter sequence
• The MEA alignment is found by finding the highest summing path through the matrix
Mxy[i, j] = P(xi is aligned to yj | x, y)
• We just need to compute these terms!
• Can use dynamic programming

z
x
y
xi
yj yj’
zk
PROBCONS – PROBABILISTIC CONSISTENCY

• Compute P(xi is aligned to yj | x, y)
P(xi is aligned to yj | x, y, z)
• We can re-estimate Mxy as (Mxz)•(Mzy) where z is a third sequence to which x and y are aligned
• Mxy[i,j] = ∑ Mxz[i.k] • Mzy(k,j), where n is the length of z
• We follow the alignment from position i of x, to position j of y, through all intermediate positions k of a third sequence z
PROBCONS – PROBABILISTIC CONSISTENCY
k = 1
n

• A straightforward generalization– sum-of-pairs– tree-based progressive alignment– iterative refinement
ABRACA-DABRAAB-ACARDI---ABRA---DABI-
AB-ACARDI---ABRA---DABI-
ABRACADABRAABRA--DABI-
ABRACA-DABRAAB-ACARDI---
ABRACA-DABRAAB-ACARDI---ABRA---DABI-
ABACARDIABRACADABRA
ABRACA-DABRAAB-ACARDI---
ABRADABI
ABRACA-DABRAAB-ACARDI---ABRA---DABI-
ABACARDI
ABRACADABRAABRA--DABI-
ABRACA-DABRAAB-ACARD--I-ABRA---DABI-
PROBCONS – MULTIPLE ALIGNMENT

PROBCONS – THE ALGORITHM
Step 1: Computation of posterior-probability matrices
• For every pair of sequences x, y compute the probability that letters xi yj are paired in a*, an alignment of x and y that is randomly generated by the model
Step 2: Computation of expected accuracies
• Define the expected accuracy of a pairwise alignment axy to be the expected number of correctly aligned pairs of letters divided by the length of the shorter sequence
• Compute the alignment axy that maximizes expected accuracy E(x,y) using dynamic programming

PROBCONS – THE ALGORITHM
Step 3: Probabilistic consistency transformation
• Re-estimate the scores with probabilistic consistency transformation by incorporating similarity of x and y to other sequences into the pairwise comparison of x and y
• Computed efficiently using sparse matrix multiplication ignoring all entries smaller than some threshold
Step 4: Computation of a guide tree
• Construct a tree by hierarchical clustering using E(x, y).
• Cluster similarity is defined by a weighted average of pairwise similarities between the clusters

PROBCONS – THE ALGORITHM
Step 5: Progressive Alignment
• Align sequence groups hierarchically according to the order specified in the guide tree
• Score using a sum of pairs function in which the aligned residues are scored according to the match quality scores and the gap penalties are set to 0
Step 6: Iterative Refinement
• Randomly partition alignment into two groups of sequences and realign.
• May be repeated as necessary

PROBCONS
Results:
• Best results so far
• Longer in running time due to the computation of posterior probability matrices (Step 1)
• Doesn’t incorporate biological information
• Could provide improved accuracy in DNA multiple alignment
• http://probcons.stanford.edu

PROBCONS

CONCLUSION
• Protein multiple alignment is a current research problem (both papers published in 2004)
• Many applications including evolutionary and phylogenetic studies, protein structure and classification
• Currently, there is some collaboration between the authors of MUSCLE and PROBCONS to create a new program which will combine the speed of MUSCLE-based tree construction and the accuracy that comes from using MEA and probabilistic consistency

REFERENCES
• Do, C.B., Brudno, M., and Batzoglou, S. PROBCONS: Probabilistic Consistency-based Multiple Alignment of Amino Acid Sequences. Submitted.
• Edgar, Robert C. (2004), MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Research 32(5), 1792-97.
• Some of these slides were adapted from Tom Do’s ISMB presentation on PROBCONS.

THANKS!


















