
Management of Transitional Cell Carcinoma of the … of Transitional Cell Carcinoma of the Lacrimal...
1
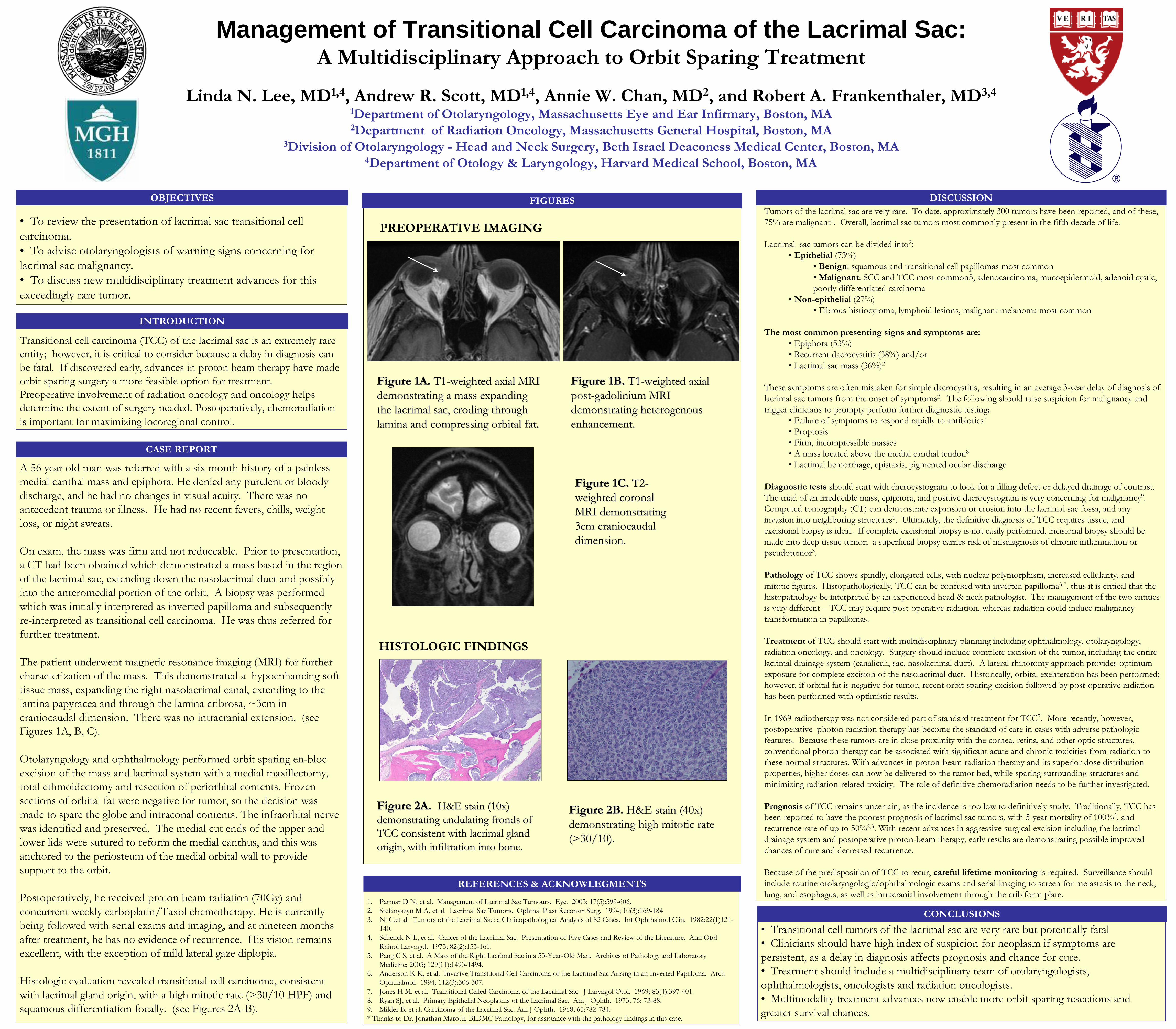
Management of Transitional Cell Carcinoma of the Lacrimal Sac: A Multidisciplinary Approach to Orbit Sparing Treatment Linda N. Lee, MD 1,4 , Andrew R. Scott, MD 1,4 , Annie W. Chan, MD 2 , and Robert A. Frankenthaler, MD 3,4 1 Department of Otolaryngology, Massachusetts Eye and Ear Infirmary, Boston, MA 2 Department of Radiation Oncology, Massachusetts General Hospital, Boston, MA 3 Division of Otolaryngology - Head and Neck Surgery, Beth Israel Deaconess Medical Center, Boston, MA 4 Department of Otology & Laryngology, Harvard Medical School, Boston, MA HISTOLOGIC FINDINGS A 56 year old man was referred with a six month history of a painless medial canthal mass and epiphora. He denied any purulent or bloody discharge, and he had no changes in visual acuity. There was no antecedent trauma or illness. He had no recent fevers, chills, weight loss, or night sweats. On exam, the mass was firm and not reduceable. Prior to presentation, a CT had been obtained which demonstrated a mass based in the region of the lacrimal sac, extending down the nasolacrimal duct and possibly into the anteromedial portion of the orbit. A biopsy was performed which was initially interpreted as inverted papilloma and subsequently re-interpreted as transitional cell carcinoma. He was thus referred for further treatment. The patient underwent magnetic resonance imaging (MRI) for further characterization of the mass. This demonstrated a hypoenhancing soft tissue mass, expanding the right nasolacrimal canal, extending to the lamina papyracea and through the lamina cribrosa, ~3cm in craniocaudal dimension. There was no intracranial extension. (see Figures 1A, B, C). Otolaryngology and ophthalmology performed orbit sparing en-bloc excision of the mass and lacrimal system with a medial maxillectomy, total ethmoidectomy and resection of periorbital contents. Frozen sections of orbital fat were negative for tumor, so the decision was made to spare the globe and intraconal contents. The infraorbital nerve was identified and preserved. The medial cut ends of the upper and lower lids were sutured to reform the medial canthus, and this was anchored to the periosteum of the medial orbital wall to provide support to the orbit. Postoperatively, he received proton beam radiation (70Gy) and concurrent weekly carboplatin/Taxol chemotherapy. He is currently being followed with serial exams and imaging, and at nineteen months after treatment, he has no evidence of recurrence. His vision remains excellent, with the exception of mild lateral gaze diplopia. Histologic evaluation revealed transitional cell carcinoma, consistent with lacrimal gland origin, with a high mitotic rate (>30/10 HPF) and squamous differentiation focally. (see Figures 2A-B). CASE REPORT DISCUSSION 1. Parmar D N, et al. Management of Lacrimal Sac Tumours. Eye. 2003; 17(5):599-606. 2. Stefanyszyn M A, et al. Lacrimal Sac Tumors. Ophthal Plast Reconstr Surg. 1994; 10(3):169-184 3. Ni C,et al. Tumors of the Lacrimal Sac: a Clinicopathological Analysis of 82 Cases. Int Ophthalmol Clin. 1982;22(1)121- 140. 4. Schenck N L, et al. Cancer of the Lacrimal Sac. Presentation of Five Cases and Review of the Literature. Ann Otol Rhinol Laryngol. 1973; 82(2):153-161. 5. Pang C S, et al. A Mass of the Right Lacrimal Sac in a 53-Year-Old Man. Archives of Pathology and Laboratory Medicine: 2005; 129(11):1493-1494. 6. Anderson K K, et al. Invasive Transitional Cell Carcinoma of the Lacrimal Sac Arising in an Inverted Papilloma. Arch Ophthalmol. 1994; 112(3):306-307. 7. Jones H M, et al. Transitional Celled Carcinoma of the Lacrimal Sac. J Laryngol Otol. 1969; 83(4):397-401. 8. Ryan SJ, et al. Primary Epithelial Neoplasms of the Lacrimal Sac. Am J Ophth. 1973; 76: 73-88. 9. Milder B, et al. Carcinoma of the Lacrimal Sac. Am J Ophth. 1968; 65:782-784. * Thanks to Dr. Jonathan Marotti, BIDMC Pathology, for assistance with the pathology findings in this case. REFERENCES & ACKNOWLEGMENTS PREOPERATIVE IMAGING Transitional cell carcinoma (TCC) of the lacrimal sac is an extremely rare entity; however, it is critical to consider because a delay in diagnosis can be fatal. If discovered early, advances in proton beam therapy have made orbit sparing surgery a more feasible option for treatment. Preoperative involvement of radiation oncology and oncology helps determine the extent of surgery needed. Postoperatively, chemoradiation is important for maximizing locoregional control. Figure 1A. Figure 1A. T1 T1 - - weighted axial MRI weighted axial MRI demonstrating a mass expanding demonstrating a mass expanding the lacrimal sac, eroding through the lacrimal sac, eroding through lamina and compressing orbital fat. lamina and compressing orbital fat. C Figure 1B. Figure 1B. T1 T1 - - weighted axial weighted axial post post - - gadolinium MRI gadolinium MRI demonstrating heterogenous demonstrating heterogenous enhancement. enhancement. INTRODUCTION • To review the presentation of lacrimal sac transitional cell carcinoma. • To advise otolaryngologists of warning signs concerning for lacrimal sac malignancy. • To discuss new multidisciplinary treatment advances for this exceedingly rare tumor. OBJECTIVES • Transitional cell tumors of the lacrimal sac are very rare but potentially fatal • Clinicians should have high index of suspicion for neoplasm if symptoms are persistent, as a delay in diagnosis affects prognosis and chance for cure. • Treatment should include a multidisciplinary team of otolaryngologists, ophthalmologists, oncologists and radiation oncologists. • Multimodality treatment advances now enable more orbit sparing resections and greater survival chances. CONCLUSIONS FIGURES Figure 2B Figure 2B . . H&E stain (40x) H&E stain (40x) demonstrating high mitotic rate demonstrating high mitotic rate (>30/10). (>30/10). Figure 2A. Figure 2A. H&E stain (10x) H&E stain (10x) demonstrating undulating fronds of demonstrating undulating fronds of TCC consistent with lacrimal gland TCC consistent with lacrimal gland origin, with infiltration into bone. origin, with infiltration into bone. Figure 1C. Figure 1C. T2 T2 - - weighted coronal weighted coronal MRI demonstrating MRI demonstrating 3cm craniocaudal 3cm craniocaudal dimension. dimension. Tumors of the lacrimal sac are very rare. To date, approximately 300 tumors have been reported, and of these, 75% are malignant 1 . Overall, lacrimal sac tumors most commonly present in the fifth decade of life. Lacrimal sac tumors can be divided into 2 : • Epithelial (73%) • Benign: squamous and transitional cell papillomas most common • Malignant: SCC and TCC most common5, adenocarcinoma, mucoepidermoid, adenoid cystic, poorly differentiated carcinoma • Non-epithelial (27%) • Fibrous histiocytoma, lymphoid lesions, malignant melanoma most common The most common presenting signs and symptoms are: • Epiphora (53%) • Recurrent dacrocystitis (38%) and/or • Lacrimal sac mass (36%) 2 These symptoms are often mistaken for simple dacrocystitis, resulting in an average 3-year delay of diagnosis of lacrimal sac tumors from the onset of symptoms 2 . The following should raise suspicion for malignancy and trigger clinicians to prompty perform further diagnostic testing: • Failure of symptoms to respond rapidly to antibiotics 7 • Proptosis • Firm, incompressible masses • A mass located above the medial canthal tendon 8 • Lacrimal hemorrhage, epistaxis, pigmented ocular discharge Diagnostic tests should start with dacrocystogram to look for a filling defect or delayed drainage of contrast. The triad of an irreducible mass, epiphora, and positive dacrocystogram is very concerning for malignancy 9 . Computed tomography (CT) can demonstrate expansion or erosion into the lacrimal sac fossa, and any invasion into neighboring structures 1 . Ultimately, the definitive diagnosis of TCC requires tissue, and excisional biopsy is ideal. If complete excisional biopsy is not easily performed, incisional biopsy should be made into deep tissue tumor; a superficial biopsy carries risk of misdiagnosis of chronic inflammation or pseudotumor 3 . Pathology of TCC shows spindly, elongated cells, with nuclear polymorphism, increased cellularity, and mitotic figures. Histopathologically, TCC can be confused with inverted papilloma 6,7 , thus it is critical that the histopathology be interpreted by an experienced head & neck pathologist. The management of the two entities is very different – TCC may require post-operative radiation, whereas radiation could induce malignancy transformation in papillomas. Treatment of TCC should start with multidisciplinary planning including ophthalmology, otolaryngology, radiation oncology, and oncology. Surgery should include complete excision of the tumor, including the entire lacrimal drainage system (canaliculi, sac, nasolacrimal duct). A lateral rhinotomy approach provides optimum exposure for complete excision of the nasolacrimal duct. Historically, orbital exenteration has been performed; however, if orbital fat is negative for tumor, recent orbit-sparing excision followed by post-operative radiation has been performed with optimistic results. In 1969 radiotherapy was not considered part of standard treatment for TCC 7 . More recently, however, postoperative photon radiation therapy has become the standard of care in cases with adverse pathologic features. Because these tumors are in close proximity with the cornea, retina, and other optic structures, conventional photon therapy can be associated with significant acute and chronic toxicities from radiation to these normal structures. With advances in proton-beam radiation therapy and its superior dose distribution properties, higher doses can now be delivered to the tumor bed, while sparing surrounding structures and minimizing radiation-related toxicity. The role of definitive chemoradiation needs to be further investigated. Prognosis of TCC remains uncertain, as the incidence is too low to definitively study. Traditionally, TCC has been reported to have the poorest prognosis of lacrimal sac tumors, with 5-year mortality of 100% 3 , and recurrence rate of up to 50% 2,3 . With recent advances in aggressive surgical excision including the lacrimal drainage system and postoperative proton-beam therapy, early results are demonstrating possible improved chances of cure and decreased recurrence. Because of the predisposition of TCC to recur, careful lifetime monitoring is required. Surveillance should include routine otolaryngologic/ophthalmologic exams and serial imaging to screen for metastasis to the neck, lung, and esophagus, as well as intracranial involvement through the cribiform plate.
Transcript of Management of Transitional Cell Carcinoma of the … of Transitional Cell Carcinoma of the Lacrimal...
Management of Transitional Cell Carcinoma of the Lacrimal Sac:A Multidisciplinary Approach to Orbit Sparing Treatment
Linda N. Lee, MD1,4, Andrew R. Scott, MD1,4, Annie W. Chan, MD2, and Robert A. Frankenthaler, MD3,4
1Department of Otolaryngology, Massachusetts Eye and Ear Infirmary, Boston, MA2Department of Radiation Oncology, Massachusetts General Hospital, Boston, MA
3Division of Otolaryngology - Head and Neck Surgery, Beth Israel Deaconess Medical Center, Boston, MA4Department of Otology & Laryngology, Harvard Medical School, Boston, MA
HISTOLOGIC FINDINGS
A 56 year old man was referred with a six month history of a painless medial canthal mass and epiphora. He denied any purulent or bloody discharge, and he had no changes in visual acuity. There was noantecedent trauma or illness. He had no recent fevers, chills, weight loss, or night sweats.
On exam, the mass was firm and not reduceable. Prior to presentation, a CT had been obtained which demonstrated a mass based in the region of the lacrimal sac, extending down the nasolacrimal duct and possibly into the anteromedial portion of the orbit. A biopsy was performed which was initially interpreted as inverted papilloma and subsequently re-interpreted as transitional cell carcinoma. He was thus referred for further treatment.
The patient underwent magnetic resonance imaging (MRI) for further characterization of the mass. This demonstrated a hypoenhancing soft tissue mass, expanding the right nasolacrimal canal, extending to the lamina papyracea and through the lamina cribrosa, ~3cm in craniocaudal dimension. There was no intracranial extension. (see Figures 1A, B, C).
Otolaryngology and ophthalmology performed orbit sparing en-bloc excision of the mass and lacrimal system with a medial maxillectomy, total ethmoidectomy and resection of periorbital contents. Frozen sections of orbital fat were negative for tumor, so the decision was made to spare the globe and intraconal contents. The infraorbital nerve was identified and preserved. The medial cut ends of the upper and lower lids were sutured to reform the medial canthus, and this was anchored to the periosteum of the medial orbital wall to providesupport to the orbit.
Postoperatively, he received proton beam radiation (70Gy) and concurrent weekly carboplatin/Taxol chemotherapy. He is currently being followed with serial exams and imaging, and at nineteen months after treatment, he has no evidence of recurrence. His vision remains excellent, with the exception of mild lateral gaze diplopia.
Histologic evaluation revealed transitional cell carcinoma, consistent with lacrimal gland origin, with a high mitotic rate (>30/10 HPF) and squamous differentiation focally. (see Figures 2A-B).
CASE REPORT
DISCUSSION
1. Parmar D N, et al. Management of Lacrimal Sac Tumours. Eye. 2003; 17(5):599-606.2. Stefanyszyn M A, et al. Lacrimal Sac Tumors. Ophthal Plast Reconstr Surg. 1994; 10(3):169-1843. Ni C,et al. Tumors of the Lacrimal Sac: a Clinicopathological Analysis of 82 Cases. Int Ophthalmol Clin. 1982;22(1)121-
140.4. Schenck N L, et al. Cancer of the Lacrimal Sac. Presentation of Five Cases and Review of the Literature. Ann Otol
Rhinol Laryngol. 1973; 82(2):153-161.5. Pang C S, et al. A Mass of the Right Lacrimal Sac in a 53-Year-Old Man. Archives of Pathology and Laboratory
Medicine: 2005; 129(11):1493-1494.6. Anderson K K, et al. Invasive Transitional Cell Carcinoma of the Lacrimal Sac Arising in an Inverted Papilloma. Arch
Ophthalmol. 1994; 112(3):306-307.7. Jones H M, et al. Transitional Celled Carcinoma of the Lacrimal Sac. J Laryngol Otol. 1969; 83(4):397-401.8. Ryan SJ, et al. Primary Epithelial Neoplasms of the Lacrimal Sac. Am J Ophth. 1973; 76: 73-88. 9. Milder B, et al. Carcinoma of the Lacrimal Sac. Am J Ophth. 1968; 65:782-784.* Thanks to Dr. Jonathan Marotti, BIDMC Pathology, for assistance with the pathology findings in this case.
REFERENCES & ACKNOWLEGMENTS
PREOPERATIVE IMAGING
Transitional cell carcinoma (TCC) of the lacrimal sac is an extremely rare entity; however, it is critical to consider because a delay in diagnosis can be fatal. If discovered early, advances in proton beam therapy have made orbit sparing surgery a more feasible option for treatment.Preoperative involvement of radiation oncology and oncology helps determine the extent of surgery needed. Postoperatively, chemoradiation is important for maximizing locoregional control.
Figure 1A. Figure 1A. T1T1--weighted axial MRI weighted axial MRI demonstrating a mass expanding demonstrating a mass expanding the lacrimal sac, eroding through the lacrimal sac, eroding through lamina and compressing orbital fat.lamina and compressing orbital fat.
C
Figure 1B. Figure 1B. T1T1--weighted axial weighted axial postpost--gadolinium MRI gadolinium MRI demonstrating heterogenous demonstrating heterogenous enhancement.enhancement.
INTRODUCTION
• To review the presentation of lacrimal sac transitional cell carcinoma.• To advise otolaryngologists of warning signs concerning for lacrimal sac malignancy.• To discuss new multidisciplinary treatment advances for this exceedingly rare tumor.
OBJECTIVES
• Transitional cell tumors of the lacrimal sac are very rare but potentially fatal• Clinicians should have high index of suspicion for neoplasm if symptoms are persistent, as a delay in diagnosis affects prognosis and chance for cure.• Treatment should include a multidisciplinary team of otolaryngologists, ophthalmologists, oncologists and radiation oncologists. • Multimodality treatment advances now enable more orbit sparing resections and greater survival chances.
CONCLUSIONS
FIGURES
Figure 2BFigure 2B.. H&E stain (40x) H&E stain (40x) demonstrating high mitotic rate demonstrating high mitotic rate (>30/10).(>30/10).
Figure 2A. Figure 2A. H&E stain (10x) H&E stain (10x) demonstrating undulating fronds of demonstrating undulating fronds of TCC consistent with lacrimal gland TCC consistent with lacrimal gland origin, with infiltration into bone. origin, with infiltration into bone.
Figure 1C. Figure 1C. T2T2--weighted coronal weighted coronal MRI demonstrating MRI demonstrating 3cm craniocaudal 3cm craniocaudal dimension.dimension.
Tumors of the lacrimal sac are very rare. To date, approximately 300 tumors have been reported, and of these, 75% are malignant1. Overall, lacrimal sac tumors most commonly present in the fifth decade of life.
Lacrimal sac tumors can be divided into2:• Epithelial (73%)
• Benign: squamous and transitional cell papillomas most common• Malignant: SCC and TCC most common5, adenocarcinoma, mucoepidermoid, adenoid cystic, poorly differentiated carcinoma
• Non-epithelial (27%) • Fibrous histiocytoma, lymphoid lesions, malignant melanoma most common
The most common presenting signs and symptoms are:• Epiphora (53%)• Recurrent dacrocystitis (38%) and/or • Lacrimal sac mass (36%)2
These symptoms are often mistaken for simple dacrocystitis, resulting in an average 3-year delay of diagnosis of lacrimal sac tumors from the onset of symptoms2. The following should raise suspicion for malignancy and trigger clinicians to prompty perform further diagnostic testing:
• Failure of symptoms to respond rapidly to antibiotics7
• Proptosis• Firm, incompressible masses• A mass located above the medial canthal tendon8
• Lacrimal hemorrhage, epistaxis, pigmented ocular discharge
Diagnostic tests should start with dacrocystogram to look for a filling defect or delayed drainage of contrast. The triad of an irreducible mass, epiphora, and positive dacrocystogram is very concerning for malignancy9. Computed tomography (CT) can demonstrate expansion or erosion into the lacrimal sac fossa, and any invasion into neighboring structures1. Ultimately, the definitive diagnosis of TCC requires tissue, and excisional biopsy is ideal. If complete excisional biopsy is not easily performed, incisional biopsy should be made into deep tissue tumor; a superficial biopsy carries risk of misdiagnosis of chronic inflammation or pseudotumor3.
Pathology of TCC shows spindly, elongated cells, with nuclear polymorphism, increased cellularity, and mitotic figures. Histopathologically, TCC can be confused with inverted papilloma6,7, thus it is critical that the histopathology be interpreted by an experienced head & neck pathologist. The management of the two entities is very different – TCC may require post-operative radiation, whereas radiation could induce malignancy transformation in papillomas.
Treatment of TCC should start with multidisciplinary planning including ophthalmology, otolaryngology, radiation oncology, and oncology. Surgery should include complete excision of the tumor, including the entire lacrimal drainage system (canaliculi, sac, nasolacrimal duct). A lateral rhinotomy approach provides optimum exposure for complete excision of the nasolacrimal duct. Historically, orbital exenteration has been performed; however, if orbital fat is negative for tumor, recent orbit-sparing excision followed by post-operative radiation has been performed with optimistic results.
In 1969 radiotherapy was not considered part of standard treatment for TCC7. More recently, however, postoperative photon radiation therapy has become the standard of care in cases with adverse pathologic features. Because these tumors are in close proximity with the cornea, retina, and other optic structures, conventional photon therapy can be associated with significant acute and chronic toxicities from radiation to these normal structures. With advances in proton-beam radiation therapy and its superior dose distribution properties, higher doses can now be delivered to the tumor bed, while sparing surrounding structures and minimizing radiation-related toxicity. The role of definitive chemoradiation needs to be further investigated.
Prognosis of TCC remains uncertain, as the incidence is too low to definitively study. Traditionally, TCC has been reported to have the poorest prognosis of lacrimal sac tumors, with 5-year mortality of 100%3, and recurrence rate of up to 50%2,3. With recent advances in aggressive surgical excision including the lacrimal drainage system and postoperative proton-beam therapy, early results are demonstrating possible improved chances of cure and decreased recurrence.
Because of the predisposition of TCC to recur, careful lifetime monitoring is required. Surveillance should include routine otolaryngologic/ophthalmologic exams and serial imaging to screen for metastasis to the neck, lung, and esophagus, as well as intracranial involvement through the cribiform plate.















![1 Tumors of Urinary Tract. 2 Urinary Tract Neoplasm KidneyRenal Cell Carcinoma [ adult], Transitional cell carcinoma [ adult], Wilms Tumor [children]](https://static.fdocuments.net/doc/165x107/56649f315503460f94c4c62c/1-tumors-of-urinary-tract-2-urinary-tract-neoplasm-kidneyrenal-cell-carcinoma.jpg)


