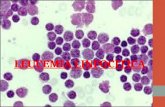
leucemia linfocitica cronica
78
-Caso clínico - • -Integrantes- Gabriel Cedillo García #48 Alejandra Leyva Flores #49 Javier Valencia Montes #47 Leticia Velázquez Fregoso #51 Luis Enrique Villanueva -Departamento de Clínica Aplicada- Universidad Autónoma de Guadalajara
-
Upload
letty-velazquez -
Category
Documents
-
view
6.209 -
download
3
description
Transcript of leucemia linfocitica cronica

-Caso clínico -
•-Integrantes- Gabriel Cedillo García #48Alejandra Leyva Flores #49Javier Valencia Montes #47
Leticia Velázquez Fregoso #51Luis Enrique Villanueva Martel
#52
-Departamento de Clínica Aplicada-
Universidad Autónoma de Guadalajara

-Interrogatorio-
Paciente masculino de 68 años de edad el cual desarrollo dolor abdominal severo en la región costal derecha el cual era continuo y no se aliviaba con paracetamol.
No tenia tos, o expectoración, y se encontraba perplejo de cual seria la causa del dolor ya que él se había sentido bien anteriormente y no había presentado ningún trauma en su tórax

Continuación…
Dos días después del inicio del dolor, el desarrollo un rash sobre la región costal derecha y decidió contactar a su medico general.
El negó la aparición de cualquier otro síntoma pero menciono que había notado aparición de una pequeña masa en el hueco axilar derecho, desde hace 4 meses. Aunque últimamente se había agrandado, no le causaba ninguna molestia y por eso no había buscado atención medica.

Continuación ….
El paciente no fuma, no toma, no ingiere drogas, trabajo por 40 años en una fabrica, actualmente esta jubilado.

-Exploracion Física-
Se encuentra paciente delgado, afebril el cual se encuentra refiriendo un dolor sobre un rash vesicular y eritematoso sobre el dermatoma T6 del lado derecho.
Había linfadenopatias generalizadas en región axilar e inguinal bilateral, indoloras.
Su frecuencia cardiaca era de 75 por minuto, regular, su presión sanguínea de 150/90 mmHg.

-Exploracion fisica-
Ruidos cardiacos normales, no soplos. Campos pulmonares limpios y bien ventilados.
En la exploración abdominal se encuentra bazo palpable 2cm por debajo del margen costal, no doloroso. Resto de la exploración normal.



-FOTOGRAFIAS-


-LABORATORIO Y GABINETE-

-Biometría Hemática-
Hb 10.7
VCM 93
HCM 34
Linfocitos 93%
Linfocitos segmentados 3%
Leucocitos 180 000
Basófilos 2%
Plaquetas 284 000

-Frotis Sangre Periférica-

Biopsia Medular Ósea: Muestra gran infiltrado linfocitico

-Inmunoglobulinas Séricas-
IgG 5.2 g/l (8-14)
IgM 0.3 g/l (0.5-2.5)
IgA 0.7 g/l (0.9-3.5)

• Pruebas funcionales hepáticas: Normales
• ES: Normales


-Leucemia linfocítica crónica /Herpes Zoster-

Etiología y epidemiología
Leticia Elizabeth Velázquez Fregoso #51
Universidad Autónoma de Guadalajara

-Definición-
Leucemia linfocítica crónica
Es una enfermedad en que los linfocitos morfológicamente maduros pero inmunológicamente incompetentes se acumulan progresivamente a nivel de sangre periférica, medula ósea y órganos linfoides.

-Etiología-
Cambios en los cromosomas
Por supresiones perdida de
alguna parte del cromosoma
Cromosoma: 13, 11 y 17
Cromosoma 12 adicional trisomía 12
Aun no esta claro los genes
que involucran ni exactamente
como producen leucemia
• Se desconoce la causa exacta

-Epidemiología-
• Tipo de leucemia mas frecuente en los países occidentales
• Europa y EUA .8% de todas las neoplasias y el 30% de las leucemias
• Frecuencia 6 veces menor en México a diferencia de las poblaciones caucásicas
• 40% individuos mayores de 65 años
• Edad media de presentación: 65 y 70 años
• Hombres/Mujeres 2:1

-Factores de Riesgo-
Ciertas exposiciones a sustancias químicas: agente naranja, actividades agrícolas y exposición a largo plazo a pesticidas
Antecedentes familiares 2 a 4 veces mas riesgo de LLC
Ser hombre, Raza étnica

Fisiopatología
•Luis Enrique Villanueva Martel #52
Universidad Autónoma de Guadalajara

• 95% de casos –LLC es una neoplasia de células B,
• las manifestaciones clínicas se deben a la infiltración progresiva:
• la médula ósea,
• ganglios linfáticos
• y otros tejidos por estos linfocitos inmunocompetentes


los linfocitos se acumulan en gran parte debido a una inhibición de la apoptosis secundaria a la sobreexpresión del gen BCL-2, aún cuando el reordenamiento de este gen es poco frecuente en esta enfermedad.
En la LLC se han observado altos niveles de la proteína BCL-2 hasta en más del 85 % de los casos.
Sin embargo, en un estudio reciente el gen estaba reordenado en sólo el 12 % de los enfermos.
Estos datos hacen pensar en diversos mecanismos genéticos, hasta el momento sin esclarecer, que pueden intervenir en la desregulación de este oncogén en la LLC


• Las células de la LLC expresan altos niveles de la proteína BCL-2
usualmente en ausencia.
• lo que las hace resistentes a la apoptosis y pueden facilitar su
acumulación progresiva, además baja expresión de Fas y ausencia de la proteína C-MYC.
También se ha señalado que ellas se encuentran detenidas en la fase Go de su ciclo celular.

• Son de pequeño tamaño, aspecto maduro y genotipo B (inmaduros)
• Se presenta hipogamaglobulinemia por afeccion de la extirpe linfatica y esto ocaciona suceptibilidad a organismos oportunistas
• . 5% de LLC – tumores de células T


• Las variaciones de los reguladores del ciclo celular: p53, p15, p16, se asocian generalmente con formas clínicas agresivas.
• o con la transformación en linfomas de células grandes.
• La inactivación del gen oncosupresor p53 se ha encontrado en un
pequeño porcentaje de pacientes (10-20 %)

Se han identificado dos tipos de LLC: -Tipo I pregerminal, en la que no se ha
producido una mutación de los genes de las cadenas pesadas de las inmunoglobulinas.
-Tipo II postgerminal, en el que se ha producido la mutación de los genes de las cadenas pesadas de las inmunoglobulinas y que tiene un claro mejor pronóstico


Fisiopatología de la anemia en la LLC
La fisiopatología de la anemia en los enfermos con LLC debe estudiarse desde varios puntos de vista.
• En los pacientes recién diagnosticados de LLC la anemia puede atribuirse a
diversas circunstancias entre las que cabe destacar:
• la hemólisis autoinmune (3 y el 37%)• la infiltración medular• el hiperesplenismo• La deficiencia o interferencia con factores hematopoyéticos • trastornos del metabolismo del hierro.

Hemólisis autoinmune
Aunque no está completamente aclarada la patogenia de dichos trastornos autoinmunes, se considera que la célula B neoplásica podría contribuir al desarrollo de los autoanticuerpos, que serían producidos por linfocitos B no leucémicos tras cooperación con linfocitos T autoreactivos específicos para el antígeno eritrocitario

-Hemólisis autoinmune-
Diversos estudios han establecido la relación entre la anemia hemolítica autoinmune y el tratamiento con análogos de las purinas. La prevalencia de anemia hemolítica autoinmune en pacientes de LLC tratados con fl udarabina oscila entre el 2% de los pacientes no tratados previamente y el 20% de los demás

Infiltración medular
La sustitución de la médula ósea normal por el acúmulode los linfocitos tumorales ha sido considerada tradicionalmente como un importante mecanismo etiopatogénico en el desarrollo de la anemia asociada a la LLC.
La explicación de este hecho puede derivarse de que la proporción de progenitores eritroides (BFU-E) en la médula ósea de los pacientes con LLC es significativamente más baja que la de los controles sanos,aunque puede volver a la normalidad en los pacientes que han recibido tratamiento quimioterápico


Esplenomegalia e Hiperesplenismo
La esplenomegalia con hiperesplenismo asociado se observa frecuentemente en los pacientes con LLC. Un estudio de casos y controles emparejó 55 pacientes que iban a ser esplenectomizados con 55 tratados con fludarabina.
Aunque no se observaron diferencias entre ambos grupos en cuanto a supervivencia, se observó una respuesta mayor en la cifra de hemoglobina en el 38% de los enfermos con anemia, siendo el peso del bazo el único factor predictivo para los incrementos de hemoglobina o de los neutrófilos.

-Esplenomegalia y Hepatomegalia-
La mortalidad en el tiempo periquirúrgico fue del 9%, por complicaciones sépticas que se asociaron a un mal estado general preoperatorio del paciente

Interferencia con factores hematopoyéticos
Con frecuencia el síndrome anémico en los pacientesdiagnosticados de LLC también se atribuye a la anemia de lasenfermedades crónicas o a la denominada “anemia secundaria alcáncer”. Ambas comparten buena parte de sus mecanismos fi siopatológicos, que estarían iniciados por la liberación de citoquinasespecífi cas como la interleuquina 1α (IL-1α), la interleuquina 1β(IL-1β), el factor de necrosis tisular (TNF), el interferón γ (INF-γ),el interferón β y la interleuquina 6. Varias de estas citoquinasse han observado aumentadas en el suero de los pacientes conLLC25.
Su efecto primario resulta en el acúmulo de los linfocitosneoplásicos en la médula ósea, pero también juegan un papel enla inhibición de la eritropoyesis por distintos mecanismos.

Así, la IL-1, el INF-γ y el TNF son capaces de inhibir el crecimiento de lasBFU-E y CFU-E. Al igual que en la anemia de las enfermedadescrónicas, el hierro queda bloqueado a nivel del sistema reticuloendotelial,impidiéndose la síntesis de hemoglobina merced alaumento de esas citoquinas. Por otra parte, la síntesis de eritropoyetina(EPO) queda seriamente disminuida por la IL-1, el INF-γy el TNF. Finalmente, el TNF induce cambios diseritropoyéticosen la médula ósea y favorece la eritrofagocitosis, acortando asíla supervivencia media de los hematíes.

Alejandra Leyva Flores #49
Universidad Autónoma de Guadalajara
-Cuadro clínico y diagnóstico-

50% pacientes asintomáticos
80% linfadenopatí
as
50% hepatomegali
a / esplenomegal
ia
Inespecíficos: cansancio
fácil, anorexia, mal
estado general
Episodios hemorrágicos
Infecciones frecuentes
5% Síndrome de Richter
McPhee Stephen J et al. “Diagnóstico Clínico y Tratamiento”. LANGE. Editorial Mc Grax Hill. 46° Edición. 2007. Pág 522 – 523

-Cuadro clínico-
Fiebre, sudoración y pérdida de peso no son habituales.
Dolor óseo Anemia hemolítica autoinmune o
trombocitopenia autoinmunitaria (5% a 10%)
En los estadios más avanzados puede además producirse la infiltración de órganos no linfoides (próstata, el hígado, tubo digestivo, pulmón, la pleura o el sistema nervioso central)

-Cuadro clínico-
Infiltración cutánea es rara y cuando se presenta, es más frecuente que sea reactiva que infiltrativa.
En raras ocasiones se ha comprobado la coexistencia de LLC y síndrome nefrótico, por glomerulonefritis membranosa

-Herpes zoster-
Dolor intenso que puede preceder a la aparición del exantema
Desarrollo de maculopápulas y evolución a vesículas y pústulas
Las lesiones siguen la distribución de cualquier raíz nerviosa (torácicas y lumbares más afectadas).
Dermatoma unilateral


Frecuentemente de manera
incidental
LINFOCITOSIS en sangre periférica y médula ósea
Marcadores: CD19, CD20, CD24, T CD5
Trombocitopenia (menos
frecuente)
Neutropenia
Anemia normocítica
normocrómica
Inmunocitoquímica
Inmunogenética
DIAGNÓSTICO

-Diagnóstico-

-Diagnóstico-

Diagnóstico de herpes zoster
Clínico Preparación de Tzanck: se
observan células gigantes multinucleadas.
Anticuerpos IgM específicos del VVZ PCR Cultivo del virus en líneas
celulares susceptibles para aislar las partículas víricas

Diagnostico diferenciales y laboratorio
Héctor Gabriel Cedillo García #48
Universidad Autónoma de Guadalajara

-Diagnóstico diferenciales-
LEUCEMIA PROLINFOCITICA B EXAMEN MORFOLÓGICO E INMUNOFENOTIPO
DOBLE POBLACION CELULAR DE LINFOCITOS + CELS. PROLINFOCITOIDES VS CUADRO MONOMÓRFICO + PREDOMINANCIA DE PROLINFOCITOS + INMUNOFENOTIPO CON ELEVADA EXPRESION DE Ig DE SUPERFICIE CD79b Y FMC7

-Diagnósticos diferenciales-
REACCION LEUCEMOIDE MENOR CANTIDAD DE BLASTOS,
NO HAY HIPOGAMMAGLOBULIEMIA Y LA BIOPSIA DE MEDULA OSEA NO MUESTRA ALTERACIONES

-Laboratorio// examen hematológico -
LEUCOCITOSIS (>15x109/L) LINFOCITOSIS SUPERIOR AL 75%
PUEDE HABER ELEVACION DE AC. URICO Y LDH
HIPOGAMMAGLOBULINEMIA MUY FRECUENTE (20% - 60%)
PLACA SIMPLE DE TORAX

ASPIRADO DE MEDULA OSEA CON INFILTRADO LINFOIDE SUPERIOR AL 30%
Los linfocitos son de pequeño tamaño, con un núcleo redondeado, cromatina condensada en grumos y escaso citoplasma, puede haber sombras nucleares de Gumprecht

Javier Valencia Montes #47
Tratamiento y pronóstico
Universidad Autónoma de Guadalajara

-Tratamiento-
Estadios 0 a II sin marcadores de mal pronosticoPuede mantenerse en observación, y cuando la enfermedad avanza inician el tratamiento.
Estadios 0 a II, uno o más factores de mal pronóstico, estadios III a IV, enfermedad avanzada Tratamiento

-Indicaciones para iniciar tratamiento-
1. Evidencia de insuficiencia de la médula ósea progresiva
2. Esplenomegalia masiva o progresiva
3. Enfermedad con adenopatía voluminosa o adenopatía e incremento
4. Linfocitosis progresiva con un incremento de más del 50%
5.Anemia hemolítica o trombocitopenia autoimmunitaria con escasa respuesta a corticoesteroides
6. Tener al menos 1 de los siguientes síntomas: • pérdida de peso no intencional ≥ 10% dentro de los seis meses
previos • fatiga significativa • fiebre ≥ 38°C por 2 semanas sin signos de infección • diaforesis nocturna sin indicios de infección

-Opciones de tratamiento-
Fármacos: -Clorambucil -Fludarabina o fludarabina con
ciclofosfamida -Fludarabina con ciclofosfamida y rituximab -Alemtuzumab
Trasplante de médula alógena
Trasplante de células progenitoras hematopoyéticas

Fludarabina o fludarabina con ciclofosfamida
Se administra vía IV Dosis de 25 mg/m2 DSC diario durante 5 días Tasa de remisión completa de 15% Si se combina con ciclofosfamida, varían del
30 al 40% fludarabina durante 3 días, y ciclofosfamida 250 mg/m2/día por vía IV por 3 días
El esquema se repite cada 4 semanas durante 6 ciclos.
*citopenias puede obligar a la suspensión temporal o definitiva del tx

Fludarabina con ciclofosfamida y rituximab(FCR)
En dosis de 25 mg/m2 de superficie corporal, por vía IV diaria durante 3 días
Rituximab en dosis de 375 mg/m2de superficie corporal IV, en el primer día del esquema en cada ciclo
Ciclofosfamida en dosis de 250 mg /m2 de superficie corporal IV al día por 3 días.
El rituximab esquemas 1era y 2da linea
La respuesta es superior

Clorambucil
Agente alquilante Tasas de remisión completa <10%,
alta tendencia a la recurrencia. Se da combinado con prednisona En no tolerancia a la vía IV Presentación: tabletas de 2 mg Dosis recomendada:0.1 mg/kg/día
por 4, 5 o 7 días cada mes durante 6 u 8 ciclos

Alemtuzumab
El anticuerpo monoclonal humanizado anti-CD52 es uno de los agentes más promisorios
Tasa de respuesta global cercana al 80% con remisiones duraderas
Destruye los linfocitos B y T
*se acompaña de un deterioro inmunitario intenso

Trasplante de médula alógena
Jóvenes, sin respuesta o con recaída temprana Recaída en un lapso de 24
meses después de haber respondido al tratamiento combinado de análogos de purinas o trasplante autólogo
Pacientes con anomalías en p53 que requieren tratamiento
*El único tratamiento con potencial curativo

Criterios de respuesta al tratamiento
La respuesta es completa cuando se normaliza la citometría hemática y desaparecen los síntomas y signos de la enfermedad.
La médula ósea debe ser normocelular y tener menos de 30% de linfocitos.
Fracaso: enfermedad estable, enfermedad sin respuesta, la enfermedad progresiva y la muerte

Terapia de apoyo
Aciclovir y TMP sulfametoxazol Herpes zoster o Pneumocystis
carinii(oportunistas) Estar alerta ante otras posibles
infecciones oportunistas 1. La inmunoglobulina G intravenosa 2. Eritropoyetina 3. Factores estimulantes de colonias
de granulocitos Las citopenias autoinmunitarias se
manejan con glucocorticoides

Tratamiento del herpes zoster
Abreviar el curso clínico.
Aliviar el dolor. Prevenir
complicaciones. Reducir la
incidencia de neuralgia posherpética.
Los antivirales acortan el curso, disminuyen el dolor, reducen las complicaciones y protegen a un individuo inmunocomprometido.

AINE Y Narcotic
osAciclovir Cortico esteroid
es
Ribavirina
Antidepresivos
tricíclicos
OTROS: anticonvulsivantes, fototerapia, oxigenación hepervarica.

Acetaminofeno adultos a razón de 325-650 mg VO cada 6 horas, o 1000 mg dos veces al día, sin exceder los 4 g diarios.
Niños: <12 años: 10-15 mg/kg/dosis VO cada 4 a 6 horas hasta unmáximo de 2,6 g diarios; >12 años:650 mg, cada 4 horas hasta un máximo de 5 dosis, en 24 horas.
La capsaicina tópica
al 0.025% o al 0.075%, tres veces al día.
Compresas conacetato de aluminio al 5% (solución deBurow) para reducir el dolor durante30 a 60 minutos
4 a 6 veces al día.Los baños de avena coloidal, bañoso lociones de fécula y loción de calamina
ayudan a aliviar la picazóny la molestia.

AciclovirEl aciclovir tópico no se recomiendaya que su efectividad es mínima.
Adultos: 800 mg VO cada 4 horas(5 dosis x día) durante 7 a 10 días.
Niños: 250-600 mg/m2/dosis VO 4a 5 veces al día durante 7 a 10 días.
Prednisona: reducir la inflamación y elriesgo de neuralgia posherpética.
Dosis en adultos: 60 mg/día reducidagradualmente durante tres semanas
Niños: 0,05 a 2 mg/kg VO fraccionadaen dos dosis, reducida gradualmenteen dos semanas
Amitriptilina en dosis de 50 a 75 mg al día

-Clasificación y Pronóstico-
Estadio de Rai Características Mediana de supervivencia desde el diagnóstico (meses)
0 Bajo riesgo Sólo linfocitosis (en sangre y médula)
Mayor de 150 meses
I Riesgo intermedio Linfocitos más crecimiento de ganglios
101
II Linfocitosis más crecimiento del bazo, del hígado o de ambos, con o sin crecimiento ganglionar
71
III Alto riesgo Linfocitosis más anemia (hemoglobina < 11 g/L), con o sin crecimiento en ganglios, bazo e hígado
19
IV Linfocitosis más trombocitopenia (plaquetas < 100 X 109/L, con o sin anemia o crecimiento en ganglios, bazo e hígado
19

Estadio de
Binet
Definición Mediana de supervivencia
(meses)
A Pacientes con menos de tres áreas linfoides implicadas (cervical, axilar, inguinal y unilateral o bilateral para bazo e hígado), con valores de Hb > 10 g/dL y plaquetas de > 100 x 109/L
115
B Al menos tres áreas linfoides implicadas y con Hb > 10 g /dL y plaquetas de > 100 x 109/L
84
C Hb < 10 g/dL o plaquetas < 100 x 109/L, o ambos, independientemente de las áreas linfoides implicadas
24

Factores para establecer el pronóstico
Estadio 0-I de Rai: 10
años
El estado no mutado del gen de las
cadenas pesadas en su región variable (IgVH) es un factor
adverso
La proteína asociada a 70-kD zeta, o ZAP-70 es un marcador
pronóstico vinculado con el estado no
mutado.
Citometría de flujo y en muestras de sangre
periférica o médula ósea es el indicador
más práctico del pronóstico

-Bibliografía-
Jules L.Dienstag, Kurt J.Isselbache, Medicina Interna de Harrison, Ed.Mc Graw Hill, capitulo 97. neoplasias malignas de las celulas linfoides
Raúl Cano Castellanos, Martha Alvarado Ibarra, Emma Álvarez Pantoja , “Primer consenso en leucemia linfocítica crónica de la Agrupación Mexicana para el Estudio de la Hematología: epidemiología, diagnóstico y tratamiento”, Medicina Universitaria 2009 Volumen 10, Núm. 40
Porfirio Hernández Ramírez. “Leucemia linfoide crónica. Aspectos clínicos y biológicos”. Revista Cubana de Hematología, Inmunología y Hemoterapia. Volumen 15. Número 1 Ciudad de la Habana. 1999
McPhee Stephen J et al. “Diagnóstico Clínico y Tratamiento”. LANGE. Editorial Mc Grax Hill. 46° Edición. 2007. Pág 1384

GRACIAS!!!


















