Lecture 1 BNFO 601 Usman Roshan. Course overview Perl progamming language (and some Unix basics)...
26
Lecture 1 BNFO 601 Usman Roshan
-
date post
19-Dec-2015 -
Category
Documents
-
view
219 -
download
0
Transcript of Lecture 1 BNFO 601 Usman Roshan. Course overview Perl progamming language (and some Unix basics)...

Lecture 1
BNFO 601
Usman Roshan

Course overview
• Perl progamming language (and some Unix basics)– Unix basics– Intro Perl exercises– Dynamic programming and Viterbi algorithm in Perl
• Sequence analysis– Algorithms for exact and heuristic pairwise alignment– Hidden Markov models– BLAST– Short read and genome alignments
• Genome-wide association studies – SNPs– Population structure– Case control studies

Overview (contd)
• Grade: Two 25% exams, 35% final exam, and two 15% projects
• Exams will cover Perl and bioinformatics algorithms
• Recommended Texts:– Introduction to Bioinformatics Algorithms by Pavel
Pevzner and Neil Jones– Biological sequence analysis by Durbin et. al.– Introduction to Bioinformatics by Arthur Lesk– Beginning Perl for Bioinformatics by James Tisdall– Introduction to Machine Learning by Ethem Alpaydin
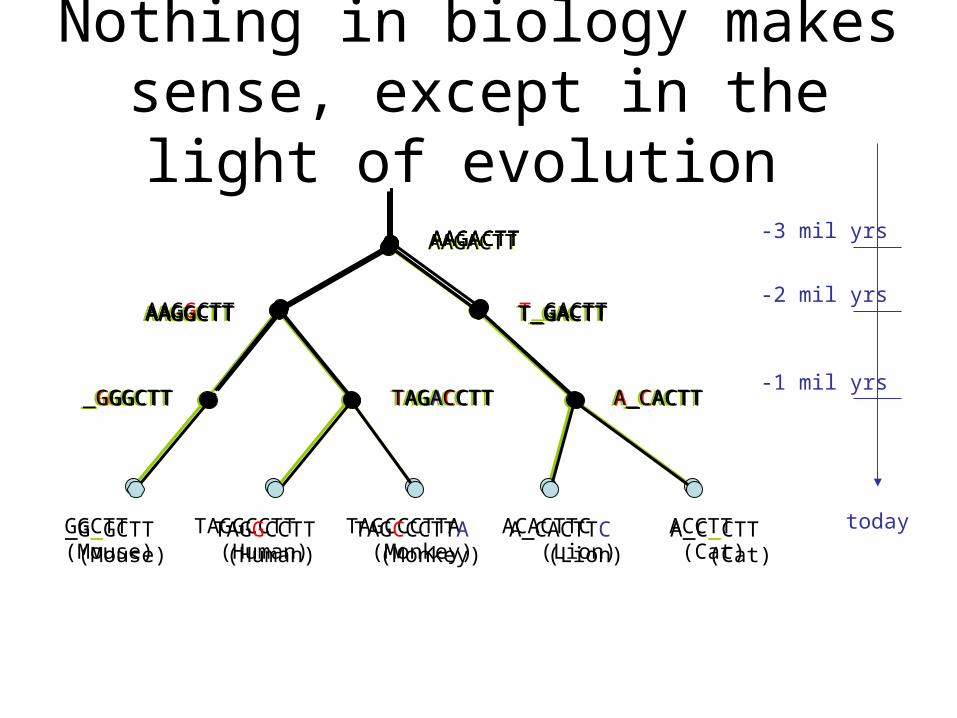
Nothing in biology makes sense, except in the light of evolution
AAGACTT -3 mil yrs
-2 mil yrs
-1 mil yrs
today
AAGACTT
T_GACTTAAGGCTT
_GGGCTT TAGACCTT A_CACTT
ACCTT (Cat)
ACACTTC (Lion)
TAGCCCTTA (Monkey)
TAGGCCTT (Human)
GGCTT(Mouse)
T_GACTTAAGGCTT
AAGACTT
_GGGCTT TAGACCTT A_CACTT
AAGGCTT T_GACTT
AAGACTT
TAGGCCTT (Human)
TAGCCCTTA (Monkey)
A_C_CTT (Cat)
A_CACTTC (Lion)
_G_GCTT (Mouse)
_GGGCTT TAGACCTT A_CACTT
AAGGCTT T_GACTT
AAGACTT

Representing DNA in a format manipulatable by computers
• DNA is a double-helix molecule made up of four nucleotides:– Adenosine (A)– Cytosine (C)– Thymine (T)– Guanine (G)
• Since A (adenosine) always pairs with T (thymine) and C (cytosine) always pairs with G (guanine) knowing only one side of the ladder is enough
• We represent DNA as a sequence of letters where each letter could be A,C,G, or T.
• For example, for the helix shown here we would represent this as CAGT.
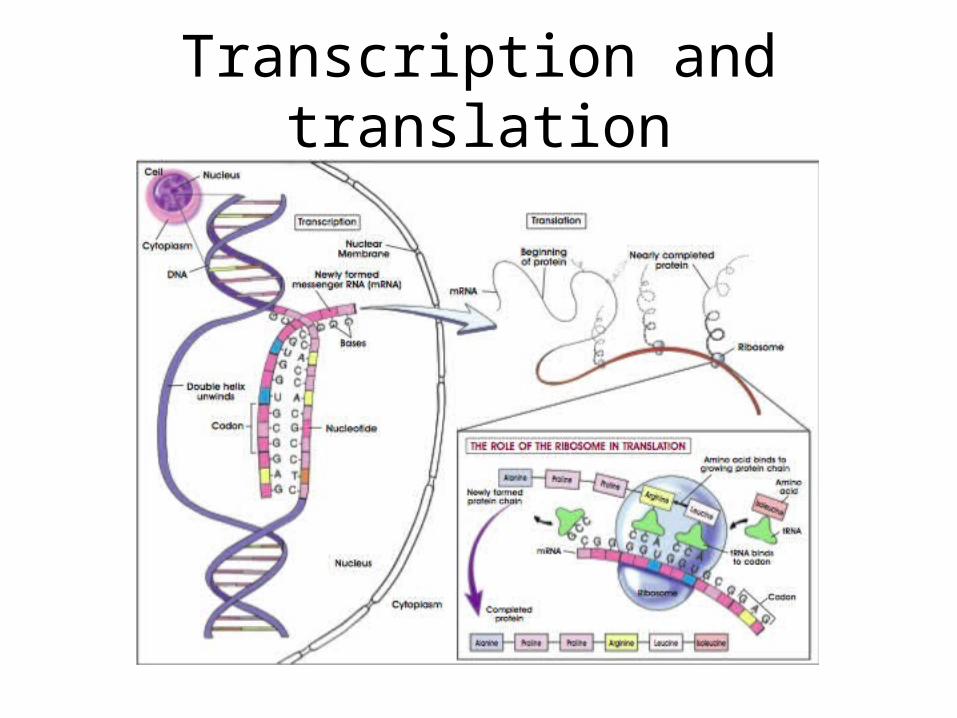
Transcription and translation
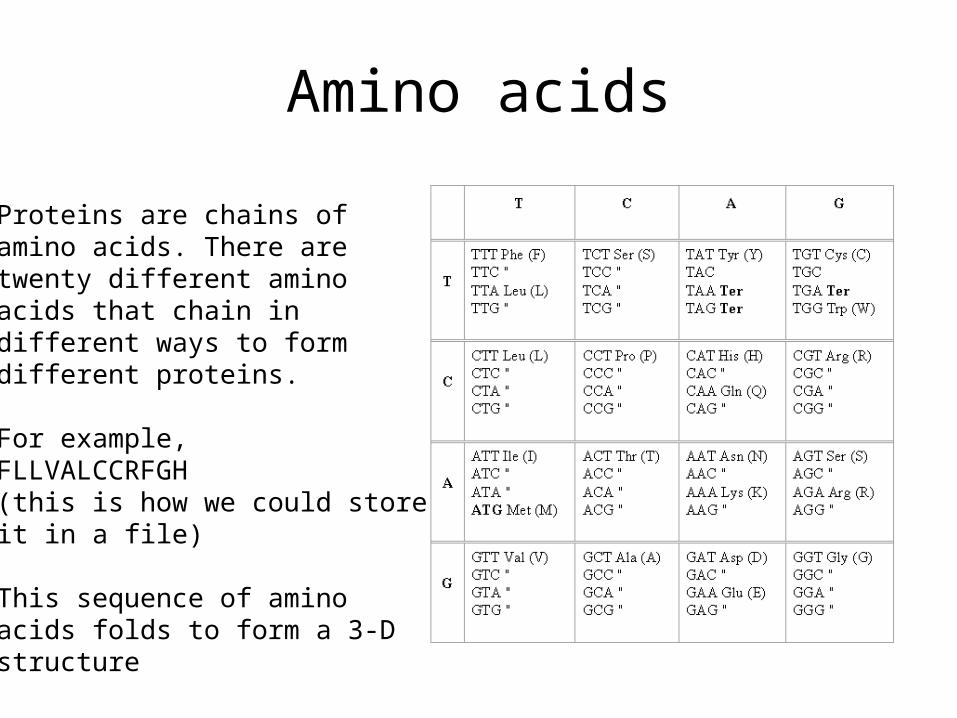
Amino acids
Proteins are chains ofamino acids. There aretwenty different aminoacids that chain indifferent ways to formdifferent proteins.
For example,FLLVALCCRFGH (this is how we could storeit in a file)
This sequence of aminoacids folds to form a 3-Dstructure
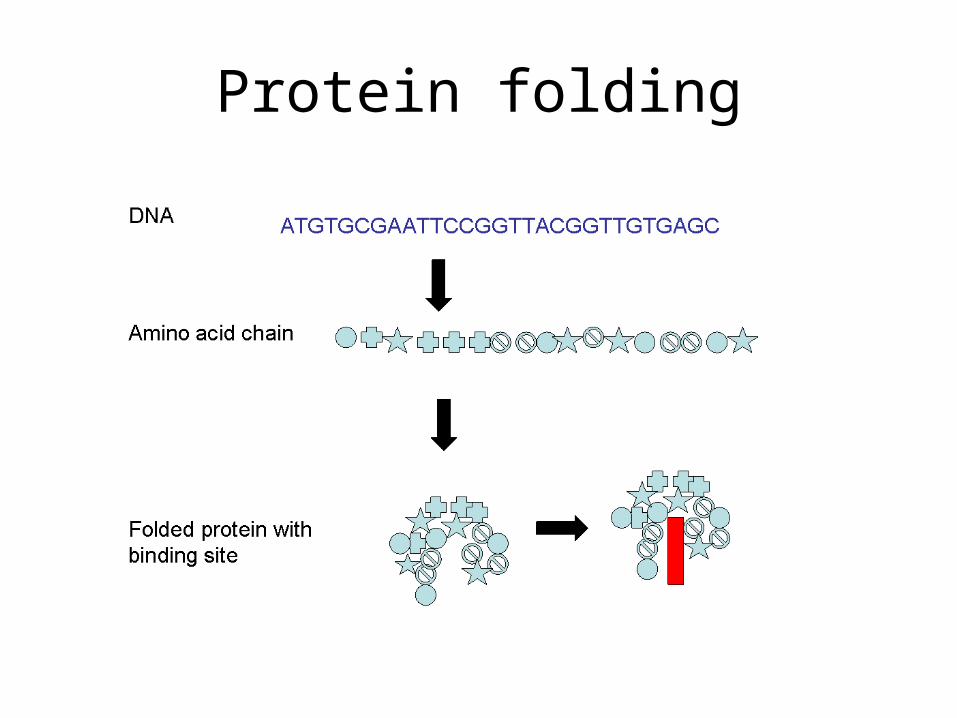
Protein folding

Protein folding
• The protein foldingproblem is to determinethe 3-D protein structurefrom the sequence.• Experimental techniquesare very expensive. • Computational are cheap but difficult to solve. • By comparing sequences we can deduce the evolutionary conserved portions which are also functional (most of the time).
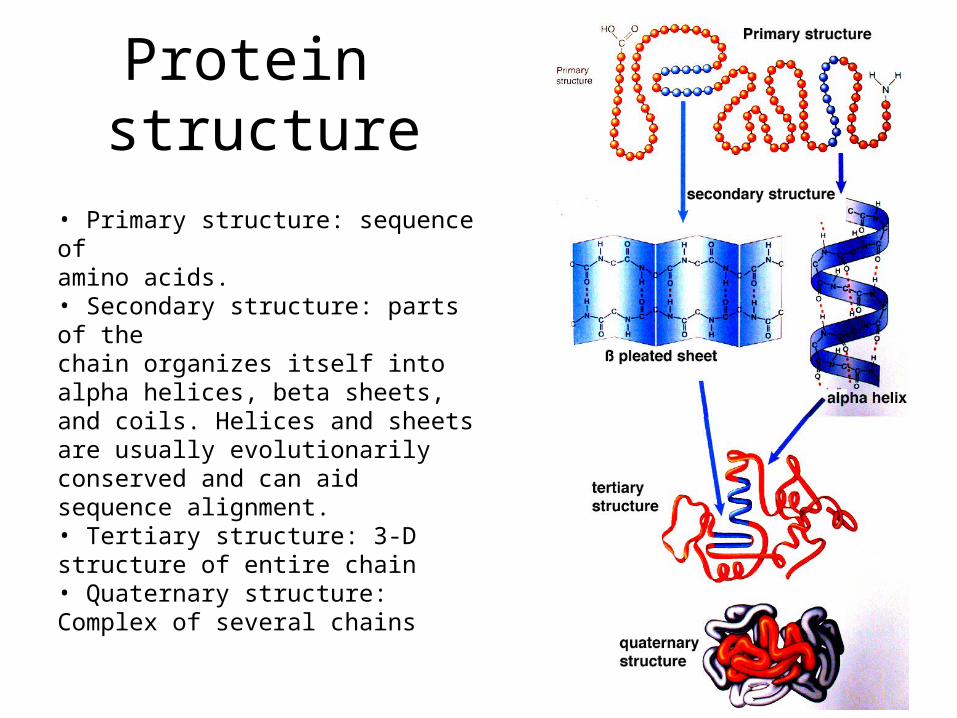
Protein structure
• Primary structure: sequence ofamino acids.• Secondary structure: parts of thechain organizes itself into alpha helices, beta sheets, and coils. Helices and sheets are usually evolutionarily conserved and can aid sequence alignment.• Tertiary structure: 3-D structure of entire chain• Quaternary structure: Complex of several chains

Key points
• DNA can be represented as strings consisting of four letters: A, C, G, and T. They could be very long, e.g. thousands and even millions of letters
• Proteins are also represented as strings of 20 letters (each letter is an amino acid). Their 3-D structure determines the function to a large extent.

Pairwise sequence alignment
• How to align two sequences?

Pairwise alignment
• How to align two sequences?• We use dynamic programming• Treat DNA sequences as strings over the
alphabet {A, C, G, T}

Pairwise alignment

Dynamic programming
Define V(i,j) to be the optimal pairwise alignment score between S1..i and T1..j (|S|=m, |T|=n)

Dynamic programming
Time and space complexity is O(mn)
Define V(i,j) to be the optimal pairwise alignment score between S1..i and T1..j (|S|=m, |T|=n)

How do we understand this dynamic programming algorithm?
• Let’s first look at some example alignments
• Let’s look at gaps. How do we know where to insert gaps
• Let’s look at the structure of an optimal alignment of two sequences x and y and how it relates optimal alignments of subsequences of x and y

How do we pick gap parameters?

Structural alignments
• Recall that proteins have 3-D structure.

Structural alignment - example 1
Alignment of thioredoxins fromhuman and fly taken from theWikipedia website. This proteinis found in nearly all organismsand is essential for mammals.
PDB ids are 3TRX and 1XWC.

Structural alignment - example 2
Computer generated aligned proteins
Unaligned proteins.2bbm and 1top areproteins from fly andchicken respectively.
Taken from http://bioinfo3d.cs.tau.ac.il/Align/FlexProt/flexprot.html

Structural alignments
• We can produce high quality manual alignments by hand if the structure is available.
• These alignments can then serve as a benchmark to train gap parameters so that the alignment program produces correct alignments.

Benchmark alignments
• Protein alignment benchmarks– BAliBASE, SABMARK, PREFAB,
HOMSTRAD are frequently used in studies for protein alignment.
– Proteins benchmarks are generally large and have been in the research community for sometime now.
– BAliBASE 3.0

Biologically realistic scoring matrices
• PAM and BLOSUM are most popular
• PAM was developed by Margaret Dayhoff and co-workers in 1978 by examining 1572 mutations between 71 families of closely related proteins
• BLOSUM is more recent and computed from blocks of sequences with sufficient similarity

PAM
• We need to compute the probability transition matrix M which defines the probability of amino acid i converting to j
• Examine a set of closely related sequences which are easy to align---for PAM 1572 mutations between 71 families
• Compute probabilities of change and background probabilities by simple counting

Next week
• Basics of Unix
• Perl programming– Basics– Exercises– Dynamic programming alignment solution in
Perl