
KUOPION YLIOPISTON JULKAISUJA G. - A.I. VIRTANEN ... · PDF fileIn addition, tangential flow...
102
Lentiviral Vectors for Gene Therapy Targeting, Imaging and Baculovirus Mediated New Method for Lentivirus Production Doctoral dissertation To be presented by permission of the Faculty of Medicine of the University of Kuopio for public examination in Auditorium, Tietoteknia building, University of Kuopio, on Friday 22 nd May 2009, at 12 noon Department of Biotechnology and Molecular Medicine A.I. Virtanen Institute for Molecular Sciences University of Kuopio HANNA LESCH JOKA KUOPIO 2009 KUOPION YLIOPISTON JULKAISUJA G. - A.I. VIRTANEN -INSTITUUTTI 73 KUOPIO UNIVERSITY PUBLICATIONS G. A.I. VIRTANEN INSTITUTE FOR MOLECULAR SCIENCES 73
Transcript of KUOPION YLIOPISTON JULKAISUJA G. - A.I. VIRTANEN ... · PDF fileIn addition, tangential flow...

Lentiviral Vectors for Gene Therapy
Targeting, Imaging and Baculovirus MediatedNew Method for Lentivirus Production
Doctoral dissertation
To be presented by permission of the Faculty of Medicine of the University of Kuopio
for public examination in Auditorium, Tietoteknia building, University of Kuopio,
on Friday 22nd May 2009, at 12 noon
Department of Biotechnology and Molecular MedicineA.I. Virtanen Institute for Molecular Sciences
University of Kuopio
HANNA LESCH
JOKAKUOPIO 2009
KUOPION YLIOPISTON JULKAISUJA G. - A.I. VIRTANEN -INSTITUUTTI 73KUOPIO UNIVERSITY PUBLICATIONS G.
A.I. VIRTANEN INSTITUTE FOR MOLECULAR SCIENCES 73

Distributor : Kuopio University Library P.O. Box 1627 FI-70211 KUOPIO FINLAND Tel. +358 40 355 3430 Fax +358 17 163 410 http://www.uku.fi/kirjasto/julkaisutoiminta/julkmyyn.shtml
Series Editors: Professor Olli Gröhn, Ph.D. Department of Neurobiology A.I . Virtanen Institute for Molecular Sciences
Professor Michael Courtney, Ph.D. Department of Neurobiology A.I . Virtanen Institute for Molecular Sciences
Author’s address: Department of Biotechnology and Molecular Medicine A.I . Virtanen Institute for Molecular Sciences University of Kuopio P.O. Box 1627 FI-70211 KUOPIO FINLAND E-mail : [email protected]
Supervisors: Professor Seppo Ylä-Herttuala, M.D., Ph.D. Department of Biotechnology and Molecular Medicine A.I . Virtanen Institute for Molecular Sciences University of Kuopio
Professor Kari Airenne, Ph.D. Department of Biotechnology and Molecular Medicine A.I . Virtanen Institute for Molecular Sciences University of Kuopio Reviewers: Docent Jarmo Wahlfors, Ph.D. Academic Development Unit University of Tampere
Docent David Mottershead, Ph.D. Robinson Institute University of Adelaine, Australia
Opponent: Professor Markku Kulomaa, Ph.D. MSK, ETH Zürich, Biologisch-Orientierte Materialwissenschaft Zürich, Switzerland Institute of Medical Technology University of Tampere
ISBN 978-951-27-1132-1ISBN 978-951-27-1113-0 (PDF)ISSN 1458-7335
KopijyväKuopio 2009Finland

Lesch, Hanna. Lentiviral vectors for gene therapy: Targeting, imaging and baculovirus mediated new method for lentivirus production. Kuopio University Publications G. - A.I. Virtanen Institute for Molecular Sciences 73. 2009. 99 p. ISBN 978-951-27-1132-1 ISBN 978-951-27-1113-0 (PDF) ISSN 1458-7335 ABSTRACT
In gene therapy, no optimal strategies are yet available to achieve targeted in vivo gene delivery. Lentiviruses are promising tools for gene therapy due to their unusual property of efficiently transducing both dividing and non-dividing cells and achieving long term expression of therapeutic genes. In this study, we describe versatile lentiviruses for targeting and imaging purposes. Lentivirus vectors displayed (strept)avidin fused to the transmembrane anchor of VSV-G on the virus envelope. The vectors were conjugated by biotinylated ligands and antibodies selectively binding to receptors expressed at high levels on cancer cells. In the in vitro targeting studies, the target cell-specific transduction of lentivirus was increased. In addition, using human ferritin as a marker, the streptavidin-displaying lentivirus could be simultaneously imaged for virus biodistribution by single photon emission computed tomography (SPECT) with expression monitored by non-invasive magnetic resonance imaging (MRI).
Another approach examined in this study was to utilize avidin fused to the endocytotic low density lipoprotein receptor in pretargeting experiments. In order to further extend the flexibility of the system, an avidin fusion protein expressing lentivirus was prepared. The results demonstrated that the virus transduced efficiently cells in vitro and in vivo, were safe to use and the receptor bound biotinylated ligands. However, an immune response was detected against the virus and transgene.
Production of replication defective lentiviral vectors on the clinical scale is challenging. As the first step towards scalable lentiviral production system, four recombinant baculoviruses were constructed. The production of lentiviruses using baculovirus technology was achieved and the production conditions were further optimized. These lentiviruses transduced cells efficiently in vitro and showed sustained GFP expression. In addition, tangential flow filtration (TFF) in lentivirus purification was tested and the results showed that TFF purification decreased DNA and protein contaminants. In order to further improve the baculovirus system, we developed a new recombinant baculovirus carrying an EGFP expression cassette in its genome (F-bacmid). This novel baculovirus vector enabled an easy and efficient virus generation, high throughput screening and fast titering in a 96-well format.
In conclusion, the study demonstrates the usefulness of lentivirus and avidin fusion proteins for several targeted therapy applications offering also a versatile tool for imaging purposes. Baculoviruses could be considered as a feasible option for the production of lentiviruses on a clinical scale. National Library of Medicine Classification: QU 55.4, QU 195, QU 300, WL 307, QU 475, QZ 52, QU 470, QW 168.5.H6, QU 55.7, WN 185, QY 60.R6, QZ 380 Medical Subject Headings: Avidin; Baculoviridae; Biotin; Cell Line; Cells, Cultured; Choroid Plexus; Drug Delivery Systems; Gene Expression; Gene Targeting; Gene Therapy; Gene Transfer Techniques; Transduction, Genetic; Genetic Vectors; Glioma; HIV-1; Lentivirus; Membrane Glycoproteins; Magnetic Resonance Imaging; Rats; Receptors LDL; Brain; Tomography, Emission-Computed, Single-Photon; Transcription, Genetic; Viral Envelope Proteins; Viral Fusion Proteins


To my dear husband
Kimi and To our two lovely daughters Oona and Aada


ACKNOWLEDGEMENTS This thesis was carried out in the Department of Biotechnology and Molecular Medicine, A. I. Virtanen Institute for Molecular Sciences, University of Kuopio, during the years 2002-2009. I wish to express my gratitude to my supervisor Professor Seppo Ylä-Herttuala, MD, PhD. for allowing me to be involved in such an exciting research group and sharing his knowledge of science, and to Professor Kari Airenne, PhD. for introducing me to the field of baculoviruses and sharing in discussions through these years. I owe my sincere thanks to the reviewers of this thesis, Docent Jarmo Wahlfors, PhD. and Docent David Mottershead, PhD. for their careful revision and valuable comments improving the thesis. For the language revision, I wish to thank Ewen MacDonald, PhD. During these years when I was completing my thesis I have had the honor to share many important moments with a number of people. Especially, I want to express my warmest thanks to all of my co-authors. The contribution to the studies made by Minna Kaikkonen has been beyond compare and I also appreciate all the meaningful discussions and support during these years. Together with Hanna-Riikka Kärkkäinen, we worked with baculoviruses, but the coffee breaks have been also unforgettable. I wish to thank Jere Pikkarainen for his invaluable input to the animal studies. Moreover, I am thankful to the three of you for the friendship we have created. In addition, I am grateful for Sanna Turpeinen, Einari Niskanen, Miia Roschier, Antti Määttä, Miia Taavitsainen for their important contributions to the molecular cloning. To Anssi Mähönen, I owe my sincere thanks for introducing me to the field of gene therapy and baculoviruses. In addition, I owe thanks to Olli Laitinen for his invaluable knowledge of avidins, to Jani Räty for his input in avidin displaying viruses, to Ale Närvänen and Tuulia Huhtala for SPECT/CT studies, to Olli Gröhn, Teemu Laitinen and Pasi Tuunanen for help with the MRI studies and to Pyry Toivanen for the VEGFs. I wish to acknowledge also Haritha Samaranayake and Taina Vuoria for their input in the animal studies and the immunostainings, Ann-Marie Määttä, Pauliina Lehtolainen-Dalcilic and Thomas Wirth for their contributions to the Lodavin project, Lauri Laitinen and Mikko Karjalainen for sharing the purification process with me and Minna Nokelainen for introducing me to the industrial side of gene therapy. The working spirit at the Ark during these years has been unique. Without such an inspiring and supporting environment, I would have given up. I would like the mention Emilia Makkonen, Diana Schenkwein, Tytteli Turunen, Roseanne Girnary, Vesa Turkki, Tommi Heikura, Tiina Nieminen and Mikko Turunen for making the atmosphere so warm. I also thank Diana and Vesa for kindly providing me with Figure 3B. I am also grateful for the people from SYH-group; I have always been able to obtain help when I had needed it. In addition, I am thankful to Ville Harjulampi, Eero Paanenen and Risto Feodoroff for helping

me with the IT-issues, and Marja Poikolainen, Helena Pernu, Saija Paukkunen, Johanna Pirinen, Anne Kainulainen, Katri Nikkinen and Jenni Tuovinen for their secretarial help. Without excellent technical help, I could not have completed this study. I would like to thank Tarja Taskinen, Juha Ruuskanen, Siiri Väistö, Joonas Malinen, Erik Peltomaa, Jaana Siponen, Katja Salminen, Anne Martikainen, Anneli Miettinen, Mervi Nieminen, Seija Sahrio for excellent technical assistance. In my memory, there is also place for Riikka Eisto, who I will always be missing. If I had not been working in this group, I would never have met my two close colleagues, Sari Kukkonen and Mervi Riekkinen. I would like to thank these dear friends for being my supporting pillars and for sharing in my laughs and tears. In addition, I wish to thank all my friends and Kimi´s family members for the shared moments. Finally, I dedicate my warmest thanks to my family. My parents, Riitta and Arvi, for their love and endless support and also for giving us a helping hand when our family needed it. The most heartfelt thanks I want to give to my husband Kimi for standing there by my side all these years, sharing the highs and lows of this work. Thank you for loving me and being there for me. Our lovable daughters, Oona and Aada, you have brought sunshine and meaning into my life and I will always love you. Kuopio, May 2009 Hanna Lesch This study was supported by Ark Therapeutics Oy, Academy of Finland, EU Clinigene (LSHB-CT-2006-018933) Consortium Flexibility Funds, European Union (LHSB-CT-2006-037541), the Finnish Foundation for Cardiovascular Research and the Finnish Cultural Foundation, North Savo Regional Fund. Thanks also to Perkin Elmer, Lappeenrannan PT Rakennus Oy, Biofellows Oy, Immuno Diagnostics Oy and Oligomer Oy.

ABBREVIATIONS AAV Adeno-associated virus AcMNPV Autographa californica multiple nuclear polyhedrovirus ADA Adenosine deaminase ADA-SCID Adenosine deaminase deficiency leading to severe combined immunodeficiency AIDS Acquired immunodeficiency syndrome ALS Amyotrophic lateral sclerosis AVD Avidin ALV Avian leukosis virus AP Alkaline phosphatase BIV Bovine immunodeficiency virus BPL Biotin protein ligase BV Budded virus CAEV Caprine Arthritis/Encephalitis Virus CAG The CMV early enhancer/chicken �-actin promoter CAR Coxsackie-adenovirus receptor CD Cytosine deaminase CGD Chronic granulomatous disease CMV Cytomegalovirus CNS Central nervous system cPPT Central polypurine track CT Computed tomography DAF Decay-accelerating factor DMEM Dulbecco's Modified Eagle's Medium DTPA Diethylene triamine pentaacetic acid EGF Epidermal growth factor EGFP Enhanced green fluorescent protein EGFR Epidermal growth factor receptor EIAV Equine Infectious Anemia Virus ELISA Enzyme-linked immunosorbent assay EMEA European Medicines Evaluation Agency FACS Fluorescence-activated cell sorting FDA Food and Drug Administration FIV Feline Immunodeficiency Virus GALV Gibbon ape leukemia virus GFP Green fluorescent protein GDNF Glial-derived neurotrophic factor GMP Good Manufacturing Practice GP64 The major envelope glycoprotein of the AcMNPV HEK 293 Human embryonic kidney cell line hHF Human heavy chain ferritin HIV Human immunodeficiency virus hPGK Human phosphoglycerate kinase promoter

HRP Horseradish peroxidase HSV-1 Herpes simplex virus type 1 i.c. Intracerebral (administration route) IE genes Immediate early genes Ifu Infectious units IRES Internal ribosome entry site i.v. Intravenous (administration route) Kb Kilo base pairs LDLR Low density lipoprotein receptor LTR Long terminal repeats MOI Multiplicity of infection MRI Magnetic resonance imaging MLV Murine leukemia virus ODV Occlusion derived virus PBS Phosphate buffered saline PCR Polymerase chain reaction PEG Polyethylene glycol PEI Polyethylenimine PET Positron emission tomography PIC Pre-integration complex Polh Polyhedrin promoter PRE Post-transcriptional regulatory element qPCR Quantitative polymerase chain reaction RCL Replication competent lentivirus RD114 Feline endogenous virus RT Reverse transcriptase SA Streptavidin SCID Severe combined immunodeficiency SCID-X1 X-linked combined immunodeficiency disease shRNA Short hairpin RNA molecules SIV Simian immunodeficiency virus SPECT Single photon emission computed tomography TA cloning Cloning of a linearized DNA with complementary 3' thymidine overhangs TFF Tangential flow filtration TK Thymidine kinase TU Transducing unit USPIO Ultra-small superparamagnetic particles of iron oxide VEGF Vascular endothelial growth factor VSV-G Vesicular stomatitis virus glycoprotein G VP Viral particles WPRE Woodchuck hepatitis virus post-transcriptional regulatory element

LIST OF ORIGINAL PUBLICATIONS This study is based on the following articles, which are referred to in the body of the text by the corresponding Roman numerals (I-IV): I Kärkkäinen HR, Lesch HP, Määttä AI, Mähönen AJ, Roschier MM, Toivanen PI,
Airenne KJ, Laitinen OH and Ylä-Herttuala S. A 96-well format for a high-throughput baculovirus generation, fast titering and recombinant protein production. BMC Res Notes. 2009 Apr 23;2(1):63. In Press
II Lesch HP, Turpeinen S, Niskanen E, Mähönen AJ, Roschier M, Airenne KJ, Ylä-
Herttuala S. Generation of lentivirus vectors using recombinant baculoviruses. Gene Ther. 2008 Sep;15(18):1280-6.
III Kaikkonen MU*, Lesch HP*, Räty JK, Pikkarainen J, Närvänen A, Gröhn O,
Airenne KJ, Ylä-Herttuala S. (Strept)avidin-displaying Lentiviruses as Versatile Tools for Targeting and Dual-imaging of Gene Delivery. Gene Therapy. 2009. In press.
IV Lesch HP *, Pikkarainen J*, Kaikkonen MU*, Taavitsainen M, Lehtolainen P,
Samaranayake H, Vuorio T, Määttä AM, Wirth T, Airenne KJ, Ylä-Herttuala S. Avidin fusion protein expressing lentiviral vector for targeted drug delivery. Human Gene Therapy. 2009. Accepted for publication
*Equal contribution. This thesis contains also unpublished data.


TABLE OF CONTENTS
1. INTRODUCTION ...................................................................................................... 15 2. REVIEW OF THE LITERATURE ......................................................................... ...16 2.1. Gene Therapy ........................................................................................................... 16 2.2. Viral vectors ............................................................................................................. 17 2.2.1. Retroviruses ........................................................................................................... 18 2.2.2. Lentiviruses ........................................................................................................... 19
2.2.2.1. Vector development........................................................................................ 20 2.2.2.2. Lentivirus production ..................................................................................... 22 2.2.2.3. Lentivirus purification .................................................................................... 23 2.2.2.4. Lentivirus titering and analysis of replication competent lentiviruses ........... 24 2.2.2.5. Lentiviral gene therapy applications............................................................... 25
2.2.3. Baculoviruses ........................................................................................................ 26 2.2.3.1. Baculovirus vector development .................................................................... 27 2.2.3.2. Baculovirus production, purification and titering .......................................... 29 2.2.3.3. Baculovirus applications ................................................................................ 29
2.2.4. Other viral vectors ................................................................................................. 31 2.2.4.1. Adenovirus ..................................................................................................... 31 2.2.4.2. Poxviruses....................................................................................................... 32 2.2.4.3. Herpes simplex virus ...................................................................................... 32 2.2.4.4. Adeno-associated virus................................................................................... 33
2.2.5. Non-viral vectors ................................................................................................... 34 2.3. Targeted therapy .................................................................................................... ..36 2.3.1 Pretargeting therapy of cancer................................................................................ 37 2.3.2. Gene-directed enzyme prodrug therapy ................................................................ 37 2.3.3. Physical targeting techniques ................................................................................ 38 2.3.4. Viral vector targeting............................................................................................. 38
2.3.4.1. Genetic targeting concept ............................................................................... 39 2.3.4.2. Pseudotyping .................................................................................................. 40 2.3.4.3. Serotype switch .............................................................................................. 41 2.3.4.4. Adaptor targeting concept .............................................................................. 42
2.4 Avidin-Biotin technology.......................................................................................... 44 2.4.1. Avidin and Streptavidin..................................................................................... 44 2.4.2. Biotin and biotinylation ..................................................................................... 45 2.4.3. Avidin and biotin in pretargeting ...................................................................... 46 2.4.4. Avidin-biotin technology in gene therapy applications..................................... 48
2.5. Imaging of viral biodistribution and transgene expression ...................................... 49 2.5.1. Magnetic Resonance Imaging (MRI) ................................................................ 50 2.5.2. PET and SPECT ................................................................................................ 50 2.5.3. Other imaging techniques.................................................................................. 51

4. AIMS OF THE STUDY ............................................................................................. 53 5. MATERIALS AND METHODS................................................................................ 54 5.1. Methods .................................................................................................................... 54 5.2. Plasmids.................................................................................................................... 55 5.3. Viral vectors ............................................................................................................. 56 5.4 Cell lines .................................................................................................................... 57 5.5. Purification of lentiviruses ....................................................................................... 57 5.6. Animal experiments.................................................................................................. 58 5.7. Primary Antibodies and ligands ............................................................................... 58 5.8. Secondary Antibodies............................................................................................... 59 6. THE RESULTS AND DISCUSSION ........................................................................ 60 6.1. Improved method for high-throughput titering and generation of baculoviruses and recombinant protein production in a 96-well plate format (I) ......................................... 60 6.2. Lentiviral vector production using recombinant baculoviruses (II) ......................... 63
6.2.1. The production of lentiviral vectors .................................................................. 63 6.2.2. The characterization of produced lentiviral vectors .......................................... 64 6.2.3. Future aspects of baculovirus mediated production .......................................... 66
6.3. Removal of residual baculoviruses from the purified lentivirus preparation ......... 67 6.4. Lentiviral vector purification by tangential flow filtration....................................... 68 6.5. (Strept)avidin displaying lentiviral vectors for different gene therapy applications (III) .................................................................................................................................. 70
6.5.1 Construction and characterization of the (strept)avidin displaying lentivirus.... 71 6.5.2. SPECT/CT imaging of streptavidin displaying lentiviral vectors for biodistribution (III) ...................................................................................................... 73 6.5.3. MRI of streptavidin displaying lentiviral vectors for transgene expression(III)74 6.5.4. Targeting of (strept)avidin displaying lentiviral vector (III) ............................. 75
6.6. Avidin fusion-protein expressing lentivirus for targeted therapy (IV)..................... 76 6.6.1 Titering of the avidin fusion protein expressing lentivirus................................. 77 6.6.2. Analysis of avidin fusion protein expressing lentivirus in vitro........................ 78 6.6.3. Antibody response against the lentivirus and transgene.................................... 80 6.6.4. In vivo expression .............................................................................................. 81
7. SUMMARY AND CONCLUSIONS ......................................................................... 82
8. REFERENCE LIST………………………...………………………………………..84

15
1. INTRODUCTION The aim of gene therapy is to efficiently transfer new genetic material into the cells of a patient. For clinical use, an optimal gene delivery vehicle must be both safe and efficient. The development of this new generation of therapy began two decades ago but to a large extent it is still at the experimental level. Though disorders arising from a single mutation are the prime candidates for gene therapy, multigene or multifactoral disorders are also potential targets for gene therapy.
Over the course of evolution, viruses have devised very efficient techniques for delivering their own nucleic acids into cells. Scientists have taken advantage of this feature and today viruses are utilized as a tool in gene therapy. Different viruses have their own characteristics, such as duration of gene expression, transduction mechanism or cell tropism. Furthermore, different diseases have their own requirements for treatment. Consequently, a single viral vector will not suffice for all gene therapy applications, and therefore, it is essentiall to develop several vectors. One concern raised by the use of viral vectors is the possible development of an immune response against the vector. Furthermore, targeting the therapy to the desired locations within the patient poses its own challenges.
In this work, lentiviral vectors utilizing avidin-biotin technology were developed for use in cancer treatment. Both targeted lentivirus delivery and targeted drug delivery after pretargeting by lentivirus were developed and tested. The surface display of avidin enabled the simultaneous imaging of viral biodistribution and the quantification of transgene expression. In addition, baculovirus technology was further improved and a novel production method for lentiviral vectors was developed. These results expand the potential use of lentiviral vectors in human gene therapy.

16
Cardiovascular diseases 9.3 % (n=137)
Monogenic diseases 8.2 % (n=120)
Gene marking 3.4 % (n=50)Other diseases 2.2 % (n=32) Healthy volunteers 2.2 % (n=32)
Ocular diseases 0.8 % (n=12)Neurological diseases 1.2 % (n= 17)
Infectious diseases 7.6. % (n= 112)
Cancer diseases 65.2 % (n=960)
2. REVIEW OF THE LITERATURE
2.1. Gene Therapy It is hoped that gene therapy will revolutionize the treatment of many diseases,
especially those caused by a single mutation. The first approved gene therapy trial was performed in 1989. In that case, the children suffered a rare disorder called severe combined immunodeficiency (SCID) caused by adenine deaminase deficiency. These young patients were treated with gene therapy and after the treatment, they were able to live a normal life. Since that time, the gene therapy field has expanded to treat also complex diseases that arise from the actions of multiple genes. Despite some clinical successes, the technology is still in its infancy. The immune responses against the vector, the possibility of causing cancer by insertional mutagenesis and the lack of targetability are the main safety concerns of gene therapy. Researchers all over the world are striving to improve the safety of these systems. In order to monitor the safety of the patients, the gene therapy trials are strictly controlled by national medicine agencies. In addition, gene therapy needs to meet the tight requirements and legislations as demanded of other drugs according to the Food and Drug Administration (FDA) and the European Medicines Evaluation Agency (EMEA). These include that there is adherence to the principals of current Good Manufacturing Practice (GMP) in the production of the vectors. Thus far, almost 1500 clinical trials in humans have been conducted (September 2008) (http://www.wiley.co.uk/genetherapy/clinical/) and the number is rising. Cancer related diseases account for the largest number (65%) of all clinical trials treated by gene therapy (figure 1). Cardiovascular diseases are the most common cause of death in western countries and 9% of all the clinical trials have been directed against these diseases and 8 % against monogenic diseases.
Figure 1. Indication addressed by gene therapy clinical trials (September 2008) modified from (http://www.wiley.co.uk/genetherapy/clinical/).

17
2.2. Viral vectors
Viruses interact with the host cell surface and transfer their genetic material into the cells where the viral genes are transcribed as a part of the viral replication cycle. In gene therapy, all or a portion of the viral genes have been replaced with the therapeutic gene. Viruses are divided into two classes depending on their replication cycle; the lytic and non-lytic viruses. Non-lytic viruses, such as retroviruses or lentiviruses, infect and exploit the machinery of cells without killing them. In contrast, infection by lytic viruses, such as herpes simplex viruses or Semliki Forest viruses, leads to eventual cell death. Another approach is to employ non-viral vectors, which means that the gene is delivered simply as a naked nucleic acid or as a nucleic acid complexed to a carrier. The most commonly used gene transfer vehicles in clinical trials have been adenovirus or retrovirus vectors. However, the delivery of naked DNA has also been widely used in gene therapy trials (Figure 2). The first gene-based products for sale in China were Gendicine™ by Shenzhen SiBiono Genetech Co.,Ltd. in 2003 and Oncorine™ by Shanghai Sunway Biotech in 2006. Both of these are based on adenovirus vectors. However, at present, EMEA and FDA have not granted marketing permission for any gene therapy products in western countries. The following chapters will review the commonly used gene delivery vehicles, with a focus on the viruses relevant to this thesis. The final section will summarize the main points in targeted therapy. Figure 2. Vectors used in gene therapy clinical trials (September 2008) modified from (http://www.wiley.co.uk/ genetherapy/clinical/)
adenovirus 24.9 % (n=367)
Retrovirus 21.7 % (n=320)
RNA transfer 1.4 % (n=21)
Lentivirus 1.2% (n=18)
Other categories 4.1 % (n=60)
Herpes simplex virus 3.2 % (n=47)
Unknown 3.3 % (n=41)
Naked/ Plasmid DNA 18.3% (n=270)
Vaccinia Virus 8.2 % (n=120)
Lipofection 7.1 % (n=105)
Poxvirus 6.1% (n=90)
Adeno-associated virus 4.1 % (n= 60)

18
2.2.1. Retroviruses
Retroviruses belong to the large Retroviridae family. The virions are roughly 100 nm in diameter, spherical and the outer layer is a lipid envelope displaying viral glycoproteins. Each particle contains two copies of the linear viral RNA genome (approximately 10 kb) which contains three essential genes, gag, pol and env. The pol gene encodes three viral enzymes: the protease, reverse transcriptase, and integrase. The gag gene encodes the structural proteins: the capsid, matrix, and nucleocapsid. Proteins are generated by proteolytic cleavage of the gag-pol precursor. The Env gene encodes the envelope glycoproteins of the virus. After the retrovirus enters the target cell, the viral genome is converted into the double-stranded DNA form by reverse transcriptase (RT). Proviral genome is then integrated into the genome of the target cell by the integrase. Viral long terminal repeats (LTRs) are important for the initiation of viral DNA synthesis, integration and regulation of viral transcription (Goff.S.P., 2001).
The most extensively used group of retroviruses in gene transfer used has been the oncogenic retroviruses. These include murine leukemia virus (MLV), spleen necrosis virus, Rous sarcoma virus, and avian leukosis virus, with the MLV being the most commonly used. Lentiviruses represent more complex retroviruses and are discussed in more details in the next chapter. The third group of retroviruses is the spumaviruses. These have been used less extensively in gene therapy (Coffin et al., 1997). Over 300 clinical trials for retroviruses had been registered by 2008 (Journal of Gene Medicine, http://www.wiley.co.uk/genetherapy/clinical/).
The ability of the retrovirus to integrate and achieve long term expression has made them attractive tools for gene therapy (Tolstoshev, 1992). Changing of the natural envelope glycoprotein into another heterologous protein (pseudotyping) has been used for altering of vector tropism (more discussed in the chapter 2.3.4.2) (Russell and Cosset, 1999). Considerable attention has been paid to the safety of the retroviral vector, in particular, the possibility of formation of replication competent viruses through recombination. This has lead to the development of three or four plasmid production systems with minimal sequence homology (Cannon and Anderson, 2004). In general, retroviral vector production yields low titers (106 TU/ml before concentration) (Merten, 2004). One limitation of most of the retroviruses is that they are only able to transduce dividing cells. Another concern is insertional mutagenesis as a consequence of random integration of viral genome into the host cell genome. Typically the MLV provirus integrates near the transcription start regions and CpG islands and may thus have an effect on the action of important host cell genes (Wu et al., 2003b).
Three hematopoietic disorders in humans, adenosine deaminase (ADA) deficiency leading to severe combined immunodeficiency (ADA-SCID), X-linked combined immunodeficiency disease (SCID-X1) and X-linked chronic granulomatous disease (X-linked CGD) have been successfully treated by retrovirus-mediated gene therapy (Aiuti et al., 2002; Hacein-Bey-Abina et al., 2002; Cavazzana-Calvo et al., 2000). Subsequently, during retrovirus therapy for SCID, leukemia developed in five of the treated children.

19
Thus, the FDA had to re-consider the regulatory requirements for the use of retroviruses in clinical trials and it came to decision that retroviral gene therapy trials would only be allowed in the treatment of life-threatening diseases and the trials had to be accomparied with the appropriate safety features (Check, 2005).
2.2.2. Lentiviruses Lentiviruses represent promising vectors for gene therapy. After the virus internalization into the target cell, the pre-integration complex (PIC) is composed of the viral genome with the viral and host proteins. PIC is then actively transported to the nucleus of the target cell, thus allowing also the transduction of non-dividing cells, such as stem cells, lymphocytes, dendritic and nerve cells (Gilbert and Wong-Staal, 2001; Cockrell and Kafri, 2007). AIDS (Acquired immunodeficiency syndrome)-causing human immunodeficiency virus type 1 (HIV-1) is a typical lentivirus and remains the most studied lentiviral vector to date. Lentiviral vectors based on primate HIV-2 and simian immunodeficiency virus (SIV) are also being developed. In addition, non-primate lentiviruses, such as feline immunodeficiency virus (FIV), equine infectious anemia virus (EIAV), caprine arthritis encephalitis virus (CAEV), and bovine immunodeficiency virus (BIV) have been described as gene therapy vectors (Goff.S.P., 2001).
Figure 3. (A) Structure of wild type lentivirus (http://www.niaid.nih.gov/factsheets/how hiv.htm). Genetic material (two ssRNAs) is inside the nucleocapsid covered by a capsid. The main capsid protein is p24. The capsid is inside the matrix formed by Gag protein p17. The outer layer is the lipid membrane from the host cell and is covered by gp41 and gp120 surface proteins. Lentivirus contains also viral enzymes: integrase, protease and reverse transcriptase. A more detailed explanation is provided in the text. (B) Electron microscopy pictures of a budding lentivirus from a producing cell (by Vesa Turkki and Diana Schenkwein).
A B

20
Lentiviruses contain more complex genome compared to oncogenic retroviruses. For example, the HIV-1 genome not only encodes for the three structural proteins (Gag, Pol and Env) but an additional six regulatory proteins (Vif, Vpr, Vpu, Nef, Rev and Tat). The Gag precursor Pr55Gag is cleaved into the matrix, capsid p24, nucleocapsid and p6 proteins. The Pr160Gag-Pol Gag-Pol polyprotein is cleaved into the protease, reverse transcriptase and intergrase proteins (figure 3) (Felder and Sutton, 2009). The accessory genes, Vif, Vpr, Vpu and nef are critical for in vivo replication and pathogenesis. Rev regulates alternative splicing of viral genes and Tat functions as a transactivator of transcription (Seelamgari et al., 2004). The natural envelope of HIV is composed of two subunits: the 41 kDa transmembrane and the 120 kDa surface proteins (figure 3.). Wild type HIV infects human immune cells, specifically CD4+ T cells, macrophages and dendritic cells (Freed and Martin, 2001).
2.2.2.1. Vector development A major effort has been placed into the progressive removal of all the non-essential HIV-1 sequences for viral replication from both transfer vectors and packaging constructs. Lentiviral replication is mediated by cis-acting sequences that do not encode any proteins. The trans-acting sequences encode the structural, regulatory and accessory proteins and these proteins are provided by the packaging plasmids. In other words, this means that viral proteins are transcribed in producing cells and only genetic material incorporated into the vector is the transgene cassette with non-coding viral elements (at the minimum LTRs, packaging signal, Rev responsible element (RRE)) (figure 4). Since the lentivirus is a complex virus, the vectors and packaging systems have been difficult to develop and often lead to low titers (Delenda, 2004).
In the first generation lentiviral vectors, viral elements were split into three separate constructs. These included the packaging construct (after major deletions of the packaging signal), the env gene (a heterologous envelope plasmid for pseudotyping) and the transfer vector RNA without any viral genes (Naldini et al., 1996b). The redundant genes for gene transfer (Vif, Vpr, Vpu and Nef) were deleted from the packaging construct resulting in the second generation of lentiviral vectors (figure 4) (Zufferey et al., 1997). The most commonly used envelope plasmid for lentivirus pseudotyping has been the vesicular stomatitis virus glycoprotein G (VSV-G) (Burns et al., 1993). Pseudotyping has broadened the transduction range and strengthened the otherwise fragile lentivirus. Pseudotyping is discussed in chapter 2.3.3.2. The biosafety of third generation lentiviruses has been further increased by substituting Tat-dependent transcription with an alternative heterologous promoter (Kim et al., 1998; Miyoshi et al., 1998; Dull et al., 1998) and splitting the original viral genome so that rev is expressed from a separate construct (Gasmi et al., 1999). After the integration, viral 3´LTR can have an effect on genes near the integration site because of promoter activity or through an enhancer effect. The development of the self-inactivating (SIN) vector was achieved by creating deletions in the 3´LTR. This abolished the

21
transcriptional activity of the LTR, thus minimizing the risk to form RCL and reduced the risk to interfere with endogenous genes (Miyoshi et al., 1998; Zufferey et al., 1998; Iwakuma et al., 1999).
Figure 4. Wild type lentivirus genome (top) and the third generation lentiviral vector system (constructs 1-4). All vector plasmids (1-4) are contransfected to the producing cells (Sinn et al., 2005). Psi: packaging signal, RRE: rev responsive element
One approach towards safer lentivirus production has been to develop codon-
optimized systems to reduce the risk of recombination. These are based on the synthesized complete codon-optimized HIV-1 gag-pol gene the expression of which is not rev-dependent (Kotsopoulou et al., 2000; Koldej et al., 2005). Another alternative has been to split the gag/gag-pol gene into several parts (Wu et al., 2000).
A minimal transgene expression cassette contains the transgene, a heterologous promoter, a packaging signal and LTRs (figure 4). The central polypurine tract (cPPT) has been shown to increase the nuclear localization and the total amount of genome integrated into the DNA of the target cells (Follenzi et al., 2000). In addition, incorporation of the post-transcriptional regulatory element from woodchuck hepatitis virus (WPRE) into the 3´end of the transfer vector has been shown to increase the total amount of mRNA and improve transgene expression (Zufferey et al., 1999). Some gene therapy applications may need expression of more than one gene. This has been achieved using bicistronic expression cassettes containing an internal ribosome entry site (IRES) (Yu et al., 2003) or a self-cleaving peptide 2A (Szymczak et al., 2004). Then two genes are transcribed as a single mRNA and the separation of two proteins occurs during the translation. To avoid transcriptional silencing of the viral vector in vivo by adjacent host genetic elements, the use of heterologous cis-elements, such as insulators has shown promising results (Ramezani et al., 2003).

22
One safety concern of using an integrating gene therapy vector is its capacity for insertional mutagenesis. Insertional mutagenesis is more risky when integration occurs near the regulatory area of genes. In contrast to MLV retroviruses, lentiviruses do not show any preference for integration near transcriptional start sites (Liu et al., 2006a) or CpG islands (Wu et al., 2003b). However, the integration profile may be cell type specific and can be different in dividing and nondividing cells. Several attempts towards site-specific integration have been made but random integration still dominates. These methods have been based on fusing different DNA-binding proteins like Zinc-finger zif268 (Bushman and Miller, 1997), LexA (Goulaouic and Chow, 1996) or E2C (Tan et al., 2006) into the viral integrase. The ability of the lentivirus to transduce non-dividing cells has also led to the development of integrase-defective lentiviral vectors. Expression of these vectors is transient because the viral genome is lost during the subsequent cell divisions. Nonetheless, long-term expression was achieved in non-dividing cells in vivo (Philippe et al., 2006; Philpott and Thrasher, 2007).
Other safety modifications include the use of tissue-specific promoters to target transgene expression to a selected tissue, or to use regulated expression systems (Miller and Whelan, 1997) to control the transgene expression. Currently, the tetracycline-based regulatory systems are the most widely used and both Tet-ON- and Tet-OFF-based tetracycline-dependent systems have been used to attain inducible lentivirus expression (Goverdhana et al., 2005).
2.2.2.2. Lentivirus production
Lentiviral vectors are commonly produced by cotransfecting adherent HEK 293T cells with several different plasmid constructs (Follenzi and Naldini, 2002; Tiscornia et al., 2006). Typically, these plasmids are a self-inactivating transfer vector plasmid encoding the transgene expression cassette, a packaging plasmid encoding gag-pol, a rev plasmid and an envelope glycoprotein plasmid which usually encodes VSV-G (figure 4). The first clinical lentiviral vector production was also based on a transient system. However, the commonly used three or four plasmid system was modified to a two-plasmid system in order to make the process easier to perform and more cost effective (Lu et al., 2004). The most commonly used reagent in plasmid transfection is calcium phosphate (Tiscornia et al., 2006; Follenzi and Naldini, 2002; Reiser, 2000; Koldej et al., 2005; Naldini et al., 1996a; Sena-Esteves et al., 2004). Less plasmid is needed when more efficient reagents, like an activated dendrimer-based Superfect (Coleman et al., 2003) or N,N-bis (2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) (Karolewski et al., 2003), have been used. Polyethylenimine (PEI)-mediated transfection in serum-free conditions has also gained interest (Kuroda et al., 2008). Transient transfection method is straightforward to perform, versatile and avoids the time-consuming development of stable cells lines. It also allows easy and rapid testing of various transgenes or pseudotypes (Sena-Esteves et al., 2004). Large scale production has been achieved using 10 layer cell factories (Geraerts et al., 2005; Slepushkin et al., 2003). Lentiviral titers are typically 106-107 TU/ml prior to

23
concentration and the best titers achieved have been 1010 TU/ml after concentration (Cockrell and Kafri, 2007).
The first successful lentivirus production process in suspension cells was published recently. Segura et al. were able to produce high titer lentiviral vectors in a 3-L bioreactor with 293-based cells being grown in serum-free medium. The transient transfection was performed with polyethylenimine (PEI) (Segura et al., 2007). This kind of suspension production method was reported to be easy to scale up and thus it is very practical.
Since the transient transfection system allows virus production only for a short time and cotransfection may increase the risk of recombination between the plasmids, several attempts have been made to develop stable packaging cell lines for the production of lentiviral vectors (Kafri et al., 1999; Farson et al., 2001; Pacchia et al., 2001; Xu et al., 2001). The generation of stable cell lines is time-consuming and needs to be developed for each vector type separately. Stable packaging cells lines lessen variation between vector stocks, reduce the likelihood of generating helper virus and allow reproducible propagation of viral vectors. However, the toxicity of lentiviral protease (Haselhorst et al., 1998) and the fusogenic envelope protein VSV-G (Cronin et al., 2005; Burns et al., 1993), have prohibited constitutive vector production. The only convenient option has been to use inducible packaging cell lines. Inducible production has been controlled either with tetracycline-inducible (Kafri et al., 1999; Xu et al., 2001; Ni et al., 2005; Farson et al., 2001; Stewart et al., 2009) or ecdysone inducible (Pacchia et al., 2001) systems. Alternatively, the toxic envelope protein VSV-G has been replaced with a less-toxic glycoprotein, such as envelopes from other retroviruses (GALV, amphotopic MLV, cat endogenous retrovirus RD114) (Strang et al., 2004).
The latest production methods are based on hybrid virus systems. In these, all the necessary elements needed for viral production are expressed from one or more hybrid viruses. Lentivirus production based on a hybrid adenovirus has yielded up to 106 TU/ml titers (Kubo and Mitani, 2003) and also a hybrid vaccinia virus (Konetschny et al., 2003), or alphavirus (Wahlfors and Morgan, 2002) was able to produce retroviral vectors.
2.2.2.3. Lentivirus purification Lentiviral vectors are most commonly concentrated for preclinical studies by ultracentrifugation without any preceding purification step (Follenzi and Naldini, 2002; Tiscornia et al., 2006; Burns et al., 1993). The disadvantage of the ultracentrifugation method is that it concentrates unwanted contaminants, such as proteins, DNA and inhibitors of transduction in addition to the final product. The contaminants increase the toxicity, inflammatory and immune responses (Baekelandt et al., 2003; Baekelandt et al., 2002). Therefore several purification methods for lentiviral vectors have been developed. The instability of lentiviral vectors has complicated the developments of the purification process because lentiviral vectors lose their infectivity rather easily. It is possible to strengthen the otherwise unstable virus by choosing the optimal pseudotype (Burns et al., 1993).

24
An optimal purification process should be capable of being scaled up, maintaining a maximal level of infective viral particles. Several methods have been used for purification of lentiviruses i.e. sucrose gradient (Baekelandt et al., 2003), anion exchange chromatography (Yamada et al., 2003; Sena-Esteves et al., 2004) or affinity chromatography (Segura et al., 2007; Scherr et al., 2002). In addition, ultrafiltration (Reiser, 2000; Miyake et al., 2007) and tangential flow filtration (TFF) (Geraerts et al., 2005) in lentivirus filtration and purification have been used. A typical downstream process in clinical use for retroviral vectors starts with a clarification step to remove the cell debris from the viral supernatant, followed by concentration by ultrafiltration, benzonase digestion of DNA (Sastry et al., 2004), a chromatographic purification process, diafiltration and final sterile filtration (Slepushkin et al., 2003; Transfiguracion et al., 2003; Rodrigues et al., 2007).
2.2.2.4. Lentivirus titering and analysis of replication competent lentiviruses Validation and safety tests are required before one can use of lentiviral vectors in human. These include assays for replication competent lentiviruses (RCL), sterility tests, determination of viral titer and potency. The titration methods are based on assessing the number of non-functional viral particles, or functional viruses. The measurement of reverse transcriptase (RT) activity by product-enhanced RT assay, the viral capsid by p24 ELISA, or RNA amount in viral supernatant by real time quantitative polymerase chain reaction (qPCR) defines non-functional viral particles. The number of integrated provirus by real time qPCR provides the titer of functional viral particles. Other functional titering methors rely on determining transduced cells by analyzing transgene expression either in mRNA level by RT-qPCR, or assaying the protein level by determining marker protein or using primary antibodies against the transgene protein (Delenda and Gaillard, 2005; Geraerts et al., 2006; Sastry et al., 2002). Viral titration methods may not be accurate and depending on the method, they usually over- or underestimate the titers. There are several sources of errors e.g. empty viral particles, contaminating plasmids or non-integrated LTR-circles. Therefore, different comparative methods should be used in order to obtain the most reliable results.
The primary concern for lentiviral vectors is the potential generation of RCL. The generation of RCL may happen during the virus production between the transfer construct, packaging or envelope plasmids, or after the introduction of lentiviral vector to target cells between viral vector and endogenous retroviral elements. By minimizing the sequence homology in plasmids, the risk has been reduced during the production phase. A challenge in the development of a RCL test has been the lack of an appropriate positive control. The current methods mainly involve PCR protocols (Sastry et al., 2003; Sastry et al., 2005; Delenda and Gaillard, 2005) but standard p24 ELISA methods (Sastry et al., 2003; Segall et al., 2003) and reporter gene activity (marker rescue) assays (Segall et al., 2003) have also been used. At present, no RCL has been associated with third generation lentiviral vectors.

25
2.2.2.5. Lentiviral gene therapy applications Lentiviral vectors have a unique ability to transduce dividing and non-dividing cells and to stably integrate into the host cell genome and achieve a long term transgene expression invitro and in vivo (Naldini et al., 1996b; Kafri et al., 1997). Most of the potential therapeutic targets for lentiviral vectors have been in the central nervous system, the hematopoietic system, liver, heart, ocular tissue and pancreas (figure 5). Successful preclinical studies have been carried out for treating Alzheimer`s, Parkinson´s and Huntington´s diseases or for correction of genetic disorders including immunodeficiency and hemoglobin disorders (Wiznerowicz and Trono, 2005; Cockrell and Kafri, 2007). RNA interference has been identified as a tool to downregulate the expression of a specific gene. Lentiviral vectors carrying polymerase-III-directed short hairpin RNA molecules (shRNA) have induced the efficient downregulation of cellular genes (Sumimoto and Kawakami, 2007). In addition, gene therapy lentiviruses have been utilized in transgenesis to generate transgenic animals (Pfeifer et al., 2002). One definite advantage of their lentiviral vector is the low immunogenicity. However, some immune response may be directed against the heterologous envelope, such as VSV-G (Baekelandt et al., 2003).
Figure 5. Lentiviral vector applications. Lentiviral vectors have been widely used in the downregulation of disease genes by RNAi, or in the delivery of therapeutic genes mainly to the central nervous system, liver or bone marrow. Lentiviral vectors have been widely used also for the transgenesis (Wiznerowicz and Trono, 2005).

26
The first clinical study using a lentiviral vector was conducted in 2002 for anti-HIV
therapy. Since then, a total of 18 clinical trials with lentivirus vectors have been performed. So far, all trials were based on ex vivo vector delivery. Nine of the trials were for the treatment of HIV, six for treatment of monogenic diseases (X linked Adrenoleukodystrophy, thalassemia, Wiskott Aldrich syndrome, Mucopolysaccharidosis Type VII, Fanconi Anemia complementation Group A) and three trials were focused on cancer treatment (September 2008) (Journal of Gene Medicinehttp://www.wiley.co.uk/ genetherapy/ clinical/).
2.2.3. Baculoviruses Baculoviruses belong to the family Baculoviridae, which consists of over 600 members (Martignoni and Iwai, 1986; Airenne et al., 2009). Baculoviruses are enveloped viruses and infect only permissive arthropod hosts. Baculoviruses are divided into two genera: Nuclear polyhedrosis-viruses (NPV) and Granulosis-viruses (GV). The genome is a circular double-stranded DNA of 80-200 kb in size, which is incorporated inside the rod-shaped viral capsid. Baculoviruses exist in two forms in their natural life cycle: occlusion derived virus (ODV) to transmit the infection between insect hosts and budded virus (BV) to spread the infection within the individual insect. ODV incorporates several viral capsids covered by polyhedron matrix derived from the nuclear membrane of the host cell. BV includes only one capsid surrounded by a lipid membrane of the host cells. The major envelope glycoproteins are p74 and gp64 for ODV and BV, respectively (figure 6). The baculovirus life cycle begins with infection of the host insect midgut cells by ODV (early phase) leading to extensive viral DNA replication, late gene expression and BV production (late phase). Infection leads finally to the production of ODV, cell lysis and often host death (very late phase). The most extensively studied baculovirus is the BV form Autographa californica nuclear multiple polyhedrovirus (AcMNPV) (Miller, 1997). AcMNPV has been used not only in protein production, but also in gene therapy and vaccination studies.

27
Figure 6. (A) Schematic diagram of baculovirus budded virus (BV) and occlusion derived virus (ODV). In the BV form, circular double-strand DNA viral genome is inside the capsid. The major capsid protein is vp39. The capsid is covered by lipid membrane and peplomers (envelope glycoprotein Gp64s). ODV incorporates several viral capsids covered by polyhedron matrix derived from nuclear membrane of host cells. The major envelope of ODV is p74. (by Dwight Lynn, http://www.answers.com/topic/nucleopolyhedrovirus-jpg-1).
2.2.3.1. Baculovirus vector development Insertion of transgenes into the large AcMNPV genome (134 kb) was first performed by homologous recombination. Transfer plasmid DNA and baculovirus genomic DNA were cotransfected into insect cells in which the homologous recombination was to take place. The identification of recombinant clones was accomplished by a tedious plaque assay (Smith et al., 1983) and thus one goal was to enhance the efficiency of this process. The first remarkable improvement was to linearize the baculovirus genome to obtain a much higher frequency of recombination (Kitts and Possee, 1993). Altogether, construction of the plasmid, plaque purification, cotransfection and virus stock production required several weeks (O'reilly et al., 2004). Despite the large size of the virus genome, direct cloning was also attempted as an alternative to the recombination (Ernst et al., 1994). The next major development was to replace homologous recombination with a faster method, based on transposition. The transgene from the donor plasmid was cloned into a baculovirus shuttle vector (bacmid) under the polyhedron promoter by site-specific transposition in

28
Escherichia coli. The recombinant bacmid was isolated and used in the transfection of the insect cells. Pure recombinant viruses were achieved in 7-10 days (Luckow et al., 1993). The method is commercially known as the Bac-to-Bac system (figure 7) (Invitrogen life technologies, Carlsbad, USA).
An addition of a lethal gene into the donor plasmid reduced the background of contaminating parent virus in E. coli, and the use of the tetra-promoter containing chicken �-actin, T7lac, p10 and pPolh promoters, allowed subsequent expression of proteins in mammalian, bacterial and insect cells (Airenne et al., 2003; Laitinen et al., 2005). The recently commercialized BaculoDirect system uses a purified recombinase in transposition and a negative selection marker to eliminate non-recombinant viruses in insect cells (Invitrogen life technologies, Carlsbad, USA).
Figure 7. Baculovirus production using Bac-to-Bac system. The recombinant donor plasmid containing the gene of interest is cloned and purified, and then transformed into DH10BacTM E.coli for transposition into the bacmid. Colonies containing the recombinant bacmid is identified using blue/white selection. Recombinant bacmid DNA is isolated and transfected into insect cells. Produced baculoviruses can be amplified, titered, or use for recombinant gene expression (Invitrogen life technologies, Carlsbad, USA).

29
2.2.3.2. Baculovirus production, purification and titering
The generation of a pure baculovirus stock requires only a small amount of starting material which is produced after the transfection of the bacmid into the host insect cells. Large virus stocks are amplified simply by infecting insect cell cultures and subsequently harvesting the culture medium (O'reilly et al., 2004). For protein production in insect cells, no further concentration is required. On the other hand, baculoviruses have emerged as a promising new vector for human gene therapy which has triggered demands to develop viral purification methods.
The current purification and concentration methods for baculovirus vectors are based on ultracentrifugation. In addition, some improved chromatographic protocols have been developed. Barsoum et al. first introduced a cation exchange chromatograpic method (Barsoum, 1999), whereas latest developments have included ion exchange membrane chromatography (Wu et al., 2007) and size exclusion chromatography (Transfiguracion et al., 2007). Vicente et al. published recently a complete downstream process for clinical grade baculoviruses comprising depth filtration, ultra/diafiltration and membrane sorption (Vicente et al., 2009).
Traditionally, titer determination of recombinant virions is performed by detecting morphological changes in the infected cells using plaque formation and end-point dilution assays (O'reilly et al., 2004) but these methods are time-consuming and laborious. Several advanced protocols for virus titering have been described. Rapid immunological assays can be utilized for detecting viral protein from the infected cells by monoclonal antibodies (Kitts and Green, 1999; Kwon et al., 2002; Mulvania et al., 2004). In addition, infected cells can be detected by measuring increased cell-diameter following infection (Janakiraman et al., 2006) or expression of fluorescent protein (Philipps et al., 2005; Cha et al., 1997). Furthermore, a protocol omitting any cell culture process is based on a quantitative PCR to measure genome copy number (Hitchman et al., 2007; Lo and Chao, 2004). Viral particles in the culture supernatant have been analyzed also by FACS after staining the viral DNA (Shen et al., 2002). These last two methods are fast but they do not reveal the amount of non-infective fragments versus the infective virus particles.
2.2.3.3. Baculovirus applications
Baculoviruses have been utilized as a biopesticide over a period of many years and their biology is well characterized. Since the 80´s baculoviruses have been used in the production of thousands of recombinant proteins. There are several features, which make this system so popular. The baculovirus system is safe and easy to use. Suspension cell cultures in serum-free conditions are easy to scale up. The virus production is helper-virus independent and the baculovirus vector can accommodate very large inserts (Miller, 1997).
Protein production in insect cells has been the most widely used application of baculovirus vectors. The expression of recombinant protein in insect cells commonly occurs at a high level and is usually driven by the polyhedrin promoter of the baculovirus.

30
Insect cells are able to carry out complex post-transcriptional modifications (O'reilly et al., 2004; Kost et al., 2005; Airenne et al., 2009). Furthermore, protein expression levels can be enhanced with several methods. Co-expression of chaperones together with recombinant protein has improved the solubility of the target protein thus enhancing the production yield (Yokoyama et al., 2000). Other applications have been to add various DNA elements, such as an optimized Kozak sequence and A-rich regions, to the promoter region of the gene in order to increase the level of transcription of a target gene (Sano et al., 2002). Overall the baculovirus-insect cell expression system is very versatile, and it has been adapted all the way from a miniaturized well plate format (Bahia et al., 2005) to large scale fully controlled bioreactor systems (Elias et al., 2007). Small scale production has served as a practical method in optimization of the virus production, titering or conditions of protein expression (Philipps et al., 2005) whereas in bioreactors, protein production can be performed in an efficient, reproducible and robust manner (Elias et al., 2007).
Transgene expression in mammalian cells was achieved when a suitable vertebrate promoter, such as cytomegalovirus (CMV) immediate early promoter, was used (Hofmann et al., 1995). This opened up the possibility to produce proteins in mammalian cells but also to develop baculoviruses towards gene transfer applications. Baculoviruses are suitable for short term therapy applications because transgene expression is transient and is lost within two weeks. Baculoviruses are capable of transducing most mammalian cell lines (Hu, 2006; Airenne et al., 2009) and efficient transduction has been achieved even in large scale suspension cell cultures under serum-free conditions (Scott et al., 2007). Successful in vivo gene transfer has been achieved in the rabbit carotic artery (Airenne et al., 2000), rodent brain (Lehtolainen et al., 2002b; Sarkis et al., 2000), mouse skeletal muscle (Pieroni et al., 2001) and eye (Haeseleer et al., 2001). An important safety aspect of baculoviruses is that they are not able to replicate in vertebrate cells. Further, no cytotoxicity is usually detected even with high MOIs (Andersson et al., 2007). Unfortunately, baculovirus-mediated gene therapy suffers two drawbacks, the inactivation of the vector by serum-derived complement factors and the low transfer efficacy in vivo (Hofmann and Strauss, 1998). Serum inactivation can be overcome or at least minimized by displaying complement blocking proteins or by attempting to avoid blood contact during the virus inoculation. The transduction efficiency has been enhanced by attaching heterologous envelope protein, such as VSV-G onto the virus surface. Still, nuclear transport may be the most critical step (Kukkonen et al., 2003). The promoter choice has an influence on transgene expression (Airenne et al., 2004; Hu, 2006) but also DNA elements, like WPRE, can be used to obtain higher expression levels in mammalian cells (Mahonen et al., 2007).
In addition to the previously mentioned applications baculoviruses have been extensively exploited as vaccination vectors. Several viral-like particles (VLP), such as hepatitis VLP (Chen et al., 2005) or influenza VLP (Galarza et al., 2005) produced by the baculovirus system have been used in vaccination. VLPs have been used also in studying virus-cell interactions and not only VLPs, but also intact viruses have been produced using hybrid baculoviruses (Cheshenko et al., 2001; Poomputsa et al., 2003). In particular, the production of the AAV vector in insect cells by baculoviruses has gained much interest due

31
to difficulties in producing sufficient AAV required for clinical trials by conventional plasmid transfection methods (Urabe et al., 2002; Huang et al., 2007).
2.2.4. Other viral vectors
2.2.4.1. Adenovirus Adenoviruses are non-enveloped viruses with a diameter of 70-100 nm, an icosahedral conformation and a linear dsDNA genome of approximately 36 kb (figure 8). The family Adenoviridae consists of over 50 different human serotypes and numerous non-human adenoviruses. The viral life cycle is divided into early and late phases, with the viral DNA replication taking place in the middle of the cycle (Shenk.T.E., 2001). The most commonly used adenovirus serotypes in gene therapy are 2 (Ad2) and 5 (Ad5). The high affinity receptor for most of adenoviruses is the coxsackie-adenovirus receptor (CAR) (Hackett and Crystal, 2004). Altogether, adenoviral vectors are the most frequently used vector (24.9% of all 1472 trials) in gene therapy clinical trials (Journal of Gene Medicine http://www.wiley. co.uk/ genetherapy /clinical/). The target diseases for adenovirus treatments have been cancer, vascular disease and monogenic disorders (Hackett and Crystal, 2004). Adenoviruses infect both dividing and quiescent cells having broad tropism and transient transgene expression. The viral particles are rather stable and easy to manipulate. In addition, adenoviral vectors can be produced in very high titers (up to 1013 viral particles/ml). One major concern in the use of adenoviruses is the recipient’s immune reaction. Toxicity is dose dependent, occurs in phases and varies with administration routes, cell types or species (St George, 2003).
Figure 8. The schematic diagram of the adenoviral particle. Adenoviral genome, linear dsDNA, is inside the icosahedral nucleocapsid composed of hexons and pentons. On the surface of the capsid there are protein structures called fibers that facilitate the attachement of the virus to its receptor (Glasgow et al., 2009).

32
Vector development has been directed towards safe production with a minimal
possibility to generate replication-competent adenoviruses. The first modification was to delete the E1 region, whose product is responsible for the virus replication and DNA synthesis. The next modification was to remove the early E2 and E4 regions which also contributed to efficient DNA replication (Gao et al., 1996; Lusky et al., 1998). Third generation vectors lack all the viral coding sequences and consist of merely the cis-elements. Helper adenoviruses must be used during viral production to provide all the necessary functions in trans. These “gutless” vectors have a high packaging capacity, up to ~36 kb and highly reduced immunogenicity with prolonged expression (Kochanek et al., 2001). Most of the gene therapy strategies rely on replication incompetent viral vectors. However, the goal in cancer therapy is to eliminate tumor cells and the use of lytic viruses mediating cell lysis and death can be justified. This application is known as oncolytic virotherapy. For example, the deletion of the E1B region in the virus restricted the replication to tumor cells lacking a normal p53 protein (Bischoff et al., 1996). Oncolytic vectors have been used in clinical trials and encouraging results were gained when they were combined with chemotherapy (e.g. ONYX-015 by (Barker and Berk, 1987)).
2.2.4.2. Poxviruses
Poxviruses and herpes simplex virus have been used in 6.1% and 3.2% of clinical trials, respectively (September 2008) (http://www.wiley.co.uk/genetherapy/clinical/). Poxviruses are classified into the chordopoxvirinae (vertebrate poxviruses) and entomopoxvirinae (insect pox viruses) subfamilies. Poxviruses are enveloped viruses containing linear dsDNA genome of 130-230 kb size (Moss, 2001). One prototype of the poxviruses is the vaccinia virus (8.2% of clinical trials), well known for its use as a vaccine for smallpox. Since then the vector has proven not only to have potential in vaccination, but also for the expression of foreign genes like antigen, cytokines and immunostimulatory molecules (McCart and Barlett, 2004). Vaccinia virus has also shown selective replicate in tumor cells, making the virus an attractive agent for oncolytic virotherapy (Puhlmann et al., 2000). The vaccinia vector has a broad tropism, a very high level of transgene expression and a large insert capacity. In addition, its episomal replication makes it safe for use because no risk of insertional mutagenesis exists. However, the virus can evoke a significant immune response (McCart and Barlett, 2004).
2.2.4.3. Herpes simplex virus Herpes viruses belongs to the family of Herpesviridae and are disseminated in nature by infecting most animal species (Roizman and Pellet, 2001). Herpes simplex virus type 1 (HSV-1) is the most extensively engineered herpes virus used in gene therapy. The HSV-1 core contains 152 kb of linear dsDNA, covered by a capsid and surrounded by a lipid envelope with several glycoproteins embedded in it. The genome is complex with more

33
than 80 genes but many genes can be deleted allowing the incorporation of large inserts (up to 30-40 kb). In addition, HSV has a broad tropism, efficient transgene expression and long term expression can be achieved especially in neurons if the latency-activated LAP2-promoter system is used. Limitations of HSV include possible cytotoxicity, and targeting of the virus poses its own challenges (Wolfe et al., 2004). HSV infection can be either lytic or latent. Lytic infection was prevented by deleting five immediate early (IE) genes (Samaniego et al., 1998). These vectors were not cytotoxic and allowed high expression of transgenes. Another approach is to utilize the lytic cycle of infection in oncolytic virotherapy. The first deletion towards tumor-selective replication was analogous to vaccinia virus when the viral tymidine kinase was deleted (Martuza et al., 1991). However, neurotoxicity occurred in healthy neurons in addition to those around the site which were infected (Boviatsis et al., 1994). Therefore, new mutants are still under development to produce a less neurovirulent HSV. Today, HSV is considered an attractive tool in the treatment of neuropathological disorders, cancer, pain, autoimmune syndrome and metabolic diseases (Wolfe et al., 2004).
2.2.4.4. Adeno-associated virus
The non-lytic adeno-associated virus (AAV) has evolved into a gene therapy tool of considerable importance since it has been used in a wide range of gene therapy approaches. About 4 % of gene therapy clinical trials have been conducted with AAV (http://www.wiley.co.uk/ genetherapy/clinical/) and that number is increasing. AAV belongs to Parvoviridae family and is one of the smallest virus (20 nm) used in gene therapy. The non-enveloped virus has an icosahedral capsid and contains a linear single-stranded 4.7 kb DNA genome (Coura and Nardi, 2007). Several naturally occurring serotypes and variants from different species have been isolated, from which the serotype 2 (AAV2) is the best characterized and most commonly used (Wu et al., 2006). The replication of AAV requires a helper virus, generally an adenovirus, but also herpes simplex virus, human papilloma virus and vaccinia virus can serve as a helper (Tenenbaum et al., 2003). In the absence of a helper virus, AAV established a latent infection within the cell (Coura and Nardi, 2007). Wild type AAV integrates site specifically into human chromosome 19. However, in the absence of a helper virus, recombinant AAV infection can lead to random integration but more commonly, the viral genome can persist as an episomal plasmid. The transgene expression from AAV is long term. AAV has a broad host and cell type tropism and it transduces both dividing and nondividing cells. The biggest limitation is the size limit of the transgene cassette and most adults have antibodies against the AAV (Tenenbaum et al., 2003). This virus is reported to be a suitable gene transfer vehicle for cystic fibrosis, hemophilia, muscular dystrophy, CNS and ocular diseases, as well as for cancer (Carter et al., 2004).

34
2.2.5. Non-viral vectors Viral-based gene therapy has its own advantages but also some associated pitfalls. Safer non-viral systems are based on delivering naked DNA into the target cells. Non-viral vectors are easy to produce on a large scale, rather safe to use, they have large insert capacity and low host immunogenicity. The two main disadvantages are the low transfection efficacy and the short term expression (Schmidt-Wolf and Schmidt-Wolf, 2003). Clinical trials to date have evaluated side effects of naked DNA and the results have indicated good tolerance of this type of therapy (Comerota et al., 2002). The most widely studied area is vaccination by naked DNA but so far, the results have been disappointing in term of immunogenicity, and much effort has been put into enhancing these vaccines (Liu et al., 2006b).
The simplest delivery technique is direct injection of naked plasmid DNA into the target tissue. Transfection efficacy can be enhanced by physical manipulations like the electroporation, “gene-gun” technique (DNA is bound to gold particles and shot into cells), ultrasound or hydrodynamic (high pressure) injection (Wells, 2004). DNA can be more efficiently delivered into the target tissue/cells with the help of cationic carriers, such as cationic polymers, dendrimers, cell-penetrating peptides or liposomes. One novel system is to pack DNA inside nanoparticles (Schmidt-Wolf and Schmidt-Wolf, 2003; Niidome and Huang, 2002). Short term expression can be overcome by using a transposon approach. Transposon-based vectors are capable of stable integration in the host cell genome and can achieve long term transgene expression. Transposons are natural DNA elements which have the ability to move and replicate within the host genome. Transposable elements are divided into retrotransposons and DNA transposons. One of the most commonly used DNA transposons is Sleeping Beauty, an active vertebrate element from a “dead” transposon fossil found in fish genomes. The Sleeping Beauty system consists of the desired DNA sequence flanked by the recognition sites of the transposase and the transposase gene. Transposition is a cut-and paste process resulting in integration of the Sleeping Beauty element into human chromosomes (Izsvak and Ivics, 2004). Other non-viral vector systems are based on the Frog Prince from amphibian genomes, Streptomyces phage FC31 (Ivics and Izsvak, 2006) or PiggyBac from the cabbage looper moth Trichoplusia ni (Wilson et al., 2007). In addition to the plasmids, also bacteria and phages have been used in non-viral gene delivery. Bacteria and phages are easy to manipulate and produce. They can enter into a variety of cell types, and have shown some promising results in delivering DNA into mammalian cells (Higgins and Portnoy, 1998; Johnson and Chiu, 2007). In addition, the use of whole cells to carry otherwise challenging “payloads” is a relatively new development level (Roth et al., 2008). Advantages and disadvantages of commonly used gene therapy vectors are summarized in Table 1.

35
Table 1. Characteristics of the most commonly used gene transfer vectors Characteristics Retroviruses (MLV) High transduction efficiency
Integrates to the host cell genome Long term transgene expression but transgene silencing may exist Insert capacity up to 10 kb Restricted transduction only to dividing cells Risk of insertional mutagenesis or RCR Low titers More suitable for ex vivo application
Lentivirus Transduces dividing and non-dividing cells Integrates to the host cell genome, long term expression Difficult to produce in large quantities Inset capacity up to 10 kb Transient production protocol HIV brings safety concerns
Baculovirus Safe, non-pathogenic Easy to produce to high titers Large insert capacity Transient expression Broad tropism, transduces dividing and non-dividing cells Inefficient in vivo, inactivation by serum complement
Adenovirus Efficiently transduces dividing and non-dividing cells, broad tropism Very high efficiency Easy to produce in high titers Transient expression Strong antiviral immune response, repeated administration is problematic
AAV Sustained transgene expression Transduces dividing and non-dividing cells, broad tropism Low insert capacity Non-pathogenic Simple genome
Vaccinia virus Broad tropism Large insert capacity Transient expression High transgene expression Difficult to produce in large quantities
Herpes simplex virus Restricted tropism, transduces only dividing cells Transient expression High titers May induce an immune response Toxicity in many cell types Large inset capacity ~40 kb
Non-viral vector Safe Unlimited insert capacity Easy to produce Inefficient in vivo Transient expression (long term expression with transposons) Low immunogenicity

36
2.3. Targeted therapy Targeted drug delivery is the most important challenge in pharmaceutical research. At the turn of the 20th century, Paul Enrich proposed the idea of “magic bullets” in targeting a compound to eradicate a disease. It took 50 years to achieve the first implementation of the targeted antibody-conjugated radionuclide by Pressman and Korngold (Pressman and Korngold, 1953). The goal in drug targeting is to increase the concentration of the drug in the vicinity of the cells responsible for disease without affecting healthy cells. Many approaches in cancer treatment are limited because of their broad range of unwanted side effects on healthy cells. The results of preliminary studies have been encouraging but so far a method that allows efficient, easy, robust and especially systemic delivery is still under development.
Targeting can reduce the dose and volume of the drug at the same time minimizing the concentration of the freely circulating drug in the patient. Targeting improves the therapeutic window and decreases the accompanying drug toxicity all of which contribute to an improved response (Goodwin and Meares, 2001). The characterization of the disease markers at the molecular level is essential in developing targeting strategies as are approaches to predict the clinical behavior of the affected tissue. Natural or artificial ligands have been designed to bind to the altered membrane-associated proteins on the cell surface (Sergeeva et al., 2006). Sometimes coupling therapeutics to carriers, such as liposomes or synthetic polymers, are needed to protect the drug during the transport (Petrak, 2005).
Therapeutic agents (e.g. drug, viral vector, radionuclide) can be targeted either by direct or indirect methods. In direct methods, the therapeutic agent is linked directly to the targeting ligand (e.g. antibody) (Paganelli et al., 1990). Pretargeting is an indirect method where the target site is first pretargeted followed by administration of the therapeutic agent which then accumulates in the pretargeted area. Direct targeting is a straightforward one step approach whereas pretargeting requires two or more steps but has been shown to increase tumor-to-nontumor targeting ratios (Boerman et al., 1999).
Antibodies, commonly immunoglobulin G (IgG), are most often used in targeting because of their specific binding selectively towards antigen on the surface of the target cells, usually cancer cells (Goldenberg et al., 2006). 131Indium (131In) and 90Yttrium (90Y) are frequently used radioconjugates in antibody labeling (Goldenberg et al., 2006). This therapy is called radioimmunotherapy. One drawback in radioimmunotherapy is the toxicity caused by the circulating radionuclide labeled antibody but one way to overcome the toxicity of radionuclide to the blood cells is by administering the labeled antibody locally (Goetz et al., 2003). Many clinical trials with directly-radioconjugated antibodies are in progress. So far, radioimmunotherapy has proved to be most efficient in the treatment of non-Hodgkin’s lymphoma (Sharkey and Goldenberg, 2005). The first approved radiolabelled antibodies for the treatment of cancer were antibodies directed to CD20

37
labeled either by 131In (Tositumomab, Bexxar, Corixa and GlaxoSmithKline Corporations) or 90Y (Ibritumomab tiuxetan, Zevalin, IDEC Pharmaceuticals Corporation). There are other targets for antibody targeting in addition to cancer cells, like tumor vessels (Ruoslahti, 2002) or to deliver drugs to the brain via targeting the blood brain barrier (Pardridge, 2002). Quite often in cancer treatment, antibody-based targeting alone is not efficient enough and a combination with non-targeted therapy (surgery, radiotherapy, chemotherapy and immunotherapy) is needed (Goldenberg et al., 2006).
2.3.1 Pretargeting therapy of cancer
The pretargeting method was proposed by Goodwin et al. over 20 year ago (Goodwin et al., 1986). In antibody-based pretargeting typically a monoclonal antibody is administered to distribute throughout the body and to bind the tumor cells expressing the antigen. When the free antibody has cleared from the circulation, the radionuclide is administered to home on on the site of antibody. Small radionuclides are cleared faster from the circulation than the larger labeled antibodies so this kind of separate administration can reduce the toxicity to the blood cells (Goldenberg et al., 2006). Monoclonal antibody based interactions are one alternative bridge between the targeting moiety and therapeutic compound. A second type of interaction was introduced by Paganelli et al. in which the avidin-biotin system is utilized in pretargeting (Paganelli et al., 1990; Paganelli et al., 1991). The (Strept)avidin-biotin system is discussed in the chapter 2.4. In addition, an antibody having a bivalent binding specificity has been shown to enhance the efficacy of the tumor targeting (figure 9). The bispecific antibodies have one binding site directed to a target and another one to a specific ligand (Boerman et al., 2003; Chatal et al., 1995). Currently, bispecific antibodies for pretargeting for radioimmunotherapy have been tested in several clinical trials (Bardies et al., 1996; Kraeber-Bodere et al., 1999; Gruaz-Guyon et al., 2001). These studies showed the capability of the technique since substantially lower amounts of radioactivity was used while increased tumor uptake of the radiolabel was achieved.
2.3.2. Gene-directed enzyme prodrug therapy One way to achieve targeted therapy is to use gene-directed enzyme prodrug therapy. A prodrug is an inactive pharmaceutical substance, which undergoes metabolic changes to become a cytotoxic compound. Firstly, the prodrug is introduced to the target site and the therapeutic effect can be achieved after the metabolism of the substance to the active form (Schepelmann and Springer, 2006). A commonly used example of a gene therapy-based prodrug is the herpes simplex virus thymidine kinase (HSV-Tk) enzyme (Reardon, 1989; Moolten, 1986) which converts ganciclovir into the ganciclovir monophosphate, which is further converted to toxic ganciclovir triphosphate by host cell leading finally to cell death. Adenovirus-mediated HSV-Tk (Cerebro,Ark Therapeutics Group, Plc, London, UK)

38
increased the survival of malignant glioma patients in a clinical trial after the local injection to the tumor bed followed by ganciclovir treatment (Immonen et al., 2004). Another early approach in suicide gene therapy was the use of E.coli cytosine deaminase (CD) enzyme, which has been assessed in clinical trials. Adenovirus expressing CD enzyme was administered into metastases of colorectal carcinoma (Crystal et al., 1997). CD converts 5-fluorocytosine after a series of reactions into cytotoxic metabolites. The effect on tumor growth inhibition was further increased when a bifunctional fusion protein CD:uracil phosphoribosyltransferase was used (Chung-Faye et al., 2001).
2.3.3. Physical targeting techniques Several physical targeting techniques are available to deliver gene therapy vectors directly to the correct location, increasing the concentration of the vector at the target site. For the treatment of malignant gliomas, an adenovirus-HSV-Tk was administered by local injection to the tumor bed (Immonen et al., 2004). In the treatment of a retinal disorder, an adeno-associated viral vector expressing RPE65 was administered locally to the retina (Buch et al., 2008). Gene transfer to the inner organs without surgery can be locally performed using catheters (Turunen et al., 2005; Rutanen et al., 2004). Target cells, like CD34+ cells, can be extracted, and transduced outside the body to increase the accessibility of the vector to the cell. After transduction, the cells are returned back to the same subject (Cavazzana-Calvo et al., 2000). This Ex vivo approach minimizes the vector leakage into the circulation and reduces the immune response but the method is only possible for those cells which can be isolated and grown in vitro. Other physical targeting techniques are the use of different delivery systems to retain the virus in the target, such as various collars (Laitinen et al., 1997). There can be enhanced delivery of naked DNA by electroporation or ultrasound. Less effective methods are focused laser, magnetic fields and ballistic gene delivery (gene gun) (Wells, 2004) but for example ballistic gene delivery has shown its applicability to deliver DNA vaccines to the skin (Trimble et al., 2003). However, in terms of viral gene delivery, these preceding methods have been less intensively investigated. Nonetheless, Chen et al (Chen et al., 2003) have reported that ultrasound mediated vector delivery could enhance adenoviral gene transfer to the heart using a liposome microbubble.
2.3.4. Viral vector targeting
After systemic administration, viral vectors are present in the blood circulation. The affinity of the vector to the target cell is based on the interaction between the viral receptor and the viral surface proteins. After internalization, most viral genomes need to travel to the nucleus where their transcription takes place. During this lifecycle, the vector has to pass several obstacles and thus also targeting of the viral vector can be dealt with at different levels. By modifying the surface, viruses can be targeted to specific cells or tissues. This

39
transductional targeting can be achieved by displaying a suitable ligand to bind the specific receptor through genetic engineering or through adaptor system (Waehler et al., 2007). In addition, viral tropism can be altered through pseudotyping (Cronin et al., 2005). Transcriptional targeting uses promoters to target transgene expression to the selected cells/tissues (Miller and Whelan, 1997). This exogenous control of expression improves the safety of the therapy and is important for protecting healthy cells in suicide gene strategies. Alternatively, integration might be targeted to a special locus of the chromosome (Bushman and Miller, 1997; Goulaouic and Chow, 1996). The next section will discuss the virus surface modifications in viral vector targeting. Table 2. summarizes the different targeting approaches with their advantages and disadvantages (Waehler et al., 2007; Verhoeyen and Cosset, 2004; Campos and Barry, 2007).
2.3.4.1. Genetic targeting concept
Direct targeting involves genetic engineering of the capsid or envelope protein in order to incorporate the targeting ligands (polypeptide, small peptide motif, or antibody) onto the viral surface. This approach was first introduced with retroviruses in 1993 when a single-chain antibody was displayed on the surface of MLV (Russell et al., 1993). Since then, the approach has been successfully applied for several vectors, such as AAV (Yang et al., 1998), adenovirus (Hedley et al., 2006), baculovirus (Mottershead et al., 2000) and herpes simplex virus (Menotti et al., 2006). For example, a successful in vivo study was performed also with an oncolytic measles virus, which was fully retargeted to tumor cells by the surface display of single-chain antibody fragments. The treatment reduced the tumor size and enhanced the survival of the treated mice (Nakamura et al., 2005). The advantage of the genetic targeting concept is that only a single production of viral vector with homogenous high titer is required and in addition, there is no risk of ligand dissociation (Waehler et al., 2007). Several techniques have been developed to genetically incorporate the targeting ligand to the virus surface. However, the genetic engineering is not always straightforward and the success of this putative single-component system depends on the context of the ligand and envelope protein. The targeting ligand can replace the natural receptor-binding domain in the surface protein (Benedict et al., 1999). An improved system was achieved by attaching an additional binding domain onto the surface protein to increase the transduction efficacy (MoMLV displaying collagen-binding domain) (Hall et al., 2000; Hall et al., 1997). Fusion to the N-terminus of the natural envelope glycoprotein has been successful with several viruses. For example “early-acting cytokine”-displaying lentiviruses achieved an improved transduction efficiency to the immature hematopoietic cells (Verhoeyen et al., 2005) or avidin-displaying baculoviruses have also exhibited enhanced transduction abilites (Raty et al., 2004). Funke et al. described recently targeted lentiviral vectors pseudotyped with envelope glycoproteins from measles virus, namely the hemagglutinin (H) and fusion protein (F), responsible for receptor recognition and fusion, respectively. The native receptor binding activity was destroyed and replaced by EGF or a single-chain antibody

40
directed against CD20 and gene transfer into cells expressing the targeted receptors was significantly improved (Funke et al., 2008).
The incorporation of a heterologous ligand may have an unexpected influence on behavior of the engineered virus. In some cases, the displayed moiety interferes with the protein fusion ability of the envelope protein (Russell and Cosset, 1999; Benedict et al., 1999), leading to a block in the virus entry (e.g. retrovirus displaying a CD33-specific single-chain antibody) (Zhao et al., 1999). It is also possible that the displayed ligand directs the virus into inappropriate cellular pathways during the production process(Magnusson et al., 2002) or that the virus is attached to the correct target cell but is directed to a non-functional entry pathway (Larochelle et al., 2002). The incorporation of a binding moiety onto the surface of a non-enveloped virus can be even more difficult because it may disturb the folding or trimerization of a virion protein, such as has been described for adenoviruses (Campos and Barry, 2007).
Viral vectors infect cells quite promiscuously. Two promising methods have been developed for targeting of the viral vector by host-range restriction, known as inverse and protease targeting. The inverse targeting selectively destroys the ability of the viral vector to infect non-target cells. The strategy exploits receptor-mediated virus neutralization as reported for retroviral vectors displaying EGF. These vectors were able to transduce EGFR-negative cells but were sequestered to lysosomes by EGFR-positive cells (Cosset et al., 1995). The concept of protease targeting is based on the narrowing of the infectivity to those cells expressing a targeted protease. Protease targeting is achieved by inserting a protease-cleavable blocking domain onto the viral envelope protein such that the infectivity of the virus is restored after the protease cleavage (Russell and Cosset, 1999). For example, a protease targeted retroviral vector achieved a selective transduction to matrix-metalloprotease-rich target cells in vivo (Peng et al., 1999).
2.3.4.2. Pseudotyping
The entry of an enveloped virus into a host cell is determined by the characteristics of the virus envelope glycoprotein and the interaction with its specific receptor. The host cell tropism of a viral vector can be expanded or altered through pseudotyping by changing the viral envelope glycoprotein with a heterologous envelope glycoprotein from another virus (Cronin et al., 2005; Russell and Cosset, 1999). Pseudotyping is considered as a part of genetic targeting. Pseudotyping is achieved during the viral vector production by expressing the alternative envelope protein from a separate plasmid (e.g. lentiviruses) (Cronin et al., 2005) in co-transfection, from the producing cell line (retroviruses) in trans (Russell and Cosset, 1999), or alternatively by genetically incorporating into the viral genome (adenoviruses) (Campos and Barry, 2007). Typically pseudotyping enhances the transduction efficiency but it does not necessarily provide any major degree of specificity if the vectors are administered systemically. On the contrary, pseudotyping can be used also to restrict the host range.

41
Enveloped viruses, such as retroviruses including lentiviruses, are highly permissive for the incorporation of different glycoproteins (Cronin et al., 2005). For example, the transduction efficacy of retro- or lentiviruses could be enhanced by changing the natural virus envelope for the feline endogenous retrovirus (RD114) or the gibbon ape leukaemia virus (GalV) 10A1 (Porter et al., 1996; Uckert et al., 2000) envelope proteins. Glycoproteins from alphaviruses, lyssaviruses or baculoviruses (Strang et al., 2004; Strang et al., 2005; Sena-Esteves et al., 2004; Kumar et al., 2003) have also been used. The incorporation of the envelope glycoprotein may be very inefficient and may require the modification of the envelope protein cytoplasmic tail (Sandrin et al., 2002).
The VSV-G has been the most widely used envelope glycoprotein in pseudotyping (Russell and Cosset, 1999; Barsoum et al., 1997). It has been used for retrovirus (Burns et al., 1993), lentivirus (Naldini et al., 1996b; Reiser et al., 1996), baculovirus (Barsoum et al., 1997), herpes simplex virus (Anderson et al., 2000) and also for adenovirus pseudotyping (Yun et al., 2003). VSV-G has broadened the transduction range (Coil and Miller, 2004), enhanced the transduction efficiency (Barsoum et al., 1997), strengthened fragile viruses and made possible concentration by ultracentrifugation (Burns et al., 1993). However, some toxicity of VSV-G has been observed after virus transduction with very high virus concentrations (Watson et al., 2002; Park et al., 2000). In addition, VSV-G has shown toxicity also to the packaging cells (Burns et al., 1993; Ory et al., 1996) thus hindering the development of production cell lines constitutively expressing VSV-G. So far, the only suitable alternatives are to use either inducible packaging cell lines for lentiviruses (Pacchia et al., 2001; Farson et al., 2001) or to replace the toxic VSV-G protein with less-toxic glycoproteins (Kumar et al., 2003). Another limitation of VSV-G has been the immune response. It evokes after systemic administration and furthermore the viruses are inactivated by human serum complement (DePolo et al., 2000; Schauber-Plewa et al., 2005). Many mutation studies have been performed in order to identify the important regions of the protein so that smaller parts of the protein could be used for specific purposes (Doms et al., 1988; Li et al., 1993). The membrane-proximal stem region of the VSV-G protein along with the transmembrane and cytoplasmic domains can promote membrane fusion activity when expressed together with a heterologous viral fusion protein (Jeetendra et al., 2002). In one example, only the 21-amino acid ectodomain with the transmembrane and cytoplasmic tail domains of VSV-G (VSV-GED) improved the transduction of baculovirus (Kaikkonen et al., 2006). The shorter VSV-G fragment has been utilized in the development of surface display vectors (Ojala et al., 2004).
2.3.4.3. Serotype switch
Serotype switching is included in pseudotyping. A fiber of a non-enveloped virus, such as adenoviruses and AAV, can be replaced by that of another serotype or simply by modifying the knob domain. However, it is far from straightforward to incorporate a protein into a capsid (Campos and Barry, 2007; Kwon and Schaffer, 2008). For example adenovirus Ad5 entry into host cells is mediated by binding to cell surface coxsackievirus B and adenovirus

42
receptor (CAR), but pseudotyped vectors may use alternative intracellular trafficking pathways (Campos and Barry, 2007). However, the use of a different serotype in pseudotyping should allow readministration of the same vector while avoiding an immune response.
2.3.4.4. Adaptor targeting concept
An alternative method in viral vector targeting is to use adaptors or molecular bridges between the virus and the cell surface. The adaptor design consists of a virus binding moiety fused to the molecule to bind the receptor which is expressed in the target cells. The adaptor can be a receptor-ligand fusion, monoclonal antibody, bispecific antibody or it can be based on the avidin-biotin system (Waehler et al., 2007). The avidin-biotin technology is discussed in detailed in the next section. An example of the receptor-ligand system is the retroviral vector carrying the avian leukosis virus (ALV) envelope which was successfully targeted to specific cells by fusing a soluble form of the ALV receptor to a cell-binding domain (Boerger et al., 1999). Antibody-based applications have been designed for all most frequently used gene therapy vectors: adenoviral, retroviral, AAV as well as for lentiviral vectors (Waehler et al., 2007). The monoclonal antibody approach requires genetical modification of the virus to express an Ig-binding domain to which the antibody binds (Tai et al., 2003; Volpers et al., 2003). Unfortunately, competition between circulating blood antibodies and the targeting antibodies hinders in vivo applications and this method may be more practical for ex vivo work. A bispecific antibody consists of two antibodies fused together having specificity for the virus and the target (Bartlett et al., 1999). However, the bond of bispecific antibodies is also generally considered as a weak interaction and this can hinder in vivo use. The last adaptor system is based on chemical modification e.g. using polyethylene glycol, to couple ligands to the virus surface (Lanciotti et al., 2003). The clear advantage of this system is that modification can protect the virus from complement inactivation and adaptor dissociation does not occur.
In conclusion, adaptor systems have proven to be versatile, absolute knowledge of the viral structure is not obligatory and this approach allows easy testing of several target receptors. The disadvantages include possible dissociation of the ligand in vivo, batch-to-batch variation of the produced adaptors and furthermore, this approach is more complicated.
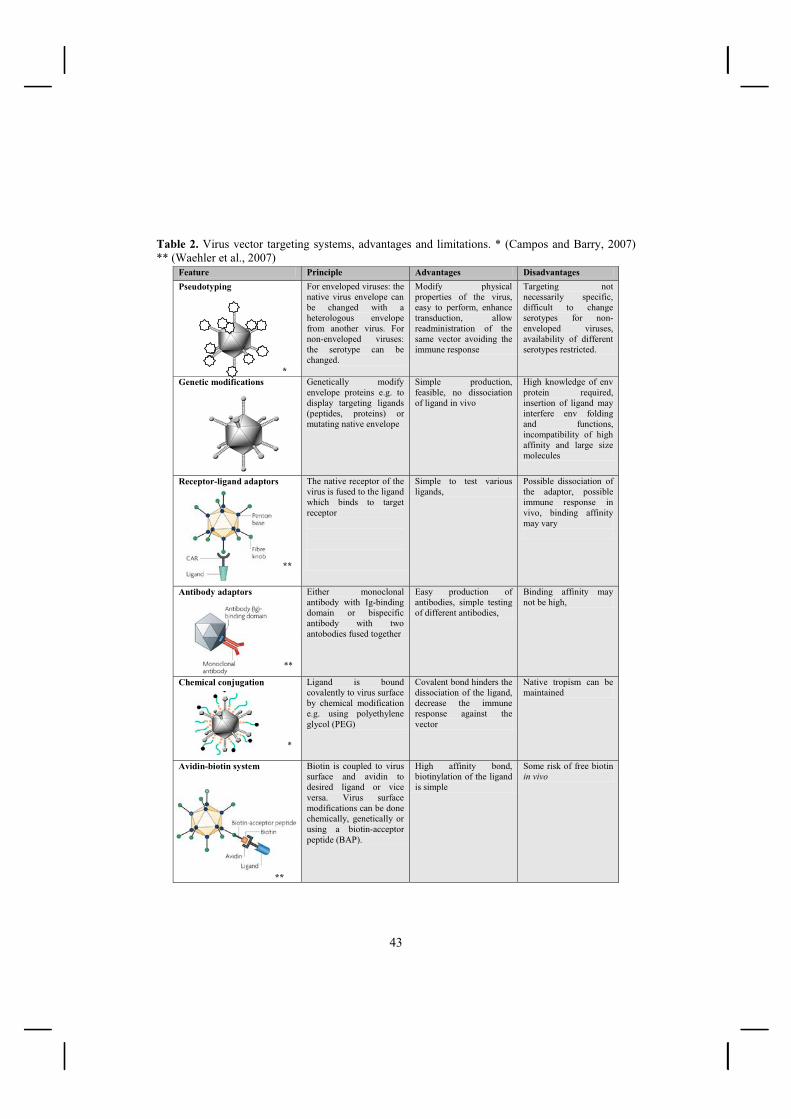
43
Table 2. Virus vector targeting systems, advantages and limitations. * (Campos and Barry, 2007) ** (Waehler et al., 2007)
Feature Principle Advantages Disadvantages Pseudotyping
*
For enveloped viruses: the native virus envelope can be changed with a heterologous envelope from another virus. For non-enveloped viruses: the serotype can be changed.
Modify physical properties of the virus, easy to perform, enhance transduction, allow readministration of the same vector avoiding the immune response
Targeting not necessarily specific, difficult to change serotypes for non-enveloped viruses, availability of different serotypes restricted.
Genetic modifications
Genetically modify envelope proteins e.g. to display targeting ligands (peptides, proteins) or mutating native envelope
Simple production, feasible, no dissociation of ligand in vivo
High knowledge of env protein required, insertion of ligand may interfere env folding and functions, incompatibility of high affinity and large size molecules
Receptor-ligand adaptors **
The native receptor of the virus is fused to the ligand which binds to target receptor
Simple to test various ligands,
Possible dissociation of the adaptor, possible immune response in vivo, binding affinity may vary
Antibody adaptors **
Either monoclonal antibody with Ig-binding domain or bispecific antibody with two antobodies fused together
Easy production of antibodies, simple testing of different antibodies,
Binding affinity may not be high,
Chemical conjugation *
Ligand is bound covalently to virus surface by chemical modification e.g. using polyethylene glycol (PEG)
Covalent bond hinders the dissociation of the ligand, decrease the immune response against the vector
Native tropism can be maintained
Avidin-biotin system
**
Biotin is coupled to virus surface and avidin to desired ligand or vice versa. Virus surface modifications can be done chemically, genetically or using a biotin-acceptor peptide (BAP).
High affinity bond, biotinylation of the ligand is simple
Some risk of free biotin in vivo

44
2.4 Avidin-Biotin technology Avidin , originally isolated from chicken egg and its bacterial analogue, streptavidin from Streptomyces avidinii, have extremely high affinity for biotin (Kd ~ 10-13-10-15 M) (Green, 1990). This unique feature is the basis of avidin-biotin technology. To date the versatile (strept)avidin-biotin technology has been used extensively for different applications in the field of molecular biology and biotechnology such as labeling, chromatographic applications, immunoassays, targeting, probing or diagnostic purposes (Figure 10) (Diamandis and Christopoulos, 1991; Laitinen et al., 2007; Laitinen et al., 2006). Figure 10. Schematic diagram of (strept)avidin-biotin technology. (Strept)avidin and biotin are used as a bridge between the different target molecules (antibodies, probe, cytotoxic compound, radiolabel, color reagent ect.). The versatile technology has been utilized in many kinds of biotechnological applications. B: Biotin moiety. Attached molecule can be e.g. fluorescent label, radiolabel or drug.
2.4.1. Avidin and Streptavidin Avidin, which is a tetrameric glycoprotein, constitutes 0.05 % of the hen egg mass. Each avidin monomer is 15 600 Da and the glycosylated tetramer is approximately 62 400 Da. One monomer binds one biotin molecule and this interaction has the highest affinity known between a protein and an organic ligand (Green, 1975; Green, 1990). The bacterial protein, streptavidin, shares many similarities with avidin. The streptavidin monomer is 16 500 Da and the size of tetrameric protein is c. 60 000 Da (Green, 1990). Both proteins consist of four identical monomers and most of the binding residues are conserved. Avidin and
(Strept)avidin-biotin
technology
Attached molecule
Avidin
Target
BB
Binder (antibory etc.)
•Drug targeting
•Affinity targeting
•Probing
•Microscopy applications
•Histochemistry
•Signal amplification
•Isolation studies
•Imaging
•Blotting technology
•Chromatographic applications
•Diagnostic
•Flow cytometer
•Immobilisation
(Strept)avidin-biotin
technology
Attached molecule
Avidin
TargetTarget
BB
Binder (antibory etc.)
•Drug targeting
•Affinity targeting
•Probing
•Microscopy applications
•Histochemistry
•Signal amplification
•Isolation studies
•Imaging
•Blotting technology
•Chromatographic applications
•Diagnostic
•Flow cytometer
•Immobilisation

45
streptavidin have only 60% amino acid sequence identity but their secondary, tertiary and quaternary structures are rather similar (DeLange, 1970; Argarana et al., 1986). The proteins are very stable against heat, denaturants, pH changes or proteolytic enzymes (Wilchek et al., 2006). Although avidin and streptavidin share many similarities, there are also some differences in their characteristics. Avidin is highly glycosylated, comprising mainly of mannose and N-acetylglucosamine residues, the isoelectric point is 10.5 and avidin is positively charged (DeLange, 1970; Bruch and White, III, 1982). Streptavidin is not glycosylated and has a pI of 5-6. Due to these differences, the proteins display different behaviors when administered into the circulation (Schechter et al., 1990; Rosebrough, 1993). Avidin is rapidly cleared from the circulation and accumulates in the liver. This feature has been exploited when avidin has been used as a clearing agent of biotinylated molecules from the circulation (Klibanov et al., 1988). Modifications such as deglycosylation or neutralization, lengthen the circulation time of avidin. Streptavidin remains for a longer time in the circulation as it has lower nonspecific binding to most tissues compared to the avidin, however it does accumulate in the kidneys (Schechter et al., 1990; Rosebrough, 1993). Both proteins have some tumor specificity. Avidin was shown to accumulate to intraperitoneal tumors when it was administered intraperitoneally into mice (Yao et al., 1998). In this study, streptavidin did not show tumor specific accumulation whereas radiolabelled streptavidin improved tumor/normal tissue ratios in xenografted colon cancer nude mice (Hnatowich et al., 1993). Avidin or streptavidin are not found in mammalian tissues providing specificity for their use. However, both proteins are immunogens which may prevent their repeated use (Hytonen et al., 2003; Knox et al., 2000).
In attempts to improve the usability of the (strept)avidin technology, the proteins involved have been modified both chemically and genetically. Genetic modification of (strept)avidin can change the binding activity to biotin, stability, alter the physio-chemical properties, structure or function of the proteins (Laitinen et al., 2007). Examples of modifications are adjustable pH-dependent binding of avidin to biotin (Nordlund et al., 2003), a monomeric form of avidin (Laitinen et al., 2003) or in order to change binding properties of streptavidin to biotin by conjugating stimuli-responsive polymers to streptavidin e.g. to respond to light or temperature (Stayton et al., 2004).
2.4.2. Biotin and biotinylation Biotin (244 Da) is an essential vitamin, known as vitamin H or B7, functioning as a coenzyme in carboxylation reactions. In mammalian cells, active biotin is covalently conjugated to biotin-dependent proteins by biotin protein ligase (BPL), also known as holocarboxylase synthetase. Bacteria on the other hand, use BirA biotin ligase in biotinylation reactions (Chapman-Smith and Cronan, Jr., 1999). In avidin-biotin systems, one component must be always labeled with biotin (=biotinylation). Fortunately, biotin contains a carboxylic acid moiety, and the rest of the molecule is relatively inert. Therefore,

46
the conjugation of the molecule to the target protein can be achieved without losing avidin binding activity (figure 11).
The biotinylation can be a chemical or biological process and several strategies have been developed. The coupling of biotin to a protein or to an antibody can be performed with the use of different biotinylation reagents. The most commonly used reagent is sulfosuccinimidyl o-(biotinamido) hexanoate (NHS-LC-biotin) (Hnatowich et al., 1987). In chemical biotinylation, biotin is easily attached to amino acid side chains covalently in non-specific manner thus requiring a purification step after the procedure. Chemical biotinylation is not site-specific and sometimes can lead to the inactivation of the protein (Stolz et al., 1998). Metabolical biotinylation was developed to overcome the problems associated with chemical biotinylation. The first biological biotinylation was described for a bacterial and yeast expression system (Cronan, Jr., 1990). Since then, the technology has been adapted to mammalian cells requiring genetic engineering of the protein to display a biotin acceptor peptide (BAP) which is then covalently biotinylated by cellular biotin protein ligase during the translation in the living cells (Parrott and Barry, 2000). Metabolic biotinylation allows the simple production of biotinylated viruses, vectors, proteins and vaccines ready to use for targeting or purification (Barry et al., 2003). Other advantages of the avidin-biotin system are the four binding sites of avidin to biotin conferring the potential for amplification of the signal, the small size of the biotin assisting the penetration of the compounds into tissue and the rapid clearing of free biotin by the kidneys.
Figure 11. The chemical structure of biotin
2.4.3. Avidin and biotin in pretargeting Since 1988 avidin-biotin technology has been used in pretargeting (Paganelli et al.,
1988) Paganelli et al. demonstrated a two-step strategy to target ovarian cancer cells using biotinylated antifolate receptor monoclonal antibody and radioactive 111In-labelled streptavidin (Paganelli et al., 1992). Subsequently, several different pretargeting approaches have been introduced. The basic principle of the 3-step avidin-biotin pretargeting method is to first target the tumor with a biotinylated antibody, followed by the injection of avidin (or streptavidin). The third step is then the injection of the biotinylated therapeutic compound which binds to the avidinylated tumor cells (Paganelli et al., 1990; Paganelli et al., 1999).

47
Avidin undergoes quite a rapid clearance from the circulation. It been has been utilized as a second step for the clearance of non-bound antibody from the blood flow. On the following day, streptavidin was injected and since streptavidin has a longer half-life, it has more time to bind to the biotinylated antibody localized at the tumor site (figure 12) (Magnani et al., 2000). The avidin-biotin system can be exploited also in the reverse way. For example, in a clinical trial Shen et al. targeted a streptavidinylated antibody to gastrointestinal cancer cells, followed by clearing of this compound from the circulation by treatment with a galactosylated-biotinylated clearing agent, and finally by the administration of the radiolabelled biotin (Shen et al., 2005). The pretargeting reduces the level of free radiolabeled agent and thus improves the exposure ratio between healthy and tumor tissue. Avidin-biotin based pretargeting strategies have already been used successfully in several clinical trials for diagnostic and treatment against several different cancer types without significant toxicity (Paganelli et al., 1999; Kalofonos et al., 1990; Knox et al., 2000; Crippa et al., 1995). The property of avidin to accumulate into sites of inflammation has also been used for imaging purposes (Yao et al., 1998; Shoup et al., 1994).
Figure 12. Three step pretargeting system. First, the tumor is targeted with a biotinylated antibody, followed by the injection of avidin and streptavidin. The third step is then the injection of the biotinylated therapeutic compound, which binds to streptavidin-coated tumor cells. (Goldenberg et al., 2006).

48
Another pretargeting approach has been to deliver an avidin gene into the target
tissue (Walker et al., 1996). In one example, the avidin and the endocytotic LDL receptor fusion gene were delivered to tumor cells using recombinant viral vectors and were expressed on the surface of the target cells. This fusion protein was capable of high-affinity binding of biotinylated molecules (Lehtolainen et al., 2003). Another comparable fusion protein is Scavidin, which consists of a fusion protein between avidin and the macrophage scavenger receptor. This fusion protein has also showed binding and endocytosis of biotinylated ligands (Lehtolainen et al., 2002a). This molecule has also been used in imaging applications (Mantyla et al., 2006).
2.4.4. Avidin-biotin technology in gene therapy applications
In gene therapy, the (strept)avidin-biotin technology has been utilized in several viral vector targeting applications. One of the first avidin-biotin adaptor-based targeting systems was published utilizing retroviral vectors in 1989 by Roux et al., where streptavidin was used as a bridge between antibodies against the viral envelope protein, and antibodies against specific cell membrane markers (Roux et al., 1989). Since then, many different avidin-biotin systems have been developed. Most commonly these techniques rely on coating the viral vector with biotin chemically or metabolically. Conversely, the viral vector can be coated by avidin chemically or genetically but these approaches are not so common.
The first chemically biotinylated adenoviral vectors examined by Smith et al. showed promising results in the targeting of the biotinylated virus to specific cells. In that study, biotinylated ligand for the c-Kit receptor was used as a targeting moiety where avidin worked as a bridge (Smith et al., 1999). An increased transduction to target cells by a similar approach of chemical biotinylation has been achieved with a vaccinia vector (Purow and Staveley-O'Carroll, 2005) and AAV (Ponnazhagan et al., 2002). Many of the commonly used viral vectors, such as adenovirus (Campos et al., 2004; Parrott et al., 2003; Pereboeva et al., 2007) AAV (Arnold et al., 2006; Stachler and Bartlett, 2006), lentivirus (Chan et al., 2005; Nesbeth et al., 2006) and baculovirus (Kaikkonen et al., 2008) have been metabolically biotinylated. Metabolic biotinylation has proven to be a way to achieve a degree of purification since biotinylation takes place already during the production and different purification methods such as monomeric avidin (Parrott et al., 2003) or (strept)avidin-coated magnetic particles (Chan et al., 2005; Kaikkonen et al., 2008) are available. Most of the published studies have demonstrated functionality in vitro but so far there is no impressive in vivo data available. Pereboeva et al. reported in vivo proof-of-principle targeting of metabolically biotinylated adenovirus vectors with epidermal growth factor (EGF)-Streptavidin to EGF receptor (EGFR) expressing cells (Pereboeva et al., 2007).
The opposite approach is to incorporate avidin onto the virus surface. Genetically modified baculoviral vectors displaying avidin enhanced viral transduction and efficiently

49
targeted viruses (Raty et al., 2004). In addition, avidin has been utilized in viral biodistribution imaging by magnetic resonance imaging (MRI) and single photon emission computed tomography (SPECT) in vivo by using biotinylated ligands (Raty et al., 2006; Raty et al., 2007a). Not only the viral vectors, but also streptavidin-fusion protein expressing cells have been used in cell extraction (Gotoh and Matsumoto, 2007) and biotinylated (Carpenter and Minchin, 1998) or (strept)avidinylated non-viral DNA vectors (Schwarzenberger et al., 1996; Wojda and Miller, 2000) have been used for targeting. In conclusion, the presence of biotin or avidin on the vector surface allows easy exchange of the targeting ligand, and thus the same construct can be applied in several applications which broadens the usefulness of this approach.
2.5. Imaging of viral biodistribution and transgene expression Imaging is important not only as a diagnostic tool, but also in monitoring the progress of therapy. Molecular imaging provides an opportunity to study diseases in a non-invasive manner of the cellular or even the genetic level. The progress towards safer gene therapy treatments requires the understanding of molecular aspects of vector delivery but also of the expression of the exogenous genes. No single technique alone possesses the complete spectrum of specification with which to image different applications from research to the clinic (Weissleder et al., 2000). Thus, several high resolution non-invasive molecular imaging techniques, including single photon emission tomography (SPECT), positron emission tomography (PET), magnetic resonance imaging (MRI), and optic imaging are available. These imaging techniques have differences in terms of resolution, availability, cost and contrast agent and often a combination of the techniques is needed (Massoud and Gambhir, 2003; Glunde et al., 2007).
To improve safety in the gene therapy field, it would be of great importance to know if an administered vector has reached the target, did the vector cause any toxicity, and then where and at what level was the transgene expressed (Bogdanov, Jr., 2003). Noninvasive imaging provides answers to these questions but both the imaging of viral biodistribution and gene expression needs to be carefully addressed. The imaging of viral biodistribution is achieved by coating viruses with suitable ligands (Min and Gambhir, 2004). Gene expression imaging is achieved by specific probes or contrast agents that allow either direct or indirect evaluation of gene expression. Direct evaluation is possible if the contrast agent binds directly to a specific target. On the contrary, the indirect method requires the use of specific substrate probes for the target enzyme. In addition, reporter/marker genes may be part of the indirect evaluation (Blasberg, 2002). Molecular imaging technologies have been utilized for monitoring the success of gene therapy of cancer, cardiovascular, neurological, musculoskeletal, hepatitic and inherited diseased (Min and Gambhir, 2004). Table 3. summarizes the differences in the commonly used imaging techniques.

50
2.5.1. Magnetic Resonance Imaging (MRI) MRI is a non-invasive imaging method based on the interaction of nuclear spins
within a strong external magnetic field. Protons (hydrogen nuclei) are excited by the radio-frequency pulse under the influence of a high magnetic field. When the excited spins undergo a relaxation process back to the initial energy level, a signal can be detected and an image or spectral information about the molecular properties of the tissue may be determined (Meikle et al., 2006). The realignment of nuclei with the magnetic field is termed T1 relaxation whereas T2 and T2* relaxation occurs when the nuclei dephase as a result of the presence of subtle local field inhomogeneties induced by changes in tissue composition surrounding the nuclei (Massoud and Gambhir, 2003). The most commonly used T1 contrast agent for MRI is gadolinium and a typical T2 contrast agent consists of iron oxide nanoparticles (Raty et al., 2007b). By coupling these two specific probes, e.g. to proteins, receptor ligands, enzyme substrates or monoclonal antibodies, they provide the possibility to study key biological processes. MRI provides exquisite contrast between the different soft tissues and between the hard and soft tissues of the body. The spatial resolution of MRI imaging is very high (4-200 �m) and does not involve the use of a radiation source. However, the technology is expensive and it has a relatively poor sensitivity for labeled probes (Meikle et al., 2006; Massoud and Gambhir, 2003). It has proved feasible to utilize MRI in gene expression studies by using cell surface receptors with suitable contrast agents. An expressed transferrin receptor was visualized by targeting superparamagnetic nanoparticles into cells (Weissleder et al., 2000). Tyrosine aminoacid (Weissleder et al., 1997) or expression of proteins, such as �-galactosidase -enzyme (Louie et al., 2000) are detectable after altering the biological properties of the contrast agent used. Gene expression can be visualized by MRI also without any external contrast agents using a cellular receptor, like ferritin which accumulates iron into cells (Cohen et al., 2005; Genove et al., 2005). MRI has been used less frequently to study viral biodistribution, though there is a report of coupling the virus surface with USPIO particles (Raty et al., 2006). MRI has been also used in tracking the in vivo distribution of cell vectors, like macrophages, glia cells or stem cells (Rome et al., 2007).
2.5.2. PET and SPECT Single photon emission computed tomography (SPECT) and positron emission
tomography (PET) are highly sensitive nuclear imaging techniques that require the administration of radionuclides. In SPECT imaging, a gamma camera detects gamma rays emitted by the radioisotope, like 111In, 123I or 99mTc. These radionuclides emit only a single photon and have a longer decay time than PET isotopes. In PET imaging, the positron-emitting isotopes, like 11C, 15O, 13N or 18F are used as radiotracers (Meikle et al., 2006; Raty et al., 2007b). The emission of the positron is annihilated in the tissue with an electron which produces two high-energy (511 keV) photons detected by the PET detector array and this is used to reconstruct the cross-sectional images (Raty et al., 2007b). PET has higher

51
sensitivity for detecting labeled probes compared to SPECT but it is also more expensive (Massoud and Gambhir, 2003). Both devices have 1-2 mm spatial resolution (table 3). The labeling of a wide range of endogenous molecules (peptides, antibodies and other large proteins) with SPECT isotopes is relatively straightforward. PET is better suited for imaging labelled drugs. The PET probes tend to have short half lives whereas SPECT isotopes can have half-lives of several days. SPECT allows the simultaneous detection of two or more radio-labeled probes based on the energy difference of the radiolabels (table 3) (Meikle et al., 2006; Min and Gambhir, 2004; Rome et al., 2007).
Several viruses have been radiolabeled and the virus biodistribution has been studied after different administration routes. 111In labelled herpes simplex virus was used to study the viral biodistribution (Schellingerhout et al., 1998) and also to monitor the success of viral accumulation into a gliosarcoma tumor after different administration routes (Schellingerhout et al., 2000). The results showed that 71% of the virus remained in the tumor 24 h after the treatment. 99mTc labelled adenovirus provides a possibility to evaluate the liver uptake of the virus (Zinn et al., 1998). Recently, an avidin displaying baculovirus was coupled to biotinylated 99mTc and the influence of different administration routes in viral biodistribution was evaluated (Raty et al., 2007a). It was concluded that the virus was capable of spreading via the lymphatic network and had strong tropism to kidneys and spleen (Raty et al., 2007a).
Transgene expression has been commonly monitored via marker genes using either intracellular enzymes or cell-surface receptors. The most widely used marker gene is the Herpes Simplex Virus thymidine kinase (HSV-Tk) (Tjuvajev et al., 1998) and its variants (Gambhir et al., 2000). HSV-Tk selectively metabolizes by phosphorylation the radiolabelled probes leading to the entrapment and accumulation of the probe in the Tk-expressing cells. In addition, a number of other reporter genes, such as cytosine deaminase (Haberkorn et al., 1996) have been developed. An alternative method is receptor-mediated imaging, where the cell surface dopamine type 2 (D2R) receptor can be targeted with a ligand-labeled radiotracer (MacLaren et al., 1999).
2.5.3. Other imaging techniques Optical imaging refers to fluorescence imaging and luminometric imaging. Fluorescence imaging uses external light to excite the fluorochrome (e.g. GFP, DsRed) which then emits light at a different wavelength detected with a low-light charge-coupled device (CCD). Different resolutions and depths can be imaged but the tissue penetration hinders the detection of the light signal. In luminometric imaging, the emission of visible photons is detected in an energy-dependent reaction catalyzed by luciferase enzymes (Golzio et al., 2004). This is very sensitive and specific technique but the maximum depth of luminometric imaging is only 2-3cm (Massoud and Gambhir, 2003). In gene therapy, optical imaging has provided a practical tool to quantify the expression of the reporter gene and has been widely used with many viral vectors (De et al., 2003; Wu et al., 2003a; Lipshutz et al., 2001; Pfeifer et al., 2001; Yang et al., 2000).

52
In ultrasound imaging, acoustic waves are transmitted through tissues and images are obtained when waves are reflected from surfaces between different tissues. This technique has high spatial resolution, is quite inexpensive and is highly suitable for observing perfusion and anatomical characteristics (Min and Gambhir, 2004; Rome et al., 2007). The technique can be utilized in monitoring the response to gene therapy, for example after viral mediated vascular endothelial growth factor gene transfer (Rissanen et al., 2005).
In computed tomography (CT), the image is obtained when X-rays pass through the body and they are differentially absorbed depending on the composition of the tissue. CT provides high resolution anatomical data, but has relatively poor soft-tissue contrast (Massoud and Gambhir, 2003). CT imaging is commonly combined with PET and SPECT techniques (Mittra and Quon, 2009). Table 3. Comparison of the molecular imaging technologies. Modified from (Meikle et al., 2006; Glunde et al., 2007; Massoud and Gambhir, 2003) Advantages Limitations CT High spatial resolution (50-200 �m)
No depth limit Suitable for anatomical imaging
Exposition to radiation Limited soft tissue resolution
MRI Excellent soft tissue contrast High spatial resolution (4-200 �m) No use of ionizing radiation No depth limit Suitable for receptor-ligand and gene expression studies
Expensive and technically challenging Relatively poor sensitivity to labelled probes Long scans and processing times Large size of contrast agents/reporters
SPECT High sensitivity Many probes available Less expensive Simultaneous multiple probe imaging No depth limit Suitable for receptor-ligand and gene expression studies
Low spatial resolution (1-2 mm) Lower sensitivity that PET Involves ionizing radiation Long –lived isotopes
PET Highest sensitivity for labeled probes Short-lived isotopes Isotopes can substitute natural atoms No depth limit Quantitative
Low spatial resolution (1-2 mm) Expensive cyclotron required for generating some isotopes Technically demanding Involves ionizing radiation
Optical High sensitivity imaging Cost effective Quick, relatively high-throughput Suitable for reporter/gene expression,cell trafficking
Low resolution (3-5 mm) Restricted depth detection (1-2 cm) Only limited ability to scale to human imaging
Ultrasound High spatial resolution (>40 �m) Cost effective Suitable for morphological studies Real-time Low cost
Few probes available Depth limit to centimeters

53
4. AIMS OF THE STUDY The general aim of this thesis was to improve the properties of lentiviral vectors for gene therapy with special emphasis on improving the targetability of the vectors. Targeted treatment was based on two different avidin fusion proteins displayed on the lentiviral surface or expressed by the lentivirus. Also, baculovirus methodology was developed. These goals were more specifically divided into aims as follows:
I To adapt baculovirus generation, titering and protein production into a high throughput 96-well plate format.
II To develop a novel production method for lentiviral vectors based on recombinant
baculoviruses.
III To construct lentivirus which display (strept)avidin on the viral surface for targeting and for non-invasive dual imaging of viral biodistribution and transgene expression
IV To construct an avidin fusion protein expressing lentiviral vector for targeted
therapy, study the level and efficacy of its expression and evaluate any potential toxicity and immunogenicity of the virus and transgene in vitro and in vivo.

54
5. MATERIALS AND METHODS
5.1. Methods Basic molecular cloning methods were used in all the studies. Primary baculoviral
vectors and lentiviral vectors were produced by plasmid transfection and secondary baculoviral vectors by primary virus infection. The produced viruses were titered and tested in vitro. In addition, some of the produced lentiviruses were also used in vivo. Methods are listed in table 4. and more detailed descriptions are found in the original publications. Table 4. Methods used in these studies Method Description Used in DNA cloning Plasmid construction I-IV Integration site determination Inverted PCR , TA-cloning, sequencing I Production of baculoviral vectors
Transient bacmid transfection Secondary baculovirus infection
I-II
Analysis of baculoviral vectors
End-point dilution Flow cytometer analysis SDS-PAGE and immunoblotting
I-II I I-II
Production of lentiviral vectors
Transient plasmid transfection Baculovirus transduction Concentration by ultracentrifugation
II-IV II III-IV
Analysis of lentiviral vectors Transduction and flow cytometic analysis p24 ELISA SDS-PAGE and immunoblotting Biotin binding ELISA RCL tests based on p24 ELISA
II-IV II-IV II II II, IV
In vitro experiments Transduction efficiency: Flow cytometry Fluorescent microscopy Immunofluorescence labelling, flow cytometry Immunofluorescence labelling, confocal microscopy Cell targeting with ligands Cell viability assay, Cell apoptosis assay Magnetic resonance imaging RNA extraction, RT-PCR
II-IV I-II IV IV III IV III III,IV
In vivo experiments Anesthesia, sacrificing, tissue sample collection Stereotactic injections of lentiviruses into rat brain Intravenous administration of lentiviruses SPECT/CT imaging MRI imaging Blood sample collection
III, IV III, IV IV III III IV
Histochemical anaysis �-galactosidase staining Antibody staining Iron staining
III III, IV III

55
5.2. Plasmids
Table 5. Details of the plasmids used in this study. The original plasmids used as a source of nucleic acid for further cloning of new plasmids and plasmids, which were used without any modification.
Plasmid Description Detailed purpose of the use Reference Used in pEntranceposon (camr)
MuA Transposon plasmid
Integration of the EGFP cassette into baculovirus genome
Finnzymes, Espoo, Finland
I
pEGFP-N1 Source of EGFP EGFP insertion into baculovirus genome
Clontech, Palo Alto, USA
I
pBVboostFG- VEGF-D�N�C
Source of VEGF-D�N�C
The generation of VEGF-D�N�C expressing baculovirus
I
pBVboostFG-avidin
Source of avidin expression cassette
The generation of avidin expressing baculovirus
(Laitinen et al., 2005)
I
pGEM®-T easy Target for TA-cloning
Integration site analysis Promega, Madison, WI, USA
I
pFastBac-1 Backbone for baculovirus constructs
Baculovirus production Invitrogen, Carlsbad, CA, USA
I, II
pBacSurf-1 Source of Gp64 Pseudotyping lentivirus Novagen, Madison, WI, USA
III
Baavi Source of the Avidin and LacZ
Avidin displaying lentivirus, LacZ transgene
(Raty et al., 2004) III
pGEM-streptavidin
Source of streptavidin
Streptavidin displaying lentivirus
M.Kulomaa, University of Tampere, Finland
III
pCMV-VSV-G Source of VSV-G Pseudotyping lentivirus Construction of BAC-VSV-G
T.Friedmann, UCSD, La Jolla, CA, USA
III, IV II
pRSV-Rev Source of rev Lentivirus production Construction of BAC-Rev
Addgene, Cambridge, MA, USA
III, IV II
pMDLg/pRRE Source of Gag and Pol proteins
Lentivirus production Construction of BAC-Gag-Pol
Addgene, Cambridge, MA, USA
III, IV II
pLV-GFP Lentivirus transfer plasmid with the GFP transgene
Lentivirus production Construction of BAC-transfer
(Makinen et al., 2006)
III, IV II
pBOB-CAG Backbone for lentivirus constructs
Lentivirus production I.Verma, Salk Institute, La Jolla, CA, USA
III, IV
pENTRTM221-Ferritin
Source of Ferritin gene
Transgene for lentivirus RZPD German Resource Center fro Genome Research, Berlin, Germany
III
pScav-2 Source of avidin fusion protein gene
Avidin fusion protein expressing lentivirus
(Lehtolainen et al., 2003)
IV

56
5.3. Viral vectors The generated baculoviral vectors and lentiviral vectors are summarized in table 6. and 7. Table 6. Baculoviral vectors used in this study Vector name Description More details Used in FG-VEGF-D�N�C
Expresses the VEGF-D�N�C transgene Generated by traditional bacmid transposition
I
F-bacmid- VEGF-D�N�C
Expresses the VEGF-D�N�C transgene Generated by using new bacmid expressing EGFP form the baculovirus genome
I
FG-avidin Expresses the avidin transgene Generated by traditional bacmid transposition
I
F-bacmid-avidin Expresses the avidin transgene Generated by using new bacmid expressing EGFP form the baculovirus genome
I
BAC-transfer Expresses the lentiviral transfer construct containing lentiviral elements and GFP expression cassette
For lentivirus production II
BAC-Gag-Pol Expresses Gag and Pol For lentivirus production II BAC-Rev Expresses Rev for the LV- production For lentivirus production II BAC-VSV-G Expresses the VSV-G envelope protein
for the LV- production For lentivirus production II
BAP-283-gp64 Biotinylated baculovirus (Kaikkonen et al., 2008)
For purification of baculoviruses from lentiviruses
Un-published
Table 7. Lentiviral vectors used in this study Vector name Promoter-transgene Pseudotype / surface modifications Used in LV-GFP hPGK-GFP VSV-G II Gp64 hPGK-GFP Gp64 III AVD-Gp64 hPGK-GFP Displaying avidin fused to VSV-GED
Gp64 pseudotype III
SA/Gp64-GFP hPGK-GFP Displaying streptavidin fused to VSV-GED Gp64 pseudotype
III
SA/Gp64-hHF CAG-human ferritin heavy chain Displaying streptavidin fused to VSV-GED Gp64 pseudotype
III
SA/Gp64-LacZ CAG-LacZ Displaying streptavidin fused to VSV-GED Gp64 pseudotype
III
Mono/Gp64 hPGK-GFP Displaying monomeric avidin fused to VSV-GED Gp64 pseudotype
III
LV-Lodavin CAG or hPGK-Lodavin VSV-G IV

57
5.4 Cell lines Baculoviral vectors (table 6.) were produced in Sf9 cells and lentiviral vectors (table 7.) in 293T cells. The produced viruses were tested in vitro by transducing different cells lines (table 8.) and analyzed by the methods listed in table 4. Table 8. Cell lines used in this study Cell line Description Reference Used in Sf9 Spodoptera frugiperda IPLB-Sf-21-AE
cells Invitrogen, Carlsbad, CA, USA I, II
293T Called as 293T/17, Human embryonic kidney cells (clone 17) expressing T antigen for plasmid episomal replication
ATCC: CRL-11268 II-IV
BT4C Rat glioma cells (Sandmair et al., 2000) I, III, IV Skov-3 Rat ovarian carcinoma cells ATCC:HTB-77 I, III HepG2 Human hepatocarcinoma cells ATCC:HB-8065 I, III Hela Human cervical cancer cells ATCC: CCL-2 II-IV A549 Human lung adenocarcinoma cells ATCC: CCL-185 III D54 Human glioblastoma cells (Bigner et al., 1981) III U118MG Human glioblastoma ATCC: HTB-15 III U87MG Human gliobastoma ATCC: HTB-14 IV
5.5. Purification of lentiviruses Produced lentiviruses were purified from contaminating baculoviruses by three different types of streptavidin coated magnetic particles and one streptavidin affinity membrane (table 9.). Tangential flow filtration in purification of lentivirus from cell culture medium was also tested by two different hollow fiber columns (table 9.) (unpublished data). Table 9. The purification material used in this study.
Purification material Description Reference Streptavidin MagneSphere Paramagnetic particles
Streptavidin coated magnetic particles
Promega Corporation, Madison, WI, USA
Dynabeads MyOne C1 Streptavidin coated magnetic particles
Dynal Biotech ASA, Oslo, Norway
Dynabeads M-280 Streptavidin coated magnetic particles
Dynal Biotech ASA, Oslo, Norway
MidiKros hollow fiber 0.02 μm 0.02 μm pore size Spectrum laboratories, Rancho Dominguez, CA, USA
MidiKros hollow fiber 0.05 μm 0.05 μm pore size Spectrum laboratories, Rancho Dominguez, CA, USA

58
5.6. Animal experiments Some of the produced lentiviruses were tested also in vivo. These experiments are listed in table 10. Injection point in rat brain after intracerebral injections is shown in Figure 13. Table 10. Animal experiments in this study Rats n Virus Administration
route Treatment Used in
Wistar male rat 22 SA/Gp64-hHF SA/Gp64-LacZ
Intracerebral Single III
BDIX male rats 33 LV-Lodavin Intravenous Intracerebral
Single or Repeated once at 3 weeks time point
IV
BDIX male rats 4 LV-Lodavin Intracerebral Repeated once IV
5.7. Primary Antibodies and ligands Primary antibodies used in immunostaining of the transduced cells or tissue section of the samples of the animals are listed in table 11. Targeting studies were performed by using primary antibodies and ligands. These antibodies and ligands are also listed in table 11. Table 11. Primary antibodies and ligands used in this study Antibody Description Reference Used in Anti-Gp64 Gp64 detection eBioscience, San Diego, USA I-III Anti-avidin Avidin detection (Laitinen et al., 2002) I, III Anti-avidin D Avidin detection Vector Laboratories, Burlingame,
CA, USA IV
Anti-avidin-Fluorescein
Avidin detection Vector Laboratories, Burlingame, CA, USA
IV
Anti-hVEGF-D VEGF-D�N�C detection R&D Systems, Minneapolis, MN, USA
I
Anti-streptavidin Streptavidin detection (Bayer et al., 1986) III Anti-VSV-G pAb VSV-G and VSV-GED
detection Bethyl Laboratories Inc., Montgomery, TX, USA
III
Anti-TfR mAb Targeting to transferrin receptor Ancell Corporation, Bayport, MN, USA
III
EGFR mAb (Cetuximab)
Targeting to EGFR receptor Merck, Daemstadt, Germany III
Anti CD46 mAb Targeting to CD46 receptor Exbio, Prague, Czech republic III Transferrin Targeting to transferrin receptor Molecular Probes, Invitrogen,
Carlsbad, CA, USA III
EGF Targeting to EGFR receptor Molecular Probes, Invitrogen, Carlsbad, CA, USA
III
Anti-Ferritin rH02 Human heavy chain ferritin detection
Paolo Arosio, University of Brescia, Milan, Italy
III

59
5.8. Secondary Antibodies In immunostainings, primary antibodies were detected by the labelled secondary antibodies listed in table 12. These secondary antibodies were conjugated to alkaline phosphatase (AP), horseradish peroxidase (HRP) or Alexa Fluor 647. Table 12. Secondary antibodies used in this study. Anti-rabbit IgG AP Detection of rabbit antibodies Bio-rad, Richmond, CA, USA III-IV Anti-mouse IgG AP Detection of mouse antibodies Bio-rad, Richmond, CA, USA III Anti-mouse IgG-HRP Detection of mouse antibodies Silenus Laboratories, Hawthorn,
Australia III
Anti-goat Alexa Fluor 647
Detection of goat antibodies Invitrogen, Carlsbad, CA, USA IV
Anti-rat HRP Detection of rat antibodies Abcam, Cambridge, UK IV

60
6. THE RESULTS AND DISCUSSION
6.1. Improved method for high-throughput titering and generation of baculoviruses and recombinant protein production in a 96-well plate format (I) Rapid screening of recombinant proteins is one of the major challenges in proteomic research. A bacterial expression systems have traditionally been used but these lack the post-translational processing machinery of eukaryotic cells (Marston, 1986). Insect cell expression systems have an advantage over bacterial expression because insect cells have eukaryotic expression machinery with the ability to utilize many post-translational processes (Jarvis, 1997). Baculoviruses have therefore become one of the most widely used systems for recombinant protein production (Kost et al., 2005). However, the disadvantages of the system are the time consuming process steps, e.g. generation of the virus stock, titer determination and optimization of the protein expression conditions (Philipps et al., 2005). Even though there is a clear need for true high-throughput screening methods, there are no reports of successful suspension cell based 96-well adaptation of baculovirus generation or protein production (Bahia et al., 2005). For these reasons, the aim of the current study was to develop a rapid and advanced miniaturized process for the generation, titering and protein expression of baculoviral vectors in a 96-well format for high throughput screening purposes.
Several improved methods have been developed to enhance the isolation and quantification of the recombinant viruses (Kost et al., 2005). A recent improvement from our group was to the BVboost system to achieve an efficient baculovirus production. The production method was based on Tn7-mediated transposition, which allowed improved selection of cloned baculoviral constructs with a SacB gene (Airenne et al., 2003). The next improvement consisted of gene expression regulated by mammalian, bacterial and insect cell promoters which enabled easy screening of libraries (Laitinen et al., 2005). The cloning strategy was based on efficient site-specific recombination by bacteriophage lambda (Gateway, Invitrogen). We wanted to further improve this pBVboost system by adding an EGFP expression cassette into the baculovirus genome by using a random MuA transposase reaction (figure 14). The new construct was refered to as the F-bacmid. The expression of EGFP during the progression of the viral infection enabled easy monitoring of the process.
It was important to be sure that the integration of the EGFP expression cassette had not affected viral function. Therefore the integration site was determined and found be in the middle of the ODV- E66 -gene. This gene encodes for a structural protein (Hong et al., 1994) which is essential only in a baculovirus ODV-life cycle. Thus it was concluded that this integration event had not influenced the budded (BV) form of the baculovirus. ODV-gene “elimination” may actually enhance the safety of the system by preventing the possible formation of occlusion derived viruses.

61
Figure 14. F-bacmid technology in a 96-well plate format. EGFP expression cassette was integrated into the baculovirus genome by MuA recombinase to ease the monitoring of the infection process and to facilitate the titering. The cloning of new baculovirus constructs was performed by site-specific recombination. The generation and titering of the baculoviruses and recombinant protein expression in insect cells was miniaturized to be suitable for a 96 well plate format.
Baculovirus production was tested in a 96-well format and the EGFP reporter enabled detection of infection by fluorescence microscopy (I/figure 4c) and flow cytometer (I/figure 4a,b). When analyzed, 88-100% of the wells of the 96-well plate contained viruses after the isolated bacmid DNA transfection (I/figure 4a). EGFP expression in those wells increased when measured daily and overall there were 70-85 % EGFP positive cells 7 days after the transfection (I/figure 4b). These generated primary virus stocks could be used for virus amplification or protein production. Prior to this, all the attempts to adapt the baculovirus suspension concept into a 96 well format have failed because of problems related to poor cell growth (Bahia et al., 2005). The critical steps in successful virus generation and protein production in a 96-well format were found to be the shape of the wells (square) and vigorous shaking to provide sufficient aeration and to keep the cells in suspension. These findings may be the reasons for earlier failuredue to wrong shape wells used with insufficient shaking.
An accurate titer determination is a critical step to achieve consistent and reliable results both to express recombinant proteins in insect cells and especially for the transduction of vertebrate cells in gene therapy studies. Laborious and time consuming titer determination usually consists of the detection of morphological changes linked to infection (O'reilly et al., 2004). Several improved protocols for virus titering have been described. These are based on immunological assays (Kwon et al., 2002), a quantitative real-time PCR
Baculovirus generation
Baculovirus titering
Recombinant protein expression
Baculovirus genome
Tn7 Tn7Tn7
CAG-tT7-p10-Gateway cassette
Tn7
Ppolh-EGFP
pPolh
-M
CS PolyA
pBVBoostFG
Transgene cloning by site-specific recombination
Recombinant virus generation by Tn7 transposition
Baculovirus generation
Baculovirus titering
Recombinant protein expression
Baculovirus genome
Tn7 Tn7Tn7
CAG-tT7-p10-Gateway cassette
Tn7
Ppolh-EGFP
pPolh
-M
CS PolyA
pBVBoostFG
Transgene cloning by site-specific recombination
Recombinant virus generation by Tn7 transposition

62
method (Hitchman et al., 2007), analyzing differences in cell size (Janakiraman et al., 2006) or the expression of a marker protein (Mulvania et al., 2004; Shen et al., 2002; Philipps et al., 2005). The existing rapid methods are based on complicated protocols and reagents or demand special reference materials and facilities. A fast flow cytometry-based titering methods for detecting GFP positive cells has been introduced, but the GFP cassette had to be cloned every time together with the transgene into new baculovirus construct, thus complicating the virus generation process (Philipps et al., 2005; Mulvania et al., 2004).
This current study demonstrated a fast (18 h) titering method for F-bacmid viruses in the 96 well plates without any need to carry the EGFP reporter in the donor vector during the cloning steps. A fast titering method was performed by transducing insect cells with different dilutions and analyzing EGFP positive cells 18 h post-transduction in a flow cytometer. The titers were calculated from a dilution containing 1-10% EGFP positive cells after normalization. The phase of the cell cycle (mitotic phase) affects the infections. During titer determination and normalization, this is taken into account by dividing the average pergantage of GFP positive cells by the maximum percentage achieved from the transduction with high virus amount (Mulvania et al., 2004). The 10% limit is based on the idea that theoretically only one virus has transduced one cell (Logan et al., 2004).The following formula was used in the calculations:
Titer (ifu/ml) =Average EGFP % of duplicate samples x Dilution factor x Cells per ml
Maximum EGFP %
The produced viruses were titered by flow cytometer and also by the traditional end-point titering. There were no statistically significant differences in the titers between the two methods (I/Figure 3) and a good correlation coefficienct was obtained (0.9924) (I/figure 4d). However, when flow cytometer analysis was performed 24 h after the infection, a four times higher titer was achieved, this being probably due to secondary infections (I/figure 3c). This is in line with the previous study (Mulvania et al., 2004) and this later time point is not recommended. Consequently, results from titering studies demonstrated that this flow cytometry based titer determination of f-bacmid baculoviruses was substantially faster and easier than could be achieved with the traditional methods (O'reilly et al., 2004). In addition, in comparison to the existing method, our method of integrating the EGFP expression cassette directly into the virus genome simplified the construction of the new virus and the need to transform a marker gene in every cloning of a new virus was eliminated (Philipps et al., 2005). Successful production and titering of baculoviruses by monitoring EGFP expression did not fully prove the functionality of these viruses. Therefore, the ability of these viruses to express recombinant proteins was tested by comparing F-bacmid derived pBVboostFG viruses with the traditional BVboost derived viruses. Two separate transgenes were cloned into both viruses: either avidin or a modified vascular endothelial growth factor D (VEGF-D�N�C), and protein production levels in insect (Sf9) and in mammalian cells (HepG2,

63
BT4C, Skov-3) were studied (I/figure 2). No significant difference was seen in the protein production levels. It was thus concluded that EGFP expression did not have any influence on the generation of virions or the level of protein production. Finally, the protein production using the F-bacmid was performed in the 96 well format by infecting Sf9 cells with the avidin or VEGF-D�N�C expressing F-bacmid virus and protein production was demonstrated 7 days after the infection using western blot analysis (I/figure 4d).
In this study, a 96 well plate format was described for baculovirus generation, titering and protein production. The fast flow cytometry based titering method was accurate and rapid to perform. The miniaturization of virus production and protein expression allows extensive and rapid optimization of multiple expression conditions, saving resources before initiation of large-scale production. It also enables high-throughput screening of the viruses, recombinant proteins or genome libraries, and can be developed for automization.
6.2. Lentiviral vector production using recombinant baculoviruses (II) Lentiviral vectors have shown their capabilities in gene therapy since they transduce both dividing and non-dividing cells and achieve long term transgene expression (Naldini et al., 1996b). However, lentiviral vector production has proved to be challenging (Merten, 2004). Partly because of the toxicity related to some viral proteins and the VSV-G envelope protein (Burns et al., 1993), which has complicated the development of stable production cell lines (Ory et al., 1996). Another complicating reason is potential safety issue related to the use of HIV based vectors, which has meant that the only suitable production method has been the three or four plasmid transient transfection method (Miyoshi et al., 1998; Dull et al., 1998). Earlier studies have proved baculoviruses to be versatile tools for many kinds of biotechnological applications, such as the production of proteins and vaccines (Airenne et al., 2004). The latest applications have been the production of viral vectors, especially AAV (Huang et al., 2007; Sollerbrant et al., 2001; Meghrous et al., 2005; Negrete and Kotin, 2007).
6.2.1. The production of lentiviral vectors
The aim of this study was to use baculoviruses in the production of third generation lentiviral vectors for gene therapy applications. All the necessary elements required for the production of the lentiviral vectors were cloned into the four baculovirus donor plasmids and subsequently four high titer baculovirus stocks were produced in insect Sf9 cells. The BAC-transfer baculovirus expressed GFP as a transgene with minimal lentiviral cis-elements (LTR, packaging signal, RRE). The BAC-gag-pol expressed lentiviral structural proteins and protease, integrase and reverse transcriptase enzymes. The BAC-VSV-G expressed the envelope protein VSV-G for lentiviral pseudotyping and the fourth baculovirus was called BAC-Rev, which expressed the rev protein for complete splicing of the viral RNA (Figure 15). Lentiviral vectors were produced by transducing adherent 293T

64
cells with various concentrations (MOI 50-1000) of the baculoviruses (figure 15). In order determine the optimal condition for the transduction, two different mediums, Dulbecco's Modified Eagle's Medium (DMEM) and RPMI 1640 Medium, were used and the transduction was performed either for 4 h or overnight after which the fresh medium was changed.
Figure 15. A schematic diagram of baculovirus-mediated lentivirus production. The necessary elements for lentiviral production were cloned into baculovirus donor plasmids, to form BAC-transfer, BAC-gag-pol, BAC-rev and BAC-VSV-G. The baculoviruses were produced in insect cells and used for transduction of 293T cells. Lentivirus containing medium was collected 48 h after the baculovirus transduction.
6.2.2. The characterization of produced lentiviral vectors
The yield of the produced lentiviruses was determined by analyzing virus particles by titering. Lentivirus titering can be based on analyzing non-functional or functional, also known as infective, particles (Geraerts et al., 2006). The non-functional particle methods are based on the amount of viral capsid protein p24 analyzed by ELISA, viral genome copy number by qRT-PCR or by assaying endogenous RT activity. When one wishes to analyze infective particles, the methods are based on measuring transgene expression by flow cytometer or by qPCR or determining the number of integrated proviral genomes. None of these titering methods gives absolutely correct results and therefore more than one titering method is recommended (Sastry et al., 2002). In this study, two of the most commonly used titering methods were applied to assess vector production: p24 ELISA for non-functional titering method and a flow cytometric method for analyzing the number of virus particles able to transduce Hela cells.
When the four baculoviruses were used at MOI 250 the highest infective titers were 2.5x106 TU/ml when baculovirus transduction was performed overnight in DMEM (Figure 16). When RPMI medium was used in transduction, the highest titers were 5.9x105 TU/ml already achieved at MOI 50. The titers from the baculovirus production were comparable to the titers obtained from the production by plasmid transfection (t-test P<0.05). Differences in the ratios of baculoviruses did not affect the titers. When transduction was performed in
CMV LTR hPGK-GFP LTR
CMV Gag-Pol
CMV VSV-G
RSV Rev
Elements in baculoviruses for the production of lentiviral vectors
293T cell transduction
Baculovirus production in insect cells
Collection of medium containing lentiviruses
CMV LTR hPGK-GFP LTR
CMV Gag-Pol
CMV VSV-G
RSV Rev
Elements in baculoviruses for the production of lentiviral vectors
293T cell transduction
Baculovirus production in insect cells
Collection of medium containing lentiviruses

65
serum free DMEM for 4 hours, the highest titers were an average of 1.2x106 TU/ml at a baculovirus amount yielding MOI 750 (figure 16). As a negative control, no lentiviral vectors were generated when one of the baculoviruses, either BAC-gag-pol, or BAC-VSV-G or BAC-REV, was absent from the system.
Figure 16. Highest titers (TU/ml) achieved when the lentivirus production was optimized by varying the medium, baculovirus MOI and time of baculovirus transduction (mean ± SD).
In addition to the infective particle titers, also viral capsid protein p24 concentration
was determined from the lentiviral preparations by ELISA assay. The p24 concentrations were on average of 191±105 ng/ml, which is in line with the values for the representative virus preparations (Follenzi and Naldini, 2002) and the results showed a good correlation (r=0.598; P<0.01) between infective titer and capsid protein amounts (II/figure 3.) .
Fresh medium was added to the transduced 293T cells after the transduction to remove the most of the baculoviruses still in the medium. The titers of baculoviruses in the collected lentivirus containing medium were evaluated by end-point titering and it was noticed, that there was still 0.1–0.5% of baculoviruses left in the medium after the washing. Because of these residual baculoviruses, it was important to be sure that the detected GFP expression was truly due to the lentivirus and the presence of baculovirus did not skew the titering results. Lentivirus integrates into the host cell genome and accomplishes a long term transgene expression (Naldini et al., 1996b) whereas baculovirus expression is transient (Airenne et al., 2004). Due to the difference in expression duration, Hela cells were transduced with three different virus batches and the GFP expression was followed by flow cytometry and fluorescent microscopy. The transduction led to sustained GFP expression proving the transgene expression was from lentiviral vectors while the expression of the control baculovirus was lost in 17 days (II/figure 4). Even though remaining baculoviruses did not have any influence on the final lentivirus titers, it was observed that an efficient downstream process will be required before the production methods can be adapted for clinical studies. Further discussion of the lentivirus purification procedure is provided in section 6.3.

66
Another concern with the use of lentiviral vectors is the safety issue related to the potential for production of replication competent lentiviruses. Thus, the elements for the production of the lentiviral vectors have been divided into four separate constructs with minimal overlapping sequences (Dull et al., 1998). Replication competent lentiviruses were tested by p24 ELISA assay (Sastry et al., 2003) and no indication of any ongoing viral replication was detected during several weeks of culturing. This is evidence that baculovirus-mediated production of lentiviruses does not give rise to any safety concerns relaited to RCL formation.
6.2.3. Future aspects of baculovirus mediated production
The scaling up of the adherent cell virus production is challenging and large scale production requires the use of cell factories (Geraerts et al., 2005; Slepushkin et al., 2003) where the maximum volume is limited to eight liters. In addition, they are heavy to handle requiring accessory equipments and all layers can not be viewed with a common microscopes. The most practical method for large scale production is the use of a suspension of cells which necessitates the use of bioreactors in viral vector production (Schoofs et al., 1998; Negrete and Kotin, 2007; Negrete and Kotin, 2008; Meghrous et al., 2005). Bioreactors are scalable, efficient, easy to handle and have a low cost of operation. Recently, a successful transient method for producing lentiviral vectors in suspension cultures was published. This study was based on PEI-mediated transfection (Segura et al., 2007). In addition, the transduction of HEK293 suspension cells by baculoviruses was efficient (Scott et al., 2007). Our preliminary results support this approach and demonstrated also a high transduction efficiency of baculoviruses for suspension-adapted HEK293 cells in serum-free conditions. In summary, these results give rise to serious expectations in the future for the production of lentiviruses in suspension cultures by baculoviruses providing for a good possibility to scale the system up.
The production and scale up of AAV has been adjusted for insect cells, allowing for the mass production of the vector (Urabe et al., 2002; Huang et al., 2007). The baculovirus/insect cell system is very efficient, but the production of lentiviral vectors in insect cells may be a challenge. Previously, lentiviral-like particles have been produced in insect cells by baculoviruses (Nermut et al., 1994) but no functional viruses were generated. Such production in this instance led to the formation of immature viral particles in the presence of the Gag-Pol-gene (Gheysen et al., 1989). Another possible disadvantage is the insect cell lipid layer, which would cover the lentiviral capsid. Baculoviral nucleocapsid is covered by this lipid layer (Miller, 1997) and it evokes an innate immune response (Facciabene et al., 2004; Abe et al., 2003) which is not desired for gene therapy vectors.
Lentiviral vectors have been commonly produced by transfecting adherent cells with several plasmids (Follenzi and Naldini, 2002; Tiscornia et al., 2006; Coleman et al., 2003; Karolewski et al., 2003). The transient transfection method is easy to perform, versatile and it avoids the time-consuming development of stable cells lines. It also allows easy and rapid testing of various transgenes or pseudotypes (Sena-Esteves et al., 2004).

67
Nonetheless, baculoviruses do have some advantages over the plasmid system. The production of plasmids without antibiotic selection for clinical studies is expensive and laborious. Baculoviruses offer a feasible alternative to plasmids since their production is fast and cheaper even in antibiotic-free and serum-free conditions. In addition, baculoviruses do not replicate in mammalian cells, which is an important safety aspect. Overall, the low risk of biosafety profile and an easy and cheap production procedure are the main advantages of the baculovirus system (Kost and Condreay, 2002). The baculovirus system has even been approved by the FDA and EMEA for the production of vaccines.
In conclusion, baculoviruses offer a new alternative method for the production of lentiviral vectors. The baculovirus system is functional, safe and has some potential advantages over the multiplasmid approach for lentiviral vector production. The production of baculoviruses is easy and rapid, their use is safe and they have low levels of cytotoxicity.
6.3. Removal of residual baculoviruses from the purified lentivirus preparation
Though baculoviruses are safe to use, do not replicate in mammalian cells and are not cytotoxic (Airenne et al., 2004), even low levels of baculoviruses in the lentivirus preparations are not desirable. The residual baculoviruses in the lentivirus containing medium was evaluated and the results showed that 0.1-0.5% of baculoviruses used in the transduction was still in the medium. It may be possible to reduce the baculovirus amounts simply by adding an extra washing step into the process, but it is likely that a further purification step will be obligatory (Rodrigues et al., 2007). Lentiviruses and baculoviruses are both enveloped viruses since the nucleic acid of the virus is surrounded by a capsid covered by a lipid envelope. Therefore the basic characteristics of the viruses are quite similar (Miller, 1997; Felder and Sutton, 2009). In addition, the viruses have approximately the same size (lentivirus 100nm round shaped and baculovirus 30-60 nm wide and 250-300 nm long). These features may complicate the separation of the viruses. When AAV was produced in insect cells, the conventional purification method has been the use of a CsCl gradient ultracentrifugation. The more sophisticated downstream process included filtration after harvest and cell lysis, ion exchange chromatography, second filtration step and another chromatography step, e.g. hydrophobic interaction chromatography (Chahal et al., 2007). Both lentiviruses and baculoviruses have been purified by several different chromatographic methods (Transfiguracion et al., 2007; Barsoum, 1999; Rodrigues et al., 2007; Segura et al., 2006) so the downstream process of a lentivirus-baculovirus system would probably be rather similar to the AAV-baculovirus system considering the features of the viruses.
Avidin-biotin technology has been utilized in different viral purification methods (Barry et al., 2003). Biotinylated baculoviruses were recently published by Kaikkonen and coworkers (Kaikkonen et al., 2008). Biotin on the virus surface was successfully used in viral vector purification. In this context, a preliminary clearance test was performed by

68
mixing biotinylated baculoviruses and unmodified lentiviruses in the same ratio as the baculovirus-mediated lentivirus preparations after the baculovirus-mediated production. Baculoviruses were removed using one of three different streptavidin coated magnetic particles and one streptavidin affinity membrane. The most successful removal of baculoviruses was achieved with Dynabeads C1 magnetic particles and only 8 % of baculovirus were left after the purification while the lentivirus recovery was 53% (unpublished data). This test proved that the avidin-biotin system could be used as an alternative purification step for lentiviruses after baculovirus-mediated production, even though some optimization is still needed. The changeover to the strategy of using biotinylated baculoviruses would require the cloning of the new baculoviruses displaying a small biotin acceptor peptide (BAP) which would be biotinylated during vector production by coexpression of biotin ligase (BirA) (Kaikkonen et al., 2008).
6.4. Lentiviral vector purification by tangential flow filtration
Overall, the purification of the final lentiviral vector product has been challenging because of the fragile structure of the virus. The viruses routinely used in the research are concentrated only by ultracentrifugation without any following purification steps (Follenzi and Naldini, 2002; Burns et al., 1993). Ultracentrifugation step also concentrates host cell derived contaminants into the final product. These include cell debris, host cell DNA and proteins, the plasmids used in the transfection and components from the spend cell culture media. The contaminants and the precence of the most commonly used envelope protein, vesicular stomatitis virus glycoprotein G (VSV-G) increases the likelihood of lentivirus-mediated toxicity, inflammation and the triggering of an immune response (Baekelandt et al., 2002; Baekelandt et al., 2003).
Several purification methods, such as anion exchange chromatography (Yamada et al., 2003; Sena-Esteves et al., 2004), affinity chromatography (Segura et al., 2007) or sucrose gradient sedimentation (Baekelandt et al., 2003) have been developed for lentiviral vectors. In addition, the suitability and scalability of ultrafiltration in lentivirus purification has been reported (Reiser, 2000; Miyake et al., 2007). TFF, also called crossflow filtration, is based on the gentle tangential filtration pressure. In the current study, the crossflow ultrafiltration method was evaluated in the lentivirus purification using two different hollow fibre modules (20 and 50 nm pore size) and the removal of DNA and protein impurities was examined. The efficiency of benzonase treatment on DNA removal during the purification was also evaluated. The results obtained by crossflow ultrafiltration were also compared to a centrifugation-based concentration method and a commercially available purification kit.
Genomic DNA and plasmids are significant sources of contaminants in lentivirus production. It was found that 43.0% of the initial DNA (786.7 μg of 1828.4 μg) was still present after the crossflow ultrafiltration with a 20 nm pore size. However, only 1.6 % (28.3 μg) of the initial DNA was detected in the material (figure 17) when it was digested with benzonase before filtration and 0.1 % (1.4 μg) was left after the final

69
ultracentrifugation. In other words, the removals of DNA were 98.6 and 99.9 % respectively, using these approach. These results showed that digestion of DNA before crossflow filtration is efficient. In contrast, 11.4% of the DNA was still present when the lentivirus was concentrated by purification kit and 9.1% when it was concentrated by ultracentrifugation.
Figure 17. A comparison of the level of contaminants after lentiviral purification by various methods. (A) Total DNA and (B) total protein concentrations measured before and after the different concentration/purification methods (mean ± SD).
The lowest total protein concentration was determined in the purified material after
the filtration with the 50 nm membrane and a final centrifugation step, when < 0.2% of the total protein was left in the product. There were no significant differences detected between filtration with a 20nm pore size membrane, a commercial kit or the ultracentrifugation method (Fig 17.).
The current work demonstrated the purity of the final product. However, during the crossflow filtration procedure, loss of viral titers was revealed. Infective particles were lost to the permeate fraction during the crossflow procedure (Table 12.). The crossflow filtration system can be scaled up to purify and concentrate large volumes (Geraerts et al., 2005). Therefore, larger volumes of the feed solution should be used to achieve a higher titer. The final centrifugation step substantially reduced the recovery and thus is not recommended. This is in line with previously reported results that the concentration procedure is more efficient when serum components are present during the

70
ultracentrifugation step (Baekelandt et al., 2003; Geraerts et al., 2005). Benzonase did not have any effect on infective titers, as expected (Sastry et al., 2004).
Table 12. The effect of purification processes on the lentiviral titers. The amount of the viral capsid protein p24 and the level of transducing units were measured before purification, after different TFF purification protocols and compared with the effect of further ultracentrifugation, or purification with a commercial kit or ultracentrifugation alone. Data is represented as an average of at least three independent determinations. Sample Total p24 (μg) TU/ml Total TU Before conc. 209,8 1,08E+07 2,15E+09 After benzonase 187,5 1,35E+07 3,00E+09 TFF (20 nm) 93,9 8,17E+06 4,66E+08 TFF (50 nm) 18,7 1,93E+06 1,10E+08 TFF (50 nm + benzonase) 29,7 6,26E+06 1,58E+08 Permeate after TFF (20 nm) 32,5 2,46E+02 1,73E+03 Permeate after TFF (50 nm) 25,2 2,71E+03 3,60E+05 Permeate after TFF (50 nm + benz.) 23,9 3,30E+03 4,38E+05 TFF (20 nm) + ultrafug. 10,5 6,93E+07 1,39E+07 TFF (50 nm) + ultrafug. 5,4 6,26E+06 1,25E+06 TFF (50 nm + benz.) + ultrafug. 7,8 6,54E+07 1,31E+07 Purification kit 16,2 2,31E+06 1,15E+08 Ultracentrifugation 46,1 8,58E+08 1,97E+08
In conclusion, the results showed that crossflow ultrafiltration is a useful
purification method to achieve better purified lentiviral vectors. It is a scalable procedure and easy to perform. The removal of protein impurities was achieved and the removal of contaminating DNA occured when benzonase digestion was included in the process. The purification process increases considerably the safety of the viral product. However, recoveries were still quite low and further optimization is needed.
6.5. (Strept)avidin displaying lentiviral vectors for different gene therapy applications (III)
Modification of a lentivirus surface for every new gene therapy application may be laborious and time-consuming. Every time it requires new cloning, production and safety evaluation of the virus. It would be ideal if one could develop one virus platform for use in a wide variety of different purposes. To meet this challenge, a versatile lentivirus was developed based on the avidin-biotin system allowing easy exchange of the ligand on the lentivirus envelope. This was achieved by displaying an avidin or a streptavidin molecule on the lentivirus surface for use in both targeting and imaging applications.

71
6.5.1 Construction and characterization of the (strept)avidin displaying lentivirus
The extremely high affinity of avidin for biotin (Kd ~10-15) (Green, 1990) has been
the main reason behind the wide-ranging use of this system in many kinds of biotechnological applications. This system has also been exploited in gene therapy for several viral vector studies, especially for targeting and purification purposes (Barry et al., 2003; Waehler et al., 2007). At present, the most commonly used method has been to coat viruses by biotinylating them either chemically or metabolically. One recent approach that has been used was to coat the virus with avidin (Raty et al., 2004). Our first attempts to create the avidin-displaying lentivirus were based on this preceding study with baculoviruses (Raty et al., 2004) and the avidin was fused to the N-terminus of the Gp64 envelope protein but the transduction capacity of the virus was impaired (data not shown). This was probably due to an incorrect conformation of the gp64 within the fusion protein (Guibinga et al., 2008; Kaikkonen et al., 2008). Also vectors displaying (strept)avidin-VSV-GED together with VSV-G were constructed but no transduction resulted (data not shown). This might be because of heterodimerization of the (strept)avidin-VSV-GED protein with the wild type VSV-G, leading to display of nonfunctional VSV-G molecules.
Figure 18. Avidin and streptavidin were fused to the transmembrane anchor of VSV-G. The gp64 pseudotyped lentiviral vector displaying avidin or streptavidin on the virus surface allowed for the easy binding of the biotinylated ligand for targeting and imaging purposes.
In the current study, a novel (strept)avidin displaying lentivirus was successfully
constructed by fusing avidin or a streptavidin to the transmembrane anchor of VSV-G (VSV-G EctoDOmain, VSV-GED) (Kaikkonen et al., 2006) on the gp64-pseudotyped lentiviral vector surface (figure 18). If this construct were to be used in targeting purposes, the targeting moiety would need to be separated from the envelope glycoprotein so that binding to the target receptor would not disturb the fusion process during the entry of the
Gp64 pseudotyped lentivirus
Strept(avidin)
Biotinylated targeting or imaging moyety
GP64
(STREPT)AVIDIN TM
VSV-GED
Gp64 pseudotyped lentivirus
Strept(avidin)
Biotinylated targeting or imaging moyety
GP64
(STREPT)AVIDIN TM
Gp64 pseudotyped lentivirus
Strept(avidin)
Biotinylated targeting or imaging moyety
GP64
(STREPT)AVIDIN TM
VSV-GED

72
virus (Russell and Cosset, 1999). The glycoprotein gp64, which is responsible for the entry of the virus, is the non-cytotoxic major envelope protein of baculovirus which has been previously used for the production of the high titer lentiviral vector and facilitates efficient transduction in vitro and in vivo (Kumar et al., 2003; Schauber et al., 2004).
(Strept)avidin displaying lentiviruses were produced with five plasmid transfection and concentrated by ultracentrifugation (Follenzi and Naldini, 2002). The p24gag protein concentration from the viral preparation was measured, proving the production of high titer lentivirus preparations (III/figure 1b). The samples were normalized based on these results and immunoblot analysis was performed in order to prove the incorporation of streptavidin or avidin and gp64 into lentiviral particles (III/figure 1a). Immunoblot analysis indicated a somewhat higher level of streptavidin on the viral surface but no statistically significant difference was seen in the transduction efficiencies of the vectors. The functionality of the viruses was determined in four cell lines (Hela, BT4C, SKOV-3, HepG2) (III/figure 1b). Titers were in range of 107-108 TU/ml in BT4C, SKOV-3 and HepG2 cells and 106 TU/ml in Hela cells. Gp64 has shown a broad transduction range in previous studies (Kumar et al., 2003; Schauber et al., 2004). Even though there was no statistical difference in titers, some decrease in transductivity of SA/GP64 virus was detected when compared to the AVD/GP64. It has been reported that avidin can enhance the efficiency of gene transfer because of its positive charge (Lee et al., 2000; Raty et al., 2004). Another explanation can be that a higher amount of streptavidin on the virus surface causes more steric hindrance for gp64 during virus entry. An optimization of the plasmid ratios during the production may present one possible way to improve the transduction of SA/GP64. In all of these experiments, a Gp64 pseudotyped vector was used as a control and no statistically significant differences in the productivity or transductivity were observed between the control virus and (strept)avidin displaying viruses.
The purpose of the (strept)avidin on the virus surface is to bind the biotinylated ligands, thus it was important to evaluate the biotin binding capacity of the fusion proteins. In this test, increasing amounts of viruses were allowed to attach to the biotin on the coated ELISA plates and after washing, the amounts of bound viruses was detected by anti-gp64. The SA/GP64 virus showed three-times higher binding to the imobolised biotin compared to the AVD/GP64 in the biotin-coated ELISA test (III/figure 1c). This may be due to a higher amount of streptavidin on the virus surface. It is also possible that differences in the conformation of the displayed avidin or streptavidin explain the increased biotin binding by the streptavidin displaying virus (Inagaki et al., 2006).
Overall, these results proved the productivity and functionality of both SA/GP64 and AVD/GP64 viruses. Due to the higher amount of the streptavidin on the virus surface and its better biotin binding capacity, SA/GP64 was chosen for the subsequent imaging studies.

73
6.5.2. SPECT/CT imaging of streptavidin displaying lentiviral vectors for biodistribution (III)
Molecular in vivo imaging has been applied to the visualization of the molecular processes and monitoring the success of gene therapy. It would be of great importance to unravel how much of the administered vector reaches the target, to quantitate virus dose, to evaluate the toxicity related to the treatment and to verify the gene expression and treatment response (Bogdanov, Jr., 2003). Several non-invasive technologies such as SPECT, PET, MRI and bioluminescence imaging, are available, but they have differences in terms of resolution, availability, cost and need for a contrast agent (Massoud and Gambhir, 2003). SPECT is a practical real-time method for analyzing viral biodistribution compared to the conventional histological methods. Viral particles are labelled with radioactive compounds, imaged non-invasively and the imaging result enables the quantization of the signal. The kinetics and distribution of radiolabelled herpes simplex virus (Schellingerhout et al., 2000), adenovirus (Zinn et al., 1998; Awasthi et al., 2004) and baculovirus (Raty et al., 2007a) have been studied using SPECT.
In this study, SA/GP64-hHF was radiolabelled with 111In, conjugated with biotin-poly-Lys-DTPA, and injected into the right corpus callosum of male Wistar rats. Control rats received either SA/GP64-LacZ control-virus or 111In conjugated with biotin-poly-Lys-DTPA only. The biodistribution of the virus was followed during three days by SPECT. Since SPECT imaging does not provide acute anatomical data it is recommended to be combined with high-resolution computer tomography (CT) (Massoud and Gambhir, 2003). A clear accumulation of the signal from the labelled virus was seen in the brain area whereas the signal accumulation of the chelate alone was located in the kidneys on the first day and shifted to the bladder on day two (III/figure 4). No signal was detected elsewhere in the body i.e. the viral biodistibution was restricted to the injection area. This was also verified by RT-PCR, showing that no lentiviral vector-mediated transgene expression was detected in the liver or spleen which are the target for gp64 pseudotyped viruses (Schauber et al., 2004; Sinn et al., 2007). However, exceptional results were achieved when radiolabelled virus was accidentally injected into the circulation and a clear signal accumulation was detected in the peritoneal area (III/figure 4c,f). This demonstrated the utility of the imaging in the evaluation of the gene therapy treatment.
Overall, SPECT imaging combined with anatomical references provided specific biodistribution information after the administration of the SA/GP64 lentiviral vector. The real-time imaging of viral biodistribution could serve as a general predictor of the accumulation of the viral vector into the target organs in clinical use. In addition, distribution information will introduce an important way not only to evaluate the safety of the treatment but also the influence of side effects, such as any immune response (Min and Gambhir, 2004; Raty et al., 2007b).
The accumulation of the virus to the target area may not necessarily lead to the transgene expression in the cells. Thus, the ultimate goal in the imaging part of this study was to follow for the first time the distribution of the virus using one imaging technique and

74
then to study the transgene expression of the same virus with another imaging method. Therefore, the same animals underwent also long term MRI follow up.
6.5.3. MRI of streptavidin displaying lentiviral vectors for transgene expression (III)
The first report of long term imaging of lentiviral vector-mediated gene expression was published by Deroose et al. who followed gene expression noninvasively in rat and mice brain using bioluminescence (Deroose et al., 2006). However, the bioluminescence imaging has a low spatial resolution and has limited depth capacity (Massoud and Gambhir, 2003). In addition, bioluminescence imaging requires the injection of a luciferin substrate (Deroose et al., 2006). MRI has the advantage of high spatial resolution and it can combine morphological and functional imaging (Massoud and Gambhir, 2003) and it does not necessarily need any external contrast agent. The overexpression of the ferritin receptor results in the accumulation of iron into cells which can be visualized by MRI non-invasively without any external contrast agent (Cohen et al., 2005; Genove et al., 2005). Thus, MRI was used in this study to image SA/GP64 lentivirus mediated gene expression and the ferritin heavy chain (hHF) was chosen as a the transgene.
Before the in vivo experiments, iron accumulation by ferritin was studied in vitro. The relaxation rate R2 (R2=1/T2) was measured 47 h after the lentivirus transduction of HepG2 cells with and without iron supplemented mediums. A two-fold increase in R2 was detected by MRI in iron-supplemented mediums compared to the control cells confirming the ferritin expression and iron storage inside the cells (III/figure 3). This established that the ferritin heavy chain functioned properly.
MRI scans were performed on rats at 4.7 T on days 3, 14, 28, 94, 141 and 211. Using T2* contrast, a signal was detected along the needle track and in the injection site as a consequence of ferritin expression and iron accumulation. An increase of the signal was detected during the long term follow up (III/figure 5a). A minor signal from the hemorrhage caused by the injection was detected from the control animals receiving SA/GP64-LacZ or 111Indium labelled chelate alone at day three but this had resolved before the time of the later measurements III/figure 5b and 5c). Ferritin expression was detected by immunohistological staining of randomly selected animals five days after the viral injection by Perls´ Prussian Blue staining and human heavy chain ferritin antibody staining. The expression, and iron accumulation was detected in the corpus callosum and also along the needle track (III/figure 6a-e). The LacZ expression of SA/GP64-Lacz virus was detected by X-Gal staining along the needle track, in the corpus callosum and in the cuboidal epithelial cells of the choroid plexus (III/figure 6f-i).
For the first time gp64-pseudotyped lentivirus was used in transduction of the choroid plexus cells and corpus callosum and the tropism was demonstrated to be similar for baculoviruses (Sarkis et al., 2000; Lehtolainen et al., 2002b; Raty et al., 2006). This offers the possibility to use the gp64-pseudotyped lentivirus in the gene therapy of CNS disorders. Overall, lentiviruses have shown great potential for CNS disorders since they

75
have a broad tropism for neuronal and glial cells, are capable of stable transduction leading to long term transgene expression and evoke only a mild inflammatory or immune response (Baekelandt et al., 2003). For example, lentiviral encoded glial-derived neurotrophic factor (GDNF) has augmented dopamine function in monkeys for treatment of Parkinsons´s disease (Kordower et al., 2000) and lentivirus expressing vascular endothelial growth factor (VEGF) has offered potential for the treatment of amyotrophic lateral sclerosis (ALS) (Azzouz et al., 2004). The next development for testing this versatile streptavidin displaying lentivirus would be to construct bicistroinic viruses expressing a therapeutic gene simultaneously with ferritin since this would allow easy monitoring of the expression in conjunction with therapeutic treatment. It is also notable, that ferritin can protect cells from the oxidative effects of iron (Orino et al., 2001) and could be considered as a treatment for example for atherosclerosis (Ong and Halliwell, 2004).
Consequently, these present imaging results proved that biodistribution and transgene expression could be monitored simultaneously from the same streptavidin displaying lentivirus. SPECT demonstrated that streptavidin was capable of binding biotinylated radionuclides whereas MRI proved useful for long term expression studies. These findings would be valuable in the later development of the system to improve the monitoring of human gene therapy.
6.5.4. Targeting of (strept)avidin displaying lentiviral vector (III) An important safety aspect for gene therapy is to target the viral vector to the
correct cells without affecting healthy tissues. The possibility to exploit the versatile strept(avidin) displaying lentivirus also in targeting purposes was tested in different cell lines in vitro. Three receptors, transferrin, EGFR and CD46 are highly expressed in tumor cells (Recht et al., 1990; Herbst, 2004; Wojnicz et al., 2002) and were selected to be targeted with the (strept)avidin displaying lentivirus coated with selective biotinylated ligands or antibodies.
Rat glioma cells (BT4C) and human glioblastoma cells (D54) overexpressing the transferrin receptor were coated with a biotinylated transferrin or a biotinylated monoclonal antibody to the human transferrin receptor and lentiviruses were allowed to attach to the receptor. GFP positive cells were analyzed three days later. In both cell lines an increase in transduction efficiency was detected, 20-30 % using AVD/GP64 and 50-60% with SA/GP64 (III/figure 2a). Lung carcinoma (A549), hepatocellular carcinoma (HepG2) and human ovarian carcinoma (Skov-3) cells overexpressing the EGFR were targeted with a biotinylated EGFR antibody (Cetuximab). The non-biotinylated Cetuximab functioned as a control. In these cell lines, an increase in GFP positive cell was also observed when a biotinylated targeting molecule, Cetuximab, was used (III/figure 2b). In particular, a statistically significant difference was seen with the SA/GP64 virus (100% increase). No significant increase in the transduction was detected with the non-biotinylated Cetuximab. In Skov-3 cells, biotinylated EGF was also tested and a 50% increase in the transduction efficiency was achieved with the SA/GP64 virus whereas there was no difference with the

76
ADV/GP64 virus (III/figure 2b). Moreover, the targeting to the human neoplastic cells U118MG overexpressing CD46 was achieved with the biotinylated CD46 antibody. These results were well in line with the other in vitro targeting studies. In this cell line, a significant enhancement in the transduction of AVD/GP64 was achieved. In summary, in all cell lines, SA/GP64 showed better transductivity compared to the AVD/GP64 virus. Based on the immunoblot analysis, more streptavidin was incorporated into the viral particles and thus there were more binding sites available for ligands which may have been the reason for the enhanced SA/GP64 transductivity. In none of these studies, did GP64 control virus cause any change in the transduction efficiency after the treatment with the targeting molecules.
These results proved, that (strept)avidin displaying lentiviruses can be used in targeting. (Strept)avidin on the surface of the virus enables easy exhange of the biotinylated targeting ligands. An alternative to biotinylated ligands could be a Strept-tag II, an eight-residue minimal peptide sequence that exhibits high affinity toward stretpavidin and can be fused to different proteins (Schmidt and Skerra, 2007; Skerra and Schmidt, 1999). Earlier studies of targeting gp64 pseudotyped lentivirus were based on genetically fusing hepatitis B virus PreS1 protein to gp64 to direct virus to the liver-derived cells (Markusic et al., 2007). Nonetheless, avidin technology provides for straightforward change of the ligand, and a further advantage of this system is also an extremely high affinity of ligand to the virus surface since the binding is based on the bond between (strept)avidin and biotin (Green, 1990). For example, in targeting strategies which are based on the antibody binding to the virus surface, the weak interaction under physiological conditions may lead to the removal of the antibody from the viral surface (Waehler et al., 2007). However, the transduction of (strept)avidin displaying lentivirus was dependent on gp64 which has shown quite broad tropism to different tissue types, such as liver, lung, skin, glia, endothelium or thymus (Schauber et al., 2004) even though the tropism is more restricted than that obtained with the VSV-G. To achieve more specific transduction, it might be worthwhile to consider other glycoproteins for pseudotyping.
Altogether, the results confirmed production of high titer lentivirus displaying avidin or streptavidin on the viral surface. Dual imaging of the viral biodistribution and transgene expression was feasible. In addition, the (strept)avidin lentivirus has been able to target to specific cells thus proving that a single virus platform can be used for different applications, such as imaging and targeting. Presumably this lentivirus platform could be used in the purification of viruses from cell culture medium by attaching viruses to e.g. biotinylated magnetic particles or biotin affinity column.
6.6. Avidin fusion-protein expressing lentivirus for targeted therapy (IV) In addition to targeting of viral vectors based on avidin-biotin technology, the system has been utilized in pretargeting strategies, as discussed in 2.4.4. Both avidin and streptavidin are immunogens (Hytonen et al., 2003; Knox et al., 2000). However, it appears

77
that avidin has a lower immunogenicity compared to streptavidin, thus being more appropriate for pretargeting applications (Marshall et al., 1996; Chinol et al., 1998; Meredith and Buchsbaum, 2006). In our group, two avidin fusion proteins, Scavidin (Lehtolainen et al., 2002a) and a low-density lipoprotein-avidin fusion. LodavinTM (Lehtolainen et al., 2003) have been constructed for pretargeting intravenously administered biotinylated molecules to the desired tissues after local gene transfer (figure 20). These avidin fusion proteins are expressed on the surface of the host cell and are capable of binding biotinylated molecules. Previous studies revealed better expression on the cell surface and biotin-binding capacity of LodavinTM compared to Scavidin and therefore it was chosen for the subsequent studies. The early LodavinTM studies were performed with Semliki Forest virus (SFV). However, potential toxicity and a short expression time hindered further studies (Rheme et al., 2005; Frolov et al., 1996) and therefore lentivirus was chosen as the vector. Lentivirus has shown efficient delivery, stable integration and long term transgene expression in dividing and non-dividing cells (Naldini et al., 1996b) and has low immunogenicity (Baekelandt et al., 2003). The aim of this study was characterize in more detail the avidin-fusion protein expressing lentivirus and evaluate the safety and efficacy of the system in vitro and in vivo.
Figure 20. A diagram of a low-density lipoprotein-avidin fusion, LodavinTM on the surface of the target cell.
6.6.1 Titering of the avidin fusion protein expressing lentivirus The avidin fusion protein gene was cloned into a third generation self-inactivating lentiviral transfer plasmid, VSV-G pseudotyped viral vectors were produced by the four plasmid transfection method and titered by ultracentrifugation (Follenzi and Naldini, 2002). The production of a high titer lentiviral vector stock was demonstrated by measuring the p24gag protein concentration from the viral preparation and based on the assumption that 1 pg of p24 equals to 12 500 vp (Farson et al., 2001), the p24 results were converted to vp/ml (IV/table 1). The results varied from 8.1 x 1011- 2.5 x 1012 vector particle /ml. Analyzed RNA titers, based on genomic copy number, were between 4.3 x 108 and 1.1 x 109. P24
Avidin
Plasma membrane Low density
lipoprotein receptor
Myc-tag
Biotinylated therapeutics or imaging compound
Avidin
Plasma membrane Low density
lipoprotein receptor
Myc-tag
Biotinylated therapeutics or imaging compound

78
ELISA is one of the most commonly used methods for lentiviral titer determination but unfortunately it is also one of the least reliable techniques (Geraerts et al., 2006). Its sensitivity is limited by the variable amount of free, nonparticle-associated p24 proteins. In addition, the viral RNA RT-qPCR method has also its limitations as it underestimates the total particle count as defective particles without any RNA are present (Geraerts et al., 2006; Sastry et al., 2002). Based on these results, it is clear that neither RNA nor p24 titers can adequately predict the functional titer since the functional titer is dependent on the vector construct and the cell type used for transduction even though they do correlate strongly with the functional titer (TU/ml). Nonetheless, they can be used for normalization of vector preparation titers.
Functional lentiviral particles were titered by analyzing transduced U87MG and BT4C cells by flow cytometer after antibody staining for avidin. Titers were 1.0 x 107- 1.5 x 107 TU/ml in U87MG cells and 1.7 x 107- 5.2 x 107 TU/ml in BT4C cells. The functional titers of avidin fusion protein expressing lentiviruses were significantly lower than the total particle titers indicating a high amount of defective-viral particles. On the other hand, the method assumes that the level of expression of all integrated vectors is above the detection threshold of flow cytometry thus potentially underestimating the functional titers. Therefore a second assay detecting the functional viral particles was established. DNA titer was determined for the batch four by measuring the number of integrated proviral copies and yieled a titer of 1.2 x 108 TU/ml. The DNA titer was 12-fold higher than the FACS titer which is a consequence of the fact that not all the integrated proviral DNA is transcribed (Sastry et al., 2002). Commonly, in lentiviral preparation the ratio between functional and defective particles is quite high (1/100-1/1000) (Delenda and Gaillard, 2005), depending on the method used the ratio was between 1/60-1/200 000 with LodavinTM virus.
6.6.2. Analysis of avidin fusion protein expressing lentivirus in vitro The lentivirus-mediated expression of the Avidin fusion protein, LodavinTM, was studied in U87MG cells by anti-avidin staining after permeabilized or non-permeabilized fixing of the cells and the stained GFP positive cells were analysed using a flow cytometer. The total expression (permeabilized fixing) was compared to the expression of the surface (non-permeabilized fixing) and it was detected that only 50% of the expressed protein was on the surface (IV/figure 1a). The level of expression in the positive cells was further studied by comparing the mean fluorescence indexes. If the results were interpreted by mean fluorescence indexes, then the expression of the virus on the surface was even lower (14% of total expression) (IV/figure 1b). This same phenomenon was visible by fluorescence microscopy (IV/figure 1c-f). The lentivirus-mediated expression is in line with the SFV-mediated expression, where avidin fusion proteins were located on the cell surface as well as inside the cells (Lehtolainen et al., 2003).
The integrating lentiviral vector provided for the first time the possibility to study the long term gene expression of the avidin fusion protein, where the transduced cells were studied for more than 30 days. During that time, from 90-100% of positive cells, expression

79
was detected from approximately 20-30% cells at later time points (IV/figure 1g). When cells were sorted, the expression stabilized at around 55% GFP positive cells. One reason for the decrease in the number of fusion protein expressing cells might be the sensitivity of the method so that not all the cells expressing LodavinTM were observed in the analysis. Another explanation could be that non-transduced cells were more viable and hence they expanded more rapidly. The early studies using LodavinTM were performed with Semliki Forest virus (SFV) having a short expression time (Lehtolainen et al., 2003). Long term expression of lentivirus provided possibility to study also ligand binding ability of avidin fusion protein using biotinylated quantum dots (very small flurescence semiconductor nanocrystals) and enabled to analyse the effect of repeated delivery of the ligand. Ligand was administered at 0, 1 and 20 h timepoints and the binding capacity was measured with a flow cytometer. The ligand binding capacity was 20-50% 1 h after the first delivery and it has risen to 65-85% at the 20 h timepoint (IV/Figure 2). The restored binding capacity established that the receptor was endocytosed and then recycled back to the cell surface (Lehtolainen et al., 2003) which is typical for native LDLR (Hao and Maxfield, 2000). Consequently, readministration of a second ligand is feasible 1 h after the first treatment though it could predicted to be more effective >20 h afterwards.
Because of these results from expression studies, the influence of the constructed lentivirus on the cell viability was determined. This was performed by transducing different cell lines (Hela, BT4C, 293T and HepG2) with the avidin fusion protein expressing lentivirus and then analyzing cell viability or the levels of apoptosis using commercially available assay kits. Non-treated cells or cells transduced by a lentivirus expressing GFP were used as a controls. No caspase-3/7 apoptosis activity was detected from the transduced cells (data not shown). Furthermore, the transgene did not have any effect on the cell growth after transduction (IV/figure 3). However, when cells were transduced with on increasing concentration of the viruses, a dose was determined when the lentiviruses transduced cells showed signs of cytotoxicity (MOI 50). There were no statistical differences between the control virus and the avidin fusion protein expressing virus in 293T or BT4C cells suggesting that the toxic effect is not due to the avidin fusion protein. Avidin has been used frequently in pretargeting studies and no toxicity has been related to avidin (Goldenberg et al., 2006; Paganelli et al., 1991). In contrast, the VSV-G has been shown to be toxic to the transduced cells at high concentrations (Watson et al., 2002; Park et al., 2000; Burns et al., 1993). In addition, the GFP expressing virus showed that with high virus concentrations more toxic effect were observed in Hela transduced cells compared to cells transduced by the avidin fusion protein expressing lentivirus (IV/figure 3). This might be due to the toxicity related to GFP (Liu et al., 1999). These studies proved that under in vitro conditions, the avidin fusion protein did not have any effect on cell viability as long as the MOI remained reasonable concentration.
The functionality of the avidin fusion protein was tested in vitro by targeting biotinylated nanoparticles carrying paclitaxel (Pulkkinen et al., 2008) to the transduced cells expressing avidin fusion protein. Cell viability was measured 72 h after the treatment. Non-biotinylated particles or cells not exposed to the transduction step served as a control.

80
A significantly lower amount of cells were living when biotinylated particles were used compared to the non-biotinylated particles (IV/figure 6). This study provided evidence that the avidin fusion protein, LodavinTM, is capable of binding therapeutic compounds and a therapeutic effect can be achieved. Cancer therapy is one obvious application for LodavinTM but in reality, there are no limitations to the use of this concept for other applications, such as the therapy of non-malignant diseases or labelling of cells for imaging. Alternatively, it could be used in the extraction of transduced cells, for example by biotin affinity columns.
6.6.3. Antibody response against the lentivirus and transgene In the aftermath of Jesse Gelsinger´s death during a course of adenoviral gene
therapy tratment, it has been even more important to assess the side effects of viral vectors. The immunogenicity of the vector is an important clinical safety factors (Bessis et al., 2004). The immune response can be targeted not only against the vector but also against the transgene product and can lead to the elimination of the vector, the transduced cells or the therapeutic protein, decreasing the effecacy and duration of the treatment (Raty et al., 2008). Even though lentiviruses are considered to be mildly immunogenic (Bessis et al., 2004), an antibody response to lentiviral vectors has been detected (Follenzi et al., 2007). For these reasons we studied the immune response against the viral vector itself and the expression of the avidin fusion protein. Lentiviral vectors were administered intracerebrally (i.c.) or intravenously (i.v.) to male Wistar rats and re-administration of the vector was performed three weeks later to some of the animals to determine the influence and strength of the immune response after re-challenge. Serum samples were collected at three time points 0, 3 and 6 weeks after the virus administration. Virus infection triggers the production of antibodies to neutralize the virus (Reading and Dimmock, 2007). Firstly, neutralizing antibodies were analyzed from the serum samples by transducing cells after the incubation of GFP expressing lentivirus with serum samples collected form the animals. The GFP positive cells were analyzed three days later. The assay revealed that sera from treated animals contained anti-lentivirus-antibodies. Higher amounts of neutralizing antibodies were detected at the six weeks time point after the repeated administration of the viral vector compared to the groups receiving a single injection. However, the differences were not statistically significant (IV/figure 5). The production of antibodies was not unexpected since the VSV-G elicits the innate immune response and the VSV-G as well as for example the gp64 pseudotyped lentiviral vector particles are inactivated by human serum complement (DePolo et al., 2000; Schauber-Plewa et al., 2005). Serum from control animals neutralized to a slight extent a virus at the highest serum concentration, probably because of the complement activation. Several strategies have been explored to protect the vector particles, such as the use of other envelope pseudotypes (Cronin et al., 2005), incorporation of the complement-regulatory proteins, e.g. human decay-accelerating factor (DAF), into the viral envelope protein (Schauber-Plewa et al., 2005), using polyethylene glycol-modified (PEGylated) viruses (Croyle et al., 2004) or to enhance the transduction by

81
pharmacological inhibition of the proteosome (Santoni de Sio et al., 2006). Furthermore, the production and purification methods need to be optimized to minimize the potential for a systemic immune response and inflammation caused by the impurities in the viral preparations (Baekelandt et al., 2003; Segura et al., 2006).
Consideration needs to be given also to the immune response elicited as a result of expression of the transgene. Avidin has been widely used in pretargeting without evoking any major signs of toxicity (Goldenberg et al., 2006; Goodwin and Meares, 2001) but an immune response against avidin has been detected (Grana et al., 2002; Bubb et al., 1993; Hytonen et al., 2003). Thus, antibodies against LodavinTM were tested from the same serum samples by an avidin ELISA assay. The results demonstrated the clear formation of antibodies with the titers in the range of 104 – 106 per ml (IV/figure 4). An immune response after the readministration of the antigen is typically more rapid, longer and the titer of the antibodies is higher compared to the primary response (Feldmann, 1998). The amounts of the antibodies were higher in every group at the six week time point when compared to the three weeks time point. However, these differences were not statistically significant, neither were there any differences detected between the groups (IV/figure 4). An additional strategy to reduce transgene immune responses is to express the transgene under some tissue-specific promoter (Follenzi et al., 2007).
6.6.4. In vivo expression
Lentiviruses have demonstrated efficient transduction in several kinds of cancer cell types (Pellinen et al., 2004). Before the final conclusion of the suitability of this concept for pretargeting cancer therapy, the expression of the avidin fusion protein lentivirus was tested in the rat glioma model (Tyynela et al., 2002). A high level of expression of the avidin fusion protein was demonstrated after lentivirus mediated local gene transfer to the glioma, which is encouraging for future use of this system in pretargeted cancer therapy (IV/figure 7). This type of in vivo cancer therapy would require, first local transduction of the virus into the tumor followed by the administration of the biotinylated toxic compound, e.g. radiolabeled yttrium. The pretargeting should reduce the side effect caused by a toxic compound.
This study presented a lentivirus mediated pre-targeting strategy based on avidin-biotin technology. High level expression was demonstrated in vitro and in vivo. Ligand binding and readministration were demonstrated and the optimal time schelude for administration was evaluated. In addition, the results obtained that the avidin fusion protein was well tolerated and it did not cause any influence on cell growth nor any apoptotic signs. However, an immune response was elicited in vivo. It also facilitated the targeting of biotinylated therapeutic compouds in the in vitro studies. Pretargeting using this concept is based on the local administration of the virus. However, the system could be improved by modifying the virus envelope, as described in the article III, to achieve even more efficient lentivirus transduction. In conclusion, the results demonstrated the potential of using avidin fusion protein expressing lentiviruses for pretargeting cancer therapy.

82
7. SUMMARY AND CONCLUSIONS 1. Baculoviruses have become the most widely used system for viral based recombinant protein production. A new EGFP expressing baculovirus was developed for fast, accurate and straightforward titering based on the detection of infected cells by flow cytometry without any extra staining step. In addition, for the first time baculoviruses were generated in a 96-well plate format, which allows also high throughput screening of the recombinant proteins. This system can be automated to increase throughput and in this way, save resources. 2. A new efficient production method for lentiviral vectors was established. This method utilized baculoviruses to transiently transfer lentiviral elements into the host cells. Baculovirus technology offers an attractive possibility for scaling up the lentivirus production. 3. Biotin displayed on the baculovirus surface enabled baculovirus separation from the lentivirus preparations. The tangential flow filtration process showed promising results in lentivirus down stream processing. 4. (Strept)avidin displaying lentiviral vectors were developed. The novel surface modifications provided a versatile tool for several kinds of gene therapy applications. (Strept)avidin was coated with biotinylated ligands and targeted transduction was achieved in several cancer cells. Streptavidin was used also in non-invasive imaging of viral biodistribution by SPECT, and for the first time, the transgene expression of the same virus was simultaneously imaged by MRI using ferritin as a transgene. Consequently, a versatile lentivirus was constructed to serve in several kinds of gene therapy applications. This study also provided information on the use of gp64 pseudotyped lentiviruses in the central nervous system. 5. An avidin fusion protein, LodavinTM, expressing lentivirus was constructed for pretargeting of biotinylated therapeutic compounds. The safety and efficacy of the construct were shown in vitro and in vivo. The administration of biotinylated ligands to the transduced cells was studied and readministration of the ligand proved to be feasible. In conclusion, these results improved the potential use of lentiviral vectors in pretargeted cancer therapy.

83
In summary, this thesis provides new information about the generation of lentiviral and baculoviral vectors. Next step would be to adapt the baculovirus-mediated production of lentiviruses into the suspension cell cultures. (Strept)avidin fusion protein displaying lentiviruses demonstrated targeted transduction and could be used for noninvasive imaging of viral biodistribution and transgene expression. An avidin fusion protein expressing lentivirus holds great potential for the development of a pretargeting cancer therapy approach. In the future, it would be very interesting to test this concept in applications other than cancer therapy, such as in targeting stem cells and also combine it with (strept)avidin surface modifications.

84
8. Reference List
Abe,T., Takahashi,H., Hamazaki,H., Miyano-Kurosaki,N., Matsuura,Y., and Takaku,H. (2003). Baculovirus induces an
innate immune response and confers protection from lethal influenza virus infection in mice. J. Immunol. 171, 1133-1139.
Airenne,K.J., Hiltunen,M.O., Turunen,M.P., Turunen,A.M., Laitinen,O.H., Kulomaa,M.S., and Yla-Herttuala,S. (2000). Baculovirus-mediated periadventitial gene transfer to rabbit carotid artery. Gene Ther. 7, 1499-1504.
Airenne,K.J., Mahonen,A.J., Laitinen,O.H., and Yla-Herttuala,S. (2004). Baculovirus-mediated gene transfer: An evolving new concept in: N.S.Templeton (Ed.). Gene and Cell Therapy,Marcel Dekker, Inc., New York, NY), pp. 181-197.
Airenne,K.J., Mahonen,A.J., Laitinen,O.H., and Yla-Herttuala,S. (2009). Baculovirus-Mediated Gene Transfer: An Emerging Universal Concept. In Gene and Cell Therapy, N.Smyth Templeton, ed. (Boca Raton: CRC Press, Taylor & Francis Group), pp. 263-292.
Airenne,K.J., Peltomaa,E., Hytonen,V.P., Laitinen,O.H., and Yla-Herttuala,S. (2003). Improved generation of recombinant baculovirus genomes in Escherichia coli. Nucleic Acids Res. 31, e101.
Aiuti,A., Slavin,S., Aker,M., Ficara,F., Deola,S., Mortellaro,A., Morecki,S., Andolfi,G., Tabucchi,A., Carlucci,F., Marinello,E., Cattaneo,F., Vai,S., Servida,P., Miniero,R., Roncarolo,M.G., and Bordignon,C. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296, 2410-2413.
Anderson,D.B., Laquerre,S., Ghosh,K., Ghosh,H.P., Goins,W.F., Cohen,J.B., and Glorioso,J.C. (2000). Pseudotyping of glycoprotein D-deficient herpes simplex virus type 1 with vesicular stomatitis virus glycoprotein G enables mutant virus attachment and entry. J. Virol. 74, 2481-2487.
Andersson,M., Warolen,M., Nilsson,J., Selander,M., Sterky,C., Bergdahl,K., Sorving,C., James,S.R., and Doverskog,M. (2007). Baculovirus-mediated gene transfer and recombinant protein expression do not interfere with insulin dependent phosphorylation of PKB/Akt in human SHSY-5Y and C3A cells. BMC. Cell Biol. 8, 6.
Argarana,C.E., Kuntz,I.D., Birken,S., Axel,R., and Cantor,C.R. (1986). Molecular cloning and nucleotide sequence of the streptavidin gene. Nucleic Acids Res. 14, 1871-1882.
Arnold,G.S., Sasser,A.K., Stachler,M.D., and Bartlett,J.S. (2006). Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol. Ther. 14, 97-106.
Awasthi,V., Meinken,G., Springer,K., Srivastava,S.C., and Freimuth,P. (2004). Biodistribution of radioiodinated adenovirus fiber protein knob domain after intravenous injection in mice. J. Virol. 78, 6431-6438.
Azzouz,M., Ralph,G.S., Storkebaum,E., Walmsley,L.E., Mitrophanous,K.A., Kingsman,S.M., Carmeliet,P., and Mazarakis,N.D. (2004). VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429, 413-417.
Baekelandt,V., Claeys,A., Eggermont,K., Lauwers,E., De,S.B., Nuttin,B., and Debyser,Z. (2002). Characterization of lentiviral vector-mediated gene transfer in adult mouse brain. Hum. Gene Ther. 13, 841-853.
Baekelandt,V., Eggermont,K., Michiels,M., Nuttin,B., and Debyser,Z. (2003). Optimized lentiviral vector production and purification procedure prevents immune response after transduction of mouse brain. Gene Ther. 10, 1933-1940.
Bahia,D., Cheung,R., Buchs,M., Geisse,S., and Hunt,I. (2005). Optimisation of insect cell growth in deep-well blocks: development of a high-throughput insect cell expression screen. Protein Expr. Purif. 39, 61-70.
Bardies,M., Bardet,S., Faivre-Chauvet,A., Peltier,P., Douillard,J.Y., Mahe,M., Fiche,M., Lisbona,A., Giacalone,F., Meyer,P., Gautherot,E., Rouvier,E., Barbet,J., and Chatal,J.F. (1996). Bispecific antibody and iodine-131-labeled bivalent hapten dosimetry in patients with medullary thyroid or small-cell lung cancer. J. Nucl. Med. 37, 1853-1859.
Barker,D.D. and Berk,A.J. (1987). Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 156, 107-121.
Barry,M.A., Campos,S.K., Ghosh,D., Adams,K.E., Mok,H., Mercier,G.T., and Parrott,M.B. (2003). Biotinylated gene therapy vectors. Expert. Opin. Biol. Ther. 3, 925-940.
Barsoum,J. (1999). Concentration of recombinant baculovirus by cation-exchange chromatography. Biotechniques 26, 834-6, 838, 840.
Barsoum,J., Brown,R., McKee,M., and Boyce,F.M. (1997). Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum. Gene Ther. 8, 2011-2018.
Bartlett,J.S., Kleinschmidt,J., Boucher,R.C., and Samulski,R.J. (1999). Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab'gamma)2 antibody. Nat. Biotechnol. 17, 181-186.

85
Bayer,E.A., Ben-Hur,H., and Wilchek,M. (1986). A sensitive enzyme assay for biotin, avidin, and streptavidin. Anal. Biochem. 154, 367-370.
Benedict,C.A., Tun,R.Y., Rubinstein,D.B., Guillaume,T., Cannon,P.M., and Anderson,W.F. (1999). Targeting retroviral vectors to CD34-expressing cells: binding to CD34 does not catalyze virus-cell fusion. Hum. Gene Ther. 10, 545-557.
Bessis,N., GarciaCozar,F.J., and Boissier,M.C. (2004). Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 11 Suppl 1, S10-S17.
Bigner,D.D., Bigner,S.H., Ponten,J., Westermark,B., Mahaley,M.S., Ruoslahti,E., Herschman,H., Eng,L.F., and Wikstrand,C.J. (1981). Heterogeneity of Genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. J. Neuropathol. Exp. Neurol. 40, 201-229.
Bischoff,J.R., Kirn,D.H., Williams,A., Heise,C., Horn,S., Muna,M., Ng,L., Nye,J.A., Sampson-Johannes,A., Fattaey,A., and McCormick,F. (1996). An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274, 373-376.
Blasberg,R. (2002). Imaging gene expression and endogenous molecular processes: molecular imaging. J. Cereb. Blood Flow Metab 22, 1157-1164.
Boerger,A.L., Snitkovsky,S., and Young,J.A. (1999). Retroviral vectors preloaded with a viral receptor-ligand bridge protein are targeted to specific cell types. Proc. Natl. Acad. Sci. U. S. A 96, 9867-9872.
Boerman,O.C., Kranenborg,M.H., Oosterwijk,E., Griffiths,G.L., McBride,W.J., Oyen,W.J., de,W.M., Oosterwijk-Wakka,J., Hansen,H.J., and Corstens,F.H. (1999). Pretargeting of renal cell carcinoma: improved tumor targeting with a bivalent chelate. Cancer Res. 59, 4400-4405.
Boerman,O.C., van Schaijk,F.G., Oyen,W.J., and Corstens,F.H. (2003). Pretargeted radioimmunotherapy of cancer: progress step by step. J. Nucl. Med. 44, 400-411.
Bogdanov,A., Jr. (2003). In vivo imaging in the development of gene therapy vectors. Curr. Opin. Mol. Ther. 5, 594-602. Boviatsis,E.J., Scharf,J.M., Chase,M., Harrington,K., Kowall,N.W., Breakefield,X.O., and Chiocca,E.A. (1994).
Antitumor activity and reporter gene transfer into rat brain neoplasms inoculated with herpes simplex virus vectors defective in thymidine kinase or ribonucleotide reductase. Gene Ther. 1, 323-331.
Bruch,R.C. and White,H.B., III (1982). Compositional and structural heterogeneity of avidin glycopeptides 1. Biochemistry 21, 5334-5341.
Bubb,M.O., Green,F., Conradie,J.D., Tchernyshev,B., Bayer,E.A., and Wilchek,M. (1993). Natural antibodies to avidin in human serum. Immunol. Lett. 35, 277-280.
Buch,P.K., Bainbridge,J.W., and Ali,R.R. (2008). AAV-mediated gene therapy for retinal disorders: from mouse to ma. Gene Ther. 15, 849-857.
Burns,J.C., Friedmann,T., Driever,W., Burrascano,M., and Yee,J.K. (1993). Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U. S. A 90, 8033-8037.
Bushman,F.D. and Miller,M.D. (1997). Tethering human immunodeficiency virus type 1 preintegration complexes to target DNA promotes integration at nearby sites. J. Virol. 71, 458-464.
Campos,S.K. and Barry,M.A. (2007). Current advances and future challenges in Adenoviral vector biology and targeting. Curr. Gene Ther. 7, 189-204.
Campos,S.K., Parrott,M.B., and Barry,M.A. (2004). Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 9, 942-954.
Cannon,P.M. and Anderson,W.F. (2004). Retroviral Vectors for Gene Therapy. In Gene and Cell Therapy, N.S.Templeton, ed. (New York, NY: Marcel Dekker, Inc.: pp. 1-16.
Carpenter,D.S. and Minchin,R.F. (1998). Targeting of a cholecystokinin-DNA complex to pancreatic cells in vitro and in vivo. Gene Ther. 5, 848-854.
Carter,B.J., Burstein,H., and Peluso,R.W. (2004). Adeno-associated Virus and AAV Vectors for Gene Delivery. In Gene and Cell Therapy, N.S.Templeton, ed. (New York, NY, USA: Marcel DEKKER, inc.), pp. 71-101.
Cavazzana-Calvo,M., Hacein-Bey,S., de Saint,B.G., Gross,F., Yvon,E., Nusbaum,P., Selz,F., Hue,C., Certain,S., Casanova,J.L., Bousso,P., Deist,F.L., and Fischer,A. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288, 669-672.
Cha,H.J., Gotoh,T., and Bentley,W.E. (1997). Simplification of titer determination for recombinant baculovirus by green fluorescent protein marker. Biotechniques 23, 782-4, 786.
Chahal,P.S., Aucoin,M.G., and Kamen,A. (2007). Primary recovery and chromatographic purification of adeno-associated virus type 2 produced by baculovirus/insect cell system. J. Virol. Methods 139, 61-70.
Chan,L., Nesbeth,D., Mackey,T., Galea-Lauri,J., Gaken,J., Martin,F., Collins,M., Mufti,G., Farzaneh,F., and Darling,D. (2005). Conjugation of lentivirus to paramagnetic particles via nonviral proteins allows efficient concentration and infection of primary acute myeloid leukemia cells. J. Virol. 79, 13190-13194.

86
Chapman-Smith,A. and Cronan,J.E., Jr. (1999). In vivo enzymatic protein biotinylation. Biomol. Eng 16, 119-125. Chatal,J.F., Faivre-Chauvet,A., Bardies,M., Peltier,P., Gautherot,E., and Barbet,J. (1995). Bifunctional antibodies for
radioimmunotherapy. Hybridoma 14, 125-128. Check,E. (2005). Gene-therapy trials to restart following cancer risk review. Nature 434, 127. Chen,S., Shohet,R.V., Bekeredjian,R., Frenkel,P., and Grayburn,P.A. (2003). Optimization of ultrasound parameters for
cardiac gene delivery of adenoviral or plasmid deoxyribonucleic acid by ultrasound-targeted microbubble destruction. J. Am. Coll. Cardiol. 42, 301-308.
Chen,Y.H., Wu,J.C., Wang,K.C., Chiang,Y.W., Lai,C.W., Chung,Y.C., and Hu,Y.C. (2005). Baculovirus-mediated production of HDV-like particles in BHK cells using a novel oscillating bioreactor. J. Biotechnol. 118, 135-147.
Cheshenko,N., Krougliak,N., Eisensmith,R.C., and Krougliak,V.A. (2001). A novel system for the production of fully deleted adenovirus vectors that does not require helper adenovirus. Gene Ther. 8, 846-854.
Chinol,M., Casalini,P., Maggiolo,M., Canevari,S., Omodeo,E.S., Caliceti,P., Veronese,F.M., Cremonesi,M., Chiolerio,F., Nardone,E., Siccardi,A.G., and Paganelli,G. (1998). Biochemical modifications of avidin improve pharmacokinetics and biodistribution, and reduce immunogenicity. Br. J. Cancer 78, 189-197.
Chung-Faye,G.A., Chen,M.J., Green,N.K., Burton,A., Anderson,D., Mautner,V., Searle,P.F., and Kerr,D.J. (2001). In vivo gene therapy for colon cancer using adenovirus-mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther. 8, 1547-1554.
Cockrell,A.S. and Kafri,T. (2007). Gene delivery by lentivirus vectors. Mol. Biotechnol. 36, 184-204. Coffin,J.M., Hughes,S.H., and Varmus,H.E. (1997). Retroviruses. (Plainview, NY: Cold Spring Harbor Laboratory Press). Cohen,B., Dafni,H., Meir,G., Harmelin,A., and Neeman,M. (2005). Ferritin as an endogenous MRI reporter for
noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia. 7, 109-117. Coil,D.A. and Miller,A.D. (2004). Phosphatidylserine is not the cell surface receptor for vesicular stomatitis viru. J. Virol.
78, 10920-10926. Coleman,J.E., Huentelman,M.J., Kasparov,S., Metcalfe,B.L., Paton,J.F., Katovich,M.J., Semple-Rowland,S.L., and
Raizada,M.K. (2003). Efficient large-scale production and concentration of HIV-1-based lentiviral vectors for use in vivo. Physiol Genomics 12, 221-228.
Comerota,A.J., Throm,R.C., Miller,K.A., Henry,T., Chronos,N., Laird,J., Sequeira,R., Kent,C.K., Bacchetta,M., Goldman,C., Salenius,J.P., Schmieder,F.A., and Pilsudski,R. (2002). Naked plasmid DNA encoding fibroblast growth factor type 1 for the treatment of end-stage unreconstructible lower extremity ischemia: preliminary results of a phase I trial. J. Vasc. Surg. 35, 930-936.
Cosset,F.L., Morling,F.J., Takeuchi,Y., Weiss,R.A., Collins,M.K., and Russell,S.J. (1995). Retroviral retargeting by envelopes expressing an N-terminal binding domain. J. Virol. 69, 6314-6322.
Coura,R.S. and Nardi,N.B. (2007). The state of the art of adeno-associated virus-based vectors in gene therapy. Virol. J. 4, 99.
Crippa,F., Bolis,G., Seregni,E., Gavoni,N., Scarfone,G., Ferraris,C., Buraggi,G.L., and Bombardieri,E. (1995). Single-dose intraperitoneal radioimmunotherapy with the murine monoclonal antibody I-131 MOv18: clinical results in patients with minimal residual disease of ovarian cancer. Eur. J. Cancer 31A, 686-690.
Cronan,J.E., Jr. (1990). Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J. Biol. Chem. 265, 10327-10333.
Cronin,J., Zhang,X.Y., and Reiser,J. (2005). Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 5, 387-398.
Croyle,M.A., Callahan,S.M., Auricchio,A., Schumer,G., Linse,K.D., Wilson,J.M., Brunner,L.J., and Kobinger,G.P. (2004). PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J. Virol. 78, 912-921.
Crystal,R.G., Hirschowitz,E., Lieberman,M., Daly,J., Kazam,E., Henschke,C., Yankelevitz,D., Kemeny,N., Silverstein,R., Ohwada,A., Russi,T., Mastrangeli,A., Sanders,A., Cooke,J., and Harvey,B.G. (1997). Phase I study of direct administration of a replication deficient adenovirus vector containing the E. coli cytosine deaminase gene to metastatic colon carcinoma of the liver in association with the oral administration of the pro-drug 5-fluorocytosine. Hum. Gene Ther. 8, 985-1001.
De,A., Lewis,X.Z., and Gambhir,S.S. (2003). Noninvasive imaging of lentiviral-mediated reporter gene expression in living mice. Mol. Ther. 7, 681-691.
DeLange,R.J. (1970). Egg white avidin. I. Amino acid composition; sequence of the amino- and carboxyl-terminal cyanogen bromide peptides. J. Biol. Chem. 245, 907-916.
Delenda,C. (2004). Lentiviral vectors: optimization of packaging, transduction and gene expression. J. Gene Med. 6 Suppl 1, S125-S138.
Delenda,C. and Gaillard,C. (2005). Real-time quantitative PCR for the design of lentiviral vector analytical assays. Gene Ther. 12 Suppl 1, S36-S50.

87
DePolo,N.J., Reed,J.D., Sheridan,P.L., Townsend,K., Sauter,S.L., Jolly,D.J., and Dubensky,T.W., Jr. (2000). VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol. Ther. 2, 218-222.
Deroose,C.M., Reumers,V., Gijsbers,R., Bormans,G., Debyser,Z., Mortelmans,L., and Baekelandt,V. (2006). Noninvasive monitoring of long-term lentiviral vector-mediated gene expression in rodent brain with bioluminescence imaging. Mol. Ther. 14, 423-431.
Diamandis,E.P. and Christopoulos,T.K. (1991). The biotin-(strept)avidin system: principles and applications in biotechnology. Clin. Chem. 37, 625-636.
Doms,R.W., Ruusala,A., Machamer,C., Helenius,J., Helenius,A., and Rose,J.K. (1988). Differential effects of mutations in three domains on folding, quaternary structure, and intracellular transport of vesicular stomatitis virus G protein. J. Cell Biol. 107, 89-99.
Dull,T., Zufferey,R., Kelly,M., Mandel,R.J., Nguyen,M., Trono,D., and Naldini,L. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463-8471.
Elias,C.B., Jardin,B., and Kamen,A. (2007). Recombinant protein production in large-scale agitated bioreactors using the baculovirus expression vector system. Methods Mol. Biol. 388, 225-246.
Ernst,W.J., Grabherr,R.M., and Katinger,H.W. (1994). Direct cloning into the Autographa californica nuclear polyhedrosis virus for generation of recombinant baculoviruses. Nucleic Acids Res. 22, 2855-2856.
Facciabene,A., Aurisicchio,L., and La,M.N. (2004). Baculovirus vectors elicit antigen-specific immune responses in mice. J. Virol. 78, 8663-8672.
Farson,D., Witt,R., McGuinness,R., Dull,T., Kelly,M., Song,J., Radeke,R., Bukovsky,A., Consiglio,A., and Naldini,L. (2001). A new-generation stable inducible packaging cell line for lentiviral vectors. Hum. Gene Ther. 12, 981-997.
Felder,J.M.I. and Sutton,R.E. (2009). Lentiviral vectors. In Gene and Cell Therapy, N.Smyth, ed. (Boca Raton, Florida: CRC Press), pp. 207-244.
Feldmann,M. (1998). Cell Cooperation in the Antibody Response. In Immunology, I.Roitt, J.Brostoff, and D.Male, eds. (London, UK: Mosby International Ltd), pp. 139-153.
Follenzi,A., Ailles,L.E., Bakovic,S., Geuna,M., and Naldini,L. (2000). Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 25, 217-222.
Follenzi,A. and Naldini,L. (2002). Generation of HIV-1 derived lentiviral vectors. Methods Enzymol. 346, 454-465. Follenzi,A., Santambrogio,L., and Annoni,A. (2007). Immune responses to lentiviral vectors. Curr. Gene Ther. 7, 306-
315. Freed,E.O. and Martin,M.A. (2001). HIVs and Their Replication. In Fields Virology, D.M.Knipe and P.M.Howley, eds.
(Philadelphia: Lippincott Williams & Wilkins), pp. 1971-2041. Frolov,I., Hoffman,T.A., Pragai,B.M., Dryga,S.A., Huang,H.V., Schlesinger,S., and Rice,C.M. (1996). Alphavirus-based
expression vectors: strategies and applications. Proc. Natl. Acad. Sci. U. S. A 93, 11371-11377. Funke,S., Maisner,A., Muhlebach,M.D., Koehl,U., Grez,M., Cattaneo,R., Cichutek,K., and Buchholz,C.J. (2008).
Targeted cell entry of lentiviral vectors. Mol. Ther. 16, 1427-1436. Galarza,J.M., Latham,T., and Cupo,A. (2005). Virus-like particle vaccine conferred complete protection against a lethal
influenza virus challenge. Viral Immunol. 18, 365-372. Gambhir,S.S., Bauer,E., Black,M.E., Liang,Q., Kokoris,M.S., Barrio,J.R., Iyer,M., Namavari,M., Phelps,M.E., and
Herschman,H.R. (2000). A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc. Natl. Acad. Sci. U. S. A 97, 2785-2790.
Gao,G.P., Yang,Y., and Wilson,J.M. (1996). Biology of adenovirus vectors with E1 and E4 deletions for liver-directed gene therapy. J. Virol. 70, 8934-8943.
Gasmi,M., Glynn,J., Jin,M.J., Jolly,D.J., Yee,J.K., and Chen,S.T. (1999). Requirements for efficient production and transduction of human immunodeficiency virus type 1-based vectors. J. Virol. 73, 1828-1834.
Genove,G., DeMarco,U., Xu,H., Goins,W.F., and Ahrens,E.T. (2005). A new transgene reporter for in vivo magnetic resonance imaging. Nat. Med. 11, 450-454.
Geraerts,M., Michiels,M., Baekelandt,V., Debyser,Z., and Gijsbers,R. (2005). Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med. 7, 1299-1310.
Geraerts,M., Willems,S., Baekelandt,V., Debyser,Z., and Gijsbers,R. (2006). Comparison of lentiviral vector titration methods. BMC. Biotechnol. 6, 34.
Gheysen,D., Jacobs,E., de,F.F., Thiriart,C., Francotte,M., Thines,D., and De,W.M. (1989). Assembly and release of HIV precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59, 103-112.
Gilbert,J.R. and Wong-Staal,F. (2001). HIV-2 and SIV vector systems. Somat. Cell Mol. Genet. 26, 83-98. Glasgow,J.N., Hemminki,A., and Curiel,D.T. (2009). Modified Adenovirus for Gene Therapy. In Gene and Cell Therapy,
N.Smyth Templeton, ed. (Boca Raton: CRC Press, Taylor & Francis Group), pp. 69-86.

88
Glunde,K., Pathak,A.P., and Bhujwalla,Z.M. (2007). Molecular-functional imaging of cancer: to image and imagine. Trends Mol. Med. 13, 287-297.
Goetz,C., Riva,P., Poepperl,G., Gildehaus,F.J., Hischa,A., Tatsch,K., and Reulen,H.J. (2003). Locoregional radioimmunotherapy in selected patients with malignant glioma: experiences, side effects and survival times. J. Neurooncol. 62, 321-328.
Goff.S.P. (2001). Retroviridae: The retroviruses and Their Replication. In Fields Virology, D.M.Knipe and P.M.Howley, eds. (Philadelphia: Lippincott Williams & Wilkins), pp. 1871-1936.
Goldenberg,D.M., Sharkey,R.M., Paganelli,G., Barbet,J., and Chatal,J.F. (2006). Antibody pretargeting advances cancer radioimmunodetection and radioimmunotherapy. J. Clin. Oncol. 24, 823-834.
Golzio,M., Rols,M.P., Gabriel,B., and Teissie,J. (2004). Optical imaging of in vivo gene expression: a critical assessment of the methodology and associated technologies. Gene Ther. 11 Suppl 1, S85-S91.
Goodwin,D.A. and Meares,C.F. (2001). Advances in pretargeting biotechnology. Biotechnol. Adv. 19, 435-450. Goodwin,D.A., Mears,C.F., McTigue,M., and David,G.S. (1986). Monoclonal antibody hapten radiopharmaceutical
delivery. Nucl. Med. Commun. 7, 569-580. Gotoh,H. and Matsumoto,Y. (2007). Cell-surface streptavidin fusion protein for rapid selection of transfected mammalian
cells. Gene 389, 146-153. Goulaouic,H. and Chow,S.A. (1996). Directed integration of viral DNA mediated by fusion proteins consisting of human
immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J. Virol. 70, 37-46. Goverdhana,S., Puntel,M., Xiong,W., Zirger,J.M., Barcia,C., Curtin,J.F., Soffer,E.B., Mondkar,S., King,G.D., Hu,J.,
Sciascia,S.A., Candolfi,M., Greengold,D.S., Lowenstein,P.R., and Castro,M.G. (2005). Regulatable gene expression systems for gene therapy applications: progress and future challenges. Mol. Ther. 12, 189-211.
Grana,C., Chinol,M., Robertson,C., Mazzetta,C., Bartolomei,M., De,C.C., Fiorenza,M., Gatti,M., Caliceti,P., and Paganelli,G. (2002). Pretargeted adjuvant radioimmunotherapy with yttrium-90-biotin in malignant glioma patients: a pilot study. Br. J. Cancer 86, 207-212.
Green,N.M. (1975). Avidin. Adv. Protein Chem. 29, 85-133. Green,N.M. (1990). Avidin and streptavidin. Methods Enzymol. 184, 51-67. Gruaz-Guyon,A., Janevik-Ivanovska,E., Raguin,O., briolle-Vaylet,C., and Barbet,J. (2001). Radiolabeled bivalent haptens
for tumor immunodetection and radioimmunotherapy. Q. J. Nucl. Med. 45, 201-206. Guibinga,G.H., Song,S., Loring,J., and Friedmann,T. (2008). Characterization of the gene delivery properties of
baculoviral-based virosomal vectors. J. Virol. Methods 148, 277-282. Haberkorn,U., Oberdorfer,F., Gebert,J., Morr,I., Haack,K., Weber,K., Lindauer,M., van,K.G., and Schackert,H.K. (1996).
Monitoring gene therapy with cytosine deaminase: in vitro studies using tritiated-5-fluorocytosine. J. Nucl. Med. 37, 87-94.
Hacein-Bey-Abina,S., Le,D.F., Carlier,F., Bouneaud,C., Hue,C., De Villartay,J.P., Thrasher,A.J., Wulffraat,N., Sorensen,R., Dupuis-Girod,S., Fischer,A., Davies,E.G., Kuis,W., Leiva,L., and Cavazzana-Calvo,M. (2002). Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 346, 1185-1193.
Hackett,N.R. and Crystal,R.G. (2004). Adenovirus Vectors for Gene Therapy. In Gene and Cell therapy, N.S.Templeton, ed. (New York, NY, USA: Marcel Dekker, Inc.), pp. 17-42.
Haeseleer,F., Imanishi,Y., Saperstein,D.A., and Palczewski,K. (2001). Gene transfer mediated by recombinant baculovirus into mouse eye. Invest Ophthalmol. Vis. Sci. 42, 3294-3300.
Hall,F.L., Gordon,E.M., Wu,L., Zhu,N.L., Skotzko,M.J., Starnes,V.A., and Anderson,W.F. (1997). Targeting retroviral vectors to vascular lesions by genetic engineering of the MoMLV gp70 envelope protein. Hum. Gene Ther. 8, 2183-2192.
Hall,F.L., Liu,L., Zhu,N.L., Stapfer,M., Anderson,W.F., Beart,R.W., and Gordon,E.M. (2000). Molecular engineering of matrix-targeted retroviral vectors incorporating a surveillance function inherent in von Willebrand factor. Hum. Gene Ther. 11, 983-993.
Hao,M. and Maxfield,F.R. (2000). Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 275, 15279-15286.
Haselhorst,D., Kaye,J.F., and Lever,A.M. (1998). Development of cell lines stably expressing human immunodeficiency virus type 1 proteins for studies in encapsidation and gene transfer. J. Gen. Virol. 79 ( Pt 2), 231-237.
Hedley,S.J., uf der,M.A., Hohn,S., Escher,D., Barberis,A., Glasgow,J.N., Douglas,J.T., Korokhov,N., and Curiel,D.T. (2006). An adenovirus vector with a chimeric fiber incorporating stabilized single chain antibody achieves targeted gene delivery. Gene Ther. 13, 88-94.
Herbst,R.S. (2004). Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 59, 21-26. Higgins,D.E. and Portnoy,D.A. (1998). Bacterial delivery of DNA evolves. Nat. Biotechnol. 16, 138-139.

89
Hitchman,R.B., Siaterli,E.A., Nixon,C.P., and King,L.A. (2007). Quantitative real-time PCR for rapid and accurate titration of recombinant baculovirus particles. Biotechnol. Bioeng. 96, 810-814.
Hnatowich,D.J., Fritz,B., Virzi,F., Mardirossian,G., and Rusckowski,M. (1993). Improved tumor localization with (strept)avidin and labeled biotin as a substitute for antibody. Nucl. Med. Biol. 20, 189-195.
Hnatowich,D.J., Virzi,F., and Rusckowski,M. (1987). Investigations of avidin and biotin for imaging applications. J. Nucl. Med. 28, 1294-1302.
Hofmann,C., Sandig,V., Jennings,G., Rudolph,M., Schlag,P., and Strauss,M. (1995). Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. U. S. A 92, 10099-10103.
Hofmann,C. and Strauss,M. (1998). Baculovirus-mediated gene transfer in the presence of human serum or blood facilitated by inhibition of the complement system. Gene Ther. 5, 531-536.
Hong,T., Braunagel,S.C., and Summers,M.D. (1994). Transcription, translation, and cellular localization of PDV-E66: a structural protein of the PDV envelope of Autographa californica nuclear polyhedrosis virus. Virology 204, 210-222.
Hu,Y.C. (2006). Baculovirus vectors for gene therapy. Adv. Virus Res. 68, 287-320. Huang,K.S., Lo,W.H., Chung,Y.C., Lai,Y.K., Chen,C.Y., Chou,S.T., and Hu,Y.C. (2007). Combination of baculovirus-
mediated gene delivery and packed-bed reactor for scalable production of adeno-associated virus. Hum. Gene Ther. 18, 1161-1170.
Hytonen,V.P., Laitinen,O.H., Grapputo,A., Kettunen,A., Savolainen,J., Kalkkinen,N., Marttila,A.T., Nordlund,H.R., Nyholm,T.K., Paganelli,G., and Kulomaa,M.S. (2003). Characterization of poultry egg-white avidins and their potential as a tool in pretargeting cancer treatment. Biochem. J. 372, 219-225.
Immonen,A., Vapalahti,M., Tyynela,K., Hurskainen,H., Sandmair,A., Vanninen,R., Langford,G., Murray,N., and Yla-Herttuala,S. (2004). AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol. Ther. 10, 967-972.
Inagaki,K., Fuess,S., Storm,T.A., Gibson,G.A., Mctiernan,C.F., Kay,M.A., and Nakai,H. (2006). Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 14, 45-53.
Ivics,Z. and Izsvak,Z. (2006). Transposons for gene therapy!. Curr. Gene Ther. 6, 593-607. Iwakuma,T., Cui,Y., and Chang,L.J. (1999). Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology
261, 120-132. Izsvak,Z. and Ivics,Z. (2004). Sleeping beauty transposition: biology and applications for molecular therapy. Mol. Ther. 9,
147-156. Janakiraman,V., Forrest,W.F., Chow,B., and Seshagiri,S. (2006). A rapid method for estimation of baculovirus titer based
on viable cell size. J. Virol. Methods 132, 48-58. Jarvis,D.L. (1997). Baculovirus Expression Vectors. In The Baculoviruses, Miller L.K., ed. (new York and London:
Plenum Press), pp. 389-431. Jeetendra,E., Robison,C.S., Albritton,L.M., and Whitt,M.A. (2002). The membrane-proximal domain of vesicular
stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J. Virol. 76, 12300-12311.
Johnson,J.E. and Chiu,W. (2007). DNA packaging and delivery machines in tailed bacteriophages. Curr. Opin. Struct. Biol. 17, 237-243.
Kafri,T., Blomer,U., Peterson,D.A., Gage,F.H., and Verma,I.M. (1997). Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 17, 314-317.
Kafri,T., van,P.H., Ouyang,L., Gage,F.H., and Verma,I.M. (1999). A packaging cell line for lentivirus vectors. J. Virol. 73, 576-584.
Kaikkonen,M.U., Raty,J.K., Airenne,K.J., Wirth,T., Heikura,T., and Yla-Herttuala,S. (2006). Truncated vesicular stomatitis virus G protein improves baculovirus transduction efficiency in vitro and in vivo. Gene Ther. 13, 304-312.
Kaikkonen,M.U., Viholainen,J.I., Narvanen,A., Yla-Herttuala,S., and Airenne,K.J. (2008). Targeting and purification of metabolically biotinylated baculovirus. Hum. Gene Ther. 19, 589-600.
Kalofonos,H.P., Rusckowski,M., Siebecker,D.A., Sivolapenko,G.B., Snook,D., Lavender,J.P., Epenetos,A.A., and Hnatowich,D.J. (1990). Imaging of tumor in patients with indium-111-labeled biotin and streptavidin-conjugated antibodies: preliminary communicatio. J. Nucl. Med. 31, 1791-1796.
Karolewski,B.A., Watson,D.J., Parente,M.K., and Wolfe,J.H. (2003). Comparison of transfection conditions for a lentivirus vector produced in large volumes . Hum. Gene Ther. 14, 1287-1296.
Kim,V.N., Mitrophanous,K., Kingsman,S.M., and Kingsman,A.J. (1998). Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J. Virol. 72, 811-816.

90
Kitts,P.A. and Green,G. (1999). An immunological assay for determination of baculovirus titers in 48 hours. Anal. Biochem. 268, 173-178.
Kitts,P.A. and Possee,R.D. (1993). A method for producing recombinant baculovirus expression vectors at high frequency. Biotechniques 14, 810-817.
Klibanov,A.L., Martynov,A.V., Slinkin,M.A., Sakharov,I.Y., Smirnov,M.D., Muzykantov,V.R., Danilov,S.M., and Torchilin,V.P. (1988). Blood clearance of radiolabeled antibody: enhancement by lactosamination and treatment with biotin-avidin or anti-mouse IgG antibodies. J. Nucl. Med. 29, 1951-1956.
Knox,S.J., Goris,M.L., Tempero,M., Weiden,P.L., Gentner,L., Breitz,H., Adams,G.P., Axworthy,D., Gaffigan,S., Bryan,K., Fisher,D.R., Colcher,D., Horak,I.D., and Weiner,L.M. (2000). Phase II trial of yttrium-90-DOTA-biotin pretargeted by NR-LU-10 antibody/streptavidin in patients with metastatic colon cancer. Clin. Cancer Res. 6, 406-414.
Kochanek,S., Schiedner,G., and Volpers,C. (2001). High-capacity 'gutless' adenoviral vectors. Curr. Opin. Mol. Ther. 3, 454-463.
Koldej,R., Cmielewski,P., Stocker,A., Parsons,D.W., and Anson,D.S. (2005). Optimisation of a multipartite human immunodeficiency virus based vector system; control of virus infectivity and large-scale production. J. Gene Med. 7, 1390-1399.
Konetschny,C., Holzer,G.W., Urban,C., Hammerle,T., Mayrhofer,J., and Falkner,F.G. (2003). Generation of transduction-competent retroviral vectors by infection with a single hybrid vaccinia virus. J. Virol. 77, 7017-7025.
Kordower,J.H., Emborg,M.E., Bloch,J., Ma,S.Y., Chu,Y., Leventhal,L., McBride,J., Chen,E.Y., Palfi,S., Roitberg,B.Z., Brown,W.D., Holden,J.E., Pyzalski,R., Taylor,M.D., Carvey,P., Ling,Z., Trono,D., Hantraye,P., Deglon,N., and Aebischer,P. (2000). Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science 290, 767-773.
Kost,T.A. and Condreay,J.P. (2002). Recombinant baculoviruses as mammalian cell gene-delivery vectors. Trends Biotechnol. 20, 173-180.
Kost,T.A., Condreay,J.P., and Jarvis,D.L. (2005). Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 23, 567-575.
Kotsopoulou,E., Kim,V.N., Kingsman,A.J., Kingsman,S.M., and Mitrophanous,K.A. (2000). A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 74, 4839-4852.
Kraeber-Bodere,F., Bardet,S., Hoefnagel,C.A., Vieira,M.R., Vuillez,J.P., Murat,A., Ferreira,T.C., Bardies,M., Ferrer,L., Resche,I., Gautherot,E., Rouvier,E., Barbet,J., and Chatal,J.F. (1999). Radioimmunotherapy in medullary thyroid cancer using bispecific antibody and iodine 131-labeled bivalent hapten: preliminary results of a phase I/II clinical trial. Clin. Cancer Res. 5, 3190s-3198s.
Kubo,S. and Mitani,K. (2003). A new hybrid system capable of efficient lentiviral vector production and stable gene transfer mediated by a single helper-dependent adenoviral vector. J. Virol. 77, 2964-2971.
Kukkonen,S.P., Airenne,K.J., Marjomaki,V., Laitinen,O.H., Lehtolainen,P., Kankaanpaa,P., Mahonen,A.J., Raty,J.K., Nordlund,H.R., Oker-Blom,C., Kulomaa,M.S., and Yla-Herttuala,S. (2003). Baculovirus capsid display: a novel tool for transduction imaging. Mol. Ther. 8, 853-862.
Kumar,M., Bradow,B.P., and Zimmerberg,J. (2003). Large-scale production of pseudotyped lentiviral vectors using baculovirus GP64. Hum. Gene Ther. 14, 67-77.
Kuroda,H., Kutner,R.H., Bazan,N.G., and Reiser,J. (2008). Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J. Virol. Methods.
Kwon,I. and Schaffer,D.V. (2008). Designer gene delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm. Res. 25, 489-499.
Kwon,M.S., Dojima,T., Toriyama,M., and Park,E.Y. (2002). Development of an antibody-based assay for determination of baculovirus titers in 10 hours. Biotechnol. Prog. 18, 647-651.
Laitinen,M., Pakkanen,T., Donetti,E., Baetta,R., Luoma,J., Lehtolainen,P., Viita,H., Agrawal,R., Miyanohara,A., Friedmann,T., Risau,W., Martin,J.F., Soma,M., and Yla-Herttuala,S. (1997). Gene transfer into the carotid artery using an adventitial collar: comparison of the effectiveness of the plasmid-liposome complexes, retroviruses, pseudotyped retroviruses, and adenoviruses. Hum. Gene Ther. 8, 1645-1650.
Laitinen,O.H., Airenne,K.J., Hytonen,V.P., Peltomaa,E., Mahonen,A.J., Wirth,T., Lind,M.M., Makela,K.A., Toivanen,P.I., Schenkwein,D., Heikura,T., Nordlund,H.R., Kulomaa,M.S., and Yla-Herttuala,S. (2005). A multipurpose vector system for the screening of libraries in bacteria, insect and mammalian cells and expression in vivo. Nucleic Acids Res. 33, e42.
Laitinen,O.H., Hytonen,V.P., Ahlroth,M.K., Pentikainen,O.T., Gallagher,C., Nordlund,H.R., Ovod,V., Marttila,A.T., Porkka,E., Heino,S., Johnson,M.S., Airenne,K.J., and Kulomaa,M.S. (2002). Chicken avidin-related proteins show altered biotin-binding and physico-chemical properties as compared with avidin. Biochem. J. 363, 609-617.

91
Laitinen,O.H., Hytonen,V.P., Nordlund,H.R., and Kulomaa,M.S. (2006). Genetically engineered avidins and streptavidins. Cell Mol. Life Sci. 63, 2992-3017.
Laitinen,O.H., Nordlund,H.R., Hytonen,V.P., and Kulomaa,M.S. (2007). Brave new (strept)avidins in biotechnology. Trends Biotechnol. 25, 269-277.
Laitinen,O.H., Nordlund,H.R., Hytonen,V.P., Uotila,S.T., Marttila,A.T., Savolainen,J., Airenne,K.J., Livnah,O., Bayer,E.A., Wilchek,M., and Kulomaa,M.S. (2003). Rational design of an active avidin monomer. J. Biol. Chem. 278, 4010-4014.
Lanciotti,J., Song,A., Doukas,J., Sosnowski,B., Pierce,G., Gregory,R., Wadsworth,S., and O'Riordan,C. (2003). Targeting adenoviral vectors using heterofunctional polyethylene glycol FGF2 conjugates. Mol. Ther. 8, 99-107.
Larochelle,A., Peng,K.W., and Russell,S.J. (2002). Lentiviral vector targeting. Curr. Top. Microbiol. Immunol. 261, 143-163.
Lee,S.G., Yoon,S.J., Kim,C.D., Kim,K., Lim,D.S., Yeom,Y.I., Sung,M.W., Heo,D.S., and Kim,N.K. (2000). Enhancement of adenoviral transduction with polycationic liposomes in vivo. Cancer Gene Ther. 7, 1329-1335.
Lehtolainen,P., Taskinen,A., Laukkanen,J., Airenne,K.J., Heino,S., Lappalainen,M., Ojala,K., Marjomaki,V., Martin,J.F., Kulomaa,M.S., and Yla-Herttuala,S. (2002a). Cloning and characterization of Scavidin, a fusion protein for the targeted delivery of biotinylated molecules. J. Biol. Chem. 277, 8545-8550.
Lehtolainen,P., Tyynela,K., Kannasto,J., Airenne,K.J., and Yla-Herttuala,S. (2002b). Baculoviruses exhibit restricted cell type specificity in rat brain: a comparison of baculovirus- and adenovirus-mediated intracerebral gene transfer in vivo. Gene Ther. 9, 1693-1699.
Lehtolainen,P., Wirth,T., Taskinen,A.K., Lehenkari,P., Leppanen,O., Lappalainen,M., Pulkkanen,K., Marttila,A., Marjomaki,V., Airenne,K.J., Horton,M., Kulomaa,M.S., and Yla-Herttuala,S. (2003). Targeting of biotinylated compounds to its target tissue using a low-density lipoprotein receptor-avidin fusion protein. Gene Ther. 10, 2090-2097.
Li,Y., Drone,C., Sat,E., and Ghosh,H.P. (1993). Mutational analysis of the vesicular stomatitis virus glycoprotein G for membrane fusion domains. J. Virol. 67, 4070-4077.
Lipshutz,G.S., Gruber,C.A., Cao,Y., Hardy,J., Contag,C.H., and Gaensler,K.M. (2001). In utero delivery of adeno-associated viral vectors: intraperitoneal gene transfer produces long-term expression. Mol. Ther. 3, 284-292.
Liu,H., Dow,E.C., Arora,R., Kimata,J.T., Bull,L.M., Arduino,R.C., and Rice,A.P. (2006a). Integration of human immunodeficiency virus type 1 in untreated infection occurs preferentially within genes. J. Virol. 80, 7765-7768.
Liu,H.S., Jan,M.S., Chou,C.K., Chen,P.H., and Ke,N.J. (1999). Is green fluorescent protein toxic to the living cells?. Biochem. Biophys. Res. Commun. 260, 712-717.
Liu,M.A., Wahren,B., and Karlsson Hedestam,G.B. (2006b). DNA vaccines: recent developments and future possibilities. Hum. Gene Ther. 17, 1051-1061.
Lo,H.R. and Chao,Y.C. (2004). Rapid titer determination of baculovirus by quantitative real-time polymerase chain reaction. Biotechnol. Prog. 20, 354-360.
Logan,A.C., Nightingale,S.J., Haas,D.L., Cho,G.J., Pepper,K.A., and Kohn,D.B. (2004). Factors influencing the titer and infectivity of lentiviral vectors. Hum. Gene Ther. 15, 976-988.
Louie,A.Y., Huber,M.M., Ahrens,E.T., Rothbacher,U., Moats,R., Jacobs,R.E., Fraser,S.E., and Meade,T.J. (2000). In vivo visualization of gene expression using magnetic resonance imaging. Nat. Biotechnol. 18, 321-325.
Lu,X., Humeau,L., Slepushkin,V., Binder,G., Yu,Q., Slepushkina,T., Chen,Z., Merling,R., Davis,B., Chang,Y.N., and Dropulic,B. (2004). Safe two-plasmid production for the first clinical lentivirus vector that achieves >99% transduction in primary cells using a one-step protocol. J. Gene Med. 6, 963-973.
Luckow,V.A., Lee,S.C., Barry,G.F., and Olins,P.O. (1993). Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 67, 4566-4579.
Lusky,M., Christ,M., Rittner,K., Dieterle,A., Dreyer,D., Mourot,B., Schultz,H., Stoeckel,F., Pavirani,A., and Mehtali,M. (1998). In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 72, 2022-2032.
MacLaren,D.C., Gambhir,S.S., Satyamurthy,N., Barrio,J.R., Sharfstein,S., Toyokuni,T., Wu,L., Berk,A.J., Cherry,S.R., Phelps,M.E., and Herschman,H.R. (1999). Repetitive, non-invasive imaging of the dopamine D2 receptor as a reporter gene in living animals. Gene Ther. 6, 785-791.
Magnani,P., Fazio,F., Grana,C., Songini,C., Frigerio,L., Pecorelli,S., Mangili,G., Colombo,N., Mariani,C.D., and Paganelli,G. (2000). Diagnosis of persistent ovarian carcinoma with three-step immunoscintigraphy. Br. J. Cancer 82, 616-620.
Magnusson,M.K., Hong,S.S., Henning,P., Boulanger,P., and Lindholm,L. (2002). Genetic retargeting of adenovirus vectors: functionality of targeting ligands and their influence on virus viability. J. Gene Med. 4, 356-370.

92
Mahonen,A.J., Airenne,K.J., Purola,S., Peltomaa,E., Kaikkonen,M.U., Riekkinen,M.S., Heikura,T., Kinnunen,K., Roschier,M.M., Wirth,T., and Yla-Herttuala,S. (2007). Post-transcriptional regulatory element boosts baculovirus-mediated gene expression in vertebrate cells. J. Biotechnol. 131, 1-8.
Makinen,P.I., Koponen,J.K., Karkkainen,A.M., Malm,T.M., Pulkkinen,K.H., Koistinaho,J., Turunen,M.P., and Yla-Herttuala,S. (2006). Stable RNA interference: comparison of U6 and H1 promoters in endothelial cells and in mouse brain. J. Gene Med. 8, 433-441.
Mantyla,T., Hakumaki,J.M., Huhtala,T., Narvanen,A., and Yla-Herttuala,S. (2006). Targeted magnetic resonance imaging of Scavidin-receptor in human umbilical vein endothelial cells in vitro. Magn Reson. Med. 55, 800-804.
Markusic,D.M., Kanitz,A., Oude-Elferink,R.P., and Seppen,J. (2007). Preferential gene transfer of lentiviral vectors to liver-derived cells, using a hepatitis B peptide displayed on GP64. Hum. Gene Ther. 18, 673-679.
Marshall,D., Pedley,R.B., Boden,J.A., Boden,R., Melton,R.G., and Begent,R.H. (1996). Polyethylene glycol modification of a galactosylated streptavidin clearing agent: effects on immunogenicity and clearance of a biotinylated anti-tumour antibody. Br. J. Cancer 73, 565-572.
Marston,F.A. (1986). The purification of eukaryotic polypeptides synthesized in Escherichia coli. Biochem. J. 240, 1-12. Martignoni,M.E. and Iwai,P.J. (1986). A Catalog of Viral Diseases of Insects. (Portland, OR: US Department of
Agriculture, Forest Service, Pacific Northwest Research Station). Martuza,R.L., Malick,A., Markert,J.M., Ruffner,K.L., and Coen,D.M. (1991). Experimental therapy of human glioma by
means of a genetically engineered virus mutant. Science 252, 854-856. Massoud,T.F. and Gambhir,S.S. (2003). Molecular imaging in living subjects: seeing fundamental biological processes in
a new light. Genes Dev. 17, 545-580. McCart,J.A. and Barlett,D.L. (2004). Vaccinia Viral Vectors. In Gene and Cell Therapy, N.S.Templeton, ed. (New York,
NY, USA: Marcel Dekker, Inc.), pp. 165-180. Meghrous,J., Aucoin,M.G., Jacob,D., Chahal,P.S., Arcand,N., and Kamen,A.A. (2005). Production of recombinant adeno-
associated viral vectors using a baculovirus/insect cell suspension culture system: from shake flasks to a 20-L bioreactor. Biotechnol. Prog. 21, 154-160.
Meikle,S.R., Beekman,F.J., and Rose,S.E. (2006). Complementary molecular imaging technologies: High resolution SPECT, PET and MRI. Drug Discov. Today: Technologies 3, 187-194.
Menotti,L., Cerretani,A., and Campadelli-Fiume,G. (2006). A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 80, 5531-5539.
Meredith,R.F. and Buchsbaum,D.J. (2006). Pretargeted radioimmunotherapy. Int. J. Radiat. Oncol. Biol. Phys. 66, S57-S59.
Merten,O.W. (2004). State-of-the-art of the production of retroviral vectors. J. Gene Med. 6 Suppl 1, S105-S124. Miller,L.K. (1997). The Baculoviruses. (New York: Plenum Press). Miller,N. and Whelan,J. (1997). Progress in transcriptionally targeted and regulatable vectors for genetic therapy. Hum.
Gene Ther. 8, 803-815. Min,J.J. and Gambhir,S.S. (2004). Gene therapy progress and prospects: noninvasive imaging of gene therapy in living
subjects. Gene Ther. 11, 115-125. Mittra,E. and Quon,A. (2009). Positron emission tomography/computed tomography: the current technology and
applications. Radiol. Clin. North Am. 47, 147-160. Miyake,K., Miyake,N., and Shimada,T. (2007). Development of targeted gene transfer into human primary T lymphocytes
and macrophages using high-titer recombinant HIV vectors. J. Biotechnol. 129, 532-538. Miyoshi,H., Blomer,U., Takahashi,M., Gage,F.H., and Verma,I.M. (1998). Development of a self-inactivating lentivirus
vector. J. Virol. 72, 8150-8157. Moolten,F.L. (1986). Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a
prospective cancer control strategy. Cancer Res. 46, 5276-5281. Moss,B. (2001). Poxviridae: The Viruses and Their Replication. In Fields Virology, D.M.Knipe, P.M.Howley,
D.E.Griffin, M.A.Martin, R.A.Lamb, B.Roizman, and S.E.Straus, eds. (Philadelphia, PA, USA: Lippicott Williams & Wilkins), pp. 2849-2885.
Mottershead,D.G., Alfthan,K., Ojala,K., Takkinen,K., and Oker-Blom,C. (2000). Baculoviral display of functional scFv and synthetic IgG-binding domains. Biochem. Biophys. Res. Commun. 275, 84-90.
Mulvania,T., Hayes,B., and Hedin,D. (2004). A flow cytometric assay for rapid, accurate determination of baculovirus titers. BioProcessing Journal 3, 47-53.
Nakamura,T., Peng,K.W., Harvey,M., Greiner,S., Lorimer,I.A., James,C.D., and Russell,S.J. (2005). Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 23, 209-214.

93
Naldini,L., Blomer,U., Gage,F.H., Trono,D., and Verma,I.M. (1996a). Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A 93, 11382-11388.
Naldini,L., Blomer,U., Gallay,P., Ory,D., Mulligan,R., Gage,F.H., Verma,I.M., and Trono,D. (1996b). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263-267.
Negrete,A. and Kotin,R.M. (2007). Production of recombinant adeno-associated vectors using two bioreactor configurations at different scales. J. Virol. Methods 145, 155-161.
Negrete,A. and Kotin,R.M. (2008). Large-scale production of recombinant adeno-associated viral vectors. Methods Mol. Biol. 433, 79-96.
Nermut,M.V., Hockley,D.J., Jowett,J.B., Jones,I.M., Garreau,M., and Thomas,D. (1994). Fullerene-like organization of HIV gag-protein shell in virus-like particles produced by recombinant baculovirus. Virology 198, 288-296.
Nesbeth,D., Williams,S.L., Chan,L., Brain,T., Slater,N.K., Farzaneh,F., and Darling,D. (2006). Metabolic biotinylation of lentiviral pseudotypes for scalable paramagnetic microparticle-dependent manipulation. Mol. Ther. 13, 814-822.
Ni,Y., Sun,S., Oparaocha,I., Humeau,L., Davis,B., Cohen,R., Binder,G., Chang,Y.N., Slepushkin,V., and Dropulic,B. (2005). Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J. Gene Med. 7, 818-834.
Niidome,T. and Huang,L. (2002). Gene therapy progress and prospects: nonviral vectors. Gene Ther. 9, 1647-1652. Nordlund,H.R., Hytonen,V.P., Laitinen,O.H., Uotila,S.T., Niskanen,E.A., Savolainen,J., Porkka,E., and Kulomaa,M.S.
(2003). Introduction of histidine residues into avidin subunit interfaces allows pH-dependent regulation of quaternary structure and biotin binding. FEBS Lett. 555, 449-454.
O'reilly,D.R., Miller,L.K., and Luckov,V.A. (2004). Baculovirus expression vectors. A laboratory Manual. New York: Oxford University Press).
Ojala,K., Koski,J., Ernst,W., Grabherr,R., Jones,I., and Oker-Blom,C. (2004). Improved display of synthetic IgG-binding domains on the baculovirus surface. Technol. Cancer Res. Treat. 3, 77-84.
Ong,W.Y. and Halliwell,B. (2004). Iron, atherosclerosis, and neurodegeneration: a key role for cholesterol in promoting iron-dependent oxidative damage?. Ann. N. Y. Acad. Sci. 1012, 51-64.
Orino,K., Lehman,L., Tsuji,Y., Ayaki,H., Torti,S.V., and Torti,F.M. (2001). Ferritin and the response to oxidative stress. Biochem. J. 357, 241-247.
Ory,D.S., Neugeboren,B.A., and Mulligan,R.C. (1996). A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U. S. A 93, 11400-11406.
Pacchia,A.L., Adelson,M.E., Kaul,M., Ron,Y., and Dougherty,J.P. (2001). An inducible packaging cell system for safe, efficient lentiviral vector production in the absence of HIV-1 accessory proteins. Virology 282, 77-86.
Paganelli,G., Belloni,C., Magnani,P., Zito,F., Pasini,A., Sassi,I., Meroni,M., Mariani,M., Vignali,M., Siccardi,A.G., and . (1992). Two-step tumour targetting in ovarian cancer patients using biotinylated monoclonal antibodies and radioactive streptavidin. Eur. J. Nucl. Med. 19, 322-329.
Paganelli,G., Grana,C., Chinol,M., Cremonesi,M., De,C.C., De,B.F., Robertson,C., Zurrida,S., Casadio,C., Zoboli,S., Siccardi,A.G., and Veronesi,U. (1999). Antibody-guided three-step therapy for high grade glioma with yttrium-90 biotin. Eur. J. Nucl. Med. 26, 348-357.
Paganelli,G., Pervez,S., Siccardi,A.G., Rowlinson,G., Deleide,G., Chiolerio,F., Malcovati,M., Scassellati,G.A., and Epenetos,A.A. (1990). Intraperitoneal radio-localization of tumors pre-targeted by biotinylated monoclonal antibodies. Int. J. Cancer 45, 1184-1189.
Paganelli,G., Riva,P., Deleide,G., Clivio,A., Chiolerio,F., Scassellati,G.A., Malcovati,M., and Siccardi,A.G. (1988). In vivo labelling of biotinylated monoclonal antibodies by radioactive avidin: a strategy to increase tumor radiolocalization. Int. J. Cancer Suppl 2, 121-125.
Paganelli,G., Stella,M., De,N.P., Magnani,P., Zito,F., Siccardi,A.G., Di,C., V, and Fazio,F. (1991). A new method for faster blood clearance in radioimmuno-guided surgery. J. Nucl. Biol. Med. 35, 88-89.
Pardridge,W.M. (2002). Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 1, 131-139.
Park,F., Ohashi,K., and Kay,M.A. (2000). Therapeutic levels of human factor VIII and IX using HIV-1-based lentiviral vectors in mouse liver. Blood 96, 1173-1176.
Parrott,M.B., Adams,K.E., Mercier,G.T., Mok,H., Campos,S.K., and Barry,M.A. (2003). Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol. Ther. 8, 688-700.
Parrott,M.B. and Barry,M.A. (2000). Metabolic biotinylation of recombinant proteins in mammalian cells and in mice. Mol. Ther. 1, 96-104.
Pellinen,R., Hakkarainen,T., Wahlfors,T., Tulimaki,K., Ketola,A., Tenhunen,A., Salonen,T., and Wahlfors,J. (2004). Cancer cells as targets for lentivirus-mediated gene transfer and gene therapy. Int. J. Oncol. 25, 1753-1762.

94
Peng,K.W., Vile,R., Cosset,F.L., and Russell,S. (1999). Selective transduction of protease-rich tumors by matrix-metalloproteinase-targeted retroviral vectors. Gene Ther. 6, 1552-1557.
Pereboeva,L., Komarova,S., Roth,J., Ponnazhagan,S., and Curiel,D.T. (2007). Targeting EGFR with metabolically biotinylated fiber-mosaic adenovirus. Gene Ther. 14, 627-637.
Petrak,K. (2005). Essential properties of drug-targeting delivery systems. Drug Discov. Today 10, 1667-1673. Pfeifer,A., Ikawa,M., Dayn,Y., and Verma,I.M. (2002). Transgenesis by lentiviral vectors: lack of gene silencing in
mammalian embryonic stem cells and preimplantation embryos. Proc. Natl. Acad. Sci. U. S. A 99, 2140-2145. Pfeifer,A., Kessler,T., Yang,M., Baranov,E., Kootstra,N., Cheresh,D.A., Hoffman,R.M., and Verma,I.M. (2001).
Transduction of liver cells by lentiviral vectors: analysis in living animals by fluorescence imaging. Mol. Ther. 3, 319-322.
Philippe,S., Sarkis,C., Barkats,M., Mammeri,H., Ladroue,C., Petit,C., Mallet,J., and Serguera,C. (2006). Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo 1. Proc. Natl. Acad. Sci. U. S. A 103, 17684-17689.
Philipps,B., Rotmann,D., Wicki,M., Mayr,L.M., and Forstner,M. (2005). Time reduction and process optimization of the baculovirus expression system for more efficient recombinant protein production in insect cells. Protein Expr. Purif. 42, 211-218.
Philpott,N.J. and Thrasher,A.J. (2007). Use of nonintegrating lentiviral vectors for gene therapy. Hum. Gene Ther. 18, 483-489.
Pieroni,L., Maione,D., and La,M.N. (2001). In vivo gene transfer in mouse skeletal muscle mediated by baculovirus vectors. Hum. Gene Ther. 12, 871-881.
Ponnazhagan,S., Mahendra,G., Kumar,S., Thompson,J.A., and Castillas,M., Jr. (2002). Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J. Virol. 76, 12900-12907.
Poomputsa,K., Kittel,C., Egorov,A., Ernst,W., and Grabherr,R. (2003). Generation of recombinant influenza virus using baculovirus delivery vector. J. Virol. Methods 110, 111-114.
Porter,C.D., Collins,M.K., Tailor,C.S., Parkar,M.H., Cosset,F.L., Weiss,R.A., and Takeuchi,Y. (1996). Comparison of efficiency of infection of human gene therapy target cells via four different retroviral receptors. Hum. Gene Ther. 7, 913-919.
Pressman,D. and Korngold,L. (1953). The in vivo localization of anti-Wagner-osteogenic-sarcoma antibodies. Cancer 6, 619-623.
Puhlmann,M., Brown,C.K., Gnant,M., Huang,J., Libutti,S.K., Alexander,H.R., and Bartlett,D.L. (2000). Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 7, 66-73.
Pulkkinen,M., Pikkarainen,J., Wirth,T., Tarvainen,T., Haapa-aho,V., Korhonen,H., Seppala,J., and Jarvinen,K. (2008). Three-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin-biotin technology: Formulation development and in vitro anticancer activity. Eur. J. Pharm. Biopharm. 70, 66-74.
Purow,B. and Staveley-O'Carroll,K. (2005). Targeting of vaccinia virus using biotin-avidin viral coating and biotinylated antibodies. J. Surg. Res. 123, 49-54.
Ramezani,A., Hawley,T.S., and Hawley,R.G. (2003). Performance- and safety-enhanced lentiviral vectors containing the human interferon-beta scaffold attachment region and the chicken beta-globin insulator. Blood 101, 4717-4724.
Raty,J.K., Airenne,K.J., Marttila,A.T., Marjomaki,V., Hytonen,V.P., Lehtolainen,P., Laitinen,O.H., Mahonen,A.J., Kulomaa,M.S., and Yla-Herttuala,S. (2004). Enhanced gene delivery by avidin-displaying baculovirus. Mol. Ther. 9, 282-291.
Raty,J.K., Lesch,H.P., Wirth,T., and Yla-Herttuala,S. (2008). Improving safety of gene therapy. Curr. Drug Saf 3, 46-53. Raty,J.K., Liimatainen,T., Huhtala,T., Kaikkonen,M.U., Airenne,K.J., Hakumaki,J.M., Narvanen,A., and Yla-Herttuala,S.
(2007a). SPECT/CT imaging of baculovirus biodistribution in rat. Gene Ther. 14, 930-938. Raty,J.K., Liimatainen,T., Unelma,K.M., Grohn,O., Airenne,K.J., and Yla-Herttuala,S. (2007b). Non-invasive Imaging in
Gene Therapy. Mol. Ther. 15, 1579-1586. Raty,J.K., Liimatainen,T., Wirth,T., Airenne,K.J., Ihalainen,T.O., Huhtala,T., Hamerlynck,E., Vihinen-Ranta,M.,
Narvanen,A., Yla-Herttuala,S., and Hakumaki,J.M. (2006). Magnetic resonance imaging of viral particle biodistribution in vivo. Gene Ther. 13, 1440-1446.
Reading,S.A. and Dimmock,N.J. (2007). Neutralization of animal virus infectivity by antibody. Arch. Virol. 152, 1047-1059.
Reardon,J.E. (1989). Herpes simplex virus type 1 and human DNA polymerase interactions with 2'-deoxyguanosine 5'-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J. Biol. Chem. 264, 19039-19044.
Recht,L., Torres,C.O., Smith,T.W., Raso,V., and Griffin,T.W. (1990). Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J. Neurosurg. 72, 941-945.

95
Reiser,J. (2000). Production and concentration of pseudotyped HIV-1-based gene transfer vectors. Gene Ther. 7, 910-913. Reiser,J., Harmison,G., Kluepfel-Stahl,S., Brady,R.O., Karlsson,S., and Schubert,M. (1996). Transduction of nondividing
cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl. Acad. Sci. U. S. A 93, 15266-15271. Rheme,C., Ehrengruber,M.U., and Grandgirard,D. (2005). Alphaviral cytotoxicity and its implication in vector
development. Exp. Physiol 90, 45-52. Rissanen,T.T., Korpisalo,P., Markkanen,J.E., Liimatainen,T., Orden,M.R., Kholova,I., de,G.A., Heikura,T., Grohn,O.H.,
and Yla-Herttuala,S. (2005). Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation 112, 3937-3946.
Rodrigues,T., Carrondo,M.J., Alves,P.M., and Cruz,P.E. (2007). Purification of retroviral vectors for clinical application: biological implications and technological challenges. J. Biotechnol. 127, 520-541.
Roizman,B. and Pellet,P.E. (2001). The Family Herpesviridae: A brief Introduction. In Fields Virology, D.M.Knipe, P.M.Howley, D.E.Griffin, R.A.Lamb, M.A.Martin, B.Roizman, and S.E.Straus, eds. (Philadelphia, PA, USA: Lippincott Williams & Wilkins), pp. 2381-2397.
Rome,C., Couillaud,F., and Moonen,C.T. (2007). Gene expression and gene therapy imaging. Eur. Radiol. 17, 305-319. Rosebrough,S.F. (1993). Pharmacokinetics and biodistribution of radiolabeled avidin, streptavidin and biotin. Nucl. Med.
Biol. 20, 663-668. Roth,J.C., Curiel,D.T., and Pereboeva,L. (2008). Cell vehicle targeting strategies. Gene Ther. 15, 716-729. Roux,P., Jeanteur,P., and Piechaczyk,M. (1989). A versatile and potentially general approach to the targeting of specific
cell types by retroviruses: application to the infection of human cells by means of major histocompatibility complex class I and class II antigens by mouse ecotropic murine leukemia virus-derived viruses. Proc. Natl. Acad. Sci. U. S. A 86, 9079-9083.
Ruoslahti,E. (2002). Drug targeting to specific vascular sites. Drug Discov. Today 7, 1138-1143. Russell,S.J. and Cosset,F.L. (1999). Modifying the host range properties of retroviral vectors. J. Gene Med. 1, 300-311. Russell,S.J., Hawkins,R.E., and Winter,G. (1993). Retroviral vectors displaying functional antibody fragments. Nucleic
Acids Res. 21, 1081-1085. Rutanen,J., Rissanen,T.T., Markkanen,J.E., Gruchala,M., Silvennoinen,P., Kivela,A., Hedman,A., Hedman,M.,
Heikura,T., Orden,M.R., Stacker,S.A., Achen,M.G., Hartikainen,J., and Yla-Herttuala,S. (2004). Adenoviral catheter-mediated intramyocardial gene transfer using the mature form of vascular endothelial growth factor-D induces transmural angiogenesis in porcine heart. Circulation 109, 1029-1035.
Samaniego,L.A., Neiderhiser,L., and DeLuca,N.A. (1998). Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72, 3307-3320.
Sandmair,A.M., Turunen,M., Tyynela,K., Loimas,S., Vainio,P., Vanninen,R., Vapalahti,M., Bjerkvig,R., Janne,J., and Yla-Herttuala,S. (2000). Herpes simplex virus thymidine kinase gene therapy in experimental rat BT4C glioma model: effect of the percentage of thymidine kinase-positive glioma cells on treatment effect, survival time, and tissue reactions. Cancer Gene Ther. 7, 413-421.
Sandrin,V., Boson,B., Salmon,P., Gay,W., Negre,D., Le,G.R., Trono,D., and Cosset,F.L. (2002). Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 100, 823-832.
Sano,K., Maeda,K., Oki,M., and Maeda,Y. (2002). Enhancement of protein expression in insect cells by a lobster tropomyosin cDNA leader sequence. FEBS Lett. 532, 143-146.
Santoni de Sio,F.R., Cascio,P., Zingale,A., Gasparini,M., and Naldini,L. (2006). Proteasome activity restricts lentiviral gene transfer into hematopoietic stem cells and is down-regulated by cytokines that enhance transduction. Blood 107, 4257-4265.
Sarkis,C., Serguera,C., Petres,S., Buchet,D., Ridet,J.L., Edelman,L., and Mallet,J. (2000). Efficient transduction of neural cells in vitro and in vivo by a baculovirus-derived vector. Proc. Natl. Acad. Sci. U. S. A 97, 14638-14643.
Sastry,L., Johnson,T., Hobson,M.J., Smucker,B., and Cornetta,K. (2002). Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther. 9, 1155-1162.
Sastry,L., Xu,Y., Cooper,R., Pollok,K., and Cornetta,K. (2004). Evaluation of plasmid DNA removal from lentiviral vectors by benzonase treatment. Hum. Gene Ther. 15, 221-226.
Sastry,L., Xu,Y., Duffy,L., Koop,S., Jasti,A., Roehl,H., Jolly,D., and Cornetta,K. (2005). Product-enhanced reverse transcriptase assay for replication-competent retrovirus and lentivirus detection. Hum. Gene Ther. 16, 1227-1236.
Sastry,L., Xu,Y., Johnson,T., Desai,K., Rissing,D., Marsh,J., and Cornetta,K. (2003). Certification assays for HIV-1-based vectors: frequent passage of gag sequences without evidence of replication-competent viruses. Mol. Ther. 8, 830-839.

96
Schauber,C.A., Tuerk,M.J., Pacheco,C.D., Escarpe,P.A., and Veres,G. (2004). Lentiviral vectors pseudotyped with baculovirus gp64 efficiently transduce mouse cells in vivo and show tropism restriction against hematopoietic cell types in vitro. Gene Ther. 11, 266-275.
Schauber-Plewa,C., Simmons,A., Tuerk,M.J., Pacheco,C.D., and Veres,G. (2005). Complement regulatory proteins are incorporated into lentiviral vectors and protect particles against complement inactivation. Gene Ther. 12, 238-245.
Schechter,B., Silberman,R., Arnon,R., and Wilchek,M. (1990). Tissue distribution of avidin and streptavidin injected to mice. Effect of avidin carbohydrate, streptavidin truncation and exogenous biotin. Eur. J. Biochem. 189, 327-331.
Schellingerhout,D., Bogdanov,A., Jr., Marecos,E., Spear,M., Breakefield,X., and Weissleder,R. (1998). Mapping the in vivo distribution of herpes simplex virions. Hum. Gene Ther. 9, 1543-1549.
Schellingerhout,D., Rainov,N.G., Breakefield,X.O., and Weissleder,R. (2000). Quantitation of HSV mass distribution in a rodent brain tumor model. Gene Ther. 7, 1648-1655.
Schepelmann,S. and Springer,C.J. (2006). Viral vectors for gene-directed enzyme prodrug therapy. Curr. Gene Ther. 6, 647-670.
Scherr,M., Battmer,K., Eder,M., Schule,S., Hohenberg,H., Ganser,A., Grez,M., and Blomer,U. (2002). Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther. 9, 1708-1714.
Schmidt,T.G. and Skerra,A. (2007). The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528-1535.
Schmidt-Wolf,G.D. and Schmidt-Wolf,I.G. (2003). Non-viral and hybrid vectors in human gene therapy: an update. Trends Mol. Med. 9, 67-72.
Schoofs,G., Monica,T.J., Ayala,J., Horwitz,J., Montgomery,T., Roth,G., and Castillo,F.J. (1998). A high-yielding serum-free, suspension cell culture process to manufacture recombinant adenoviral vectors for gene therapy. Cytotechnology 28, 81-89.
Schwarzenberger,P., Spence,S.E., Gooya,J.M., Michiel,D., Curiel,D.T., Ruscetti,F.W., and Keller,J.R. (1996). Targeted gene transfer to human hematopoietic progenitor cell lines through the c-kit receptor. Blood 87, 472-478.
Scott,M.J., Modha,S.S., Rhodes,A.D., Broadway,N.M., Hardwicke,P.I., Zhao,H.J., Kennedy-Wilson,K.M., Sweitzer,S.M., and Martin,S.L. (2007). Efficient expression of secreted proteases via recombinant BacMam virus. Protein Expr. Purif. 52, 104-116.
Seelamgari,A., Maddukuri,A., Berro,R., de la,F.C., Kehn,K., Deng,L., Dadgar,S., Bottazzi,M.E., Ghedin,E., Pumfery,A., and Kashanchi,F. (2004). Role of viral regulatory and accessory proteins in HIV-1 replication. Front Biosci. 9, 2388-2413.
Segall,H.I., Yoo,E., and Sutton,R.E. (2003). Characterization and detection of artificial replication-competent lentivirus of altered host range. Mol. Ther. 8, 118-129.
Segura,M.M., Garnier,A., Durocher,Y., Coelho,H., and Kamen,A. (2007). Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 98, 789-799.
Segura,M.M., Kamen,A., and Garnier,A. (2006). Downstream processing of oncoretroviral and lentiviral gene therapy vectors. Biotechnol. Adv. 24, 321-337.
Sena-Esteves,M., Tebbets,J.C., Steffens,S., Crombleholme,T., and Flake,A.W. (2004). Optimized large-scale production of high titer lentivirus vector pseudotypes. J. Virol. Methods 122, 131-139.
Sergeeva,A., Kolonin,M.G., Molldrem,J.J., Pasqualini,R., and Arap,W. (2006). Display technologies: application for the discovery of drug and gene delivery agents. Adv. Drug Deliv. Rev. 58, 1622-1654.
Sharkey,R.M. and Goldenberg,D.M. (2005). Perspectives on cancer therapy with radiolabeled monoclonal antibodies. J. Nucl. Med. 46 Suppl 1, 115S-127S.
Shen,C.F., Meghrous,J., and Kamen,A. (2002). Quantitation of baculovirus particles by flow cytometry. J. Virol. Methods 105, 321-330.
Shen,S., Forero,A., LoBuglio,A.F., Breitz,H., Khazaeli,M.B., Fisher,D.R., Wang,W., and Meredith,R.F. (2005). Patient-specific dosimetry of pretargeted radioimmunotherapy using CC49 fusion protein in patients with gastrointestinal malignancies. J. Nucl. Med. 46, 642-651.
Shenk.T.E. (2001). Adenoviridae: The Viruses and Their Replication. In Fields Virology, D.M.Knipe, P.M.Howley, D.E.Griffin, R.A.Lamb, M.A.Martin, B.Roizman, and S.E.Straus, eds. (Philadelphia, PA, USA: Lippincott Williams & Wilkins), pp. 2265-2300.
Shoup,T.M., Fischman,A.J., Jaywook,S., Babich,J.W., Strauss,H.W., and Elmaleh,D.R. (1994). Synthesis of fluorine-18-labeled biotin derivatives: biodistribution and infection localization. J. Nucl. Med. 35, 1685-1690.
Sinn,P.L., Goreham-Voss,J.D., Arias,A.C., Hickey,M.A., Maury,W., Chikkanna-Gowda,C.P., and McCray,P.B., Jr. (2007). Enhanced gene expression conferred by stepwise modification of a nonprimate lentiviral vector. Hum. Gene Ther. 18, 1244-1252.

97
Sinn,P.L., Sauter,S.L., and McCray,P.B., Jr. (2005). Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors--design, biosafety, and production. Gene Ther. 12, 1089-1098.
Skerra,A. and Schmidt,T.G. (1999). Applications of a peptide ligand for streptavidin: the Strep-tag. Biomol. Eng 16, 79-86.
Slepushkin,V., Chang,N., Cohen,R., Gan,Y., Jiang,B., Deausen,D., Berlinger,D., Binder,G., Andre,K., Humeau,L., and Dropulic,B. (2003). Large-scale Purification of a Lentiviral Vector by Size Exclusion Chromatography or Mustang Q Ion Exchange Capsule. BioProcessing Journal 2, 89-95.
Smith,G.E., Summers,M.D., and Fraser,M.J. (1983). Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell Biol. 3, 2156-2165.
Smith,J.S., Keller,J.R., Lohrey,N.C., McCauslin,C.S., Ortiz,M., Cowan,K., and Spence,S.E. (1999). Redirected infection of directly biotinylated recombinant adenovirus vectors through cell surface receptors and antigens. Proc. Natl. Acad. Sci. U. S. A 96, 8855-8860.
Sollerbrant,K., Elmen,J., Wahlestedt,C., Acker,J., Leblois-Prehaud,H., Latta-Mahieu,M., Yeh,P., and Perricaudet,M. (2001). A novel method using baculovirus-mediated gene transfer for production of recombinant adeno-associated virus vectors. J. Gen. Virol. 82, 2051-2060.
St George,J.A. (2003). Gene therapy progress and prospects: adenoviral vectors. Gene Ther. 10, 1135-1141. Stachler,M.D. and Bartlett,J.S. (2006). Mosaic vectors comprised of modified AAV1 capsid proteins for efficient vector
purification and targeting to vascular endothelial cells. Gene Ther. 13, 926-931. Stayton,P.S., Ding,Z., and Hoffman,A.S. (2004). Smart polymer-streptavidin conjugates. Methods Mol. Biol. 283, 37-43. Stewart,H.J., Leroux-Carlucci,M.A., Sion,C.J., Mitrophanous,K.A., and Radcliffe,P.A. (2009). Development of inducible
EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther.epud ahead of print Stolz,J., Ludwig,A., and Sauer,N. (1998). Bacteriophage lambda surface display of a bacterial biotin acceptor domain
reveals the minimal peptide size required for biotinylation. FEBS Lett. 440, 213-217. Strang,B.L., Ikeda,Y., Cosset,F.L., Collins,M.K., and Takeuchi,Y. (2004). Characterization of HIV-1 vectors with
gammaretrovirus envelope glycoproteins produced from stable packaging cells. Gene Ther. 11, 591-598. Strang,B.L., Takeuchi,Y., Relander,T., Richter,J., Bailey,R., Sanders,D.A., Collins,M.K., and Ikeda,Y. (2005). Human
immunodeficiency virus type 1 vectors with alphavirus envelope glycoproteins produced from stable packaging cells. J. Virol. 79, 1765-1771.
Sumimoto,H. and Kawakami,Y. (2007). Lentiviral vector-mediated RNAi and its use for cancer research. Future. Oncol. 3, 655-664.
Szymczak,A.L., Workman,C.J., Wang,Y., Vignali,K.M., Dilioglou,S., Vanin,E.F., and Vignali,D.A. (2004). Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589-594.
Tai,C.K., Logg,C.R., Park,J.M., Anderson,W.F., Press,M.F., and Kasahara,N. (2003). Antibody-mediated targeting of replication-competent retroviral vectors. Hum. Gene Ther. 14, 789-802.
Tan,W., Dong,Z., Wilkinson,T.A., Barbas,C.F., III, and Chow,S.A. (2006). Human immunodeficiency virus type 1 incorporated with fusion proteins consisting of integrase and the designed polydactyl zinc finger protein E2C can bias integration of viral DNA into a predetermined chromosomal region in human cells. J. Virol. 80, 1939-1948.
Tenenbaum,L., Lehtonen,E., and Monahan,P.E. (2003). Evaluation of risks related to the use of adeno-associated virus-based vectors. Curr. Gene Ther. 3, 545-565.
Tiscornia,G., Singer,O., and Verma,I.M. (2006). Production and purification of lentiviral vectors. Nat. Protoc. 1, 241-245. Tjuvajev,J.G., Avril,N., Oku,T., Sasajima,T., Miyagawa,T., Joshi,R., Safer,M., Beattie,B., DiResta,G., Daghighian,F.,
Augensen,F., Koutcher,J., Zweit,J., Humm,J., Larson,S.M., Finn,R., and Blasberg,R. (1998). Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 58, 4333-4341.
Tolstoshev,P. (1992). Retroviral-mediated gene therapy--safety considerations and preclinical studies. Bone Marrow Transplant. 9 Suppl 1, 148-150.
Transfiguracion,J., Jaalouk,D.E., Ghani,K., Galipeau,J., and Kamen,A. (2003). Size-exclusion chromatography purification of high-titer vesicular stomatitis virus G glycoprotein-pseudotyped retrovectors for cell and gene therapy applications. Hum. Gene Ther. 14, 1139-1153.
Transfiguracion,J., Jorio,H., Meghrous,J., Jacob,D., and Kamen,A. (2007). High yield purification of functional baculovirus vectors by size exclusion chromatography. J. Virol. Methods 142, 21-28.
Trimble,C., Lin,C.T., Hung,C.F., Pai,S., Juang,J., He,L., Gillison,M., Pardoll,D., Wu,L., and Wu,T.C. (2003). Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine 21, 4036-4042.
Turunen,P., Puhakka,H., Rutanen,J., Hiltunen,M.O., Heikura,T., Gruchala,M., and Yla-Herttuala,S. (2005). Intravascular adenovirus-mediated lipoprotein-associated phospholipase A2 gene transfer reduces neointima formation in balloon-denuded rabbit aorta. Atherosclerosis 179, 27-33.

98
Tyynela,K., Sandmair,A.M., Turunen,M., Vanninen,R., Vainio,P., Kauppinen,R., Johansson,R., Vapalahti,M., and Yla-Herttuala,S. (2002). Adenovirus-mediated herpes simplex virus thymidine kinase gene therapy in BT4C rat glioma model. Cancer Gene Ther. 9, 917-924.
Uckert,W., Becker,C., Gladow,M., Klein,D., Kammertoens,T., Pedersen,L., and Blankenstein,T. (2000). Efficient gene transfer into primary human CD8+ T lymphocytes by MuLV-10A1 retrovirus pseudotype. Hum. Gene Ther. 11, 1005-1014.
Urabe,M., Ding,C., and Kotin,R.M. (2002). Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 13, 1935-1943.
Verhoeyen,E. and Cosset,F.L. (2004). Surface-engineering of lentiviral vectors. J. Gene Med. 6 Suppl 1, S83-S94. Verhoeyen,E., Wiznerowicz,M., Olivier,D., Izac,B., Trono,D., Dubart-Kupperschmitt,A., and Cosset,F.L. (2005). Novel
lentiviral vectors displaying "early-acting cytokines" selectively promote survival and transduction of NOD/SCID repopulating human hematopoietic stem cells. Blood 106, 3386-3395.
Vicente,T., Peixoto,C., Carrondo,M.J., and Alves,P.M. (2009). Purification of recombinant baculoviruses for gene therapy using membrane processes. Gene Ther.epud ahead of print
Volpers,C., Thirion,C., Biermann,V., Hussmann,S., Kewes,H., Dunant,P., von der,M.H., Herrmann,A., Kochanek,S., and Lochmuller,H. (2003). Antibody-mediated targeting of an adenovirus vector modified to contain a synthetic immunoglobulin g-binding domain in the capsid. J. Virol. 77, 2093-2104.
Waehler,R., Russell,S.J., and Curiel,D.T. (2007). Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8, 573-587.
Wahlfors,J. and Morgan,R.A. (2002). Minigene-containing retroviral vectors using an alphavirus/retrovirus hybrid vector system. Production and use. Methods Mol. Med. 69, 173-186.
Walker,L., Kulomaa,M.S., Bebok,Z., Parker,W.B., Allan,P., Logan,J., Huang,Z., Reynolds,R.C., King,S., and Sorscher,E.J. (1996). Development of drug targeting based on recombinant expression of the chicken avidin gene. J. Drug Target 4, 41-49.
Watson,D.J., Kobinger,G.P., Passini,M.A., Wilson,J.M., and Wolfe,J.H. (2002). Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol. Ther. 5, 528-537.
Weissleder,R., Moore,A., Mahmood,U., Bhorade,R., Benveniste,H., Chiocca,E.A., and Basilion,J.P. (2000). In vivo magnetic resonance imaging of transgene expression. Nat. Med. 6, 351-355.
Weissleder,R., Simonova,M., Bogdanova,A., Bredow,S., Enochs,W.S., and Bogdanov,A., Jr. (1997). MR imaging and scintigraphy of gene expression through melanin induction. Radiology 204, 425-429.
Wells,D.J. (2004). Gene therapy progress and prospects: electroporation and other physical methods. Gene Ther. 11, 1363-1369.
Wilchek,M., Bayer,E.A., and Livnah,O. (2006). Essentials of biorecognition: the (strept)avidin-biotin system as a model for protein-protein and protein-ligand interaction. Immunol. Lett. 103, 27-32.
Wilson,M.H., Coates,C.J., and George,A.L., Jr. (2007). PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 15, 139-145.
Wiznerowicz,M. and Trono,D. (2005). Harnessing HIV for therapy, basic research and biotechnology. Trends Biotechnol. 23, 42-47.
Wojda,U. and Miller,J.L. (2000). Targeted transfer of polyethylenimine-avidin-DNA bioconjugates to hematopoietic cells using biotinylated monoclonal antibodies. J. Pharm. Sci. 89, 674-681.
Wojnicz,D., Bar,J., and Jankowski,S. (2002). [The role of membrane glycoproteins CD46, CD55 and CD59 in protection of tumor cells against complement lysis]. Postepy Hig. Med. Dosw. 56, 603-616.
Wolfe,D., Goins,W.F., Fink,D.J., Burton,E.A., Krisky,D.M., and Gloriso,J.C. (2004). Engineering Herpes Simplex Viral Vectors for Therapeutics Gene Transfer. In Gene and Cell Therapy, N.S.Templeton, ed. (New York, NY, USA: Marcel Dekker, Inc.), pp. 103-129.
Wu,C., Soh,K.Y., and Wang,S. (2007). Ion-exchange membrane chromatography method for rapid and efficient purification of recombinant baculovirus and baculovirus gp64 protein. Hum. Gene Ther. 18, 665-672.
Wu,J.C., Chen,I.Y., Sundaresan,G., Min,J.J., De,A., Qiao,J.H., Fishbein,M.C., and Gambhir,S.S. (2003a). Molecular imaging of cardiac cell transplantation in living animals using optical bioluminescence and positron emission tomography. Circulation 108, 1302-1305.
Wu,X., Li,Y., Crise,B., and Burgess,S.M. (2003b). Transcription start regions in the human genome are favored targets for MLV integration. Science 300, 1749-1751.
Wu,X., Wakefield,J.K., Liu,H., Xiao,H., Kralovics,R., Prchal,J.T., and Kappes,J.C. (2000). Development of a novel trans-lentiviral vector that affords predictable safety. Mol. Ther. 2, 47-55.
Wu,Z., Asokan,A., and Samulski,R.J. (2006). Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 14, 316-327.

99
Xu,K., Ma,H., McCown,T.J., Verma,I.M., and Kafri,T. (2001). Generation of a stable cell line producing high-titer self-inactivating lentiviral vectors. Mol. Ther. 3, 97-104.
Yamada,K., McCarty,D.M., Madden,V.J., and Walsh,C.E. (2003). Lentivirus vector purification using anion exchange HPLC leads to improved gene transfer. Biotechniques 34, 1074-8, 1080.
Yang,M., Baranov,E., Moossa,A.R., Penman,S., and Hoffman,R.M. (2000). Visualizing gene expression by whole-body fluorescence imaging. Proc. Natl. Acad. Sci. U. S. A 97, 12278-12282.
Yang,Q., Mamounas,M., Yu,G., Kennedy,S., Leaker,B., Merson,J., Wong-Staal,F., Yu,M., and Barber,J.R. (1998). Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum. Gene Ther. 9, 1929-1937.
Yao,Z., Zhang,M., Sakahara,H., Saga,T., Arano,Y., and Konishi,J. (1998). Avidin targeting of intraperitoneal tumor xenografts. J. Natl. Cancer Inst. 90, 25-29.
Yokoyama,N., Hirata,M., Ohtsuka,K., Nishiyama,Y., Fujii,K., Fujita,M., Kuzushima,K., Kiyono,T., and Tsurumi,T. (2000). Co-expression of human chaperone Hsp70 and Hsdj or Hsp40 co-factor increases solubility of overexpressed target proteins in insect cells. Biochim. Biophys. Acta 1493, 119-124.
Yu,X., Zhan,X., D'Costa,J., Tanavde,V.M., Ye,Z., Peng,T., Malehorn,M.T., Yang,X., Civin,C.I., and Cheng,L. (2003). Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol. Ther. 7, 827-838.
Yun,C.O., Cho,E.A., Song,J.J., Kang,D.B., Kim,E., Sohn,J.H., and Kim,J.H. (2003). dl-VSVG-LacZ, a vesicular stomatitis virus glycoprotein epitope-incorporated adenovirus, exhibits marked enhancement in gene transduction efficiency. Hum. Gene Ther. 14, 1643-1652.
Zhao,Y., Zhu,L., Lee,S., Li,L., Chang,E., Soong,N.W., Douer,D., and Anderson,W.F. (1999). Identification of the block in targeted retroviral-mediated gene transfer. Proc. Natl. Acad. Sci. U. S. A 96, 4005-4010.
Zinn,K.R., Douglas,J.T., Smyth,C.A., Liu,H.G., Wu,Q., Krasnykh,V.N., Mountz,J.D., Curiel,D.T., and Mountz,J.M. (1998). Imaging and tissue biodistribution of 99mTc-labeled adenovirus knob (serotype 5). Gene Ther. 5, 798-808.
Zufferey,R., Donello,J.E., Trono,D., and Hope,T.J. (1999). Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 73, 2886-2892.
Zufferey,R., Dull,T., Mandel,R.J., Bukovsky,A., Quiroz,D., Naldini,L., and Trono,D. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72, 9873-9880.
Zufferey,R., Nagy,D., Mandel,R.J., Naldini,L., and Trono,D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15, 871-875.


Original publications
I-IV

Kuopio University Publications G. - A.I.Virtanen Institute G 51. Keinänen, Riitta et al. (eds.). The first annual post-graduate symposium of the graduate school of molecular medicine: winter school 2007. 2007. 65 p. Abstracts. G 52. Vartiainen, Suvi. Caenorhabditis elegans as a model for human synucleopathies. 2007. 94 p. Acad. Diss. G 53. Määttä, Ann-Marie. Development of gene and virotherapy against non-small cell lung cancer. 2007. 75 p. Acad. Diss. G 54. Rautsi, Outi. Hurdles and Improvements in Therapeutic Gene Transfer for Cancer. 2007. 79 p. Acad. Diss. G 55. Pehkonen, Petri. Methods for mining data from genome wide high-throughput technologies. 2007. 91 p. Acad. Diss. G 56. Hyvönen, Mervi T. Regulation of spermidine/spermine N'-acetyltransferase and its involvement in cellular proliferation and development of acute pancreatitis. 2007. 79 p. Acad. Diss. G 57. Gurevicius, Kestutis. EEG and evoked potentials as indicators of interneuron pathology in mouse models of neurological diseases. 2007. 76 p. Acad. Diss. G 58. Leppänen, Pia. Mouse models of atherosclerosis, vascular endothelial growth factors and gene therapy. 2007. 91 p. Acad. Diss. G 59. Keinänen, Riitta et al. The second annual post-graduate symposium of the graduate school of molecular medicine: winter school 2008. 2008. 57 p. Abstracts. G 60. Koponen, Jonna. Lentiviral vector for gene transfer: a versatile tool for regulated gene expression, gene silencing and progenitor cell therapies. 2008. 71 p. Acad. Diss. G 61. Ahtoniemi, Toni. Mutant Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis: molecular mechanisms of neurotoxicity. 2008. 107 p. Acad. Diss. G 62. Purhonen, Anna-Kaisa. Signals arising from the gastointestinal tract that affect food intake. 2008. 86 p. Acad. Diss. G 63. Kaikkonen, Minna. Engineering baculo- and lentiviral vectors for enhanced and targeted gene delivery. 2008. 109 p. Acad. Diss. G 64. Gureviciene, Irina. Changes in hippocampal synaptic plasticity in animal models of age-related memory impairment. 2 2008. 106 p. Acad. Diss. G 65. Oikari, Sanna. Evaluation of phenotypic changes of Acyl-CoA binding protein / diazepam binding inhibitor overexpression in transgenic mice and rats. 2008. 79 p. Acad. Diss. G 66. Laurema, Anniina. Adenoviral gene therapy and fertility: distribution studies in reproductive organs and risk of vertical transmission in female rabbits and rats. 2008. 79 p. Acad. Diss.


















