Kinetics and irreversibility of cesium and uranium ... Alamos National Labs/General... · Kinetics...
14
ELSEVIER Available online at www.sclen<:edirect.com SCIBNCEI @DIRBCTO Applied Clay Science 26 (2004) 137-150 www.elsevier.com/locate/c1ay Kinetics and irreversibility of cesium and uranium sorption onto bentonite colloids in a deep granitic environment Abstract T. Missana *, M. Garda-Gutierrez, U. Alonso CIEMAT-Departamento de Jmpacto Ambiental de Ia Energia, Avenida Complutense. 22, 28040 Madrid, Spain Received 12 March 2003; received in revised form I 2 September 2003; accepted 15 September 2003 Available online 24 March 2004 Sorption of radionuclides onto a stable colloidal phase may significantly enhance their transport in groundwater. A key point, to be analysed to assess the relevance of colloids in the safety of a deep geological radioactive waste repository, is the irreversibility of the colloid/radionuclide bond. In this work, sorption and desorption kinetics of cesium and uranium(VI) onto bentonite colloids in a granitic reduced environment was studied by means of batch experiments, carried out in anoxic conditions under N 2 atmosphere. Sorption kinetics was followed during 18 weeks, and sorption isotherms were also carried out to get additional information on sorption mechanisms. The water used in all the experiments was an alkaline, low ionic strength (pH= 9.5 and I"' I x 10- 3 M) granitic groundwater from the NAGRA's Grimsel Test Site (GTS), Switzerland, which also presents reduced Eh (- 200 mY). In this water, bentonite colloids were shown to be stable during several months. Both cesium and uranium presented a nonlinear sorption behaviour in the range of concentration investigated. In kinetic experiments, the measured log Kd for Cs ([Cs] = I x 10- 7 M) was 3.94 ± 0.15, and this value did not show significant variations with time. However, the adsorption of cesium on bentonite colloids involves two reactions, a rapid exchange on planar sites (hours) and a slower component (days) in which cesium diffuses to less available but highly selective sites. This slow process, that can be evidenced only when very low tracer concentrations are used (<I x 10- 9 M), is most probably responsible for the fixation of a fraction of the sorbed cesium, and for the partial sorption irreversibility shown in desorption tests. Kd values measured after several desorption experiments increased significantly with the age of the sorption complex. For example, for the sample with 1-day contact time, the second desorption Kd was - 8600 mllg whereas the 5 and 8 weeks contact time samples showed second desorption Kd - 15 000 and - 30 000 mllg, respectively. The measured log Kd for U ([U(VI)] =4 x 10- 7 M) varied from 2.91 to 3.21 ( ± 0.15) during 18 weeks of the kinetic experiment. The main variation of Kd values took place in the first 4 weeks, and then a very slow increasing trend was observed, which could be probably attributed to a partial reduction of U(VI) to U(IV). In desorption tests with uranium, desorption Kds were independent on the initial contact time. Nevertheless, a certain sorption/desorption hysteresis was observed, which is most probably due to the contribution of surface complexation reactions, at the edge sites of clay colloids, to uranium sorption. Hence, U sorption is not completely reversible. © 2004 Elsevier B.V. All rights reserved. Keywords: Bentonite; Cesium; Colloids; Radioa,tive waste; Sorption; Uranium • Corresponding author. Tel.: + 34-913-46-6 I-85; fax:+ 34-913-46-65-42. E-mail address: [email protected] (T. Missana). 0169-1317/$- see front 2004 Elsevier B.V. All rights reserved. doi: 10.10 16/j.clay.2003.09.008 11111111111111111111111111111111111 14563
Transcript of Kinetics and irreversibility of cesium and uranium ... Alamos National Labs/General... · Kinetics...

ELSEVIER
Available online at www.sclen<:edirect.com
SCIBNCEI @DIRBCTO
Applied Clay Science 26 (2004) 137-150 www.elsevier.com/locate/c1ay
Kinetics and irreversibility of cesium and uranium sorption onto bentonite colloids in a deep granitic environment
Abstract
T. Missana *, M. Garda-Gutierrez, U. Alonso
CIEMAT-Departamento de Jmpacto Ambiental de Ia Energia, Avenida Complutense. 22, 28040 Madrid, Spain
Received 12 March 2003; received in revised form I 2 September 2003; accepted 15 September 2003 Available online 24 March 2004
Sorption of radionuclides onto a stable colloidal phase may significantly enhance their transport in groundwater. A key point, to be analysed to assess the relevance of colloids in the safety of a deep geological radioactive waste repository, is the irreversibility of the colloid/radionuclide bond.
In this work, sorption and desorption kinetics of cesium and uranium(VI) onto bentonite colloids in a granitic reduced environment was studied by means of batch experiments, carried out in anoxic conditions under N2 atmosphere. Sorption kinetics was followed during 18 weeks, and sorption isotherms were also carried out to get additional information on sorption mechanisms. The water used in all the experiments was an alkaline, low ionic strength (pH= 9.5 and I"' I x 10- 3M) granitic groundwater from the NAGRA's Grimsel Test Site (GTS), Switzerland, which also presents reduced Eh (- 200 mY). In this water, bentonite colloids were shown to be stable during several months.
Both cesium and uranium presented a nonlinear sorption behaviour in the range of concentration investigated. In kinetic experiments, the measured log Kd for Cs ([Cs] = I x 10- 7 M) was 3.94 ± 0.15, and this value did not show significant variations with time. However, the adsorption of cesium on bentonite colloids involves two reactions, a rapid exchange on planar sites (hours) and a slower component (days) in which cesium diffuses to less available but highly selective sites. This slow process, that can be evidenced only when very low tracer concentrations are used (<I x 10- 9 M), is most probably responsible for the fixation of a fraction of the sorbed cesium, and for the partial sorption irreversibility shown in desorption tests. Kd values measured after several desorption experiments increased significantly with the age of the sorption complex. For example, for the sample with 1-day contact time, the second desorption Kd was - 8600 mllg whereas the 5 and 8 weeks contact time samples showed second desorption Kd - 15 000 and - 30 000 mllg, respectively.
The measured log Kd for U ([U(VI)] =4 x 10- 7 M) varied from 2.91 to 3.21 ( ± 0.15) during 18 weeks of the kinetic experiment. The main variation of Kd values took place in the first 4 weeks, and then a very slow increasing trend was observed, which could be probably attributed to a partial reduction of U(VI) to U(IV).
In desorption tests with uranium, desorption Kds were independent on the initial contact time. Nevertheless, a certain sorption/desorption hysteresis was observed, which is most probably due to the contribution of surface complexation reactions, at the edge sites of clay colloids, to uranium sorption. Hence, U sorption is not completely reversible. © 2004 Elsevier B.V. All rights reserved.
Keywords: Bentonite; Cesium; Colloids; Radioa,tive waste; Sorption; Uranium
• Corresponding author. Tel.: + 34-913-46-6 I -85; fax:+ 34-913-46-65-42. E-mail address: [email protected] (T. Missana).
0169-1317/$- see front matter~ 2004 Elsevier B.V. All rights reserved. doi: 10.10 16/j.clay.2003.09.008
11111111111111111111111111111111111 14563

138 T. Missana el a/./ Applied Clay Science 26 (2004) 137-150
1. Introduction
One of the options taken into account for isolating high-level radioactive waste from the biosphere is the deep geological disposal (DGP) in a granite host rock. This kind of disposal consists of a geological barrier (host rock) and engineering barriers that aim to contain the radionuclides until they have decayed below harmful levels. The waste is generally contained in steel canisters surrounded by a layer of compacted clay such as bentonite. Bentonite has a very low permeability, which provides a hydrological barrier and good swelling properties and high plasticity that allow sealing rock fractures. In addition, bentonite has a very high sorption capability for most radionuclides.
Within the compacted bentonite (near field), the transport of radionuclides is expected to be a diffusion-controlled process retarded by sorption. The transport of radionuclides in the host rock formation (far field) is modelled considering advection in fractures, rock matrix diffusion and including sorption on the rocks or fault gouge materials. However, it has been shown recently that colloidal particles could be detached from the bentonite at the clay/granite interface (Missana et al., 1999) so that bentonite colloidmediated transport might affect radionuclide migration, above all for those radionuclides that present high sorption onto the clay.
"Colloids" are solid particles suspended in a fluid with a size ranging between I nm and 1 J,tm. The existence of colloidal species in the aqueous phases may affect the radionuclide migration because colloids can represent a mobile or immobile phase depending on their nature, size and the chemistry of the water in which they are suspended (Kim. 1994; Kim et al., 1987; McCarthy and Zachara, 1989; Lieser et al., 1990; Ryan and Elimelech, 1996; Degueldre et al., 2000).
Colloids will be important for radionuclide transport if the following conditions are fulfilled: that colloids exist in a nonnegligible concentration, they are mobile and stable in the environment of interest, and they are able to adsorb radionuclides irreversibly. If one of the above mentioned conditions is not fulfilled, their role might be negligible (Miller et al., 1994).
Recently, it has been shown that the large distances travelled by actinides in the Nevada nuclear tests areas
could be attributed to colloid-driven transport (Buddemaier and Hunt, 1988; Penrose et al., 1990; Nuttal and Long, 1993; Kersting et a!.. 1999). Therefore, in the frame of the performance assessment of a DGP, it is necessary to carry out different studies to analyse the properties of colloids that could be generated from all the subsystem of the repository, especially bentonite colloids.
The possible mechanisms of bentonite colloid generation from the surface of the bentonite backfill were discussed in a separate work where these colloids were also shown to be stable over long periods in low ionic strength and alkaline waters, as the Grimsel groundwater (Missana et al., 2003a).
At present, the effects of bentonite colloids on the migration of DGP-relevant actinides and fission products in granite fractures are being studied, in the framework of an international project (Colloids and Radionuclide Retardation [CRR] Project, Mori ct al., 2003). In this project, several in situ migration experiments with colloids and/or radionuclides were performed, in a granitic shear zone at the NAGRA's Grimsel Test Site (GTS, Switzerland), to evaluate their mobility.
FEBEX bentonite colloids have a mean size of 250 ± 50 nm. In the above mentioned migration experiments, the partial filtration of larger colloids was observed, but the smallest clay particles ( < 100 nm) were shown to be able to move unretarded along with the fracture water flow (Hauser ct al., 2002). This result showed that, in certain conditions, bentonite colloids could effectively act as carriers for sorbed radionuclides. However, the long-range colloid-mediated transport is very dependent on the irreversibility of the sorption process. Ifradionuclides readily desorb when chemical changes occur along the colloids flow path, they would have very little or no effect at all on radionuclide migration.
In a general way, the reversibility of contaminant uptake and the kinetics of sorption/desorption processes are very important issues for the understanding of contaminant transport in groundwater. Regrettably, the information on the irreversibility of sorption processes onto geological materials is scarce. Moreover, the uncertainties related to radionuclide/colloid bond irreversibility are key problems to be solved to assess the actual role of colloids in the radionuclide transport.

T. M•ssana eta/. I Applied Clay Science ]6 (1004) 137-150 139
The aim of this work is to study the uptake mechanisms of cesium and uranium onto bentonite colloids in a deep granitic environment (Grimsel water), focusing on the kinetics of the sorption/desorption processes, to evaluate the extent of sorption irreversibility. This work was carried out in the framework of the CRR project, within the experimental laboratory programme developed as support of the in situ studies. The considered scenario is the migration of colloids in a granite fracture, where the change of chemical conditions is represented by the influx of "fresh" Grimsel water.
Studies performed at a real scale and in natural environments are crucial for a better understanding of the colloid-mediated transport and to decide if it is necessary to include the colloid contribution to the performance assessment calculations. Nevertheless, in situ studies are much more complicated to carry out than laboratory experiments and, often, they are affected by severe experimental constraints. With these constraints, it may be difficult to interpret correctly all the processes involved in colloid migration and, particularly, to obtain information about the kinetics and the irreversibility of these processes. The results of the present work can be useful for the interpretation of the in situ migration experiments of radionuclides in the presence of bentonite colloids.
2. Experimental
2.1. Materials
The water used in all the experiments was the groundwater from the Grimsel Test Site (GTS, Switzerland). The water was taken at the GTS from the borehole BOMI 87.008 in a 50-1 aluminium barrel provided by FZK-INE (Germany). Container walls were coated with PTFE in order to avoid extensive sorption of trace elements. The chemical characteristics of the Grimsel water are summarised in Table 1. Eh (- 200 ±50 mV) and pH (9.5 ± 0.2) were measured during the sampling and afterwards in the laboratory.
The bentonite used in sorption tests (FEBEX bentonite) comes from the Cortijo de Archidona deposit (Almeria, Spain). This clay has a high smectite content (93 ± 2%), with quartz (2 ± 1 %), plagio-
Table I Chemical composition and main characteristics of the Grimsel water
Ion/element Concentration
Na•
Ca2
'
Mg2• K'" s?• u• Cls<Y.F-1-
Po!Fe AI Alkalinity as (CaC03)
Conductivity pH Eh
16.0-16.5 ppm 5.6-6.6 ppm <0.4 ppm 0.07-0.18 ppm 199-210 ppb 0.08 ppm 5.5-7.7 ppm 6.1-6.3 ppm 6.4-6.5 ppm 50.02 ppm <0.1 ppm <30 ppb 0.10-0.14 ppm 19.5-23.6 ppm 93-103 f.IS/cm 9.5 ± 0.2 -200±50mV
clase (3 ± 1 %), cristo halite (2 ± 1 %), potassic feldspar, calcite and tridymite as accessory minerals. Even if illite was never detected as a "pure" phase by XRD analysis, mineralogical studies showed that the smectite phase is actually made up by illite-smectite mixed layer with a 10-15% of illite layers {Cuadros and Linares, 1996; Huertas et al., 2000). Further details on this clay can be found elsewhere (Huertas eta!., 2000).
The structural formula of the FEBEX bentonite, after homo ionisation with Ca of the fraction less than 2 j.lm (which contains more than 99% smectite), is the following (Fernandez ct al., in press):
(Si7.7sAlo.22)(Al2.1sFe6j3Fe~t2 Tio.o2Mg0_81 )Ozo
x (OH)4 (C3{).soNao.osKo.u ).
The interlayer charge is + 1.19, the charge of the tetrahedral sheet is - 0.22 and that of the octahedral sheet - 0.97, which indicates the montmorillonitic character of this smectite.
The cation exchange capacity (CEC) of the FEB EX clay is 102 ± 4 meq/1 00 g (Huertas ct al., 2000).
Previously to the preparation of bentonite colloids, dry "natural" bentonite (crushed and sieved at size fraction< 64 11m) was repeatedly washed with Milli-Q deionised water. The last washing step was performed with Grimsel water, allowing the equilibration of the clay with this aqueous phase. After the last washing,

!40 T Miss ana et at. I Applied Clay Science 26 (2004) 13 7-150
the excess of water was eliminated and the solid was dried in a special container, by flushing N2, and then contacted again with Grimsel water, under N2 atmosphere, at a solid to liquid ratio of approximately 1:10. Still in the glove box, the suspension was then transferred to centrifuge tubes (that were sealed) and, after a day, it was centrifuged (5369 x g, 10 min). The supernatant, which contained the bentonite colloidal fraction conditioned to the Grimsel water, was separated from the solid in an anoxic glove box, where it was finally stored. The pH of the suspension ( ± 0.2) and the conductivity ( ± 20 fJS) were the same as those of the Grimsel water (Table I). The concentration of the bentonite colloids was determined by gravimetry, drying three 50 ml aliquots of the suspension in an oven at 105 oc for 2 days and weighting the remaining solid. The concentration of the colloidal suspension typically ranged between I and 2 g/1.
The colloidal phases were characterised by X-ray diffraction (XRD), Photon correlation spectrometry (PCS) and transmission electron microscopy (TEM) techniques. TEM samples were obtained by depositing a drop of a diluted bentonite colloid suspension onto a carbon-coated Cu grid and allowing it to dry in air. More details on the characterisation of bentonite colloids can be found elsewhere (Missana et al., 2003b).
The XRD analysis of the FEBEX bentonite colloids, previously homoionised with sodium, is consistent with an aluminium-rich dioctahedral smectite (Newman, 1987), and no other mineral phases have been found. However, the existence of illite-smectite mixed layer, as observed in the starting material, is expected.
PCS experiments were performed with a 4700 Malvern apparatus equipped with a Spectra Physics 4 W argon laser (A.=514 nm). The photomultiplier was set to a scattering angle of90° and the signal was transmitted to a Malvern 7032 digital autocorrelator.
The average diffusion coefficient (D) of colloids was determined using the cumulant methods and the mean hydrodynamic radius of the particles (r) was calculated using the Stokes- Einstein equation:
kT D=--
6nl1r
where k is the Boltzmann constant, T the absolute temperature and 11 the viscosity of the fluid. Further multimodal analyses of the PCS spectra were made, in order to determine the actual size distribution and polydispersivity. This study is based on the deconvolution of the correlograms obtained in the PCS experiment using a noncommercial Pascal compiled code (Caceci, 19R8), based on the calculation of the best nonnegatively constrained Laplace transform of the acquired data (Dahnckc, 1983). Further details on the PCS technique can be found in Holthofl' ct al. ( 1996).
2.2. Sorption/desorption experiments
Sorption and desorption experiments were carried out as batch experiments in an anoxic glove box under N 2 atmosphere (02 < 1 ppm). All the experimental procedures were carried out in the glove box with the unique exception of centrifugation. However, the perfect sealing of centrifuge tubes prevented air contamination.
The radionuclides used for the experiments were 137Cs(D and 233U(VI). The tracers used were cesium chloride and uranyl nitrate.
2.2.1. Stability of uranium in Grimsel water Uranium is a redox-sensitive element and Grimsel
groundwater has a reduced Eh, therefore, before the sorption experiments, we checked uranium stability in the conditions and time scales ofthe sorption tests. The possible uranium precipitation or colloid formation, which would indicate the reduction of the U(VI) to U(N), was therefore experimentally analysed. To do so, Grimsel water free ofbentonite colloids was spiked with U(VI) (4 x 10- 7 M) and periodically sampled. The sampling was carried out every week. The samples were centrifuged (645 000 x g, 30 min) and the activity in the upper liquid measured. According to Stoke's law, this centrifugation ensured the sedimentation of particles larger than 1 nm. A decrease of the activity with time would have been an indication of precipitation or colloid formation and therefore of the reduction of the element. After approximately 3 months, no evidence of uranium precipitation was observed.
These experimental results are not, apparently, in agreement with thermodynamic calculations ofU(VI) stability in Grimsel water that were carried out in the frame of the CRR project (Bruno et al., 2000). These
------------~-------· .. ----------

T. Missana eta/. I Applied Clay Science 26 (2004) 137-150 141
calculations showed that at pH 9.5, in the Eh range from+ 400 to - 1 00 m V, the predominant aqueous uranium aqueous species is U02(0H)3. In more strongly reducing conditions like those of Grimsel water (- 200 mV), the major thennodynamically predicted species is U(OHk The calculated solubility of uranium in Grimsel water was I x 1 o- 8 M.
The reason of this apparent contradiction is probably that the uranium reduction in Grimsel water is very slow, much slower than the time scale of our experiments (few months). This fact has to be taken into account to interpret correctly in situ and laboratory experiments.
2.2.2. S01ption experiments The kinetics of the sorption and sorption isothenns
were investigated. The radionuclide concentration used in the kinetic experiments was 1.1 x 1 o- 7 and 4.0 x 10- 7 M for cesium and uranium, respectively. The kinetic experiments lasted from 1 to 18 weeks. For the isotherm study, the radionuclide concentration varied from approximately 1 x 10- 10 to I x 10- 4 M, in the case of cesium, and from approximately 10- 7
to I o- 4 M in the case of uranium. The contact times were 15 days for cesium and I day, 15 days and 3.5 months for uranium.
In all the sorption experiments, three aliquots of the suspension (1-2 g/1) were introduced in 12.4-ml polyallomere ultracentrifuge tubes, maintained in continuous stirring for the selected contact time, then centrifuged (645000 x g, 30 min). Two aliquots of the supernatant from each tube were extracted for the analysis of their activity. The cesium activity in the supernatant was measured with a Nal -y-detector (Cobra, Camberra), whereas the uranium activity was measured by liquid scintillation with a TR-2700 Packard counter. The errors in the activity measurements for cesium and uranium are lower than 1%.
The distribution coefficients measured by sorption methods will be referred to as "sorption Kd" (Kd,.0 ,).
The sorption distribution coefficients "Kd,0 ," were calculated, as usual, by the following fonnula:
Kdsor = C; - Cr . V = Cads ~ Cr m Cr m
(E. I)
where C; is the initial activity, Cris the final activity in the liquid, Cads=(C;-Cr) is the activity in the solid
phase, m is the mass of the solid and V is the liquid volume.
2.2.3. Desorption experiments A bentonite colloid suspension (2 g/1) was spiked
with uranium (1 x 10- 7 M) or cesium (9 x 10- 8 M). In order to study the kinetics of the sorption/desorption behaviour, two aliquots were extracted and centrifuged after the selected contact time (I day, l, 2, 5 or 8 weeks). Because one of the main difficult points in desorption experiments with clays is the resuspension of the solid that remains "compacted" at the bottom of the tube, in these experiments, the samples were centrifuged at a lower rate (26 326 x g, 20 min) than that used in the sorption experiments described in the Section 2.2.2. This centrifugation ensured the sedimentation of particles larger than 5 nm.
After centrifugation, the closed tubes were transferred again into the anoxic glove box, where the solid was separated from the liquid. The supernatant was used for counting and the determination of the sorption Kd (Kd,0 r). After that, the solid was contacted again with fresh Grimsel water, maintaining the initial solid to liquid ratio (2 g/1), and suspended again.
During the "first desorption" step, the clay was maintained in contact with the fresh Grimsel water for the same amount of time used in the sorption step, to allow comparison of sorption and desorption rates. After that time, the solid was separated from the liquid and the activity in the liquid phase was measured for the first desorption Kd (Kd1-des) detennination. After this first step, the solid was contacted with fresh water and suspended again. This process was repeated for the same solid sample 4 or 5 times.
The distribution coefficients measured by desorption methods will be referred to as "desorption Kd" (Kddes). The desorption distribution coefficients ''Kddes" were calculated, by the following formula:
K -1 . _ Cads - Cr* . ~ "des - • Cr m
(E.2)
where Cads is the adsorbed radionuclide, as defined in Eq. (E.l), Ct is the final activity in the liquid upon the new equilibrium, m is the mass of the solid and V is the liquid volume.
142 T. Missana et al. I Applied Clay Science 26 (2004) J 3 7-J 50
3. Results
3.1. Bentonite colloid characterisation
The significance of colloids in radionuclide transport is absolutely dependent on their stability. As mentioned before, bentonite colloids are stable in low ionic strength and basic pH waters. The initial mean hydrodynamic size of bentonite colloids in Grimsel water, determined by PCS was 250 ± 50 nm. Fig. 1 shows the size distribution of a bentonite colloids sample prepared in Grimsel water, after 5 months. Comparing the peak of the distribution with the initial range of the colloid size, it can be noticed that the mean hydrodynamic diameter measured after 5 months (- 300 nm) did not differ significantly from the initial one, indicating that coagulation processes did not occur in the system. This is important because it means that, in Grimsel water, bentonite colloids are very stable and can act as a carrier for radionuclides.
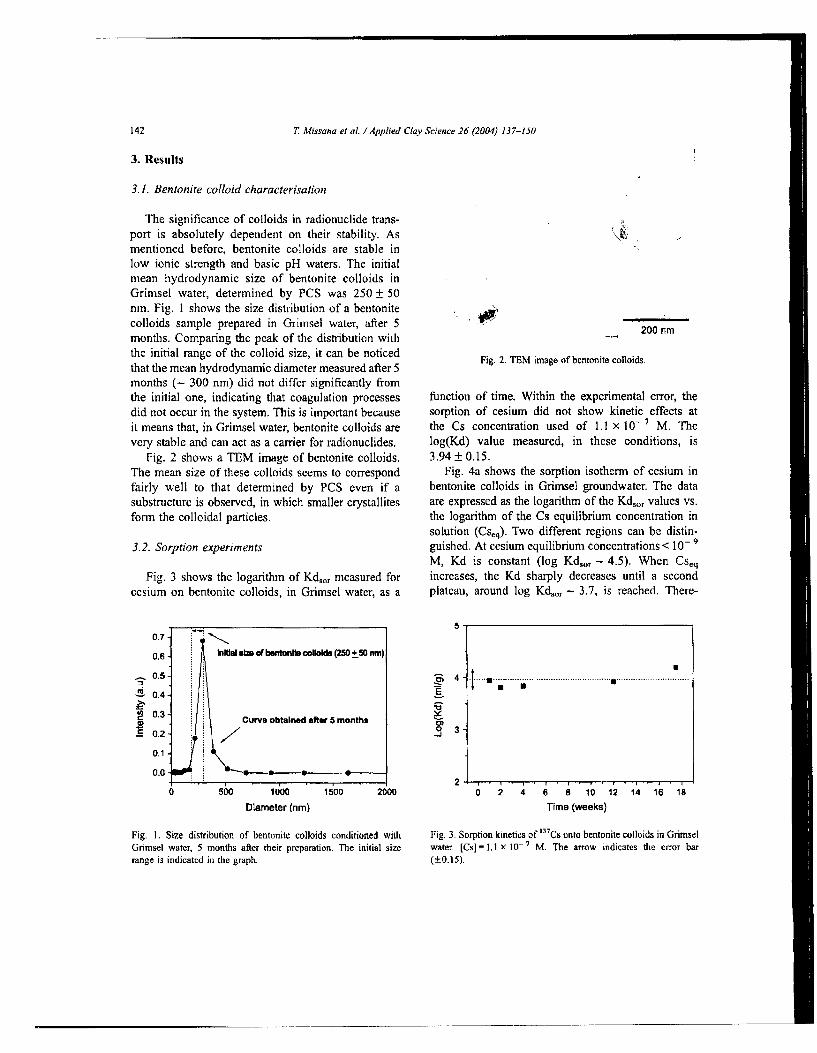
Fig. 2 shows a TEM image of bentonite colloids. The mean size of these colloids seems to correspond fairly well to that determined by PCS even if a substructure is observed, in which smaller crystallites form the colloidal particles.
3.2. Sorption experiments
Fig. 3 shows the logarithm of Kdsor measured for cesium on bentonite colloids, in Grimsel water, as a
0.7
0.6
S" 0.5
~ 0.4 ~ Ill 0.3 c ~ ..!: 0.2
0.1
0.0
0
lnlllal alza of bentonl18 colloids (250 :t_ 50 nm)
Curve obtained after 5 months
/
--.---.-----.----------~
500 1000
Diameter (nm)
1500 2000
Fig. I. Size distribution of bentonite colloids conditioned with Grimsel water, 5 months after their preparation. The initial size range is indicated in the graph.
200nm
Fig. 2. TEM image of bentonite colloids.
function of time. Within the experimental error, the sorption of cesium did not show kinetic effects at the Cs concentration used of 1.1 x 10- 7 M. The log(Kd) value measured, in these conditions, is 3.94±0.15.
Fig. 4a shows the sorption isotherm of cesium in bentonite colloids in Grimsel groundwater. The data are expressed as the logarithm of the Kdsor values vs. the logarithm of the Cs equilibrium concentration in solution (Cseq). Two different regions can be distinguished. At cesium equilibrium concentrations< I o- 9
M, Kd is constant (log Kdsor - 4.5). When Cseq increases, the Kd sharply decreases until a second plateau, around log Kdsor- 3.7, is reached. There-
5.-----------------------------~
• g; 4 ·!-····················································································· .§_ • •
~ :§1 3
0 2 4 6 8 10 12 14 16 18
Time (weeks)
Fig. 3. Sorption kinetics of 137Cs onto bentonite colloids in Grimsel water. [Cs] = 1.1 x 10- 7 M. The arrow indicates the error bar (±0.15).

T Missana eta!. I Applied Clay Science 26 (2004) /37-150 143
(a) 5.0 ..,...------------------.
~ 4.0
_g 3.5
3.0 +-~-.---~--.-~-.,..-~-...----!
(b) 5.0 ~-------------,
4.0 I ;g; .§. :0 3.0 :.:: g; ...1
2.0 v 1 MlnNaCIO, [J 0.1 MIn NaCIO,
1.0 +-~--.--~--,.-~-.---,...--.-........ ---l -12 -10 -8 -6 -2
Log(Cs..,) (MoVI) (c)
4.0..-----------------.
~
~ 3.5 g
~ J 3.0
[J
B D
1:1
···r:i········ ···c·········o··
o [Cs),1·10 .. M
1:1 [Cs)=H 0"' M
..... 0 ........................... . ---·············· & 0 0 0
2.5+---.--~..,.-....... -..--~--.-..---.---...-~ 0 2000 40()0 6000 8000 10000
Time (min)
Fig. 4. (a) Sorption isothcnn of cesium on bentonite colloids in Grimsel water. Contact time 15 days. {b) Sorption isothenn of cesium in bentonite colloids in NaCI04 (V) 1 M or (D) 0.1 M. Contact time 15 days. In the latter case, bentonite was previously homoionised with Na. The lines correspond to the modelling of data considering the selectivity coefficients ~~KscL = 1.65 and ~~KsEL =7.50 for ionic exchange in the "low" and "high" affinity sites, respectively. (c) Kinetics of cesium uptake in Nahomoionised bentonite in NaCI04 0. I M, at two different Cs concentrations: (D) [Cs]; I x 10- 9 M and (0) [CsJ= I x to- 6 M.
fore, cesium sorption is not linear over the whole range of concentrations, and the shape of the isotherm indicates that, at least, two different sites, with different affinity for Cs, are involved in sorption. The first site has high affinity and low capacity (- 3.2 x w- 8
eq/g) and the second one with much higher capacity (- 1 x 10- 3 eq/g - CEC) but lower affinity.
The sorption isothenn of cesium in bentonite colloids conditioned to Grimsel water, presented in this study, shows similar characteristics to those previously observed by Missana ct al. (2002} in the homoionised (Na or Ca) FEBEX clay. An example of the Cs isotherms, observed at two different ionic strengths (1 and 0.1 M in NaC104), in the Na-FEBEX clay, is shown in Fig. 4b. The Cs uptake as a function of time at two different Cs concentrations (1 x w- 9
and 1 X w- 6 M) in the Na-homoionised clay is also shown in Fig. 4c .
It is worth noticing that at "high" cesium concentration (low affinity) the uptake was very rapid (hours or less) whereas at "low" cesium concentration (high affinity) the uptake was significantly slower, and the sorption process was completed within days. This result shows that sorption in the two different sites might be controlled by different mechanisms.
Fig. 5 shows the logarithm of the Kdsor measured for uranium on bentonite colloids, in Grimsel water, as a function of time. The Kdsor values increase more rapidly during the first 4 weeks and, after that, a much slower increase is observed. Even after 18 weeks, the equilibrium seems not to be completely reached.
3.50
3.25
~·l t-i ~
.!?J I ~
3.00
Ci 0
....J 2.75
0 2 4 6 8 10 12 14 16 18
Time (weeks)
Fig. 5. Sorption kinetics of 233U(VI) onto bentonite colloids in Grimsel water. [U]=4.0 x 10- 7 M.

144 T Missana e/ a/. I Applied Clay Science 26 (2004) 137-150
5.0 -r.=====,------------, 1:. 1 day
1
I 4.5
;g; 4.0
.§. - 3.5 ~ :§' 3.0
2.5
& 15 days o 3.5months
I I
2.0 +-..,._,.-~,--..--.-,.......,r--"--r~---.---.--.~-l -8.5 -8.0 -7.5 ·7.0 -6.5 -6.0 ·5.5 -5.0 -4.5
Log(U00) (Mol/1)
Fig. 6. Sorption isothenns of uranium onto bentonite colloids in Grimsel water.
In similar experiments and in presence of solid phases (bentonite, granite or fracture filling materials), U concentration was observed to decrease in the Grimsel water, first more rapidly, then very slowly (Missana et al., 2003b ). Even if it has been previously mentioned that the uranium is stable in the solid-free Grimsel water, it has to be taken into account that this very slow process might be related to the reduction of the radionuclide and not to the sorption itself. The presence of a solid phase, particularly if it contains Fe(II), which is a good reductant for uranium (Ligcr et al., 1999), might buffer the Eh, favour and accelerate the actinide reduction. This stresses the importance of analysing the kinetics of all the possible processes related to radionuclide transport in deep groundwater.
Fig. 6 shows the sorption isotherm of U(VI) in bentonite colloids in Grimsel groundwater, where the data are expressed as the logarithm of the Kd,or vs. the logarithm of the equilibrium U concentration in solution (Ueq)- Sorption is not linear: there is a plateau for Ueq from 1 x 10- 7 M to approximately 6 x 10- 6 M (- 7 <log Ueq <- 5.25), but at lower u.q the distribution coefficient increases. Because the range of concentrations used for uranium was smaller than that used for cesium, it is not clear if two different linear sorption regions exist in this case as well.
The isotherms were carried out at different contact times (1 day, 15 days and 3.5 months) and sorption increased with time. The log Kdsor value obtained after 1 day in the observed plateau region is - 2.66;
after 2 weeks, it is - 3.00, in agreement with the previous series of experiments (Fig. 5). When the Ueq is increased above 6 X 1o- 6 M (Jog u.q =- 5.25), the sharp increase in the Kd values clearly indicates that precipitation is occurring (Langmuir, 1997). This concentration is approximately I order of magnitude higher than that used for the determination of Kds in sorption/desorption experiments.
However, it can be noticed that precipitation tends to occur at lower equilibrium concentrations when the contact time increases (see the curve obtained after 3.5 months of contact time in Fig. 6). Again, this could be a further indication that a slow partial reduction of uranium in the clay/water system, under the experimental anoxic/reducing conditions, might be taking place.
3.3. Desorption experiments
The results obtained for cesium in desorption experiments are shown in Fig. 7. Sorption results are plotted in the right part of the graph and desorption results on the left part. This representation of sorption/desorption data is very useful to analyse sorption reversibility at a glance. If sorption and first desorption graphs are symmetrical, then sorption is reversible. The same symbol always
35000~-------------------r---------,
30000
;g;
5"' • • ·~3"and4"DES~
(2""DES.)-J ...- (3• and 4"' DES.)
.s 25000 ~~ ~ 15000
l (2""DES.) i ' I
10000
5000
120 100 80 60 40 20 0 20 40 60
- DESOIU'TION Time (days) sDRPTJON-
Fig. 7. Sorption and desorption kinetics of Cs in bentonite colloids conditioned with Grimsel water. The same symbols refer to the same sample, in which sorption and then successive desorptions were performed. Samples are defined by their contact time in sorption experiments: (•) I day,(&) I week, (e) 2 weeks, (T) S weeks, (+) 8 weeks.

T. Missana eta/. I Applied Clay Science 26 (2004) 137-150 145
10000
8000
~ 6000
.s ""C 4000 ~
2000
0
(S"~ES.)
~ 14" DES.} · : : 1 ' '
(3°DES.)
(2'"' DES.)
120 100 80
~ 6040 20 0
Time(days)
20 40 60 ~
Fig. 8. Sorption and desorption kinetics of U in bentonite colloids conditioned with Grimsel water. The same symbols refer to the same sample, in which sorption and then successive desorptions were perfonned. Samples are defined by their contact time in sorption experiments: (•) I day, (4) I week, (e) 2 weeks,("') 5 weeks, (t) 8 weeh.
refers to the same sample, in which sorption and then successive desorptions were performed. As explained in the experimental section, during the "first desorption" step, the solid was maintained in contact with the fresh Grimsel water for the same amount of time used in the sorption step, to compare directly sorption and desorption rates.
As mentioned before, sorption of cesium at these concentrations is not expected to be affected by kinetics, so that the variation of the values can be attributed to the experimental dispersion of data. The mean value for the Kdsoo calculated for these experimental conditions and indicated in Fig. 7 as a continuous line, is approximately 8400 ml/g (log Kdsor- 3.92). The values are very similar to those obtained in the previous experiments (Figs. 3 and 4), in spite of the lower centrifugation rate used.
The mean value for the first desorption Kd, considering all the samples, is approximately 5600 mtlg (log Kd1-des- 3.75). The "first desorption" Kd values are even lower than the "sorption" Kd and, in spite of the different initial contact time, nearly constant.
By definition of a reversible sorption process, it is assumed that Kdsor "" Kddes and that sorption and desorption rates are similar. If Kd50r < K~e .. sorption cannot be considered totally reversible. Hence, taking into account only the first desorption results, cesium sorption would seem reversible. However, the K~es
values upon further washings are always higher than those of the first desorption step and kinetics starts playing an important role. For the sample that has been contacted with cesium one day, the Kddes value obtained in the second desorption experiment is lower (- 8600 ml/g, log Kdn-dcs- 3.93) than that of the samples contacted 5 or 8 weeks [- 15000 ml/g (log Kdn.des- 4.18) and - 30000 ml/g (log KdJl.dcs - 4.48), respectively]. The same occurs for the third and fourth desorption steps. In fact, for the sample that has been in contact with cesium one day, the Kd values obtained in the third/fourth desorption experiment are lower (- 28 000 ml/g, log Kdm-des - 4.45) than those of the sample contacted 8 weeks(- 32000 ml/g, log Kdm-des- 4.51). Upon the third desorption, the Kddes values do not vary significantly anymore.
Fig. 8 shows the desorption behaviour of uranium from bentonite colloids. Kdsor increases with contact time (from - 390 mg/1, log Kdsor - 2.59 to - 1300 mg/1, log Kdsor - 3.11 ), as already observed (Figs. 5 and 6).
The Kddes of the first desorption experiments ("" 1850 ml/g, log Kd1.des- 3.27) is always higher than the sorption values and almost independent on the initial contact time. Therefore, sorption is not completely reversible. When further desorptions in the same sample were carried out, the Kd values for successive desorption steps increase, as occurred for cesium. Nevertheless, the values obtained in each desorption step are substantially independent on the initial contact time, indicating that different processes than those occurring for cesium are taking place.
4. Discussion
As observed in XRD analysis, bentonite colloids are mainly formed by smectite. Smectite is a 2:1 layer silicate mineral, whose basic structure is composed of two tetrahedral (T) and one octahedral (0) sheets. An example of the smectite structure can be seen in Fig. 9 (Grim, 1968). The T -0-T layers are stacked and form the clay particles. These particles carry a permanent negative charge, due to isomorphic substitutions in the sheets (AI3 + for Si4 + in the tetrahedral sheet and bivalent ions for Atl + in the octahedral sheet). Cations are adsorbed at the surface of the clay

146 r. Missana eta/. I Applied Clay Science 26 (2004) 137-15()
Exchansoable caliona, interlaminar -fer
b)
c)
X
0 Oxigen 9 Hydroxll e AI, Fe, Mg 0 Y • Sl, ocaslonally AI
y - lntertayer
I I t==l
Fig. 9. (a) T -0-T structure of smectite layers (from Grim. 196R). The tetrahedra create a hexagonal pattern with a cavity in its centre. This is represented by the additional sketch (b) in which the spheres represent the basal oxygen atoms of the tetrahedra. The stacked TOT units fonn the smectite particles (c).
particles and in the interlayers to balance the structural negative charge.
In addition, on the edges of the layers, a pHdependent charge is originated, due to protonation/ deprotonation of the silanol (SiOH) and alumino! groups (AIOH) and it can be either positive or negative. In smectites, this charge is very small in relation to the total charge of the clay (approximately 5% in the FEBEX clay); however, the surface reactions with these groups can significantly contrib-
ute to the sorption of certain radionuclides, especially actinides.
At the external surface of the clay particles, cations can adsorb both on basal planes (negatively charged) and edges (negatively or positively charged). The other sites accessible to certain cations are in the interlayer region. Sorption of ions in "basaVinterlayer" and "edge" sites occurs predominantly as ion exchange and surface complexation, respectively.
Surface complexation processes of cesium, on the edges of the clay particles, can be discarded because cesium sorption was found to be independent on pH (Missana et al., 2002). However, it is generally accepted that in clay minerals different sites exist for cesium sorption and spectroscopic evidence of "different environments" for cesium sorbed in clays can be found in the literature (Weiss et al., 1990; Kim et a!., 1996).
Sorption of cesium on FEB EX bentonite colloids is not linear, which can be interpreted with a two-site model (see Fig. 4b ), with one ion exchange site of "high" affinity and another of "low" affinity. The nonlinearity of cesium sorption was already observed in 2: I clays (e.g., Oscarson eta!., 1987; Staunton and Roubaud, 1997), and two-site models were already successfully used to model cesium sorption data (Zacham et al., 2002).
The adsorption of cesium onto bentonite colloids seems to involve a rapid exchange reaction (hours) and a slower component (days) that seems anomalous for ion exchange reactions. This slow process, can be evidenced only when very low tracer concentration are used (< 1 x w- 9 M) due to the very low density of these sites (- 3.2 x 10- 8 eqlg) compared to the bentonite cation exchange capacity. We interpret these two processes, in connection with the high affinity and low affinity sites, as adsorption on planar sites (rapid process) and cesium diffusion to less available but highly selective sites (slow process).
The existence of "low" and "high" affinity sites for cesium sorption in clays is usually explained considering that cesium can be exchanged with hydrated cations in basal/interlayer sites (low-affinity sites) and can sorb, in a highly selective way in frayed edge sites, PES, (high-affinity sites) (Zachara et al., 2002). FES sites are not present in expanding clays like smectite but develop in weathered micas and illite.

T. Missana et af_ I Applied Clay Science 26 (2004) 137-150 147
Bradbury and Baeyens (2000) interpreted sorption on illite in terms of an equilibrium ion exchange model, with three different sites with different selectivities. Two of these sites were considered to have low capacity, but high selectivity for cesium, and the last one, representing more than the 95% of the total CEC, was attributed to the planar surface of the clay. On the other hand, Poinssot ct al. ( 1999) assumed a pure ion exchange model to quantify sorption, even if at low cesium concentration the sorption kinetics was not negligible, as observed for the bentonite colloids in the present work.
As mentioned in the Section 2, illite was not observed in the XRD analysis of the bentonite colloids but the existence of smectite-illite mixed layer may possibly lead to the existence of FES-like sorption sites in FEBEX bentonite.
Sawhcncy ( 1966) attributed the irreversibility of cesium sorption on illites to the selective adsorption at the frayed edge sites. Other authors modelled the cesium uptake onto illites considering a kinetically controlled reversible reaction with an equilibrium described by a Freundlich isotherm, followed by an irreversible first-order process (Comans and Hockley, 1991) because their experimental results suggested a noncompletely reversible sorption (Comans et a!., 1991).
From the results of the desorption experiments, it could be deduced that part of cesium is "weakly" sorbed on the clay. The fact that the first desorption Kd values are lower than the sorption Kd values, indicates that part of the cesium is reversibly sorbed on the clay. The desorption of this "weakly sorbed" cesium is not dependent on the initial contact time (first desorption Kd, Fig. 7) and this suggests that part of cesium is located in ion exchange sites and readily accessible.
However, when trying to extract more cesium from the solid, it was observed that the longer the element had been in contact with the solid, the greater the sorption/desorption hysteresis was. Because Kddes increases with the age of the sorption complex, the cesium uptake cannot be considered reversible. This result shows the importance of kinetic studies in the analysis of the irreversibility of sorption processes. The fact that the "desorption Kd" values of part of the adsorbed Cs depend on the initial contact time indicates that a "pure" ion exchange mechanism is not the
only one that controls the cesium uptake. Even if, quantitatively, the process can be explained as an exchange process (Fig. 4b ), part of the cesium has to diffuse towards more energetically stable locations in the clay structure, that are not immediately accessible. When cesium is transferred to less available sites, "fixation" of the ion may also take place (Comans ct al., 1991 ).
The cesium fixation in 2: 1 clays has been reported to occur at the hexagonal cavity of the tetrahedral surface (Fig. 9), and it is more favoured in montmorillonite-like than in beidellite-like smectites, as reported by Onodera et al. (1998).
Amongst the cations that exist in solution, those that present the highest probability of fixation are potassium, cesium, rubidium and ammonium. Larger cations, or cations with a stable hydration shell, as Ca2 +or Sr Hcannot be "fixed".
In the migration experiments at the GTS, it was found that, in the presence of bentonite colloids, a small quantity of cesium was able to move unretarded in the fracture along with colloids (Mori ct al., 2003). Interestingly, this behaviour was not observed for strontium, although it is also adsorbed by bentonite colloids. The reason why this occurred is most probably related to the selective adsorption of cesium in the high-affinity sites (not available for Sr) and to its partial fixation.
In principle, the "fixed" fraction of cesium could be exchangeable with the aqueous phase if sufficient large periods are given (Cornell, 1993), but the presence of competitive ions, like potassium, with high affinity for these sites is also required. Because the potassium content in Grimsel water is not high, part of the cesium adsorbed in bentonite colloids could remain on the solid enough time to be transported significant distances from the colloid source.
Previous sorption studies of uranium(VD in smectites indicated that multiple surface species result both from ion exchange on planar sites and surface coordination reactions at the edge sites (McKinley ct a!., 1995; Zachara and McKinley, 1993). Uranium(VI) is preferentially sorbed by ionic exchange at acidic pH (Boult et a!., 1998) and in low saline content waters. At higher pH and ionic strengths, the mechanism of surface complexation, with the formation of inner sphere complexes, will prevail (Turner et al., 1996). Several studies were made with spectroscopic techni-

148 T Missana el al. I Applied Clay Science 26 (2004) 137-150
ques in order to explain the sorption mechanisms of uranyl ion onto smectite clays. Different binding sites have been identified which are in agreement with observations in classical sorption studies. At pH< 6 and low ionic strength, the uranyl maintains its outer hydration shell so that an outer sphere mechanism (most probably cation exchange) must be responsible of the binding (Sylvester ct a!., 2000). At near neutral pH, the evidence of inner sphere complexation (Sylvester ct al., 2000) is shown. Above pH 5-6, the deprotonation of the silanol and alumino! groups provides additional sorption sites and, in addition, uranyl-hydroxide species start to form. Finally at "low" uranium concentrations, the edge seems to be most important sites for U uptake (Morris et al., 1994), with the formation of inner sphere complexes. As surface coverage increases, a less close approach of the uranyl to the surface and the formation of outer sphere complexes was observed (Chisholm-llrausc ct a!., 1994).
Taking into account all these previous studies, in Grimsel water and the experimental conditions used in these experiments (high pH, low ionic strength and trace uranyl concentration) both mechanisms (ion exchange and surface complexation) may compete.
From the uranium sorption isotherms (Fig. 6), it can be observed that the Kd is not constant for all U concentrations. Excluding the region where precipitation starts (logUeq>-5.5), the tendency of Kd is to decrease when the uranium concentration increases. The fact that the adsorption of uranium in bentonite colloids in Grimsel water is not linear over the entire range of concentration is an indication that different sorption sites or complexes exist. The information obtained from the isotherm (Fig. 6) is not enough to draw a conclusion about the existence of multiple sites for uranium sorption in Grimsel water. However, it is reasonable to assume that, despite of the very low ionic strength of the water, ion exchange is not the only mechanism controlling the uranium uptake in Grimsel water conditions. The high pH of the water and the low uranium concentration may favour the existence of inner sphere complexes with the surface silanol and alumino! groups, as observed in previous works.
The fact that the first Kddes are always higher than Kdsor (Fig. 8) indicates that the uranium sorption process in bentonite colloids is not totally reversible, which might be a further corroboration of the hypoth-
esis of the existence of surface complexation reactions because a reversible process would be expected for a purely ionic exchange. Hysteresis of sorption/desorption has been already shown for actinides surface complexation onto colloidal phases (Lu et al., !998).
Despite that uranium sorption is not linear and not fully reversible, the uranium behaviour is clearly not the same as that of cesium, as, in the second/third/ fourth desorption steps, uranium Kdctes did not depend on the initial contact time, as previously observed in the case of cesium. This is an indication that the kinetic fixation in the clay structure does not occur in the case of uranium. The possibility of the uranyl "fixation" mechanism is quite unlikely because it hardly looses the hydration water (Sposito, 1984), and hydrolysed species expected at high pH are too large to fit any structural position.
A last point to be discussed is related to the redox effects that can affect the uranium behaviour. Redox effects may compete with sorption onto colloids and affect the transport of the radionuclide in the fractured rock. Therefore, to interpret correctly in situ and laboratory experiments reduction processes have to be taken into account.
In solid-free Grimsel groundwater, the reduction of the element is thermodynamically predicted, but the kinetics of the process was shown to be much slower than the experimental time investigated in this work (months). In the presence of solid phases, a rapid and then a very slow disappearance of the radionuclide in the liquid phase was observed. This slow process might be related to the partial reduction ofU(Vl) to U(IV).
Because in situ CRR experiments are conducted under rather fast flow conditions, and last hours-days, the possible redox reactions would play a minor role in the experiment because they have been shown to be very slow. However, ifuranium reduction processes at the clay surface or (surface) precipitation of U(IV) occurs, these processes will affect the sorption reversibility when large time scales are considered.
5. Conclusions
In Grimsel water conditions, cesium and uranium are significantly sorbed on bentonite colloids. Isotherms showed a nonlinear sorption behaviour for both elements. In particular, in the case of cesium,

T Missana et al. I Applied Clay Science 26 (2004) 137-150 149
isotherms allowed identifying two different sorption sites for cesium: "low" affinity sites related to ion exchange in planar sites and "high" affinity sites whose behaviour is similar to that of illite frayed edge sites. This could be due to the fact that the smectite phase, in FEBEX bentonite, contains smectite-illite mixed layer with a I 0-15% of illite layers.
Desorption kinetics experiments showed that the sorption of both cesium and uranium onto bentonite colloids in Grimsel water conditions is not completely reversible. The main consequence of finding a noncompletely reversible sorption in the colloidal phase or a desorption rate much slower than the sorption one, is that radionuclides would be able to migrate large distances, provided that colloids are stable and mobile. Considering the long-term behaviour, the case of cesium is of particular interest; its selective fixation in the high-affinity sites represents, in fact, an irreversible process unless the geochemical conditions change. In particular, Cs displacement from these high affinity sites can be only affected by ions like potassium, rubidium or ammonium.
Finally, to interpret correctly all the experiments in deep groundwaters, the kinetics of possible redox processes, that can affect the uranium sorption/migration behaviour, has to be adequately taken into account.
Acknowledgements
This work has been partially supported by the ENRESA-ClEMAT association and was carried out within the context of the Grimsel CRR (Colloids and Radionuclide Retardation) project which is cofunded by ANDRA(F), ENRESA(E), FZK(D), JNC(J), Nagra (CH) and the USDoE (USA).
The authors greatly acknowledge J. Cuadros and two anonymous reviewers for their valuable comments on the manuscript.
References
Boult, K.A., Cowper, M.M., Haeth, T.G., Sato, H., Shibutani, T., Yui, M., 1998. Towards an understanding of the sorption of U(Vl) and Se(IV) on sodium bentonite. J. Contam. Hydro!. 35, 141-150.
Bradbury, M.H., Baeyens, B., 2000. A generalised sorption model for the concentration dependent uptake of caesium by argillaceous rocks. J. Contam. Hydro!. 42, 141-163.
Bruno, J., Duro, L., Rollin, C., Guimeni., J., 2000. Prediction of the solubility and speciation of radionuclides in FEBEX and Grimset water. Tech. CRR Rep, 200 pp.
Buddemaier, R.W., Hunt, J.R., 1988. Transport of colloidal contaminants in groundwater: radionuclide migration at the Nevada Test Site. Appl. Geochem. 3, 535.
Caceci, M., 1988. Utilisation de Ia Spectroscopic de Correlation des Photons Pour La Detennination de La Taille et de Ia Fonne des Particules. CEA-SESD/88.42-FA 6790-11.
Chisho1m-Brause, C., Conradson, S.D., Busher, C.T., Eller, P., Morris, D.E., 1994. Speciation of uranyl sorbed at multiple binding sites on monbnorillonite. Geochim. Cosmochim. Acta 58 (17), 2631-3625.
Comans, R.N., Hockley, D.E., 1991. Kinetics of cesium sorption on
illite. Geochim. Cosmochim. Acta 38, 1157-1164. Comans, R.N., Holler, M., DePreter, P., 1991. Sorption of Cs on
illite: non equilibrium behaviour and reversibility. Geochim. Cosmochim. Acta 55,433-440.
Cornell, R.M., 1993. Adsorption of cesium on minerals: a review. J. Radioanal. Nucl. Chern. 171, 483-500.
Cuadros, J., Linares, J., 1996. Experimental kinetic study of the
smectite-to-illite transfonnation. Geochim. Cosmochim. Acta 60 {3), 439-453.
Dahneke, B., 1983. Measurements of Suspended Particles by QuasiElastic Light Scattering. Wiley, New York.
Degueldre, C., Triay, 1., Kim, J.l., Vilks, P., Laaksoharju, M., Miekeley, N., 2000. Groundwater colloid properties: a global ap
proach. Appl. Geochem. 15, 1043. Fernandez, A.M., Baeyens, B., Bradbury, M., Rivas, P., 2004. Anal·
ysis of the pore water chemical composition of a Spanish compacted bentonite used in an engineered barrier. J. Geochem. Explor. (in press).
Grim, R.E., 1968. Clay Mineralogy. McGraw-Hill. New York. Hauser, W., Geckeis, H., Kim, J.l., Fiertz, T., 2002. A mobile laser
induced breakdown detection system and its application for the in-situ monitoring of colloid migration. Colloids Surf, A Physicochem. Eng. Asp. 203, 37.
Holthoff, H., Egelhaaf, S.U., Borkovec, M., Shurtenberger, P., Stieber, H., 1996. Coagulation rates of colloidal particles by simultaneous static and dynamic light scattering. Langmuir 12, 5541-5549.
Huertas, F., CantiJlana, J.L.F., Jullien, F., Rivas, P., Linares, J., Fariiia, P., Ghoreychi, M., Jockwer, N., Kickmaier, W., Martinez, M.A., Samper, J., Alonso, E., Elorza, F.J., 2000. FEBEX project final report. EUR 19147, pp. 15-21.
Kersting, A.B., Efurt, D.W., Finnegan, D.L., Rokop, D.J., Smith, D.K., Thompson, J.L., 1999. Migration of plutonium in groundwater at the Nevada test site. Nature 397, 56-59.
Kim, J.I., 1994. Actinide colloids in natural aquifer systems. MRS Bull. XIX, 47-53.
Kim, J.l., Buckau, G., Klenze, R., 1987. Natural Colloids and Generation of Actinide Pseudocolloids in Groundwater. Graham and Trotman, London.
Kim, Y., Cygan, R.T., Kirkpatrick, R.J., 1996. "'Cs NMR and XPS

!50 T. Missana et al. I Applied Clay Science 26 (2004) I 37-1 JO
investigations of Cs adsorbed on clay minerals and related phases. Geochim. Cosmochim. Acta 60 (6), 1041-1052.
Langmuir, D., 1997. Aqueous Environmental Geochemistry. Pren· lice Hall, New York.
Lieser, K.H., Ament, A., Hill, R., Singh, R.N., Sting!, U., Thy· busch, B., 1990. Colloids in groundwater and their influence on migration of trace elements and radionuclides. Radiochim. Acta 49, 83.
Liger, E., Charlet, L., Cappellen, P.V., 1999. Surface catalysis of uranium(Vl) reduction by iron(ll). Geochim. Cosmochim. Acta 63 (19 -20), 2939-2955.
Lu, N., Cotter, C.R., Kitten, H.D., Bentley, J., Triay, I.R., 1998. Reversibility of sorption of 239Pu onto hematite and goethite colloids. Radiochim. Acta 83, 167-173.
McCarthy, J.F., Zachara, J.M., 1989. Subsurface transport of contaminants. Environ. Sci. Te-chno!. 23, 496-502.
McKinley, J.P., Zachara, J.M., Smith, S.C., Turner, G.D., 1995. The influence of hydrolysis and multiple sire binding reactions on adsortpion of U(VI) onto montmorillonite. Clays Clay Miner. 43, 568-598.
Miller, W., Alexander, W.R., Chapman, N., McKinley, J., Smellie, J., 1994. Natural Analogue Studies in the Geological Disposal of Radioactive Waste. Elsevier, Amsterdam.
Missana, T., Turrero, M.J., Melon, A., 1999. Generation and char· acterisation of colloids of the bentonite/granite interface. Mater. Res. Soc. Symp. Proc. 556, 647-654.
Missana, T., Garda-Gutierrez, M., Femandez, V., Gil, P., 2002. Ap· plication of mechanistic models for the interpretation of radionuclide sorption in clays. Part (I) C!EMAT/DIAE/54610/01/03.
Missana, T., Alonso, U., Turrero, M.J., 2003a. Generation and stability of bentonite colloids at the bentonite/granite interface of a deep geological repository. J. Contam. Hydro!. 61, 17-31.
Missana, T., Gutierrez, M.G., Turrero, M.J., Alonso, U., Mingarro, M., 2003b. The role of colloids in the radionuclide transport in a deep geological repository: participation of CIEMAT in the CRR project. ENRESA Tech. Rep. 03/ 2003, 147 pp.
Miiri, A., Alexander, W., Geckeis, H., Hauser, W., Shafer, T., Eikenberg, 1., Fierz, T., Degueldre, C., Missana, T., 2003. The colloid and radionuclide retardation experiment at the grimsel test site: influence of bentonite colloids on radionuclide migra· tion in a fractured rock. Colloids Surf., A Physicochem. Eng. Asp. 217, 33--47.
Morris, D.E., Chisohlom-Brause, C.J., Barr, M.E., Conmdson, S.D., Eller, P.G., 1994. Optical spectroscopic studies of the sorption of uranyl species on a reference smectite. Geoch im. Cosmochim. Acta 58, 3626.
Newman, A.C., 1987. Chemistry of Clay and Clay Minerals. Min· eralogical Society, London.
Nuttal, H.E., Long, R.L., 1993. Mobility of radioactive colloidal particles in groundwater. Radioact. Waste Manag. Nucl. Fuel Cycle 17 (3 -4), 237-251.
Onodera, Y., Iwasaki, T., Ebina, T., Hayashi, H., Torii, K., Chatterjee, A., Mimura, H., !998. Effect of layer charge on fixation of cesium ions in smectites. J. Contam. Hydro!. 35, 131-140.
Oscarson, D.W., Watson, R.L., Miller, H.G., 1987. The interactions of trace levels of cesium with montrnorillonitic and illitic clays. Appl. Clay Sci. 2, 29-39.
Penrose, W.R., Polzer, W.L., Essington, E.H, Nefson, D.M., Orlandini, K.A., 1990. Mobility of plutonium and americium through a shallow aquifer in a semiarid region. Environ. Sci. Techno). 24, 228-234.
Poinssot, C., Baeyens, B., Bradbury, M.H., 1999. Experimental and modelling studies of caesium sorption on illite. Geochim. Cosmochim. Acta 63, 3217-3227.
Ryan, J .N., Elimelech, M., 1996. Colloid mobilisation and transport in groundwater. Colloids Surf., A Physicochem. Eng. Asp. 107, I -56.
Sawheney, B.L .• I 966. Kinetics of cesium sorption by clay minerals. Proc. Soil Sci. Soc. Am. 30, 565-569.
Sposito, G., 1984. The Surface Chemistry of Soils. Oxford Univ. Press, Oxford.
Staunton, S., Roubaud, M., 1997. Adsorption of cesium on mont· morillonite and illite: effects of charge compensating cations, ionic strength, concentration of Cs K and fulvic acids. Clays Clay Miner. 45 (2), 251-260.
Sylvester, E.R., Hudson, E.A., Allen, P.G., 2000. The structure of uranium(Vl) sorption complexes on silica alumina and montmorillonite. Geochim. Cosmochim. Acta 64 {14), 2431-2438.
Turner, G.D., Zachara, J.M., McKinley, J.P., Smith, S.C., 1996. Surface charge properties and U~ • adsorption on a subsurface smectite. Geochim. Cosmochim. Acta 60 (18), 3399-3414.
Weiss, C.A., Kirkpatrick, R.J., Altaner, S.P., 1990. The structural environment of cations adsorbed onto clays: mcs variable temperature MAS NMR spectroscopic study of hectorite. Geochim. Cosmochim. Acta 54, 1655-1669.
Zachara, J.M., McKinley, J.P., 1993. Influence of hydrolysis on the sorption of metal cations by smectite; importance of edge co· ordination reactions. Aquat. Sci. 55, 250-261.
Zachara, J.M., Smith, S.C., Liu, C., McKinley, J.P., Serne, R.J., Gassman, P.L., 2002. Sorption of Cs+ to micaceous subsurface sediments from the Hanfonl site, USA. Geochim. Cosmochim. Acta 66 (2), 193-21].