
Killing of Normal Melanocytes, Combined with Heat … 70 and CD40L Expression, Cures Large...
11
of June 4, 2018. This information is current as Melanomas Expression, Cures Large Established with Heat Shock Protein 70 and CD40L Killing of Normal Melanocytes, Combined and Richard G. Vile Rosa Maria Diaz, Jill Thompson, Jose Pulido, Alan Melcher Luis Sanchez-Perez, Timothy Kottke, Gregory A. Daniels, http://www.jimmunol.org/content/177/6/4168 doi: 10.4049/jimmunol.177.6.4168 2006; 177:4168-4177; ; J Immunol References http://www.jimmunol.org/content/177/6/4168.full#ref-list-1 , 28 of which you can access for free at: cites 58 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2006 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 4, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 4, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Killing of Normal Melanocytes, Combined with Heat … 70 and CD40L Expression, Cures Large...

of June 4, 2018.This information is current as
MelanomasExpression, Cures Large Establishedwith Heat Shock Protein 70 and CD40L Killing of Normal Melanocytes, Combined
and Richard G. VileRosa Maria Diaz, Jill Thompson, Jose Pulido, Alan Melcher Luis Sanchez-Perez, Timothy Kottke, Gregory A. Daniels,
http://www.jimmunol.org/content/177/6/4168doi: 10.4049/jimmunol.177.6.4168
2006; 177:4168-4177; ;J Immunol
Referenceshttp://www.jimmunol.org/content/177/6/4168.full#ref-list-1
, 28 of which you can access for free at: cites 58 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2006 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Killing of Normal Melanocytes, Combined with Heat ShockProtein 70 and CD40L Expression, Cures Large EstablishedMelanomas
Luis Sanchez-Perez,2*† Timothy Kottke,2* Gregory A. Daniels,* Rosa Maria Diaz,*†
Jill Thompson,* Jose Pulido,‡ Alan Melcher,§ and Richard G. Vile3*†§
Previously, we showed that nine intradermal injections of a plasmid in which the HSVtk suicide gene is expressed from amelanocyte-specific promoter (Tyr-HSVtk), combined with a plasmid expressing heat shock protein 70 (CMV-hsp70), along withsystemic ganciclovir, kills normal melanocytes and raises a CD8� T cell response that is potent enough to eradicate small, 3-dayestablished B16 tumors. We show in this study that, in that regimen, hsp70 acts as a potent immune adjuvant through TLR-4signaling and local induction of TNF-�. hsp70 is required for migration of APC resident in the skin to the draining lymph nodesto present Ags, derived from the killing of normal melanocytes, to naive T cells. The addition of a plasmid expressing CD40Lincreased therapeutic efficacy, such that only six plasmid injections were now required to cure large, 9-day established tumors.Generation of potent immunological memory against rechallenge in cured mice accompanied these therapeutic gains, as didinduction of aggressive autoimmune symptoms. Expression of CD40L, along with hsp70, increased both the frequency and activityof T cells activated against melanocyte-derived Ags. In this way, addition of CD40L to the hsp70-induced inflammatory killing ofmelanocytes can be used to cure large established tumors and to confer immunological memory against tumor cells, although aconcomitant increase in autoimmune sequelae also is produced. The Journal of Immunology, 2006, 177: 4168–4177.
M any different studies, in both preclinical and clinicaltrials, have suggested that successful immunotherapiesfor cancer are often associated with the concomitant
development of autoimmune disease against the normal tissue typefrom which the tumor derived (1–12). The association betweensuccessful anti-tumor immune responses and autoimmune mani-festations derives from the fact that many tumor-associated Ags ofmelanoma are unaltered self proteins of melanocytes (5, 6, 10).Based on these observations, we hypothesized that, if successfultumor immunotherapies can induce concomitant autoimmune re-activity, then perhaps intentional generation of autoimmune reac-tivity against cells of a certain tissue type, through inflammatory insitu killing, could successfully induce concomitant anti-tumor im-munity against tumors derived from that same tissue type (13). Ifthe tissue targeted for priming of complete, or partial, autoimmuneresponses is not essential for survival, then the intentional immunestimulatory killing of normal cells in situ may be used to primeanti-tumor immune cells without causing unacceptable toxicities tothe patient. In addition, this approach has various practical andtheoretical advantages over other strategies to generate anti-tumorimmune reactivities (14). Most normal tissues are readily accessi-
ble to delivery of vectors encoding appropriate immune stimula-tory/cytotoxic genes, and no tumor-derived materials, such as tu-mor cells, proteins, or peptides, are required to produce moreclassical tumor vaccine preparations. In addition, because normalcells share a wide repertoire of Ags with the associated tumor cells,allowing the inflammatory killing of normal cells to prime anti-tumor immunity removes the need to identify specific Ags towhich self-reactive T cell responses can be raised in vivo(14).
We validated this approach as a new paradigm for tumor im-munotherapy by using the intentional induction of pathological-like killing of normal melanocytes to generate immune responsesthat are highly effective against established melanomas (8, 13). Wedemonstrated that intradermal injections of a plasmid in which theHSV thymidine kinase (HSVtk)4 suicide gene (15) is transcrip-tionally targeted to melanocytes through the tyrosinase promoter(Tyr-HSVtk) leads to tissue specific killing of melanocytes on ad-ministration of the prodrug ganciclovir (GCV) (13). However,simple killing of melanocytes was only very poorly effective atgenerating anti-melanocyte/melanoma T cell responses in vivo(13). A combination of multiple injections of the Tyr-HSVtk plas-mid with a plasmid expressing the murine heat shock protein 70(hsp70) gene (16) (CMV-hsp70) generated localized killing of nor-mal melanocytes within a highly inflammatory environment (13).This was sufficient to generate a CD8� T cell response whichcleared 3-day established s.c. tumors, or 6-day established sys-temic tumors in the lungs (13), consistent with the known activitiesof hsp70 as a key molecule mediating the switch between tolero-genic and immunostimulatory cell killing (16–21). However, thisCD8� T cell response also was rapidly suppressed in vivo by apopulation of putative suppressor cells within the CD25� T cellpopulation (22), which prevented development of autoimmune
*Molecular Medicine Program, †Department of Immunology, and ‡Department ofOphthalmology, Mayo Clinic College of Medicine, Rochester, MN 55905; and §Can-cer Research U.K. Clinical Centre, St. James University Hospital, Leeds, UnitedKingdom
Received for publication April 17, 2006. Accepted for publication June 23, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Mayo Foundation and by National Institutes ofHealth Grants R0194180 and P50CA91956.2 L.S.-P. and T.K. contributed equally to this work.3 Address correspondence and reprint requests to Dr. Richard G. Vile, MolecularMedicine Program, Guggenheim 1836, Mayo Clinic, 200 First Street Southwest,Rochester, MN 55902. E-mail address: [email protected]
4 Abbreviations used in this paper: HSVtk, HSV thymidine kinase; GCV, ganciclovir;hsp70, heat shock protein 70; LN, lymph node; MHC-II, MHC class II.
The Journal of Immunology
Copyright © 2006 by The American Association of Immunologists, Inc. 0022-1767/06/$02.00
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

vitiligo in tumor cured mice (13). In addition, cured mice did notdevelop memory against the melanocyte Ags and were unable toreject a subsequent challenge of B16 tumor cells 6 mo after theinitial treatment regimen. However, autoimmune vitiligo could beinduced in treated animals, but only if they were not challengedwith tumor, and this autoimmune disease was significantly moreaggressive if the animals also were depleted of CD25� T cellsbefore plasmid administration (13). Presumably these mechanismsof suppression of anti-self T cell responses exist in vivo to safe-guard against the development of autoimmune disease in circum-stances where pathological killing of normal cells occurs (22).
In these previous studies, we were able to cure large percentages(70–100%) of mice carrying 3-day established tumors using a totalof nine injections of the Tyr-HSVCtk/CMV-hsp70 mixture (13).When we attempted to treat more established tumors seeded 9days previously, we observed therapeutic delays in tumor pro-gression with plasmid injections, but the percentage of curesdecreased to between 0 and 10% (23). In those mice wheretumor growth was delayed but not cured, there was a potent invivo immunoselection by a CD8� T cell response of tumor cellvariants that had lost expression of a specific subset of knownmelanocyte Ags (23).
In this study, we have investigated both the mechanisms bywhich inflammatory killing of melanocytes leads to the priming ofCD8� T cell responses against B16 melanomas and methods bywhich it can be enhanced to treat larger, more established tumors.Taken together, our data show that, by manipulating in vivo mech-anisms that normally restrain autoimmune responses, increased anti-tumor responses can be induced following the killing of normalcells.
Materials and MethodsCell lines, plasmids, and viruses.
The murine melanoma B16.F1, tumor cell line has been described previ-ously (24).
Plasmids used in these studies have been described previously (13).Briefly, the Tyr-HSVtk plasmid uses a hybrid promoter of three tandemcopies of a 200-bp element of the murine tyrosinase enhancer (25) up-stream of a 270-bp fragment of the tyrosinase promoter (26) to drive ex-pression of the HSVtk gene (27). In CMV-hsp70, the murine hsp70 gene(16) is driven by the CMV promoter in pCR3.1 (Invitrogen Life Technol-ogies). In pCD40L, the murine CD40L gene is driven by the CMVpromoter.
The adenovirus expressing murine TNF-� was a gift from Dr. ZhouXing, (McMaster University, Hamilton, Ontario, Canada).
RT-PCR
Skin samples at the site of plasmid injection were snap-frozen in liquidnitrogen. RNA was prepared with the Qiagen RNA extraction kit. Onemicrograms of total cellular RNA was reverse transcribed in a 20-�l vol-ume using oligo(dT) as a primer. A cDNA equivalent of 1 ng RNA wasamplified by PCR for a variety of murine cytokines or melanoma/melano-cyte Ags as described previously (24, 28) (details of the primers are avail-able upon request).
Splenocyte preparation
Splenocytes enriched in lymphocytes were prepared from spleens by stan-dard techniques (29) and Lympholyte-M density separation (CedarlaneLaboratories). CD8� T cells were purified from spleens using the MACSCD8a (Ly-2) Microbead magnetic cell sorting system (Miltenyi Biotec).
Ag presentation assays and tetramers
Freshly purified splenocyte populations were washed in PBS and pulsedwith 5 �M peptide for 2 h at 37oC before being incubated with purifiedCD8� T cells or splenocytes harvested from mice from the appropriatetreatment groups. Seventy-two hours later, splenocytes were subjected toFACS analysis or cell-free supernatants were tested for IFN-� by ELISA(BD Pharmingen). The synthetic, H-2Kb-restricted peptides hgp10025–33,KVPRNQDWL, TRP-2180–188 SVYDFFVWL (30) and Ova SIINFEKL
(31) were synthesized at the Mayo Foundation Core Facility (Rochester,MN). We used the altered ligand from hgp100, as opposed to the murineepitope, because it has been shown to be presented more effectively in thecontext of H-2Kb-restricted murine DC (6). Tetramers bound with theH-2Kb-restricted SIINFEKL or TRP-2 180–188 SVYDFFVWL peptides arecommercially available from Beckman Coulter.
ELISPOT analysis for IFN-� secretion
IFN-� ELISPOT assays were purchased from BD Pharmingen and used asrecommended. Splenocytes were stimulated in the presence of the appro-priate peptide in triplicate cultures at a density of 250,000 splenocytes perwell. Spot numbers were determined 72 h later by computer-assisted imageanalyzer.
In vivo studies
All procedures were approved by the Mayo Foundation Institutional Ani-mal Care and Use Committee. C57BL/10ScNJ mice have a deletion of theTlr4 gene. B6;129S6-Tnftm1Gkl/J mice are TNF deficient. C57BL/10ScNJand B6;129S6-Tnftm1Gkl/J mice were purchased from The Jackson Lab-oratory (stock nos. 003752 and 003008, respectively). C57BL/6 mice wereage- and sex-matched for individual experiments. To establish s.c. tumors,2 � 105 B16 cells were injected s.c. (100 �l) into the flank region. Animalswere examined daily until the tumor became palpable, whereafter the di-ameter, in two dimensions, was measured thrice weekly using calipers.Animals were killed when tumor size was �1.0 � 1.0 cm in two perpen-dicular directions. In all experiments, 10 mice per group were used unlessindicated otherwise in the figure legends.
Plasmid injections were conducted by intradermal injection (13, 32) ina final volume of 50 �l in PBS. For lymph node (LN)/cell tracking studies,5�-chloro-methyl-fluorescein diacetate (Cell Tracker Green; MolecularProbes) was added to the plasmid mix at a final concentration of 25 �Mbefore intradermal injections.
Tumor treatment protocols
For protocols aimed at treating established s.c. tumors, 2 � 105 B16 cellswere seeded subcutaneously in the right flank of C57BL/6 mice (day 0). Atthe appropriate day following tumor seeding, 20 or 30 �g of plasmid DNAwas injected intradermally on the contralateral flank. GCV at 50 mg/kg wasadministered i.p. In our hands, a 3-day established tumor was usually pal-pable under the skin; a 9-day established tumor was usually �0.3–0.4 cmin its longest diameter.
Statistics
Data from the animal studies were analyzed by the logrank test (33). Sta-tistical significance was determined at the level of p � 0.05.
ResultsHsp70 induces anti-tumor immunity through TLR-4-dependentsignaling
We have reported previously (13, 23) that three rounds of Tyr-HSVtk/CMV-hsp70/GCV treatment (a total of 9 intradermal plas-mid injections and 15 i.p. injections of GCV) cures 70–100% ofmice bearing 3-day established s.c. B16 tumors on the contralateralflank (Fig. 1A). Both the Tyr-HSVtk and the CMV-hsp-70 plas-mids are required for this therapy to be effective (Fig. 1A and Ref.13). Consistent with anti-tumor therapy, i.d. injection of the hsp70plasmid is required for the priming of TRP-2-specific responses inthe spleens of vaccinated mice (Fig. 1B), confirming our previousdata that TRP-2 and tyrosinase, but not gp100, are targets of thecurative T cell responses raised in vivo by inflammatory killing ofmelanocytes (13, 23).
Hsp70 has been reported to act as a chaperone of immunogenicpeptides (34–39), a cytokine (39–41), an immunogen (42–44), amaturation agent for dendritic cells (20), and as an inducer ofproinflammatory cytokines from monocytes (45) following liga-tion to TLR 2 and TLR 4 (40, 41). Therefore, to understand whichof these possible activities hsp70 is exerting in our system, wetested our protocol of inflammatory melanocyte killing in micelacking key elements of these effector responses. WhereasC57BL/6 mice bearing 3-day established B16 tumors were cured
4169The Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

by intradermal injections of Tyr-HSVtk/CMV-hsp70/GCV (Fig.1A), C57BL/10ScNJ mice, which carry a deletion of the Tlr4 gene,were completely unable to reject their tumors and died as rapidlyas control-treated C57BL/6 mice (Fig. 1C). These results suggestthat hsp70 plays a major role in the in vivo therapy by signalingthrough TLR-4 on the surface of endogenous cells.
Local expression of hsp70 induces priming of anti-melanoma/melanocyte responses through induction of TNF-�
Because of the striking dependence upon TLR-4 for hsp70-medi-ated therapy, and because we and others have reported that hsp70released from dying tumor cells induces proinflammatory cyto-kines (TNF-�, IL-1�, and IL-6) from macrophages (18, 40, 41,46), associated with anti-tumor immune responses (17, 18, 46), wescreened the site of hsp70 injection for the expression of suchcytokines. Of the seven different cytokines, we tested TNF-� cor-related consistently with the expression of hsp70 (Fig. 2A). In con-trast, hsp70 expression at the site of melanocyte killing was unableto induce local TNF-� expression in C57BL/10ScNJ mice lackingTLR-4 signaling (Fig. 2B).
Therefore, we hypothesized that hsp70 expression induces localimmune activation through TNF-� induction as a critical elementto the in vivo, CD8� T cell-mediated therapy of B16 tumors. Inthis respect, we observed that, whereas the Tyr-HSVtk/CMV-hsp70/GCV treatment effectively primed TRP-2 specific responsesin C57BL/6 mice (Figs. 1B and 2C), these effects were lost inB6;129S6-Tnftm1Gkl/J TNF-��/� mice (Fig. 2C). However, thelocal provision of TNF-� by delivery of an adenovirus expressingTNF-� at the site of plasmid injection was able to rescue the abilityof these mice to generate TRP-2 specific responses, but only ifhsp70 was provided in the plasmid injections (Fig. 2C).
Hsp70 expression induces trafficking of an APC-like populationto the draining LN
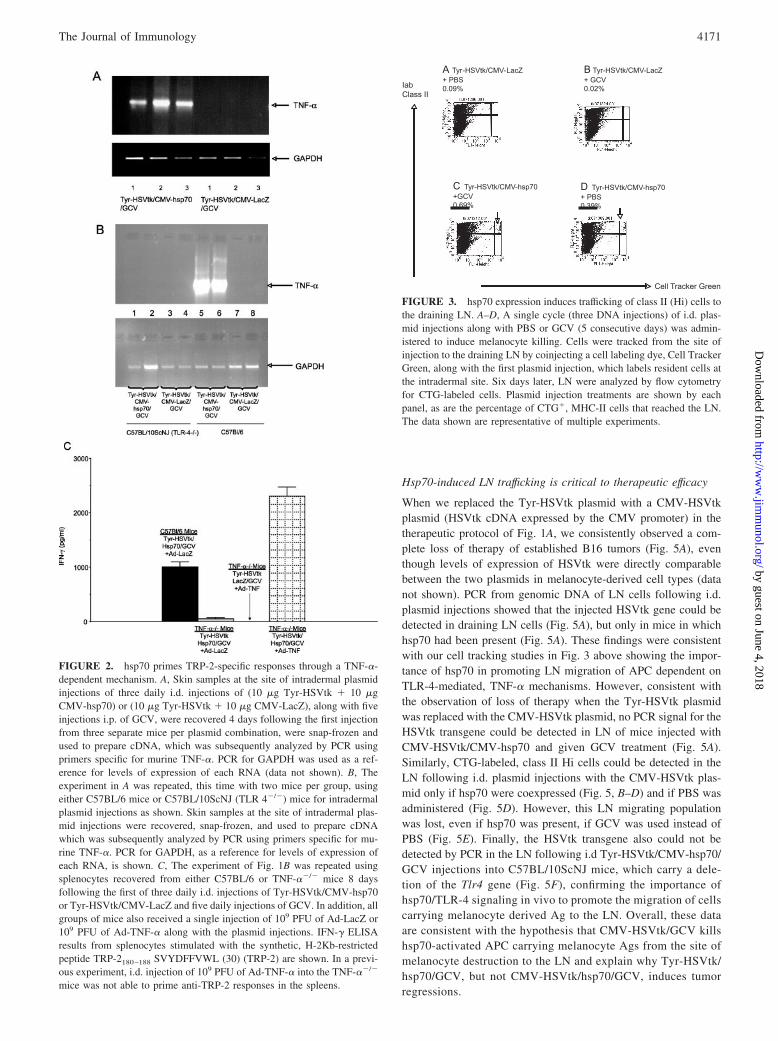
We hypothesized that local immune activation by hsp70, throughTLR-4-mediated signaling and TNF-� induction, induces migra-tion of APC from the site of plasmid injection to the LN. To testthis hypothesis, we tracked Cell Tracker Green-labeled cells fromthe site of injection to the draining LN. No CTG� cells could bedetected in draining LN following i.d. injection of CTG alone (datanot shown), or with any plasmid combination in which hsp70 wasnot present (Fig. 3, A and B). However, when hsp70 was expressedlocally, MHC class II (MHC-II)Hi, CTG� cells trafficked to theLN, irrespective of whether GCV was administered as well (redarrows Fig. 3, C and D), consistent with this being a population ofactivated APC. This CTG�/MHC-IIHi population was furthercharacterized and shown to consist of between 55 and 60%MAC3� cells (Fig. 3F) and �40% Mac3�, CD11c� cells (datanot shown).
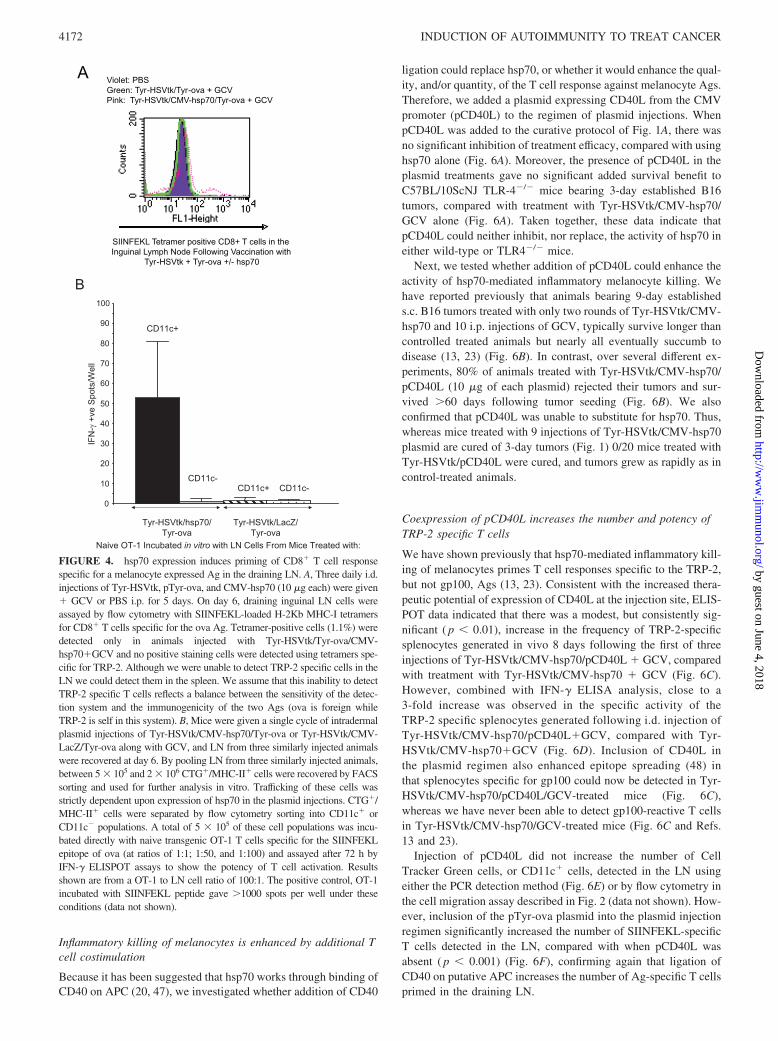
To test the functional relevance of this LN migration, we code-livered a plasmid (Tyr-ova) in which the cDNA of the model chickova Ag, expressed from the tyrosinase promoter, is only expressedin melanocytes. CD8� T cells specific for the H-2Kb-restrictedSIINFEKL peptide of ova could be detected in LN by tetrameranalysis, but only if pTyr-ova was coinjected with Tyr-HSVtk �GCV (to kill melanocytes and release ova Ag) and CMV-hsp70(consistent with migration to the LN of a putative APC population)(Fig. 4A). (We were unable to detect priming of naive T cell re-sponses to the TRP-2 Ag in these assays).
Because the Mac3 marker is not truly specific for macrophages,we also used transgenic OT-1 T cells (specific for H-2Kb-restrictedSIINFEKL) to monitor which of the Mac3�, or CD11c�, cell pop-ulations migrating to the LN are presenting the melanocyte-de-rived (ova) Ag. Fig. 4B shows that the SIINFEKL epitope of theova Ag, expressed from the melanocyte-specific tyrosinase pro-moter, was presented almost exclusively by the CD11c� popula-tion of cells, which hsp70 induces to migrate to the draining LN.
FIGURE 1. hsp70 stimulates anti-tumor immunity through TLR-4-me-diated mechanisms. A, C57BL/6 mice (10 per group) were seeded with B16tumors s.c. on day 1. On days 4–6, 11–13, and 18–20, mice were injectedon the right flank i.d. with Tyr-HSVtk � CMV-hsp70 (10 �g each) orTyr-HSVtk � CMV-LacZ plasmids. GCV was administered i.p. on days4–8, 11–15, and 18–22 as described previously (13, 23). Survival (tumorsize �1.0 cm) is shown. B, Splenocytes were recovered from naive mice,or from mice 5 days following the first of three daily i.d. injections ofTyr-HSVtk/CMV-hsp70 or Tyr-HSVtk/CMV-LacZ and 5 daily injectionsof GCV. Splenocytes from each treatment group were divided into fourseparate cultures and stimulated with either no added peptide (�) or withthe synthetic, H-2Kb-restricted peptides hgp100 25–33, KVPRNQDWL(gp100), TRP-2180–188 SVYDFFVWL (30) (TRP-2), or OVA SIINFEKL(31) (ova) at 500,000 splenocytes per well in triplicate. Seventy-twohours later, supernatants were recovered and assayed for IFN-� by ELISA.Error bars represent SDs. Results from two separate mice per plasmidtreatment are shown. C, The experiment of A was repeated using TLR-4�/�
C57BL/10ScNJ mice to seed the B16 tumors.
4170 INDUCTION OF AUTOIMMUNITY TO TREAT CANCER
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Hsp70-induced LN trafficking is critical to therapeutic efficacy
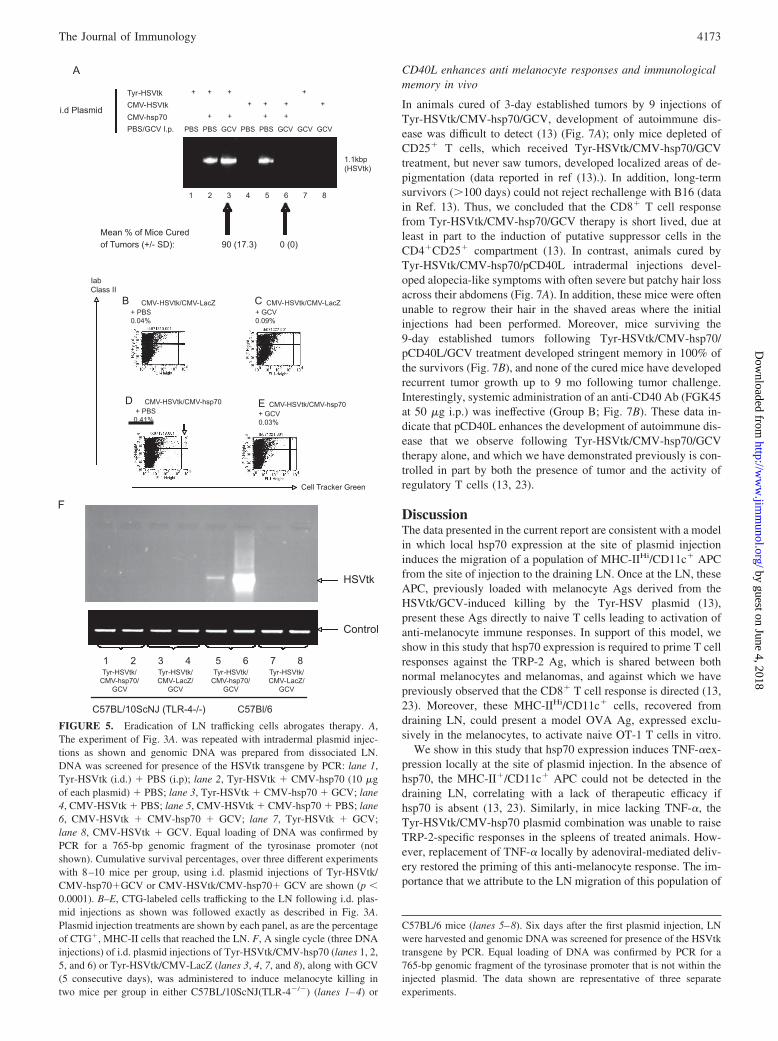
When we replaced the Tyr-HSVtk plasmid with a CMV-HSVtkplasmid (HSVtk cDNA expressed by the CMV promoter) in thetherapeutic protocol of Fig. 1A, we consistently observed a com-plete loss of therapy of established B16 tumors (Fig. 5A), eventhough levels of expression of HSVtk were directly comparablebetween the two plasmids in melanocyte-derived cell types (datanot shown). PCR from genomic DNA of LN cells following i.d.plasmid injections showed that the injected HSVtk gene could bedetected in draining LN cells (Fig. 5A), but only in mice in whichhsp70 had been present (Fig. 5A). These findings were consistentwith our cell tracking studies in Fig. 3 above showing the impor-tance of hsp70 in promoting LN migration of APC dependent onTLR-4-mediated, TNF-� mechanisms. However, consistent withthe observation of loss of therapy when the Tyr-HSVtk plasmidwas replaced with the CMV-HSVtk plasmid, no PCR signal for theHSVtk transgene could be detected in LN of mice injected withCMV-HSVtk/CMV-hsp70 and given GCV treatment (Fig. 5A).Similarly, CTG-labeled, class II Hi cells could be detected in theLN following i.d. plasmid injections with the CMV-HSVtk plas-mid only if hsp70 were coexpressed (Fig. 5, B–D) and if PBS wasadministered (Fig. 5D). However, this LN migrating populationwas lost, even if hsp70 was present, if GCV was used instead ofPBS (Fig. 5E). Finally, the HSVtk transgene also could not bedetected by PCR in the LN following i.d Tyr-HSVtk/CMV-hsp70/GCV injections into C57BL/10ScNJ mice, which carry a dele-tion of the Tlr4 gene (Fig. 5F), confirming the importance ofhsp70/TLR-4 signaling in vivo to promote the migration of cellscarrying melanocyte derived Ag to the LN. Overall, these dataare consistent with the hypothesis that CMV-HSVtk/GCV killshsp70-activated APC carrying melanocyte Ags from the site ofmelanocyte destruction to the LN and explain why Tyr-HSVtk/hsp70/GCV, but not CMV-HSVtk/hsp70/GCV, induces tumorregressions.
FIGURE 2. hsp70 primes TRP-2-specific responses through a TNF-�-dependent mechanism. A, Skin samples at the site of intradermal plasmidinjections of three daily i.d. injections of (10 �g Tyr-HSVtk � 10 �gCMV-hsp70) or (10 �g Tyr-HSVtk � 10 �g CMV-LacZ), along with fiveinjections i.p. of GCV, were recovered 4 days following the first injectionfrom three separate mice per plasmid combination, were snap-frozen andused to prepare cDNA, which was subsequently analyzed by PCR usingprimers specific for murine TNF-�. PCR for GAPDH was used as a ref-erence for levels of expression of each RNA (data not shown). B, Theexperiment in A was repeated, this time with two mice per group, usingeither C57BL/6 mice or C57BL/10ScNJ (TLR 4�/�) mice for intradermalplasmid injections as shown. Skin samples at the site of intradermal plas-mid injections were recovered, snap-frozen, and used to prepare cDNAwhich was subsequently analyzed by PCR using primers specific for mu-rine TNF-�. PCR for GAPDH, as a reference for levels of expression ofeach RNA, is shown. C, The experiment of Fig. 1B was repeated usingsplenocytes recovered from either C57BL/6 or TNF-��/� mice 8 daysfollowing the first of three daily i.d. injections of Tyr-HSVtk/CMV-hsp70or Tyr-HSVtk/CMV-LacZ and five daily injections of GCV. In addition, allgroups of mice also received a single injection of 109 PFU of Ad-LacZ or109 PFU of Ad-TNF-� along with the plasmid injections. IFN-� ELISAresults from splenocytes stimulated with the synthetic, H-2Kb-restrictedpeptide TRP-2180–188 SVYDFFVWL (30) (TRP-2) are shown. In a previ-ous experiment, i.d. injection of 109 PFU of Ad-TNF-� into the TNF-��/�
mice was not able to prime anti-TRP-2 responses in the spleens.
FIGURE 3. hsp70 expression induces trafficking of class II (Hi) cells tothe draining LN. A–D, A single cycle (three DNA injections) of i.d. plas-mid injections along with PBS or GCV (5 consecutive days) was admin-istered to induce melanocyte killing. Cells were tracked from the site ofinjection to the draining LN by coinjecting a cell labeling dye, Cell TrackerGreen, along with the first plasmid injection, which labels resident cells atthe intradermal site. Six days later, LN were analyzed by flow cytometryfor CTG-labeled cells. Plasmid injection treatments are shown by eachpanel, as are the percentage of CTG�, MHC-II cells that reached the LN.The data shown are representative of multiple experiments.
4171The Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Inflammatory killing of melanocytes is enhanced by additional Tcell costimulation
Because it has been suggested that hsp70 works through binding ofCD40 on APC (20, 47), we investigated whether addition of CD40
ligation could replace hsp70, or whether it would enhance the qual-ity, and/or quantity, of the T cell response against melanocyte Ags.Therefore, we added a plasmid expressing CD40L from the CMVpromoter (pCD40L) to the regimen of plasmid injections. WhenpCD40L was added to the curative protocol of Fig. 1A, there wasno significant inhibition of treatment efficacy, compared with usinghsp70 alone (Fig. 6A). Moreover, the presence of pCD40L in theplasmid treatments gave no significant added survival benefit toC57BL/10ScNJ TLR-4�/� mice bearing 3-day established B16tumors, compared with treatment with Tyr-HSVtk/CMV-hsp70/GCV alone (Fig. 6A). Taken together, these data indicate thatpCD40L could neither inhibit, nor replace, the activity of hsp70 ineither wild-type or TLR4�/� mice.
Next, we tested whether addition of pCD40L could enhance theactivity of hsp70-mediated inflammatory melanocyte killing. Wehave reported previously that animals bearing 9-day establisheds.c. B16 tumors treated with only two rounds of Tyr-HSVtk/CMV-hsp70 and 10 i.p. injections of GCV, typically survive longer thancontrolled treated animals but nearly all eventually succumb todisease (13, 23) (Fig. 6B). In contrast, over several different ex-periments, 80% of animals treated with Tyr-HSVtk/CMV-hsp70/pCD40L (10 �g of each plasmid) rejected their tumors and sur-vived �60 days following tumor seeding (Fig. 6B). We alsoconfirmed that pCD40L was unable to substitute for hsp70. Thus,whereas mice treated with 9 injections of Tyr-HSVtk/CMV-hsp70plasmid are cured of 3-day tumors (Fig. 1) 0/20 mice treated withTyr-HSVtk/pCD40L were cured, and tumors grew as rapidly as incontrol-treated animals.
Coexpression of pCD40L increases the number and potency ofTRP-2 specific T cells
We have shown previously that hsp70-mediated inflammatory kill-ing of melanocytes primes T cell responses specific to the TRP-2,but not gp100, Ags (13, 23). Consistent with the increased thera-peutic potential of expression of CD40L at the injection site, ELIS-POT data indicated that there was a modest, but consistently sig-nificant ( p � 0.01), increase in the frequency of TRP-2-specificsplenocytes generated in vivo 8 days following the first of threeinjections of Tyr-HSVtk/CMV-hsp70/pCD40L � GCV, comparedwith treatment with Tyr-HSVtk/CMV-hsp70 � GCV (Fig. 6C).However, combined with IFN-� ELISA analysis, close to a3-fold increase was observed in the specific activity of theTRP-2 specific splenocytes generated following i.d. injection ofTyr-HSVtk/CMV-hsp70/pCD40L�GCV, compared with Tyr-HSVtk/CMV-hsp70�GCV (Fig. 6D). Inclusion of CD40L inthe plasmid regimen also enhanced epitope spreading (48) inthat splenocytes specific for gp100 could now be detected in Tyr-HSVtk/CMV-hsp70/pCD40L/GCV-treated mice (Fig. 6C),whereas we have never been able to detect gp100-reactive T cellsin Tyr-HSVtk/CMV-hsp70/GCV-treated mice (Fig. 6C and Refs.13 and 23).
Injection of pCD40L did not increase the number of CellTracker Green cells, or CD11c� cells, detected in the LN usingeither the PCR detection method (Fig. 6E) or by flow cytometry inthe cell migration assay described in Fig. 2 (data not shown). How-ever, inclusion of the pTyr-ova plasmid into the plasmid injectionregimen significantly increased the number of SIINFEKL-specificT cells detected in the LN, compared with when pCD40L wasabsent ( p � 0.001) (Fig. 6F), confirming again that ligation ofCD40 on putative APC increases the number of Ag-specific T cellsprimed in the draining LN.
FIGURE 4. hsp70 expression induces priming of CD8� T cell responsespecific for a melanocyte expressed Ag in the draining LN. A, Three daily i.d.injections of Tyr-HSVtk, pTyr-ova, and CMV-hsp70 (10 �g each) were given� GCV or PBS i.p. for 5 days. On day 6, draining inguinal LN cells wereassayed by flow cytometry with SIINFEKL-loaded H-2Kb MHC-I tetramersfor CD8� T cells specific for the ova Ag. Tetramer-positive cells (1.1%) weredetected only in animals injected with Tyr-HSVtk/Tyr-ova/CMV-hsp70�GCV and no positive staining cells were detected using tetramers spe-cific for TRP-2. Although we were unable to detect TRP-2 specific cells in theLN we could detect them in the spleen. We assume that this inability to detectTRP-2 specific T cells reflects a balance between the sensitivity of the detec-tion system and the immunogenicity of the two Ags (ova is foreign whileTRP-2 is self in this system). B, Mice were given a single cycle of intradermalplasmid injections of Tyr-HSVtk/CMV-hsp70/Tyr-ova or Tyr-HSVtk/CMV-LacZ/Tyr-ova along with GCV, and LN from three similarly injected animalswere recovered at day 6. By pooling LN from three similarly injected animals,between 5 � 105 and 2 � 106 CTG�/MHC-II� cells were recovered by FACSsorting and used for further analysis in vitro. Trafficking of these cells wasstrictly dependent upon expression of hsp70 in the plasmid injections. CTG�/MHC-II� cells were separated by flow cytometry sorting into CD11c� orCD11c� populations. A total of 5 � 105 of these cell populations was incu-bated directly with naive transgenic OT-1 T cells specific for the SIINFEKLepitope of ova (at ratios of 1:1; 1:50, and 1:100) and assayed after 72 h byIFN-� ELISPOT assays to show the potency of T cell activation. Resultsshown are from a OT-1 to LN cell ratio of 100:1. The positive control, OT-1incubated with SIINFEKL peptide gave �1000 spots per well under theseconditions (data not shown).
4172 INDUCTION OF AUTOIMMUNITY TO TREAT CANCER
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
CD40L enhances anti melanocyte responses and immunologicalmemory in vivo
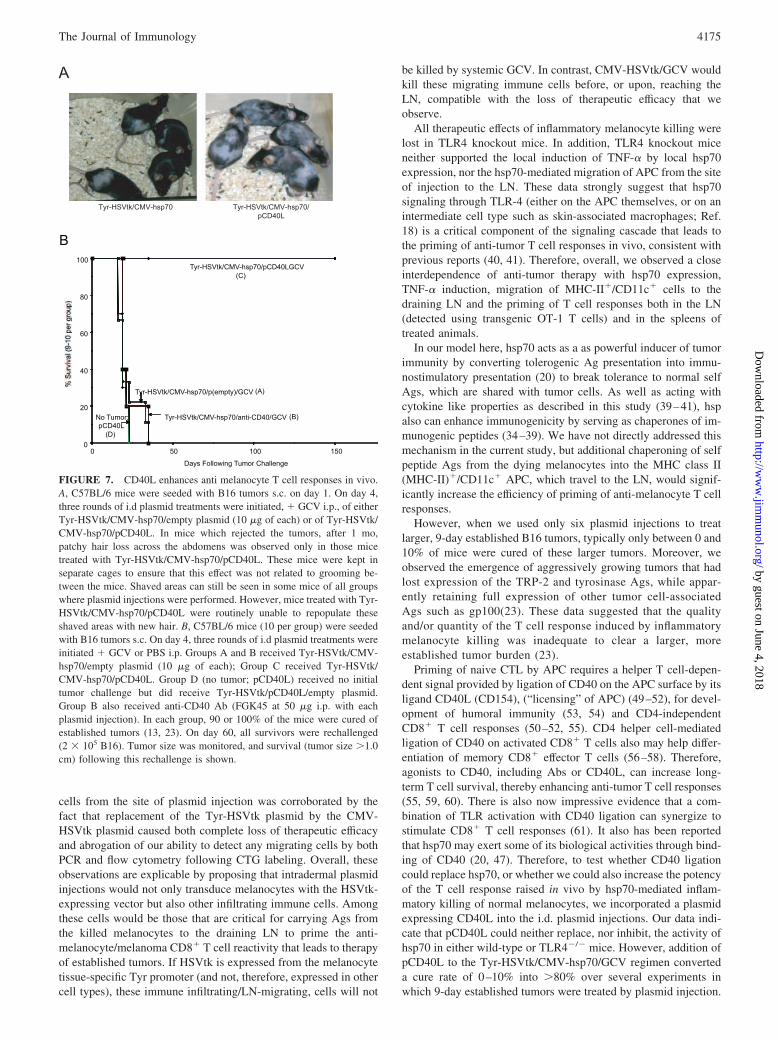
In animals cured of 3-day established tumors by 9 injections ofTyr-HSVtk/CMV-hsp70/GCV, development of autoimmune dis-ease was difficult to detect (13) (Fig. 7A); only mice depleted ofCD25� T cells, which received Tyr-HSVtk/CMV-hsp70/GCVtreatment, but never saw tumors, developed localized areas of de-pigmentation (data reported in ref (13).). In addition, long-termsurvivors (�100 days) could not reject rechallenge with B16 (datain Ref. 13). Thus, we concluded that the CD8� T cell responsefrom Tyr-HSVtk/CMV-hsp70/GCV therapy is short lived, due atleast in part to the induction of putative suppressor cells in theCD4�CD25� compartment (13). In contrast, animals cured byTyr-HSVtk/CMV-hsp70/pCD40L intradermal injections devel-oped alopecia-like symptoms with often severe but patchy hair lossacross their abdomens (Fig. 7A). In addition, these mice were oftenunable to regrow their hair in the shaved areas where the initialinjections had been performed. Moreover, mice surviving the9-day established tumors following Tyr-HSVtk/CMV-hsp70/pCD40L/GCV treatment developed stringent memory in 100% ofthe survivors (Fig. 7B), and none of the cured mice have developedrecurrent tumor growth up to 9 mo following tumor challenge.Interestingly, systemic administration of an anti-CD40 Ab (FGK45at 50 �g i.p.) was ineffective (Group B; Fig. 7B). These data in-dicate that pCD40L enhances the development of autoimmune dis-ease that we observe following Tyr-HSVtk/CMV-hsp70/GCVtherapy alone, and which we have demonstrated previously is con-trolled in part by both the presence of tumor and the activity ofregulatory T cells (13, 23).
DiscussionThe data presented in the current report are consistent with a modelin which local hsp70 expression at the site of plasmid injectioninduces the migration of a population of MHC-IIHi/CD11c� APCfrom the site of injection to the draining LN. Once at the LN, theseAPC, previously loaded with melanocyte Ags derived from theHSVtk/GCV-induced killing by the Tyr-HSV plasmid (13),present these Ags directly to naive T cells leading to activation ofanti-melanocyte immune responses. In support of this model, weshow in this study that hsp70 expression is required to prime T cellresponses against the TRP-2 Ag, which is shared between bothnormal melanocytes and melanomas, and against which we havepreviously observed that the CD8� T cell response is directed (13,23). Moreover, these MHC-IIHi/CD11c� cells, recovered fromdraining LN, could present a model OVA Ag, expressed exclu-sively in the melanocytes, to activate naive OT-1 T cells in vitro.
We show in this study that hsp70 expression induces TNF-�ex-pression locally at the site of plasmid injection. In the absence ofhsp70, the MHC-II�/CD11c� APC could not be detected in thedraining LN, correlating with a lack of therapeutic efficacy ifhsp70 is absent (13, 23). Similarly, in mice lacking TNF-�, theTyr-HSVtk/CMV-hsp70 plasmid combination was unable to raiseTRP-2-specific responses in the spleens of treated animals. How-ever, replacement of TNF-� locally by adenoviral-mediated deliv-ery restored the priming of this anti-melanocyte response. The im-portance that we attribute to the LN migration of this population of
FIGURE 5. Eradication of LN trafficking cells abrogates therapy. A,The experiment of Fig. 3A. was repeated with intradermal plasmid injec-tions as shown and genomic DNA was prepared from dissociated LN.DNA was screened for presence of the HSVtk transgene by PCR: lane 1,Tyr-HSVtk (i.d.) � PBS (i.p); lane 2, Tyr-HSVtk � CMV-hsp70 (10 �gof each plasmid) � PBS; lane 3, Tyr-HSVtk � CMV-hsp70 � GCV; lane4, CMV-HSVtk � PBS; lane 5, CMV-HSVtk � CMV-hsp70 � PBS; lane6, CMV-HSVtk � CMV-hsp70 � GCV; lane 7, Tyr-HSVtk � GCV;lane 8, CMV-HSVtk � GCV. Equal loading of DNA was confirmed byPCR for a 765-bp genomic fragment of the tyrosinase promoter (notshown). Cumulative survival percentages, over three different experimentswith 8–10 mice per group, using i.d. plasmid injections of Tyr-HSVtk/CMV-hsp70�GCV or CMV-HSVtk/CMV-hsp70� GCV are shown (p �0.0001). B–E, CTG-labeled cells trafficking to the LN following i.d. plas-mid injections as shown was followed exactly as described in Fig. 3A.Plasmid injection treatments are shown by each panel, as are the percentageof CTG�, MHC-II cells that reached the LN. F, A single cycle (three DNAinjections) of i.d. plasmid injections of Tyr-HSVtk/CMV-hsp70 (lanes 1, 2,5, and 6) or Tyr-HSVtk/CMV-LacZ (lanes 3, 4, 7, and 8), along with GCV(5 consecutive days), was administered to induce melanocyte killing intwo mice per group in either C57BL/10ScNJ(TLR-4�/�) (lanes 1–4) or
C57BL/6 mice (lanes 5–8). Six days after the first plasmid injection, LNwere harvested and genomic DNA was screened for presence of the HSVtktransgene by PCR. Equal loading of DNA was confirmed by PCR for a765-bp genomic fragment of the tyrosinase promoter that is not within theinjected plasmid. The data shown are representative of three separateexperiments.
4173The Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
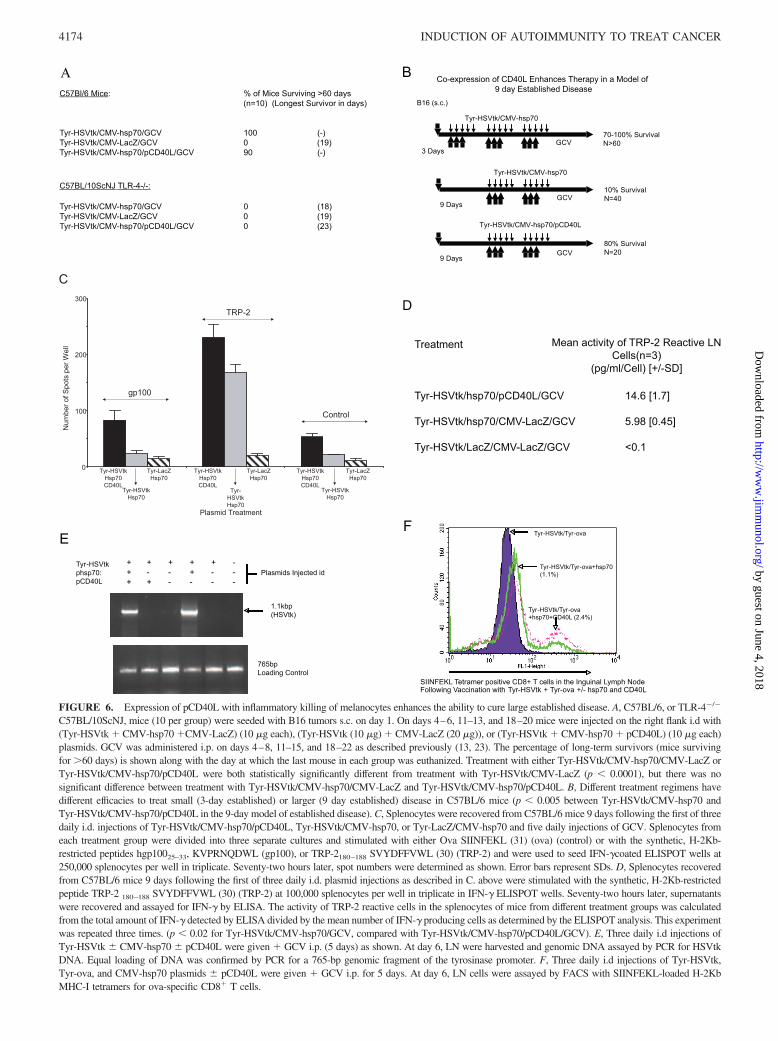
FIGURE 6. Expression of pCD40L with inflammatory killing of melanocytes enhances the ability to cure large established disease. A, C57BL/6, or TLR-4�/�
C57BL/10ScNJ, mice (10 per group) were seeded with B16 tumors s.c. on day 1. On days 4–6, 11–13, and 18–20 mice were injected on the right flank i.d with(Tyr-HSVtk � CMV-hsp70 �CMV-LacZ) (10 �g each), (Tyr-HSVtk (10 �g) � CMV-LacZ (20 �g)), or (Tyr-HSVtk � CMV-hsp70 � pCD40L) (10 �g each)plasmids. GCV was administered i.p. on days 4–8, 11–15, and 18–22 as described previously (13, 23). The percentage of long-term survivors (mice survivingfor �60 days) is shown along with the day at which the last mouse in each group was euthanized. Treatment with either Tyr-HSVtk/CMV-hsp70/CMV-LacZ orTyr-HSVtk/CMV-hsp70/pCD40L were both statistically significantly different from treatment with Tyr-HSVtk/CMV-LacZ (p � 0.0001), but there was nosignificant difference between treatment with Tyr-HSVtk/CMV-hsp70/CMV-LacZ and Tyr-HSVtk/CMV-hsp70/pCD40L. B, Different treatment regimens havedifferent efficacies to treat small (3-day established) or larger (9 day established) disease in C57BL/6 mice (p � 0.005 between Tyr-HSVtk/CMV-hsp70 andTyr-HSVtk/CMV-hsp70/pCD40L in the 9-day model of established disease). C, Splenocytes were recovered from C57BL/6 mice 9 days following the first of threedaily i.d. injections of Tyr-HSVtk/CMV-hsp70/pCD40L, Tyr-HSVtk/CMV-hsp70, or Tyr-LacZ/CMV-hsp70 and five daily injections of GCV. Splenocytes fromeach treatment group were divided into three separate cultures and stimulated with either Ova SIINFEKL (31) (ova) (control) or with the synthetic, H-2Kb-restricted peptides hgp10025–33, KVPRNQDWL (gp100), or TRP-2180–188 SVYDFFVWL (30) (TRP-2) and were used to seed IFN-�coated ELISPOT wells at250,000 splenocytes per well in triplicate. Seventy-two hours later, spot numbers were determined as shown. Error bars represent SDs. D, Splenocytes recoveredfrom C57BL/6 mice 9 days following the first of three daily i.d. plasmid injections as described in C. above were stimulated with the synthetic, H-2Kb-restrictedpeptide TRP-2 180–188 SVYDFFVWL (30) (TRP-2) at 100,000 splenocytes per well in triplicate in IFN-� ELISPOT wells. Seventy-two hours later, supernatantswere recovered and assayed for IFN-� by ELISA. The activity of TRP-2 reactive cells in the splenocytes of mice from different treatment groups was calculatedfrom the total amount of IFN-� detected by ELISA divided by the mean number of IFN-� producing cells as determined by the ELISPOT analysis. This experimentwas repeated three times. (p � 0.02 for Tyr-HSVtk/CMV-hsp70/GCV, compared with Tyr-HSVtk/CMV-hsp70/pCD40L/GCV). E, Three daily i.d injections ofTyr-HSVtk � CMV-hsp70 � pCD40L were given � GCV i.p. (5 days) as shown. At day 6, LN were harvested and genomic DNA assayed by PCR for HSVtkDNA. Equal loading of DNA was confirmed by PCR for a 765-bp genomic fragment of the tyrosinase promoter. F, Three daily i.d injections of Tyr-HSVtk,Tyr-ova, and CMV-hsp70 plasmids � pCD40L were given � GCV i.p. for 5 days. At day 6, LN cells were assayed by FACS with SIINFEKL-loaded H-2KbMHC-I tetramers for ova-specific CD8� T cells.
4174 INDUCTION OF AUTOIMMUNITY TO TREAT CANCER
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
cells from the site of plasmid injection was corroborated by thefact that replacement of the Tyr-HSVtk plasmid by the CMV-HSVtk plasmid caused both complete loss of therapeutic efficacyand abrogation of our ability to detect any migrating cells by bothPCR and flow cytometry following CTG labeling. Overall, theseobservations are explicable by proposing that intradermal plasmidinjections would not only transduce melanocytes with the HSVtk-expressing vector but also other infiltrating immune cells. Amongthese cells would be those that are critical for carrying Ags fromthe killed melanocytes to the draining LN to prime the anti-melanocyte/melanoma CD8� T cell reactivity that leads to therapyof established tumors. If HSVtk is expressed from the melanocytetissue-specific Tyr promoter (and not, therefore, expressed in othercell types), these immune infiltrating/LN-migrating, cells will not
be killed by systemic GCV. In contrast, CMV-HSVtk/GCV wouldkill these migrating immune cells before, or upon, reaching theLN, compatible with the loss of therapeutic efficacy that weobserve.
All therapeutic effects of inflammatory melanocyte killing werelost in TLR4 knockout mice. In addition, TLR4 knockout miceneither supported the local induction of TNF-� by local hsp70expression, nor the hsp70-mediated migration of APC from the siteof injection to the LN. These data strongly suggest that hsp70signaling through TLR-4 (either on the APC themselves, or on anintermediate cell type such as skin-associated macrophages; Ref.18) is a critical component of the signaling cascade that leads tothe priming of anti-tumor T cell responses in vivo, consistent withprevious reports (40, 41). Therefore, overall, we observed a closeinterdependence of anti-tumor therapy with hsp70 expression,TNF-� induction, migration of MHC-II�/CD11c� cells to thedraining LN and the priming of T cell responses both in the LN(detected using transgenic OT-1 T cells) and in the spleens oftreated animals.
In our model here, hsp70 acts as a as powerful inducer of tumorimmunity by converting tolerogenic Ag presentation into immu-nostimulatory presentation (20) to break tolerance to normal selfAgs, which are shared with tumor cells. As well as acting withcytokine like properties as described in this study (39–41), hspalso can enhance immunogenicity by serving as chaperones of im-munogenic peptides (34–39). We have not directly addressed thismechanism in the current study, but additional chaperoning of selfpeptide Ags from the dying melanocytes into the MHC class II(MHC-II)�/CD11c� APC, which travel to the LN, would signif-icantly increase the efficiency of priming of anti-melanocyte T cellresponses.
However, when we used only six plasmid injections to treatlarger, 9-day established B16 tumors, typically only between 0 and10% of mice were cured of these larger tumors. Moreover, weobserved the emergence of aggressively growing tumors that hadlost expression of the TRP-2 and tyrosinase Ags, while appar-ently retaining full expression of other tumor cell-associatedAgs such as gp100(23). These data suggested that the qualityand/or quantity of the T cell response induced by inflammatorymelanocyte killing was inadequate to clear a larger, moreestablished tumor burden (23).
Priming of naive CTL by APC requires a helper T cell-depen-dent signal provided by ligation of CD40 on the APC surface by itsligand CD40L (CD154), (“licensing” of APC) (49–52), for devel-opment of humoral immunity (53, 54) and CD4-independentCD8� T cell responses (50–52, 55). CD4 helper cell-mediatedligation of CD40 on activated CD8� T cells also may help differ-entiation of memory CD8� effector T cells (56–58). Therefore,agonists to CD40, including Abs or CD40L, can increase long-term T cell survival, thereby enhancing anti-tumor T cell responses(55, 59, 60). There is also now impressive evidence that a com-bination of TLR activation with CD40 ligation can synergize tostimulate CD8� T cell responses (61). It also has been reportedthat hsp70 may exert some of its biological activities through bind-ing of CD40 (20, 47). Therefore, to test whether CD40 ligationcould replace hsp70, or whether we could also increase the potencyof the T cell response raised in vivo by hsp70-mediated inflam-matory killing of normal melanocytes, we incorporated a plasmidexpressing CD40L into the i.d. plasmid injections. Our data indi-cate that pCD40L could neither replace, nor inhibit, the activity ofhsp70 in either wild-type or TLR4�/� mice. However, addition ofpCD40L to the Tyr-HSVtk/CMV-hsp70/GCV regimen converteda cure rate of 0–10% into �80% over several experiments inwhich 9-day established tumors were treated by plasmid injection.
FIGURE 7. CD40L enhances anti melanocyte T cell responses in vivo.A, C57BL/6 mice were seeded with B16 tumors s.c. on day 1. On day 4,three rounds of i.d plasmid treatments were initiated, � GCV i.p., of eitherTyr-HSVtk/CMV-hsp70/empty plasmid (10 �g of each) or of Tyr-HSVtk/CMV-hsp70/pCD40L. In mice which rejected the tumors, after 1 mo,patchy hair loss across the abdomens was observed only in those micetreated with Tyr-HSVtk/CMV-hsp70/pCD40L. These mice were kept inseparate cages to ensure that this effect was not related to grooming be-tween the mice. Shaved areas can still be seen in some mice of all groupswhere plasmid injections were performed. However, mice treated with Tyr-HSVtk/CMV-hsp70/pCD40L were routinely unable to repopulate theseshaved areas with new hair. B, C57BL/6 mice (10 per group) were seededwith B16 tumors s.c. On day 4, three rounds of i.d plasmid treatments wereinitiated � GCV or PBS i.p. Groups A and B received Tyr-HSVtk/CMV-hsp70/empty plasmid (10 �g of each); Group C received Tyr-HSVtk/CMV-hsp70/pCD40L. Group D (no tumor; pCD40L) received no initialtumor challenge but did receive Tyr-HSVtk/pCD40L/empty plasmid.Group B also received anti-CD40 Ab (FGK45 at 50 �g i.p. with eachplasmid injection). In each group, 90 or 100% of the mice were cured ofestablished tumors (13, 23). On day 60, all survivors were rechallenged(2 � 105 B16). Tumor size was monitored, and survival (tumor size �1.0cm) following this rechallenge is shown.
4175The Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Surprisingly, the results of Fig. 7B show that only addition of theplasmid form of CD40L, rather than the anti-CD40 Ab, conferredmemory to mice cured of 3-day disease by the Tyr-HSVtk/CMV-hsp70 treatment, which pCD40L alone was unable to achieve(Group D). We hypothesize that both temporal and spatial locationof CD40 ligation on the APC being loaded with melanocyte-de-rived Ags, which subsequently migrate to the LN, may be criticalin mediating the effects of pCD40L. Thus, it may be that the anti-CD40 Ab cannot access the local site of melanocyte killing, wherethe CD40L is expressed from the injected plasmids. Ligation ofCD40 on the APC at that site may be important before these APCreach the LN where, presumably, the anti-CD40� Ab is readilyaccessible.
Moreover, inclusion of pCD40L increased the severity of auto-immune manifestations in treated mice, which were only observedfollowing treatment with Tyr-HSVtk/CMV-hsp70 alone followingprior depletion of CD4�CD25� T reg cells (13). We observedalopecia-like autoimmune hair loss, along with occasional locallyintense vitiligo concentrated around the eyes, the top of the headand shoulders, but with no obvious ocular involvement. Suchsymptoms are reminiscent of Vogt-Koyanagi-Harada disease, con-sidered to be an autoimmune disease directed against melanocytesand specifically the TRP-1 and 2 Ags (62, 63). In addition, micecured of large established tumors by Tyr-HSVtk/CMV-hsp70/pCD40L/GCV treatment developed almost complete and long-term immunological memory against tumor rechallenge. Onceagain, such memory was never observed in mice cured of smallertumors by the Tyr-HSVtk/CMV-hsp70/GCV regimen (13).
These data increased T cell-mediated therapy, autoimmunity,and immunological memory, are all consistent with the predictedactions of CD40L on activating APC (49–52) and on differentia-tion of memory CD8� effector T cells (56–58). Thus, we observedthat, although pCD40L did not increase the numbers of MHC-II�/CD11c� APC that migrated to the LN under the influence of hsp70expression, these APC primed more naive T cells against the mel-anocyte-expressed ova Ag in vitro than did APC recovered fromLN following Tyr-HSVtk/CMV-hsp70/GCV treatment. In addi-tion, inclusion of pCD40L not only induced both greater numbersof TRP-2-specific splenocytes than Tyr-HSVtk/CMV-hsp70/GCVtreatment, but those splenocytes also secreted greater amounts ofIFN-� per cell. These results suggest that CD40L binding to CD40expressed on the APC, which migrate to the LN, licenses theseAPC to activate T cells both more potently and in higher numbers.Nonetheless, the increases in numbers of effector T cells wererather modest suggesting that CD40L expression may have addi-tional effects as well as enhancing the priming of Ag-specific Tcells in the LN. Consistent with our demonstration that the activityof the activated T cells was increased in CD40L-treated animals,we have recently observed that CD40L may be exerting potentimmunostimulatory effects through modulation of regulatory T cellactivity in vivo through mechanisms that are currently under in-vestigation in our laboratory (L. Sanchez-Perez, manuscript inpreparation).
In summary, we show in this study that CD40L-enhanced,hsp70-mediated inflammatory melanocyte killing can be used tocure large established tumors and to confer immunological mem-ory against tumor cells, although a concomitant increase in auto-immune sequelae also is produced.
AcknowledgmentsWe thank Toni Higgins for expert secretarial assistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Pardoll, D. M. 2002. Spinning Mol. Immunol. into successful immunotherapy.
Nat. Rev. Immunol. 2: 227–238.2. Dudley, M. E., J. R. Wunderlich, P. F. Robbins, J. C. Yang, P. Hwu,
D. J. Schwartzentruber, S. L. Topalian, R. Sherry, N. P. Restifo, A. M. Hubicki,et al. 2002. Cancer regression and autoimmunity in patients after clonal repopu-lation with antitumor lymphocytes. Science 298: 850–854.
3. Hodi, F. S., M. C. Mihm, R. J. Soiffer, F. G. Haluska, M. Butler, M. V. Seiden,T. Davis, R. Henry-Spires, S. MacRae, A. Willman, et al. 2003. Biologic activityof cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previouslyvaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl.Acad. Sci. USA 100: 4712–4717.
4. Phan, G. Q., J. C. Yang, R. M. Sherry, P. Hwu, S. L. Topalian,D. J. Schwartzentruber, N. P. Restifo, L. R. Haworth, C. A. Seipp, L. J. Freezer,et al. 2003. Cancer regression and autoimmunity induced by cytotoxic T lym-phocyte-associated antigen 4 blockade in patients with metastatic melanoma.Proc. Natl. Acad. Sci. USA 100: 8372–8377.
5. Engelhard, V. H., T. N. Bullock, T. A. Colella, S. L. Sheasley, and D. W. Mullins.2002. Antigens derived from melanocyte differentiation proteins: self-tolerance,autoimmunity, and use for cancer immunotherapy. Immunol. Rev. 188: 136–146.
6. Overwijk, W., M. Theoret, S. Finkelstein, D. Surman, L. de Jong,F. Vyth-Dreese, T. Dellemijn, P. Antony, P. Spiess, D. Palmer, et al. 2003. Tumorregression and autoimmunity after reversal of a functionally tolerant state ofself-reactive CD8� T cells. J. Exp. Med. 198: 569–580.
7. Parmiani, G. 1993. Tumor immunity as autoimmunity: tumor antigens includenormal self proteins which stimulate anergic peripheral T cells. Immunol. Today14: 536–538.
8. Pardoll, D. M. 1999. Inducing autoimmune disease to treat cancer. 96:5340–5342.
9. Das, P. K., R. M. van den Wijngaard, A. Wankowicz-Kalinska, andI. C. Le Poole. 2001. A symbiotic concept of autoimmunity and tumor immunity:lessons from vitiligo. Trends Immunol. 22: 130–136.
10. Bronte, V., E. Apolloni, R. Ronca, P. Zamboni, W. W. Overwijk, D. R. Surman,N. P. Restifo, and P. Zanovello. 2000. Genetic vaccination with “self” tyrosinase-related protein 2 causes melanoma eradication but not vitiligo. Cancer Res. 60:253–258.
11. Engelhorn, M. E., J. A. Guevara-Patino, G. Noffz, A. T. Hooper, O. Lou,J. S. Gold, B. J. Kappel, and A. N. Houghton. 2006. Autoimmunity and tumorimmunity induced by immune responses to mutations in self. Nat. Med. 12:198–206.
12. Gogas, H., J. Ioannovich, U. Dafni, C. Stavropoulou-Giokas, K. Frangia,D. Tsoutsos, P. Panagiotou, A. Polyzos, O. Papadopoulos, A. Stratigos, et al.2006. Prognostic significance of autoimmunity during treatment of melanomawith interferon. N. Engl. J. Med. 354: 758–760.
13. Daniels, G., L. Sanchez-Perez, T. Kottke, R. M. Diaz, J. Thompson, M. Lai,M. Gough, M. Karim, A. Bushell, H. Chong, et al. 2004. A simple method to cureestablished tumors by inflammatory killing of normal cells. Nat. Biotechnol. 22:1125–1132.
14. Ferrone, S. 2004. Immunotherapy dispenses with tumor antigens. Nat. Biotech-nol. 22: 1096–1098.
15. Moolten, F. L. 1994. Drug sensitivity (“suicide”) genes for selective cancer che-motherapy. 1: 279–287.
16. Melcher, A. A., S. Todryk, N. Hardwick, M. Ford, M. Jacobson, and R. G. Vile.1998. Tumor immunogenicity is determined by the mechanism of cell death viainduction of heat shock protein expression. Nat. Med. 4: 581–587.
17. Todryk, S., A. A. Melcher, N. Hardwick, E. Linardakis, A. Bateman,M. P. Colombo, A. Stoppacciaro, and R. G. Vile. 1999. Heat shock protein 70induced during tumor cell killing induces Th1 cytokines and targets immaturedendritic cell precursors to enhance antigen uptake. J. Immunol. 163: 1398–1408.
18. Gough, M. J., A. A. Melcher, A. Ahmed, M. R. Crittenden, D. S. Riddle,E. Linardakis, A. N. Ruchatz, and R. G. Vile. 2001. Macrophages orchestrate theimmune response to tumor cell death. Cancer Res. 61: 7240–7247.
19. Srivastava, P. K. 2003. Hypothesis: controlled necrosis as a tool for immuno-therapy of human cancer. Cancer Immun. 3: 4.
20. Millar, D. G., K. M. Garza, B. Odermatt, A. R. Elford, N. Ono, Z. Li, andP. S. Ohashi. 2003. Hsp70 promotes antigen-presenting cell function and convertsT-cell tolerance to autoimmunity in vivo. Nat. Med. 9: 1469–1476.
21. Srivastava, P., H. Udono, N. E. Blachere, and Z. Li. 1994. Heat shock prtoeinstransfer peptides during antigen processing and CTL priming. Immunogenetics39: 93–98.
22. Walker, L. S. K., and A. K. Abbas. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2: 11–19.
23. Sanchez-Perez, L., T. Kottke, R. M. Diaz, J. Thompson, S. Holmen, G. Daniels,and R. G. Vile. 2005. Potent selection of antigen loss variants of B16 melanomafollowing inflammatory killing of melanocytes in vivo. Cancer Res. 65:2009–2017.
24. Linardakis, E., A. Bateman, V. Phan, A. Ahmed, M. Gough, K. Olivier,F. Errington, K. Harrington, A. Melcher, and R. Vile. 2002. Enhancing the ef-ficacy of a weak allogeneic melanoma vaccine by viral fusogenic membraneglycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 62: 5495–5504.
25. Ganss, R., L. Montoliu, A. P. Monaghan, and G. Schutz. 1994. A cell-specificenhancer far upstream of the mouse tyrosinase gene confers high level and copynumber-related expression in transgenic mice. EMBO J. 13: 3083–3093.
26. Vile, R. G., and I. R. Hart. 1993. In vitro and in vivo targeting of gene expressionto melanoma cells. Cancer Res. 53: 962–967.
27. Vile, R. G., and I. R. Hart. 1993. Use of tissue-specific expression of the HerpesSimplex Virus thymidine kinase gene to inhibit growth of established murine
4176 INDUCTION OF AUTOIMMUNITY TO TREAT CANCER
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

melanomas following direct intratumoral injection of DNA. Cancer Res. 53:3860–3864.
28. Vile, R. G., S. C. Castleden, J. Marshall, R. Camplejohn, C. Upton, andH. Chong. 1997. Generation of an anti-tumour immune response in a non-im-munogenic tumour: HSVtk-killing in vivo stimulates a mononuclear cell infiltrateand a Th1-like profile of intratumoural cytokine expression. Int. J. Cancer 71:267–274.
29. Coligan, J. E., A. M. Kruisbeek, D. H. Margulies, E. M. Shevach, and W. Strober.1998. Isolation of mouse mononuclear cells. In Current Protocols in Immunol-ogy. Wiley, New York. pp. 3.1.2–3.2.4.
30. Dyall, R., W. B. Bowne, L. W. Weber, J. LeMaoult, P. Szabo, Y. Moroi,G. Piskun, J. J. Lewis, A. N. Houghton, and J. Nikolic-Zugic. 1998. Heterocliticimmunization induces tumor immunity. J. Exp. Med. 188: 1553–1561.
31. Hogquist, K. A., S. C. Jameson, W. R. Health, J. L. Howard, M. J. Bevan, andF. R. Carbone. 1994. T cell receptor antagonistic peptides induce positive selec-tion. Cell 76: 17.
32. Bonnotte, B., M. Gough, V. Phan, A. Ahmed, H. Chong, F. Martin, and R. Vile.2003. Intradermal injection, as opposed to subcutaneous injection, enhances im-munogenicity and suppresses tumorigenicity of tumor cells. Cancer Res. 63:2145–2149.
33. Altman, D. G. 1991. Analysis of survival times. In Practical Statistics for Med-ical Research. Chapman & Hall, New York, pp. 365–395.
34. Singh-Jasuja, H., R. E. Toes, P. Spee, C. Munz, N. Hilf, S. P. Schoenberger,P. Ricciardi-Castagnoli, J. Neefjes, H. G. Rammensee, D. Arnold-Schild, andH. Schild. 2000. Cross-presentation of glycoprotein 96-associated antigens onmajor histocompatibility complex class I molecules requires receptor-mediatedendocytosis. J. Exp. Med. 191: 1965–1974.
35. Castellino, F., P. E. Boucher, K. Eichelberg, M. Mayhew, J. E. Rothman,A. N. Houghton, and R. N. Germain. 2000. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class Iantigen presentation via two distinct processing pathways. J. Exp. Med. 191:1957–1964.
36. Tamura, Y., P. Peng, K. Liu, M. Daou, and P. K. Srivastava. 1997. Immunother-apy of tumors with autologous tumor-derived heat shock protein preparations.Science 278: 117–120.
37. Srivastava, P. K., A. Menoret, S. Basu, R. J. Binder, and K. L. McQuade. 1998.Heat shock proteins come of age: primitive functions acquire new roles in anadaptive world. 8: 657–665.
38. Suzue, K., X. Zhou, H. N. Eisen, and R. A. Young. 1997. Heat shock fusionproteins as vehicles for antigen delivery into the major histocompatibility com-plex class I presentation pathway. Proc. Natl. Acad. Sci. USA 94: 13146–13151.
39. MacAry, P. A., B. Javid, R. A. Floto, K. G. Smith, W. Oehlmann, M. Singh, andP. J. Lehner. 2004. HSP70 peptide binding mutants separate antigen deliveryfrom dendritic cell stimulation. Immunity 20: 95–106.
40. Asea, A., S. K. Kraeft, E. A. Kurt-Jones, M. A. Stevenson, L. B. Chen,R. W. Finberg, G. C. Koo, and S. K. Calderwood. 2000. HSP70 stimulates cy-tokine production through a CD14-dependent pathway, demonstrating its dualrole as a chaperone and cytokine. Nat. Med. 6: 435–442.
41. Asea, A., M. Rehli, E. Kabingu, J. A. Boch, O. Bare, P. E. Auron,M. A. Stevenson, and S. K. Calderwood. 2002. Novel signal transduction path-way utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4.J. Biol. Chem. 277: 15028–15034.
42. Lukacs, K. V., D. B. Lowrie, R. W. Stokes, and M. J. Colston. 1993. Tumor cellstransfected with a bacterial heat-shock gene lose tumorigenicity and induce pro-tection against tumors. J. Exp. Med. 178: 343–334.
43. Lukacs, K. V., A. Nakakes, C. J. Atkins, D. B. Lowrie, and M. J. Colston. 1997.In vivo gene therapy of malignant tumors with heat shock protein-65 gene. GeneTher. 4: 346–350.
44. Suzue, K., and R. A. Young. 1996. Adjuvant-free hsp70 fusion protein systemelicits humoral and cellular immune responses to HIV-1 p24. J. Immunol. 156:873–879.
45. Moroi, Y., M. Mayhew, J. Trcka, M. H. Hoe, Y. Takechi, F. U. Hartl,J. E. Rothman, and A. N. Houghton. 2000. Induction of cellular immunity byimmunization with novel hybrid peptides complexed to heat shock protein 70.Proc. Natl. Acad. Sci. USA 97: 3485–3490.
46. Gough, M. J., A. A. Melcher, M. R. Crittenden, L. Sanchez, R. Voellmy, andR. G. Vile. 2004. Induction of cell stress through gene transfer of an engineeredheat shock transcription factor enhances tumor immunogenicity. Gene Therapy11: 1099–1104.
47. Becker, T., F. U. Hartl, and F. Wieland. 2002. CD40, an extracellular receptor forbinding and uptake of Hsp70-peptide complexes. J. Cell Biol. 158: 1227–1285.
48. Ribas, A., J. M. Timmerman, L. H. Butterfield, and J. S. Economou. 2003. De-terminant spreading and tumor responses after peptide-based cancer immuno-therapy. Trends Immunol. 24: 58–61.
49. Heath, W. R., and F. R. Carbone. 2001. Cross-presentation in viral immunity andself-tolerance. Nat. Rev. Immunol. 1: 126–134.
50. Ridge, J. P., F. Di Rosa, and P. Matzinger. 1998. A conditioned dendritic cell canbe a temporal bridge between a CD4� T-helper and a T-killer cell. Nature 393:474–478.
51. Bennett, S. R., F. R. Carbone, F. Karamalis, R. A. Flavell, J. F. Miller, andW. R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40signalling. Nature 393: 478–480.
52. Schoenberger, S. P., R. E. Toes, E. I. van der Voort, R. Offringa, and C. J. Melief.1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40Linteractions. Nature 393: 480–483.
53. Renshaw, B. R., W. C. Fanslow III, R. J. Armitage, K. A. Campbell, D. Liggitt,B. Wright, B. L. Davison, and C. R. Maliszewski. 1994. Humoral immune re-sponses in CD40 ligand-deficient mice. J. Exp. Med. 180: 1889–1900.
54. Farrington, M., L. S. Grosmaire, S. Nonoyama, S. H. Fischer, D. Hollenbaugh,J. A. Ledbetter, R. J. Noelle, A. Aruffo, and H. D. Ochs. 1994. CD40 ligandexpression is defective in a subset of patients with common variable immuno-deficiency. Proc. Natl. Acad. Sci. USA 91: 1099–1103.
55. Lefrancois, L., J. D. Altman, K. Williams, and S. Olson. 2000. Soluble antigenand CD40 triggering are sufficient to induce primary and memory cytotoxic Tcells. J. Immunol. 164: 725–732.
56. Bourgeois, C., B. Rocha, and C. Tanchot. 2002. A role for CD40 expression onCD8� T cells in the generation of CD8� T cell memory. Science 297:2060–2063.
57. Shedlock, D. J., and H. Shen. 2003. Requirement for CD4 T cell help in gener-ating functional CD8 T cell memory. Science 300: 337–339.
58. Sun, J. C., and M. J. Bevan. 2003. Defective CD8 T cell memory following acuteinfection without CD4 T cell help. Science 300: 339–342.
59. Sotomayor, E. M., I. Borrello, E. Tubb, F. M. Rattis, H. Bien, Z. Lu, S. Fein,S. Schoenberger, and H. I. Levitsky. 1999. Conversion of tumor-specific CD4�
T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 5:780–787.
60. Diehl, L., A. T. den Boer, S. P. Schoenberger, E. I. van der Voort,T. N. Schumacher, C. J. Melief, R. Offringa, and R. E. Toes. 1999. CD40 acti-vation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tol-erance and augments anti-tumor vaccine efficacy. Nat. Med. 5: 774–779.
61. Ahonen, C. L., C. L. Doxsee, S. M. McGurran, T. R. Riter, W. F. Wade,R. J. Barth, J. P. Vasilakos, R. J. Noelle, and R. M. Kedl. 2004. Combined TLRand CD40 triggering induces potent CD8� T cell expansion with variable de-pendence on type I IFN. J. Exp. Med. 199: 775–784.
62. Yamaki, K., K. Gocho, K. Hayakawa, I. Kondo, and S. Sakuragi. 2000. Tyrosi-nase family proteins are antigens specific to Vogt-Koyanagi-Harada disease.J. Immunol. 165: 7323–7329.
63. Gocho, K., I. Kondo, and K. Yamaki. 2001. Identification of autoreactive Tcells in Vogt-Koyanagi-Harada disease. Invest. Ophthalmol. Vis. Sci. 42:2004 –2009.
4177The Journal of Immunology
by guest on June 4, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















