
KCNE1 enhances phosphatidylinositol 4,5-bisphosphate PIP ...jcui/publications/iks1.pdf · KCNE1...
7
KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP 2 ) sensitivity of I Ks to modulate channel activity Yang Li, Mark A. Zaydman, Dick Wu 1 , Jingyi Shi, Michael Guan, Brett Virgin-Downey, and Jianmin Cui 2 Department of Biomedical Engineering, Center for the Investigation of Membrane Excitability Disorders, Cardiac Bioelectricity and Arrhythmia Center, Washington University, St. Louis, MO 63130 Edited by Richard W. Aldrich, University of Texas at Austin, Austin, TX, and approved April 18, 2011 (received for review January 17, 2011) Phosphatidylinositol 4,5-bisphosphate (PIP 2 ) is necessary for the function of various ion channels. The potassium channel, I Ks , is important for cardiac repolarization and requires PIP 2 to activate. Here we show that the auxiliary subunit of I Ks , KCNE1, increases PIP 2 sensitivity 100-fold over channels formed by the pore-forming KCNQ1 subunits alone, which effectively amplifies current because native PIP 2 levels in the membrane are insufficient to activate all KCNQ1 channels. A juxtamembranous site in the KCNE1 C terminus is a key structural determinant of PIP 2 sensitivity. Long QT syn- drome associated mutations of this site lower PIP 2 affinity, result- ing in reduced current. Application of exogenous PIP 2 to these mutants restores wild-type channel activity. These results reveal a vital role of PIP 2 for KCNE1 modulation of I Ks channels that may represent a common mechanism of auxiliary subunit modula- tion of many ion channels. K CNQ1 α-subunits coassemble with KCNE1 β-subunits to form the cardiac slow-delayed rectifier channel, I Ks , which conducts a potassium current that is important for the termina- tion of the cardiac action potential. Although exogenous expres- sion of KCNQ1 alone is sufficient to produce a voltage-gated channel, coexpression of KCNE1 with KCNQ1 leads to changes in current properties to resemble the physiologically important cardiac I Ks current (Fig. 1A) (1, 2). These changes include in- creased current amplitude, shift of the voltage dependence of ac- tivation toward more positive potentials, slowed activation and deactivation kinetics, and suppressed inactivation, all of which are essential for the physiological role of I Ks (3). After many years of investigation, it is still poorly understood how the KCNE1 peptide exerts such a dramatic effect on the I Ks current. Loss-of- function mutations in I Ks lead to delayed cardiac cell repolariza- tion that manifests clinically as long QT (LQT) syndrome. Patients with LQT syndrome are predisposed to ventricular arrhythmia and suffer from syncope and a high risk of sudden cardiac death. At present, the molecular mechanisms of disease pathogenesis are still unknown for many LQT-associated muta- tions in I Ks . Phosphatidylinositol 4,5-bisphosphate (PIP 2 ) is a minor acidic membrane lipid found primarily in the inner leaflet of the plasma membrane. PIP 2 has been shown to be a necessary cofactor for a wide variety of ion channels, e.g., voltage-gated K þ and Ca 2þ channels, transient receptor potential channels, inward rectifying K þ channels, and epithelial Na þ channels (4). Recently, I Ks has been shown to require PIP 2 for channel activity, and several LQT- associated mutations located in KCNQ1 have been suggested to decrease the PIP 2 affinity of I Ks (5, 6). Here we provide evidence that KCNE1, as a β-subunit, alters the function of I Ks by modu- lating the interaction between PIP 2 and the heteromeric ion channel complex. Knowledge of the molecular mechanisms of channel modulation by KCNE1 is of great importance as at least 36 LQT-associated mutations reside within KCNE1 (7–9). Results KCNE1 Slows the PIP 2 -Dependent Channel Rundown. To investigate the regulation of I Ks by PIP 2 , we expressed KCNQ1 with and without KCNE1 in oocytes from Xenopus laevis and recorded currents using patch-clamp and two-electrode voltage-clamp techniques. Currents recorded from inside-out membrane patches expressing KCNQ1 immediately and rapidly decayed fol- lowing patch excision such that channel activity was completely Fig. 1. KCNE1 slows the PIP 2 -dependent channel rundown. (A) KCNQ1 and KCNQ1 þ KCNE1 currents at various times after patch excision. Voltage was stepped from a holding potential of −80 to þ80 mV and then back to the holding potential. (B) Time dependence of current amplitude after patch excision. Normalized tail current amplitude at various time, I t ∕I 0 , is plotted; I 0 is the tail current amplitude immediately following patch excision. KCNQ1 (filled circles), n ¼ 9; KCNQ1 þ KCNE1 (open circles), n ¼ 12; KCNQ1þ KCNE1 þ 10 μM PIP 2 (open squares), n ¼ 6; KCNQ1 þ KCNE1 coexpressed with Ci-VSP (open diamonds), n ¼ 6. Solid lines are monoexponential fits to data. Voltage was stepped from a holding potential of −80 mV to þ80 mV for 5 s and then back to the holding potential for 5 s. (C) Time con- stants of exponential fits and initial delay to current rundown. (D) Normal- ized tail current amplitude at various times, I t ∕I 0 , with KCNQ1: KCNE1 mRNA ratio 1∶1 (open circles), n ¼ 12; 1∶0.05 (open diamonds), n ¼ 6; 1∶0.01 (open squares), n ¼ 6; 1∶0 (filled circles), n ¼ 9.(E) Time constants of exponential fits and initial delay to current rundown. In this and other figures, the data are presented as mean SEM. Author contributions: Y.L., M.A.Z., D.W., and J.C. designed research; Y.L., M.A.Z., J.S., M.G., and B.V.-D. performed research; Y.L. performed patch–clamp experiments in Fig. 1–5; M.A.Z. and M.G. performed voltage–clamp experiments in Fig. 4. J.S., M.G., and B.V.-D. performed molecular biology; Y.L. analyzed data; and Y.L., M.A.Z., D.W., and J.C. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 Present address: Stanford Institute for Neuro-innovation and Translational Neurosciences, Stanford, CA 94305. 2 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/ doi:10.1073/pnas.1100872108/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1100872108 PNAS Early Edition ∣ 1 of 6 BIOPHYSICS AND COMPUTATIONAL BIOLOGY
Transcript of KCNE1 enhances phosphatidylinositol 4,5-bisphosphate PIP ...jcui/publications/iks1.pdf · KCNE1...
KCNE1 enhances phosphatidylinositol 4,5-bisphosphate(PIP2) sensitivity of IKs to modulate channel activityYang Li, Mark A. Zaydman, Dick Wu1, Jingyi Shi, Michael Guan, Brett Virgin-Downey, and Jianmin Cui2
Department of Biomedical Engineering, Center for the Investigation of Membrane Excitability Disorders, Cardiac Bioelectricity and Arrhythmia Center,Washington University, St. Louis, MO 63130
Edited by Richard W. Aldrich, University of Texas at Austin, Austin, TX, and approved April 18, 2011 (received for review January 17, 2011)
Phosphatidylinositol 4,5-bisphosphate (PIP2) is necessary for thefunction of various ion channels. The potassium channel, IKs, isimportant for cardiac repolarization and requires PIP2 to activate.Here we show that the auxiliary subunit of IKs, KCNE1, increasesPIP2 sensitivity 100-fold over channels formed by the pore-formingKCNQ1 subunits alone, which effectively amplifies current becausenative PIP2 levels in the membrane are insufficient to activate allKCNQ1 channels. A juxtamembranous site in the KCNE1 C terminusis a key structural determinant of PIP2 sensitivity. Long QT syn-drome associated mutations of this site lower PIP2 affinity, result-ing in reduced current. Application of exogenous PIP2 to thesemutants restores wild-type channel activity. These results reveala vital role of PIP2 for KCNE1 modulation of IKs channels thatmay represent a common mechanism of auxiliary subunit modula-tion of many ion channels.
KCNQ1 α-subunits coassemble with KCNE1 β-subunits toform the cardiac slow-delayed rectifier channel, IKs, which
conducts a potassium current that is important for the termina-tion of the cardiac action potential. Although exogenous expres-sion of KCNQ1 alone is sufficient to produce a voltage-gatedchannel, coexpression of KCNE1 with KCNQ1 leads to changesin current properties to resemble the physiologically importantcardiac IKs current (Fig. 1A) (1, 2). These changes include in-creased current amplitude, shift of the voltage dependence of ac-tivation toward more positive potentials, slowed activation anddeactivation kinetics, and suppressed inactivation, all of whichare essential for the physiological role of IKs (3). After many yearsof investigation, it is still poorly understood how the KCNE1peptide exerts such a dramatic effect on the IKs current. Loss-of-function mutations in IKs lead to delayed cardiac cell repolariza-tion that manifests clinically as long QT (LQT) syndrome.Patients with LQT syndrome are predisposed to ventriculararrhythmia and suffer from syncope and a high risk of suddencardiac death. At present, the molecular mechanisms of diseasepathogenesis are still unknown for many LQT-associated muta-tions in IKs.
Phosphatidylinositol 4,5-bisphosphate (PIP2) is a minor acidicmembrane lipid found primarily in the inner leaflet of the plasmamembrane. PIP2 has been shown to be a necessary cofactor for awide variety of ion channels, e.g., voltage-gated Kþ and Ca2þchannels, transient receptor potential channels, inward rectifyingKþ channels, and epithelial Naþ channels (4). Recently, IKs hasbeen shown to require PIP2 for channel activity, and several LQT-associated mutations located in KCNQ1 have been suggested todecrease the PIP2 affinity of IKs (5, 6). Here we provide evidencethat KCNE1, as a β-subunit, alters the function of IKs by modu-lating the interaction between PIP2 and the heteromeric ionchannel complex. Knowledge of the molecular mechanisms ofchannel modulation by KCNE1 is of great importance as at least36 LQT-associated mutations reside within KCNE1 (7–9).
ResultsKCNE1 Slows the PIP2-Dependent Channel Rundown. To investigatethe regulation of IKs by PIP2, we expressed KCNQ1 with andwithout KCNE1 in oocytes from Xenopus laevis and recorded
currents using patch-clamp and two-electrode voltage-clamptechniques. Currents recorded from inside-out membranepatches expressing KCNQ1 immediately and rapidly decayed fol-lowing patch excision such that channel activity was completely
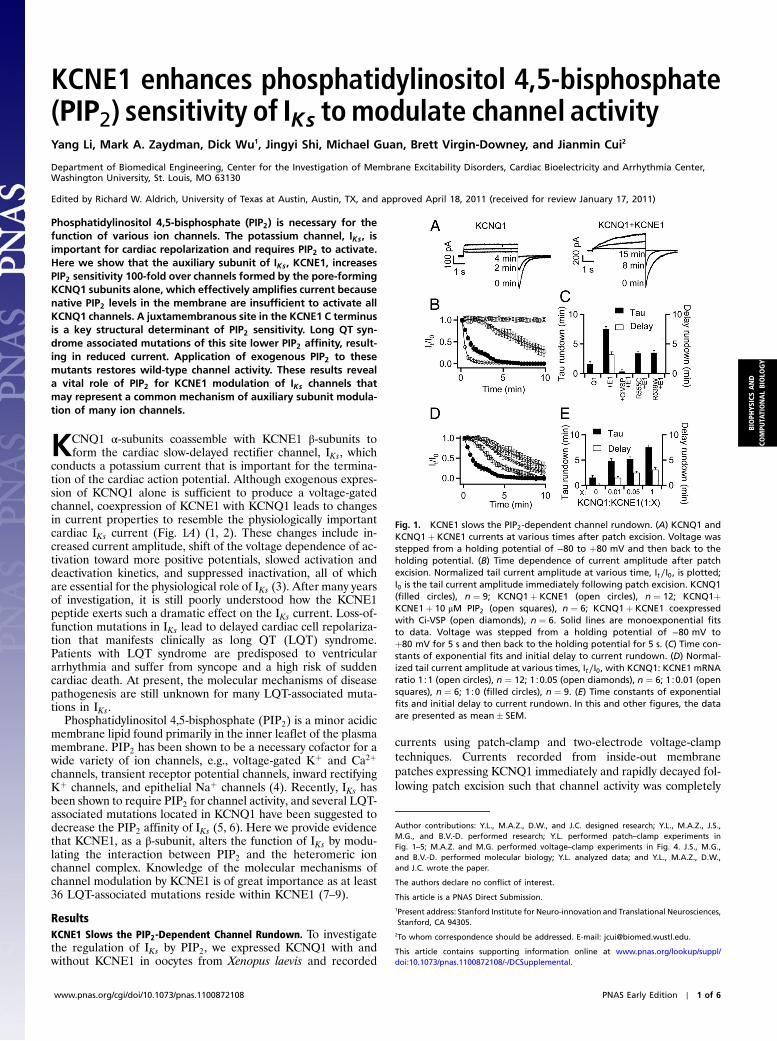
Fig. 1. KCNE1 slows the PIP2-dependent channel rundown. (A) KCNQ1 andKCNQ1þ KCNE1 currents at various times after patch excision. Voltage wasstepped from a holding potential of −80 to þ80 mV and then back to theholding potential. (B) Time dependence of current amplitude after patchexcision. Normalized tail current amplitude at various time, It∕I0, is plotted;I0 is the tail current amplitude immediately following patch excision. KCNQ1(filled circles), n ¼ 9; KCNQ1þ KCNE1 (open circles), n ¼ 12; KCNQ1þKCNE1þ 10 μM PIP2 (open squares), n ¼ 6; KCNQ1þ KCNE1 coexpressedwith Ci-VSP (open diamonds), n ¼ 6. Solid lines are monoexponential fitsto data. Voltage was stepped from a holding potential of −80 mV toþ80 mV for 5 s and then back to the holding potential for 5 s. (C) Time con-stants of exponential fits and initial delay to current rundown. (D) Normal-ized tail current amplitude at various times, It∕I0, with KCNQ1: KCNE1 mRNAratio 1∶1 (open circles), n ¼ 12; 1∶0.05 (open diamonds), n ¼ 6; 1∶0.01 (opensquares), n ¼ 6; 1∶0 (filled circles), n ¼ 9. (E) Time constants of exponentialfits and initial delay to current rundown. In this and other figures, the dataare presented as mean� SEM.
Author contributions: Y.L., M.A.Z., D.W., and J.C. designed research; Y.L., M.A.Z., J.S.,M.G., and B.V.-D. performed research; Y.L. performed patch–clamp experiments inFig. 1–5; M.A.Z. and M.G. performed voltage–clamp experiments in Fig. 4. J.S., M.G.,and B.V.-D. performed molecular biology; Y.L. analyzed data; and Y.L., M.A.Z., D.W.,and J.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1Present address: Stanford Institute for Neuro-innovation and Translational Neurosciences,Stanford, CA 94305.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1100872108/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1100872108 PNAS Early Edition ∣ 1 of 6
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
lost within 5 min (Fig. 1A). This rundown of current was fit wellwith a single exponential (Fig. 1B). However, after excisinginside-out membrane patches expressing KCNQ1þ KCNE1,currents remained stable over 3 min prior to rundown, whichwas not observed with KCNQ1 alone (Fig. 1B). The subsequentrundown of KCNQ1þ KCNE1 current was slower than thatobserved with KCNQ1 alone (Fig. 1 B and C). This rundownof IKs activity is attributed to the loss of PIP2 from the excisedmembrane patch (6). The PIP2 dependence of current rundownafter patch excision was supported by the following experi-ments. The voltage sensitive phosphatase from Ciona intestinalis(CiVSP) that dephosphorylates PIP2 upon membrane depolari-zation (10) was coexpressed with KCNQ1þ KCNE1. CiVSPhastens PIP2 loss from the membrane patch, resulting in a fasterrate of current rundown. Conversely, directly applying 10 μM ofexogenous PIP2 to the intracellular face of membrane patchesexpressing KCNQ1þ KCNE1 prevented current rundown forlonger than 10 min (Fig. 1 B and C). These results show thatPIP2 loss after patch excision leads to loss of channel activity.
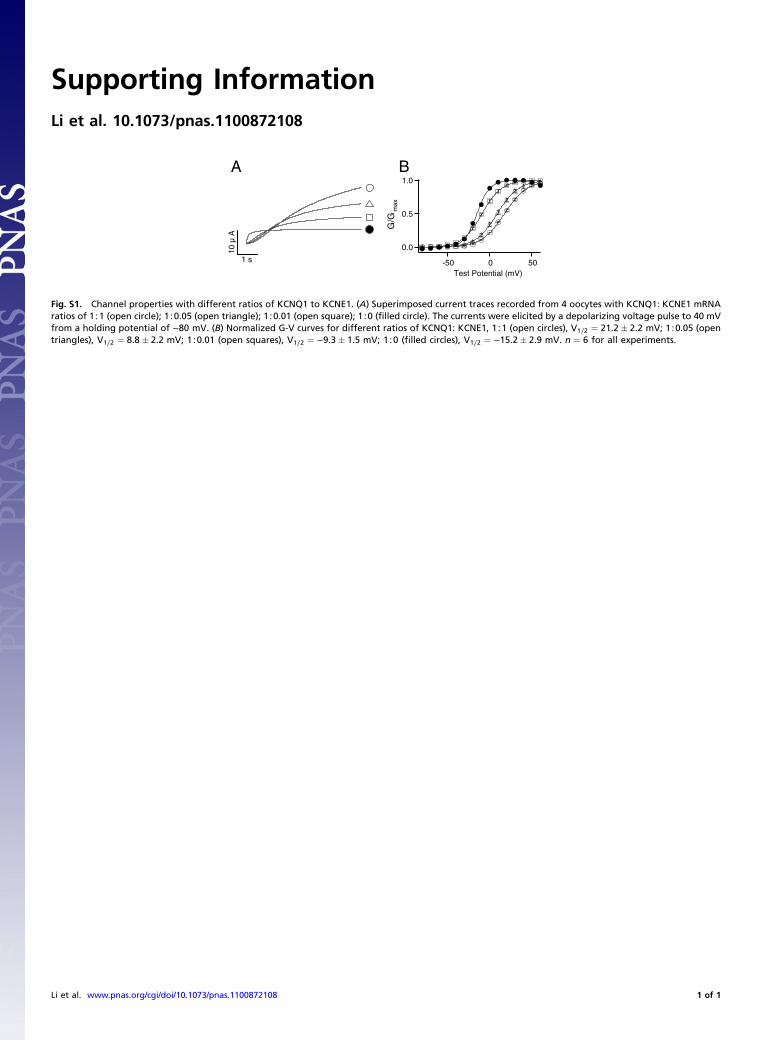
KCNQ1 activity is lost more quickly after patch excision thanKCNQ1þ KCNE1, indicating that KCNE1 slows PIP2-depen-dent rundown. To further demonstrate this effect of KCNE1,we injected oocytes with the same amount of KCNQ1 mRNAand various amounts of KCNE1 mRNAs in molar ratios of 1∶0,1∶0.01, 1∶0.05, and 1∶1. The activation kinetics and the steady-state voltage dependence of activation of the expressed channelsdepend on the KCNQ1∶KCNE1 ratio, which suggested that pore-forming KCNQ1 subunits are associated with varying numbers ofKCNE1 subunits (11–13) (Fig. S1). The time course of currentrundown became progressively longer with higher concentrationsof KCNE1 due to a longer delay and a larger time constant ofrundown (Fig. 1D). A lower molar ratio of KCNQ1∶KCNE1mRNA results in a larger portion of the expressed channelpopulation containing a higher stoichiometry of KCNE1∶KCNQ1, and these results show that channels with more KCNE1subunits have slower PIP2-dependent rundown (Fig. 1 D and E).
Why does the association of KCNE1 affect the time course ofPIP2-dependent rundown? To answer this question, we appliedPIP2 to the intracellular face of membrane patches after com-
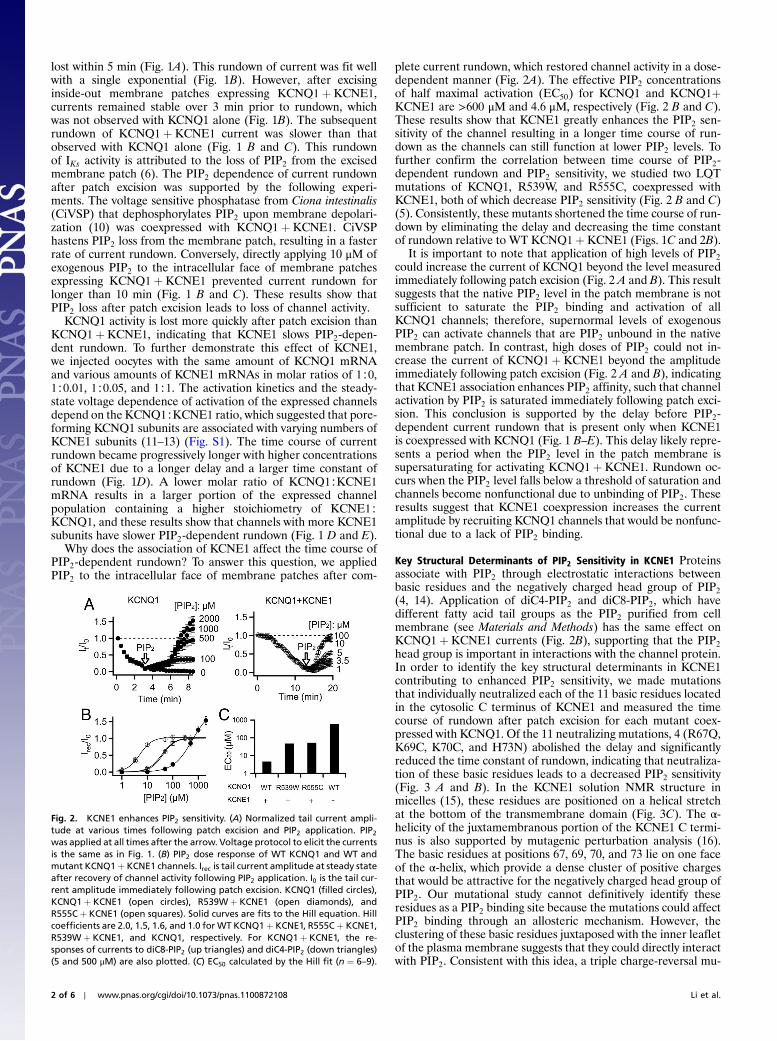
plete current rundown, which restored channel activity in a dose-dependent manner (Fig. 2A). The effective PIP2 concentrationsof half maximal activation (EC50) for KCNQ1 and KCNQ1þKCNE1 are >600 μM and 4.6 μM, respectively (Fig. 2 B and C).These results show that KCNE1 greatly enhances the PIP2 sen-sitivity of the channel resulting in a longer time course of run-down as the channels can still function at lower PIP2 levels. Tofurther confirm the correlation between time course of PIP2-dependent rundown and PIP2 sensitivity, we studied two LQTmutations of KCNQ1, R539W, and R555C, coexpressed withKCNE1, both of which decrease PIP2 sensitivity (Fig. 2 B and C)(5). Consistently, these mutants shortened the time course of run-down by eliminating the delay and decreasing the time constantof rundown relative to WT KCNQ1þ KCNE1 (Figs. 1C and 2B).
It is important to note that application of high levels of PIP2
could increase the current of KCNQ1 beyond the level measuredimmediately following patch excision (Fig. 2 A and B). This resultsuggests that the native PIP2 level in the patch membrane is notsufficient to saturate the PIP2 binding and activation of allKCNQ1 channels; therefore, supernormal levels of exogenousPIP2 can activate channels that are PIP2 unbound in the nativemembrane patch. In contrast, high doses of PIP2 could not in-crease the current of KCNQ1þ KCNE1 beyond the amplitudeimmediately following patch excision (Fig. 2 A and B), indicatingthat KCNE1 association enhances PIP2 affinity, such that channelactivation by PIP2 is saturated immediately following patch exci-sion. This conclusion is supported by the delay before PIP2-dependent current rundown that is present only when KCNE1is coexpressed with KCNQ1 (Fig. 1 B–E). This delay likely repre-sents a period when the PIP2 level in the patch membrane issupersaturating for activating KCNQ1þ KCNE1. Rundown oc-curs when the PIP2 level falls below a threshold of saturation andchannels become nonfunctional due to unbinding of PIP2. Theseresults suggest that KCNE1 coexpression increases the currentamplitude by recruiting KCNQ1 channels that would be nonfunc-tional due to a lack of PIP2 binding.
Key Structural Determinants of PIP2 Sensitivity in KCNE1 Proteinsassociate with PIP2 through electrostatic interactions betweenbasic residues and the negatively charged head group of PIP2
(4, 14). Application of diC4-PIP2 and diC8-PIP2, which havedifferent fatty acid tail groups as the PIP2 purified from cellmembrane (see Materials and Methods) has the same effect onKCNQ1þ KCNE1 currents (Fig. 2B), supporting that the PIP2
head group is important in interactions with the channel protein.In order to identify the key structural determinants in KCNE1contributing to enhanced PIP2 sensitivity, we made mutationsthat individually neutralized each of the 11 basic residues locatedin the cytosolic C terminus of KCNE1 and measured the timecourse of rundown after patch excision for each mutant coex-pressed with KCNQ1. Of the 11 neutralizing mutations, 4 (R67Q,K69C, K70C, and H73N) abolished the delay and significantlyreduced the time constant of rundown, indicating that neutraliza-tion of these basic residues leads to a decreased PIP2 sensitivity(Fig. 3 A and B). In the KCNE1 solution NMR structure inmicelles (15), these residues are positioned on a helical stretchat the bottom of the transmembrane domain (Fig. 3C). The α-helicity of the juxtamembranous portion of the KCNE1 C termi-nus is also supported by mutagenic perturbation analysis (16).The basic residues at positions 67, 69, 70, and 73 lie on one faceof the α-helix, which provide a dense cluster of positive chargesthat would be attractive for the negatively charged head group ofPIP2. Our mutational study cannot definitively identify theseresidues as a PIP2 binding site because the mutations could affectPIP2 binding through an allosteric mechanism. However, theclustering of these basic residues juxtaposed with the inner leafletof the plasma membrane suggests that they could directly interactwith PIP2. Consistent with this idea, a triple charge-reversal mu-
Fig. 2. KCNE1 enhances PIP2 sensitivity. (A) Normalized tail current ampli-tude at various times following patch excision and PIP2 application. PIP2was applied at all times after the arrow. Voltage protocol to elicit the currentsis the same as in Fig. 1. (B) PIP2 dose response of WT KCNQ1 and WT andmutant KCNQ1þ KCNE1 channels. Irec is tail current amplitude at steady stateafter recovery of channel activity following PIP2 application. I0 is the tail cur-rent amplitude immediately following patch excision. KCNQ1 (filled circles),KCNQ1þ KCNE1 (open circles), R539Wþ KCNE1 (open diamonds), andR555Cþ KCNE1 (open squares). Solid curves are fits to the Hill equation. Hillcoefficients are 2.0, 1.5, 1.6, and 1.0 for WT KCNQ1þ KCNE1, R555Cþ KCNE1,R539Wþ KCNE1, and KCNQ1, respectively. For KCNQ1þ KCNE1, the re-sponses of currents to diC8-PIP2 (up triangles) and diC4-PIP2 (down triangles)(5 and 500 μM) are also plotted. (C) EC50 calculated by the Hill fit (n ¼ 6–9).
2 of 6 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1100872108 Li et al.
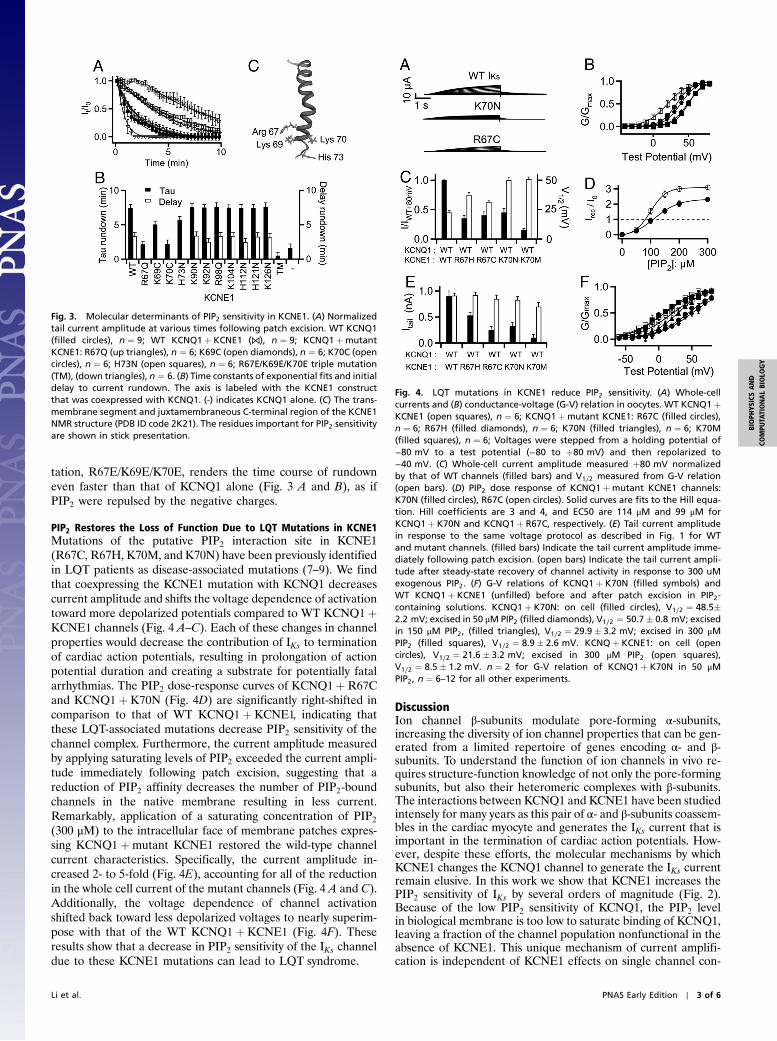
tation, R67E/K69E/K70E, renders the time course of rundowneven faster than that of KCNQ1 alone (Fig. 3 A and B), as ifPIP2 were repulsed by the negative charges.
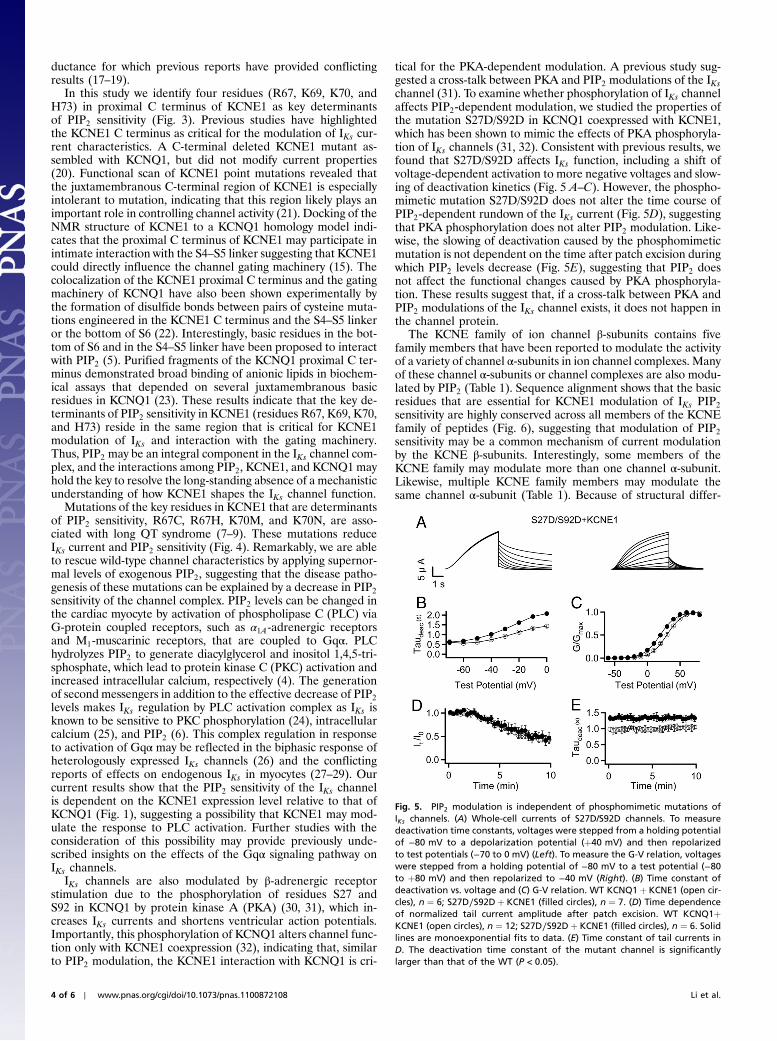
PIP2 Restores the Loss of Function Due to LQT Mutations in KCNE1Mutations of the putative PIP2 interaction site in KCNE1(R67C, R67H, K70M, and K70N) have been previously identifiedin LQT patients as disease-associated mutations (7–9). We findthat coexpressing the KCNE1 mutation with KCNQ1 decreasescurrent amplitude and shifts the voltage dependence of activationtoward more depolarized potentials compared to WT KCNQ1þKCNE1 channels (Fig. 4 A–C). Each of these changes in channelproperties would decrease the contribution of IKs to terminationof cardiac action potentials, resulting in prolongation of actionpotential duration and creating a substrate for potentially fatalarrhythmias. The PIP2 dose-response curves of KCNQ1þR67Cand KCNQ1þ K70N (Fig. 4D) are significantly right-shifted incomparison to that of WT KCNQ1þ KCNE1, indicating thatthese LQT-associated mutations decrease PIP2 sensitivity of thechannel complex. Furthermore, the current amplitude measuredby applying saturating levels of PIP2 exceeded the current ampli-tude immediately following patch excision, suggesting that areduction of PIP2 affinity decreases the number of PIP2-boundchannels in the native membrane resulting in less current.Remarkably, application of a saturating concentration of PIP2
(300 μM) to the intracellular face of membrane patches expres-sing KCNQ1þmutant KCNE1 restored the wild-type channelcurrent characteristics. Specifically, the current amplitude in-creased 2- to 5-fold (Fig. 4E), accounting for all of the reductionin the whole cell current of the mutant channels (Fig. 4 A and C).Additionally, the voltage dependence of channel activationshifted back toward less depolarized voltages to nearly superim-pose with that of the WT KCNQ1þ KCNE1 (Fig. 4F). Theseresults show that a decrease in PIP2 sensitivity of the IKs channeldue to these KCNE1 mutations can lead to LQT syndrome.
DiscussionIon channel β-subunits modulate pore-forming α-subunits,increasing the diversity of ion channel properties that can be gen-erated from a limited repertoire of genes encoding α- and β-subunits. To understand the function of ion channels in vivo re-quires structure-function knowledge of not only the pore-formingsubunits, but also their heteromeric complexes with β-subunits.The interactions between KCNQ1 and KCNE1 have been studiedintensely for many years as this pair of α- and β-subunits coassem-bles in the cardiac myocyte and generates the IKs current that isimportant in the termination of cardiac action potentials. How-ever, despite these efforts, the molecular mechanisms by whichKCNE1 changes the KCNQ1 channel to generate the IKs currentremain elusive. In this work we show that KCNE1 increases thePIP2 sensitivity of IKs by several orders of magnitude (Fig. 2).Because of the low PIP2 sensitivity of KCNQ1, the PIP2 levelin biological membrane is too low to saturate binding of KCNQ1,leaving a fraction of the channel population nonfunctional in theabsence of KCNE1. This unique mechanism of current amplifi-cation is independent of KCNE1 effects on single channel con-
Fig. 3. Molecular determinants of PIP2 sensitivity in KCNE1. (A) Normalizedtail current amplitude at various times following patch excision. WT KCNQ1(filled circles), n ¼ 9; WT KCNQ1þ KCNE1 (⋈), n ¼ 9; KCNQ1þmutantKCNE1: R67Q (up triangles), n ¼ 6; K69C (open diamonds), n ¼ 6; K70C (opencircles), n ¼ 6; H73N (open squares), n ¼ 6; R67E/K69E/K70E triple mutation(TM), (down triangles), n ¼ 6. (B) Time constants of exponential fits and initialdelay to current rundown. The axis is labeled with the KCNE1 constructthat was coexpressed with KCNQ1. (-) indicates KCNQ1 alone. (C) The trans-membrane segment and juxtamembraneous C-terminal region of the KCNE1NMR structure (PDB ID code 2K21). The residues important for PIP2 sensitivityare shown in stick presentation.
Fig. 4. LQT mutations in KCNE1 reduce PIP2 sensitivity. (A) Whole-cellcurrents and (B) conductance-voltage (G-V) relation in oocytes. WT KCNQ1þKCNE1 (open squares), n ¼ 6; KCNQ1þmutant KCNE1: R67C (filled circles),n ¼ 6; R67H (filled diamonds), n ¼ 6; K70N (filled triangles), n ¼ 6; K70M(filled squares), n ¼ 6; Voltages were stepped from a holding potential of−80 mV to a test potential (−80 to þ80 mV) and then repolarized to−40 mV. (C) Whole-cell current amplitude measured þ80 mV normalizedby that of WT channels (filled bars) and V1∕2 measured from G-V relation(open bars). (D) PIP2 dose response of KCNQ1þmutant KCNE1 channels:K70N (filled circles), R67C (open circles). Solid curves are fits to the Hill equa-tion. Hill coefficients are 3 and 4, and EC50 are 114 μM and 99 μM forKCNQ1þ K70N and KCNQ1þ R67C, respectively. (E) Tail current amplitudein response to the same voltage protocol as described in Fig. 1 for WTand mutant channels. (filled bars) Indicate the tail current amplitude imme-diately following patch excision. (open bars) Indicate the tail current ampli-tude after steady-state recovery of channel activity in response to 300 uMexogenous PIP2. (F) G-V relations of KCNQ1þ K70N (filled symbols) andWT KCNQ1þ KCNE1 (unfilled) before and after patch excision in PIP2-containing solutions. KCNQ1þ K70N: on cell (filled circles), V1∕2 ¼ 48.5�2.2 mV; excised in 50 μM PIP2 (filled diamonds), V1∕2 ¼ 50.7� 0.8 mV; excisedin 150 μM PIP2, (filled triangles), V1∕2 ¼ 29.9� 3.2 mV; excised in 300 μMPIP2 (filled squares), V1∕2 ¼ 8.9� 2.6 mV. KCNQþ KCNE1: on cell (opencircles), V1∕2 ¼ 21.6� 3.2 mV; excised in 300 μM PIP2 (open squares),V1∕2 ¼ 8.5� 1.2 mV. n ¼ 2 for G-V relation of KCNQ1þ K70N in 50 μMPIP2, n ¼ 6–12 for all other experiments.
Li et al. PNAS Early Edition ∣ 3 of 6
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
ductance for which previous reports have provided conflictingresults (17–19).
In this study we identify four residues (R67, K69, K70, andH73) in proximal C terminus of KCNE1 as key determinantsof PIP2 sensitivity (Fig. 3). Previous studies have highlightedthe KCNE1 C terminus as critical for the modulation of IKs cur-rent characteristics. A C-terminal deleted KCNE1 mutant as-sembled with KCNQ1, but did not modify current properties(20). Functional scan of KCNE1 point mutations revealed thatthe juxtamembranous C-terminal region of KCNE1 is especiallyintolerant to mutation, indicating that this region likely plays animportant role in controlling channel activity (21). Docking of theNMR structure of KCNE1 to a KCNQ1 homology model indi-cates that the proximal C terminus of KCNE1 may participate inintimate interaction with the S4–S5 linker suggesting that KCNE1could directly influence the channel gating machinery (15). Thecolocalization of the KCNE1 proximal C terminus and the gatingmachinery of KCNQ1 have also been shown experimentally bythe formation of disulfide bonds between pairs of cysteine muta-tions engineered in the KCNE1 C terminus and the S4–S5 linkeror the bottom of S6 (22). Interestingly, basic residues in the bot-tom of S6 and in the S4–S5 linker have been proposed to interactwith PIP2 (5). Purified fragments of the KCNQ1 proximal C ter-minus demonstrated broad binding of anionic lipids in biochem-ical assays that depended on several juxtamembranous basicresidues in KCNQ1 (23). These results indicate that the key de-terminants of PIP2 sensitivity in KCNE1 (residues R67, K69, K70,and H73) reside in the same region that is critical for KCNE1modulation of IKs and interaction with the gating machinery.Thus, PIP2 may be an integral component in the IKs channel com-plex, and the interactions among PIP2, KCNE1, and KCNQ1 mayhold the key to resolve the long-standing absence of a mechanisticunderstanding of how KCNE1 shapes the IKs channel function.
Mutations of the key residues in KCNE1 that are determinantsof PIP2 sensitivity, R67C, R67H, K70M, and K70N, are asso-ciated with long QT syndrome (7–9). These mutations reduceIKs current and PIP2 sensitivity (Fig. 4). Remarkably, we are ableto rescue wild-type channel characteristics by applying supernor-mal levels of exogenous PIP2, suggesting that the disease patho-genesis of these mutations can be explained by a decrease in PIP2
sensitivity of the channel complex. PIP2 levels can be changed inthe cardiac myocyte by activation of phospholipase C (PLC) viaG-protein coupled receptors, such as α1A-adrenergic receptorsand M1-muscarinic receptors, that are coupled to Gqα. PLChydrolyzes PIP2 to generate diacylglycerol and inositol 1,4,5-tri-sphosphate, which lead to protein kinase C (PKC) activation andincreased intracellular calcium, respectively (4). The generationof second messengers in addition to the effective decrease of PIP2
levels makes IKs regulation by PLC activation complex as IKs isknown to be sensitive to PKC phosphorylation (24), intracellularcalcium (25), and PIP2 (6). This complex regulation in responseto activation of Gqα may be reflected in the biphasic response ofheterologously expressed IKs channels (26) and the conflictingreports of effects on endogenous IKs in myocytes (27–29). Ourcurrent results show that the PIP2 sensitivity of the IKs channelis dependent on the KCNE1 expression level relative to that ofKCNQ1 (Fig. 1), suggesting a possibility that KCNE1 may mod-ulate the response to PLC activation. Further studies with theconsideration of this possibility may provide previously unde-scribed insights on the effects of the Gqα signaling pathway onIKs channels.
IKs channels are also modulated by β-adrenergic receptorstimulation due to the phosphorylation of residues S27 andS92 in KCNQ1 by protein kinase A (PKA) (30, 31), which in-creases IKs currents and shortens ventricular action potentials.Importantly, this phosphorylation of KCNQ1 alters channel func-tion only with KCNE1 coexpression (32), indicating that, similarto PIP2 modulation, the KCNE1 interaction with KCNQ1 is cri-
tical for the PKA-dependent modulation. A previous study sug-gested a cross-talk between PKA and PIP2 modulations of the IKschannel (31). To examine whether phosphorylation of IKs channelaffects PIP2-dependent modulation, we studied the properties ofthe mutation S27D/S92D in KCNQ1 coexpressed with KCNE1,which has been shown to mimic the effects of PKA phosphoryla-tion of IKs channels (31, 32). Consistent with previous results, wefound that S27D/S92D affects IKs function, including a shift ofvoltage-dependent activation to more negative voltages and slow-ing of deactivation kinetics (Fig. 5 A–C). However, the phospho-mimetic mutation S27D/S92D does not alter the time course ofPIP2-dependent rundown of the IKs current (Fig. 5D), suggestingthat PKA phosphorylation does not alter PIP2 modulation. Like-wise, the slowing of deactivation caused by the phosphomimeticmutation is not dependent on the time after patch excision duringwhich PIP2 levels decrease (Fig. 5E), suggesting that PIP2 doesnot affect the functional changes caused by PKA phosphoryla-tion. These results suggest that, if a cross-talk between PKA andPIP2 modulations of the IKs channel exists, it does not happen inthe channel protein.
The KCNE family of ion channel β-subunits contains fivefamily members that have been reported to modulate the activityof a variety of channel α-subunits in ion channel complexes. Manyof these channel α-subunits or channel complexes are also modu-lated by PIP2 (Table 1). Sequence alignment shows that the basicresidues that are essential for KCNE1 modulation of IKs PIP2
sensitivity are highly conserved across all members of the KCNEfamily of peptides (Fig. 6), suggesting that modulation of PIP2
sensitivity may be a common mechanism of current modulationby the KCNE β-subunits. Interestingly, some members of theKCNE family may modulate more than one channel α-subunit.Likewise, multiple KCNE family members may modulate thesame channel α-subunit (Table 1). Because of structural differ-
Fig. 5. PIP2 modulation is independent of phosphomimetic mutations ofIKs channels. (A) Whole-cell currents of S27D/S92D channels. To measuredeactivation time constants, voltages were stepped from a holding potentialof −80 mV to a depolarization potential (þ40 mV) and then repolarizedto test potentials (−70 to 0 mV) (Left). To measure the G-V relation, voltageswere stepped from a holding potential of −80 mV to a test potential (−80to þ80 mV) and then repolarized to −40 mV (Right). (B) Time constant ofdeactivation vs. voltage and (C) G-V relation. WT KCNQ1þ KCNE1 (open cir-cles), n ¼ 6; S27D∕S92Dþ KCNE1 (filled circles), n ¼ 7. (D) Time dependenceof normalized tail current amplitude after patch excision. WT KCNQ1þKCNE1 (open circles), n ¼ 12; S27D∕S92Dþ KCNE1 (filled circles), n ¼ 6. Solidlines are monoexponential fits to data. (E) Time constant of tail currents inD. The deactivation time constant of the mutant channel is significantlylarger than that of the WT (P < 0.05).
4 of 6 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1100872108 Li et al.

ences in these α- and β-subunits, the putative interactions amongKCNE β-subunits, channel α-subunits and PIP2 may vary withindifferent ion channel complexes, resulting in diverse effects ofKCNE peptides.
Materials and MethodsMutagenesis and Oocyte Preparation. KCNQ1 and KCNE1 were subclonedinto pcDNA3.1(+) (Invitrogen). All mutations were generated by using over-lap extension PCR and verified by sequencing. mRNA was transcribed in vitroby using the mMessage mMachine T7 polymerase kit (Applied Biosystems).The follicle cells were removed by using type 1A collagenase (Sigma-Aldrich).Stage IV–V Xenopus oocytes were selected and injected with 4.6 ng mRNAper oocyte. Injected oocytes were incubated in ND96 solution (in mM:96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 Hepes, pH 7.60) at 18 °C for3–5 d before recording.
Electrophysiology Macroscopic currents were recorded from inside-outpatches formed with patch pipettes of 0.5–1.0 MΩ resistance. The data wereacquired using an Axopatch 200-B patch–clamp amplifier (Axon Instruments)and pulse acquisition software (HEKA). Experiments were conducted atroom temperature (20–22 °C). The pipette solution contained (in mM)140 KMeSO3, 20 Hepes, 2 KCl, and 2 MgCl2, pH 7.2. The internal solution con-tained (in mM) 140 KMeSO3, 20 Hepes, 2 KCl, 5 EGTA, 1.5 MgATP, and PIP2 as
indicated in the text, pH 7.2. PIP2 (bovine brain, Sigma-Aldrich), diC8-, anddiC4-PIP2 (Echelon) were sonicated in ice bath for 30 min to dissolve inthe internal solution at 2 mM as stock and kept in aliquots in −80 °C beforeuse. Whole cell current recordings were obtained with the two-microelec-trode voltage–clamp technique. Microelectrodes were pulled from glass ca-pillary tubes and filled with 3 M KCl. Oocytes were constantly superfusedwith ND96. Themembrane potential was clamped using a voltage–clamp am-plifier (Dagan CA-1B; Dagan Corporation). Data acquisition was controlledusing PULSE/PULSEFIT software (HEKA).
Data Analysis The relative conductance was determined by measuring tailcurrent amplitudes at indicated voltages. The conductance-voltage (G-V)relationships were fitted with the Boltzmann equation:
GGmax
¼ 1
1þ expð−zeðV−V1∕2ÞkT Þ
; [1]
where G∕Gmax is the ratio of conductance to maximum conductance, z is thenumber of the equivalent charges, V1∕2 is the voltage at which the channel is50% activated, e is the elementary charge, k is Boltzmann’s constant, and T isthe absolute temperature. Data analysis and curve fitting were done usingthe Igor Pro software (WaveMetrics).
ACKNOWLEDGMENTS. We thank Junqiu Yang for technical help in the firstpatch–clamp recording. We thank S. Goldstein (University of Chicago,Chicago, IL) and S. Nakanishi (Kyoto University, Kyoto, Japan) for humanKCNQ1 and KCNE1 clones. The cDNA of CiVSP was kindly provided byY. Okamura (Osaka University, Osaka, Japan). This study was funded byEstablished Investigator Award 0440066N from the American HeartAssociation (AHA) and National Science Foundation of China Grant30528011 (to J.C.), AHA predoctoral fellowship 0910020G (to D.W.), and11PRE5720009 (to M.A.Z). J.C. is the Spencer T. Olin Professor of BiomedicalEngineering.
1. Sanguinetti MC, et al. (1996) Coassembly of KvLQT1 and minK (IsK) proteins to formcardiac IKs potassium channel. Nature 384:80–83.
2. Barhanin J, et al. (1996) KvLQT1 and IsK (minK) proteins associate to form the IKscardiac potassium current. Nature 384:78–80.
3. Rudy Y, Silva JR (2006) Computational biology in the study of cardiac ion channels andcell electrophysiology. Q Rev Biophys 39:57–116.
4. Suh BC, Hille B (2008) PIP2 is a necessary cofactor for ion channel function: how andwhy? Annu Rev Biophys 37:175–195.
5. Park KH, et al. (2005) Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bispho-sphate interaction underlies the long QT syndrome. Circ Res 96:730–739.
6. Loussouarn G, et al. (2003) Phosphatidylinositol-4,5-bisphosphate, PIP2, controlsKCNQ1/KCNE1 voltage-gated potassium channels: A functional homology betweenvoltage-gated and inward rectifier Kþ channels. EMBO J 22:5412–5421.
7. Lai LP, et al. (2005) Denaturing high-performance liquid chromatography screening ofthe long QT syndrome-related cardiac sodium and potassium channel genes and iden-tification of novel mutations and single nucleotide polymorphisms. J Hum Genet50:490–496.
8. Hedley PL, et al. (2009) The genetic basis of long QT and short QT syndromes: A muta-tion update. Hum Mutat 30:1486–1511.
9. Kapplinger JD, et al. (2009) Spectrum and prevalence of mutations from the first 2,500consecutive unrelated patients referred for the FAMILION long QT syndrome genetictest. Heart Rhythm 6:1297–1303.
10. Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y (2005) Phosphoinositide phospha-tase activity coupled to an intrinsic voltage sensor. Nature 435:1239–1243.
11. Cui J, Kline RP, Pennefather P, Cohen IS (1994) Gating of IsK expressed in Xenopusoocytes depends on the amount of mRNA injected. J Gen Physiol 104:87–105.
12. Wang W, Xia J, Kass RS (1998) MinK-KvLQT1 fusion proteins, evidence for multiplestoichiometries of the assembled IsK channel. J Biol Chem 273:34069–34074.
13. Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY (2010) Stoichiometry of the KCNQ1—KCNE1ion channel complex. Proc Natl Acad Sci USA 107:18862–18867.
14. Rosenhouse-Dantsker A, Logothetis DE (2007) Molecular characteristics of phosphoi-nositide binding. Pflugers Arch 455:45–53.
15. Kang CB, et al. (2008) Structure of KCNE1 and implications for how it modulates theKCNQ1 potassium channel. Biochemistry 47:7999–8006.
16. Rocheleau JM, Gage SD, KobertzWR (2006) Secondary structure of a KCNE cytoplasmicdomain. J Gen Physiol 128:721–729.
17. Romey G, et al. (1997) Molecular mechanism and functional significance of the MinKcontrol of the KvLQT1 channel activity. J Biol Chem 272:16713–16716.
18. Sesti F, Goldstein SA (1998) Single-channel characteristics of wild-type IKs channels andchannels formed with two MinK mutants that cause long QT syndrome. J Gen Physiol112:651–663.
19. Yang YS, Sigworth FJ (1998) Single-channel properties of IKs potassium channels. J GenPhysiol 112:665–678.
20. Tapper AR, George AL, Jr (2000) MinK subdomains that mediate modulation of andassociation with KvLQT1. J Gen Physiol 116:379–390.
21. Takumi T, et al. (1991) Alteration of channel activities and gating by mutations of slowIsK potassium channel. J Biol Chem 266:22192–22198.
22. Lvov A, Gage SD, Berrios VM, Kobertz WR (2010) Identification of a protein-proteininteraction between KCNE1 and the activation gate machinery of KCNQ1. J GenPhysiol 135:607–618.
23. Thomas AM, Harmer SC, Khambra T, Tinker A (2010) Characterization of a binding sitefor anionic phospholipids on KCNQ1. J Biol Chem 286:2088–2100.
Fig. 6. Partial sequence alignment of all members of the KCNE family.Numbering indicates the position on KCNE1. The filled box highlights thepredicted transmembrane domain (TMD) (53). Open boxes indicateconserved residues that are key determinants of PIP2 sensitivity.
Table 1. Kþ channel α-subunits that interact with KCNE families andshow PIP2 sensitivity
KCNE α-subunit* PIP2 sensitivity†
KCNE1 KCNQ1 (1, 2) αþ β (6)HERG (33) α (34)Kv 4.3 (35)
KCNE2 KCNQ1 (36) αþ β (37)KCNQ2 and 2∕3 (38) α (39)
HERG (40) α (34)HCN1&2 (41) α (42)
Kv4.2 (43) and Kv4.3 (35)KCNE3 KCNQ1 (44)
Kv2.1 and Kv3.1 (45) α (46)Kv3.4 (47) α (48)HERG (44) α (34)KCNQ4 (44) α (49)
KCNE4 KCNQ1 (50)KV1.1 and Kv1.3 (51) α (48)
KCNE5 KCNQ1 (52)
*Coexpression of a KCNE with the α-subunit changes the properties of thechannel.
†PIP2 sensitivity of the channel either with the α-subunits alone (α) or with theα-subunits and a KCNE coexpression (αþ β) has been reported.
Li et al. PNAS Early Edition ∣ 5 of 6
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

24. Lo CF, Numann R (1998) Independent and exclusive modulation of cardiac delayedrectifying Kþ current by protein kinase C and protein kinase A. Circ Res 83:995–1002.
25. Shamgar L, et al. (2006) Calmodulin is essential for cardiac IKs channel gating andassembly—Impaired function in long-QT mutations. Circ Res 98:1055–1063.
26. Matavel A, Lopes CM (2009) PKC activation and PIP2 depletion underlie biphasicregulation of IKs by Gq-coupled receptors. J Mol Cell Cardiol 46:704–712.
27. Habuchi Y, et al. (1992) Endothelin enhances delayed potassium current via phospho-lipase C in guinea pig ventricular myocytes. Am J Physiol 262:H345–354.
28. Ding WG, Toyoda F, Matsuura H (2004) Regulation of cardiac IKs potassium current bymembrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem 279:50726–50734.
29. Yasuda Y, et al. (2005) Regulation of the muscarinic Kþ channel by extracellularATP through membrane phosphatidylinositol 4,5-bisphosphate in guinea-pig atrialmyocytes. Br J Pharmacol 145:156–165.
30. Marx SO, et al. (2002) Requirement of a macromolecular signaling complex for βadrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science295:496–499.
31. Lopes CM, et al. (2007) Protein kinase a modulates PLC-dependent regulation andPIP2-sensitivity of Kþ channels. Channels 1:124–134.
32. Kurokawa J, Chen L, Kass RS (2003) Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci USA100:2122–2127.
33. McDonald TV, et al. (1997) A minK-HERG complex regulates the cardiac potassiumcurrent IKr . Nature 388:289–292.
34. Bian JS, Cui J, McDonald TV (2001) HERG Kþ channel activity is regulated by changes inphosphatidylinositol 4,5-bisphosphate. Circ Res 89:1168–1176.
35. Deschenes I, Tomaselli GF (2002) Modulation of Kv4. 3 current by accessory subunits.FEBS Lett 528:183–188.
36. Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J (2000) KCNE2 confers back-ground current characteristics to the cardiac KCNQ1 potassium channel. EMBO J19:6326–6330.
37. Heitzmann D, et al. (2004) Heteromeric KCNE2/KCNQ1 potassium channels in theluminal membrane of gastric parietal cells. J Physiol 561:547–557.
38. Tinel N, et al. (2000) M-type KCNQ2-KCNQ3 potassium channels are modulated by theKCNE2 subunit. FEBS Lett 480:137–141.
39. Winks JS, et al. (2005) Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels.J Neurosci 25:3400–3413.
40. Abbott GW, et al. (1999) MiRP1 forms IKr potassium channels with HERG and isassociated with cardiac arrhythmia. Cell 97:175–187.
41. Yu H, et al. (2001) MinK-related peptide 1: A beta subunit for the HCN ion channelsubunit family enhances expression and speeds activation. Circ Res 88:e84–e87.
42. Pian P, Bucchi A, Robinson RB, Siegelbaum SA (2006) Regulation of Gating andrundown of HCN hyperpolarization-activated channels by exogenous and endogen-ous PIP2. J Gen Physiol 128:593–604.
43. Zhang M, Jiang M, Tseng GN (2001) MinK-related peptide 1 associates with Kv4. 2 andmodulates its gating function: Potential role as beta subunit of cardiac transientoutward channel? Circ Res 88:1012–1019.
44. Schroeder BC, et al. (2000) A constitutively open potassium channel formed by KCNQ1and KCNE3. Nature 403:196–199.
45. McCrossan ZA, et al. (2003) MinK-related peptide 2 modulates Kv2. 1 and Kv3.1potassium channels in mammalian brain. J Neurosci 23:8077–8091.
46. Xie LH, John SA, Ribalet B, Weiss JN (2008) Phosphatidylinositol-4,5-bisphosphate(PIP2) regulation of strong inward rectifier Kir2.1 channels: Multilevel positive coop-erativity. J Physiol 586:1833–1848.
47. Abbott GW, et al. (2001)MiRP2 forms potassium channels in skeletal muscle with Kv3.4and is associated with periodic paralysis. Cell 104:217–231.
48. Oliver D, et al. (2004) Functional conversion between A-type and delayed rectifierKþ channels by membrane lipids. Science 304:265–270.
49. Zhang H, et al. (2003) PIP2 activates KCNQ channels, and its hydrolysis underliesreceptor-mediated inhibition of M currents. Neuron 37:963–975.
50. GrunnetM, et al. (2002) KCNE4 is an inhibitory subunit to the KCNQ1 channel. J Physiol542:119–130.
51. Grunnet M, et al. (2003) KCNQ1 channels sense small changes in cell volume. J Physiol549:419–427.
52. Angelo K, et al. (2002) KCNE5 induces time- and voltage-dependent modulation ofthe KCNQ1 current. Biophys J 83:1997–2006.
53. Abbott GW, Goldstein SA (2002) Disease-associated mutations in KCNE potassiumchannel subunits (MiRPs) reveal promiscuous disruption of multiple currents andconservation of mechanism. FASEB J 16:390–400.
6 of 6 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1100872108 Li et al.
Supporting InformationLi et al. 10.1073/pnas.1100872108
1.0
0.5
0.0
-50 0 50Test Potential (mV)
G/G
max
B
1 s
10 µ
A
A
Fig. S1. Channel properties with different ratios of KCNQ1 to KCNE1. (A) Superimposed current traces recorded from 4 oocytes with KCNQ1: KCNE1 mRNAratios of 1∶1 (open circle); 1∶0.05 (open triangle); 1∶0.01 (open square); 1∶0 (filled circle). The currents were elicited by a depolarizing voltage pulse to 40 mVfrom a holding potential of −80 mV. (B) Normalized G-V curves for different ratios of KCNQ1: KCNE1, 1∶1 (open circles), V1∕2 ¼ 21.2� 2.2 mV; 1∶0.05 (opentriangles), V1∕2 ¼ 8.8� 2.2 mV; 1∶0.01 (open squares), V1∕2 ¼ −9.3� 1.5 mV; 1∶0 (filled circles), V1∕2 ¼ −15.2� 2.9 mV. n ¼ 6 for all experiments.
Li et al. www.pnas.org/cgi/doi/10.1073/pnas.1100872108 1 of 1


















