
Introductoin to Bioinformatics - IISER Punefarhat/wordpress/wp-content/uploads/2011/06/... ·...
49
Introductoin to Bioinformatics Part I: Dynamic Programming, Pairwise Alignment, Multiple Alignment (1) Burkhard Morgenstern Dept. Bioinformatics Institute of Microbiology and Genetics (IMG) University of Göttingen September 20, 2011
Transcript of Introductoin to Bioinformatics - IISER Punefarhat/wordpress/wp-content/uploads/2011/06/... ·...

Introductoin to BioinformaticsPart I: Dynamic Programming,
Pairwise Alignment, Multiple Alignment (1)
Burkhard Morgenstern
Dept. BioinformaticsInstitute of Microbiology and Genetics (IMG)
University of Göttingen
September 20, 2011

Introduction
Topics of lecture
Dynamic ProgrammingPairwise Sequence AlignmentMultiple Sequence AlignmentRNA Secondary StructuresPhylogeny using Pairwise DistancesPhylogeny using Maximum Parsimony

Dynamic Programming
DP important method to solve complex optimization problems
Application in bioinformatics e.g. for
Pairwise sequence alignmentGene prediction: select optimal gene structure from list ofpotential exonsPrediction of RNA secondary structuresPhylogeny (Maximum Parsimon, maximum likelihood)Hidden Markov Models (HMM)

Dynamic Programming
Basis of DP four steps (Cormen et al):
1 Determine structure of optimal solutions of problems: optimalsolutions contain optimal solutions for sub-problems
2 Define score of optimal solution of sub-problem recursively3 Calculate score of optimal solution of sub-problem recursively4 Find optimal solution for given problem by trace back

Dynamic Programming
ExampleFind shortest path in directed graph between nodes A und B.Given
n nodes X1 . . .Xn
length d(i , j) of edge (Xi ,Xj).
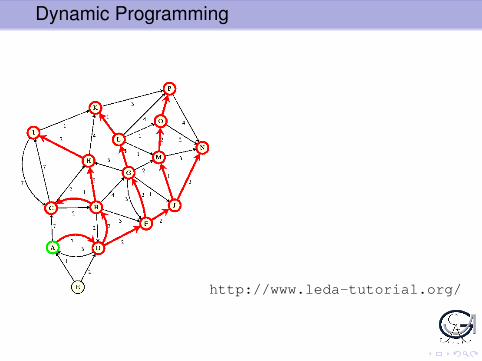
Dynamic Programming
http://www.leda-tutorial.org/

Dynamic Programming
Idea: Consider sub-problem: shortest path from A to Xi
1. Topological sort of nodes X1, . . . ,Xn, i.e. for edge (Xi , Xj) , Xi isbefore Xj in list (order not uniquely determined!)
2. Go through ordered list of nodes and calculate for each Xi lengthDi of optimal path from A to XiIdea:
I Consider all possible predecessors Xj of Xi , i.e. all Xj for whichthere is an edge (Xj ,Xi).

Dynamic Programming
I Length of shortest path through Xj to Xi :
Dj + d(j , i)
I Select best predecessor Xj ; store Xj and Di
Once B is reached, length of shortest path from A to B is known -but not shortest path itself!
3. Calculate shortest path from A to B by ‘trace back’: Findpredecessor of B, predecessor of predecessor etc.

Sequence Alignment
seq1 W T Y I V M R E A Q Y E S A Qseq2 R C L V M R E A Q E Wseq3 Y I M Q E V Q Q E R Aseq4 A L Y I A M R E V Q Y E S A
Sequence analyis based on comparison of sequencesFirst step: sequence alignment
I Assign homologous positionsI Introduce gaps into sequences
Basis of (almost) all methods in sequence analysis

Sequence Alignment
seq1 W T Y I V M R E A Q Y E S A Qseq2 - R C L V M R E A Q - E W - -seq3 - - Y I - M Q E V Q Q E R A -seq4 A L Y I A M R E V Q Y E S A -
Sequence analyis based on comparison of sequencesFirst step: sequence alignment
I Assign homologous positionsI Introduce gaps into sequences
Basis of (almost) all methods in sequence analysis

Sequence Alignment
Sequence alignment crucial for
PhylogenyGenome analysis, gene predictionProtein structureRNA secondary structureDatabase searchingetc.
⇒ If alignments wrong, all results that are based on alignments arewrong!
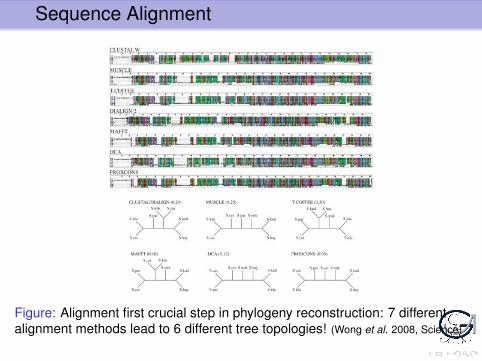
Sequence Alignment
Figure: Alignment first crucial step in phylogeny reconstruction: 7 differentalignment methods lead to 6 different tree topologies! (Wong et al. 2008, Science)

Sequence Alignment
Distinguish:
Paarwise alignment: two sequences alignedMultiple alignment: more than two sequences aligned

Pairwise sequence alignment
Example (Simple protein alignment)
seq1 A L S C V W M I Pseq2 A I S C M I P T
Input sequences

Pairwise sequence alignment
Example (Simple protein alignment)
seq1 A L S C V W M I P -seq2 A I S C - - M I P T
Possible alignment

Pairwise sequence alignment
Example (Simple protein alignment)
seq1 A L S C V W M I P -seq2 A I S C - - M I P T
Alignment as hypothesis:
1 Substitution L→ I or I→ L
3 insertions deletions of amino acid residues (V,M,T)

Pairwise sequence alignment
Example (Simple protein alignment)
seq1 A L S C V W M I P -seq2 A I S - C M - I P T
Alternative alignment (hypothesis):
3 substitutions L→ I, V→ C , W→ M
3 insertions or deletions of amino acid residues ( C,D,T)

Pairwise sequence alignment
Goal: find best possible alignment for two given sequencesTwo central questions to be solved:
Objective function: how to measure the quality of an alignment?Optimization algorithm: how to find best alignment?Problem: number of possible alignments huge!For two sequences of length n:
(2n)!
(n!)2 ≈22n√
2πn
different alignments!

Pairwise sequence alignment
Objective function for pairwise protein alignment:
(For DNA alignment similar, but simpler)Goal: find alignment that explains evolution of sequences in mostplausible way.
Define score forSubstitutions: Score s(a,b) for every possible substitution ofamino acids a→ bInsertions/deletions: penalty for every gap in alignment
Total score of an alignment:
Sum of scores for substitutions und gaps

Pairwise sequence alignment
For substitutions: use substitution matrix, e.g. PAM, BLOSUM:20× 20 matrix for all possible substitutions of amino acids.
Usual definition of substitution scores:
s(a,b) = logpa,b
qa × qb
withI pa,b probability of substitution a→ b or b → aI qa probability of a

Pairwise sequence alignment
Simples method to score gaps: linear gap penalty, i.e. gap oflength l gets penalty
g × l
for constant g.
For linear gap penalty: every gap symbol ‘-’ receives penalty g.
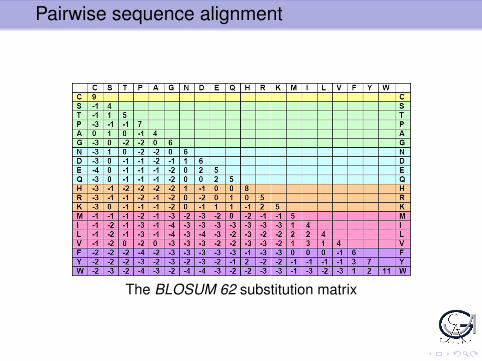
Pairwise sequence alignment
The BLOSUM 62 substitution matrix

Pairwise sequence alignment
Find optimal alignment by dynamic programming (DP).
Necessary: linear gap penalty!
For sequences X ,Y
X = X1 . . .Xm
Y = Y1 . . .Yn
of length m and n, respectively, . . .

Pairwise sequence alignment
. . . consider sub-problems:
For 0 ≤ i ≤ m and 0 ≤ j ≤ m, find optimal alignment of
X1 . . .Xi
Y1 . . .Yj
(‘prefixes’ of X and Y )

Pairwise sequence alignment
F (i , j) = Score of optimal alignment of prefixes up to i und j
Recursion to calculate F (i , j): consider 3 possibilities for last column ofprefix alignment:
Xi and Xj aligned:
... Xi
... Yj
In this case: F (i , j) = F (i − 1, j − 1) + s(Xi ,Yj)

Pairwise sequence alignment
Gap in sequence X
... −
... Yj
In this case : F (i , j) = F (i , j − 1)− gGap in sequence Y
... Xi
... −
In this case: F (i , j) = F (i − 1, j)− g

Pairwise sequence alignment
All together:
F (i , j) = max
F (i − 1, j − 1) + s(Xi ,Yj)F (i − 1, j) − gF (i , j − 1) − g
(1)
Initialize values:
F (0, j) = −j × g (alignment only consists of j gaps in sequence X )F (i ,0) = −i × g (alignment only consists of i gaps in sequence Y )
Find best alignment:Trace Back!
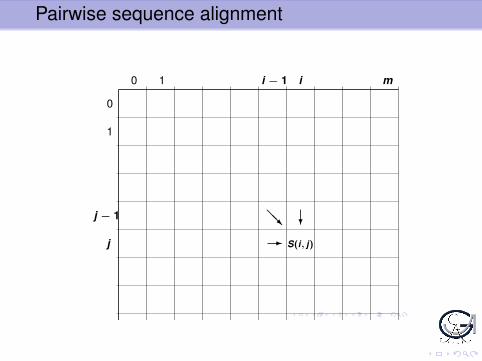
Pairwise sequence alignment
0 1 i − 1 i m
0
1
j − 1
j
n
?@@R
- S(i, j)
Abbildung: Rekursive Berechnung des Scores S(i, j) eines optimalen Alignments derPräfixe der Sequenzen X und Y bis zu den Positionen i bzw. j . Der Wert S(i, j) kannberechnet werden, wenn drei mögliche “Vorgängerwerte” S(i − 1, j − 1), S(i − 1, j)und S(i, j − 1) bekannt sind. Die Werte S(i, j) können daher schrittweise berechnetwerden, indem man die zu den Sequenzen X und Y gehörende Vergleichsmatrix vonlinks oben nach rechts unten ausfüllt.

Multiple sequence alignment
Example
S1 N G P E V R E L WS2 A R D I W AS3 Q A R E S I Y AS4 V R E S L W SS5 W Y V R D A S L W S
Protein Family, not aligned. Homologies not visible.
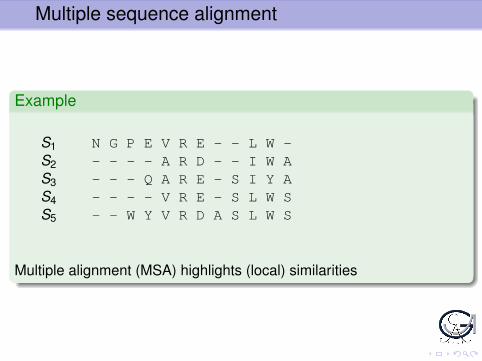
Multiple sequence alignment
Example
S1 N G P E V R E - - L W -S2 - - - - A R D - - I W AS3 - - - Q A R E - S I Y AS4 - - - - V R E - S L W SS5 - - W Y V R D A S L W S
Multiple alignment (MSA) highlights (local) similarities

Multiple sequence alignment
Goal: calculate best MSA for input sequences
S1, . . . ,Sn
automatically
To define score of multiple alignment:
For each pair of sequences Si and Sj , consider pairwise alignment
Pi,j(A)
of Si and Sj ‘contained’ in A
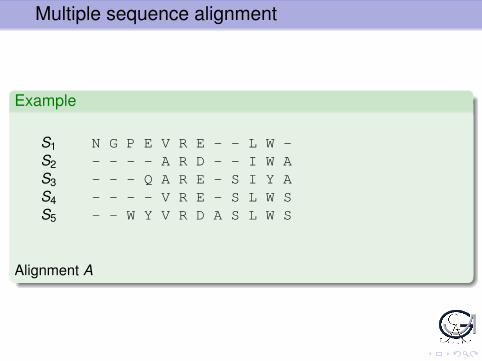
Multiple sequence alignment
Example
S1 N G P E V R E - - L W -S2 - - - - A R D - - I W AS3 - - - Q A R E - S I Y AS4 - - - - V R E - S L W SS5 - - W Y V R D A S L W S
Alignment A

Multiple sequence alignment
Example
S1S2S3 - - - Q A R E - S I Y AS4S5 - - W Y V R D A S L W S
Pairwise alignment P3,5(A) of S3 and S5 contained in A

Multiple sequence alignment
Example
S3 - Q A R E - S I Y AS5 W Y V R D A S L W S
Pairwise alignment P3,5(A) of S3 and S5 contained in A

Multiple sequence alignment
Sum-of-pairs score Sc(A) of MSA A of S1, . . . ,Sn defined as:
Sc(A) =∑i<j
Sc(Pi,j(A))
DP algorth to calculate optimal alignments can be generalized tomultiple alignment (in theory!)
But: computing time far too long, not practicable
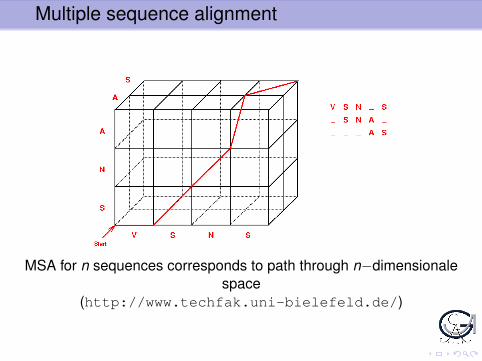
Multiple sequence alignment
MSA for n sequences corresponds to path through n−dimensionalespace
(http://www.techfak.uni-bielefeld.de/)

Multiple sequence alignment
Heuristic solutions necessary, i.e. fast algorithms, that do notnecessarily find the mathematically optimal alignment.
Most important heuristic approach: progressive alignment!
Idea for ‘progressive alignment’:Align sucessively sequences and groups of previously alignedsequences, until all sequences are aligned in one MSATwo groups G1,G2 of sequences aligned by A1,A2 can be alignedlike two single sequences if A1,A2 remain unchanged

Multiple sequence alignment
ExampleAlignment of profiles
S1 E V R E - V W -S2 - A R D - I W AS3 Q A R E S I Y AS4 - - R E S L W S
S5 R E W S L W SS6 R E Y S - - S

Multiple sequence alignment
ExampleAlignment of profiles
S1 E V R E - - V W -S2 - A R D - - I W AS3 Q A R E - S I Y AS4 - - R E - S L W S
S5 - - R E W S L W SS6 - - R E Y S - - S

Multiple sequence alignment
Procedure for progressive alignment:
Construct phylogenetic tree of input sequences S1, . . . ,Sn (‘guidetree’).Traverse T from leaves to rootAt every inner node, construct profile alignments of sequencescorresponding to daughter nodes

Multiple sequence alignment
Most important implementation: CLUSTAL W
Figure: Des Higgins, Dublin (source: IDA Ireland)
J. Thompson, D. Higgins, T. Gibson (1994), NAR 22, 4673–4680

CLUSTAL W
Construct optimal global pairwise alignments withNeedleman-Wunsch algorithm (Dynamic Programming)Calculate distance di,j between sequences Si and Sj as
di,j = 1− percentage identity in alignment100
Calculate guide tree using Neighbor-Joining with distances di,j
Determine root r as ‘center’ of guide tree (average distance ofleaves equal on both sides of root r ).

CLUSTAL W
Determine weights wi of sequences Si : Groups of closely relatedsequences get lower weightsAlign sequences in order defined by guide tree using profilealignment.Gap penalties depending on sequence compositions and onexisting gaps.
CLUSTAL W
Figure: Progressive Alignment in CLUSTAL W (Thompson et al, 1994)
CLUSTAL W
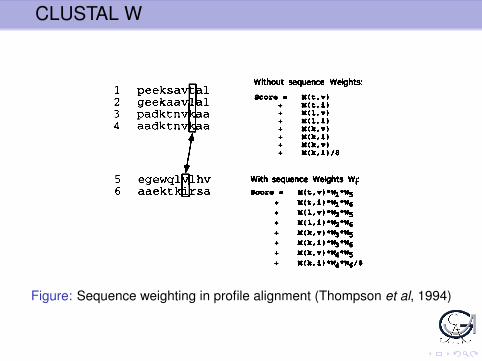
Figure: Sequence weighting in profile alignment (Thompson et al, 1994)

CLUSTAL W
Calculate weight wi of sequence Si :
Consider ‘path’ in guide tree from root r to Si
Normalize lenght ofpath to 1Edge k in path gets weight
gk =lknk
where lk length of k and nk number of sequences under k .Define
wi =∑
k
gk

CLUSTAL W
Two extreme cases:1 Length of edge leading to Si ≈ 1. No other edges branching off:
wi ≈ 1
2 Long edge k to node n (lk ≈ 1), N leaves including Sj ‘under’ n :
wj ≈1N

CLUSTAL W
Gap penalties in CLUSTAL W
Use affine-linear gap penalties with gap opening penalty (GOP)and gap extension penalty (GEP)Initial values of GOP and GEP specified by userDuring progressive alignment, GOP and GEP modified dependingon
I Substitution matrixI Similarity between sequencesI Length of sequencesI Differences in sequence lengthsI Local sequence composition (hydrophilic or hydrophobic amino
acid residues)I Existing gapsI Position in sequence: end gaps get lower penalty
CLUSTAL W
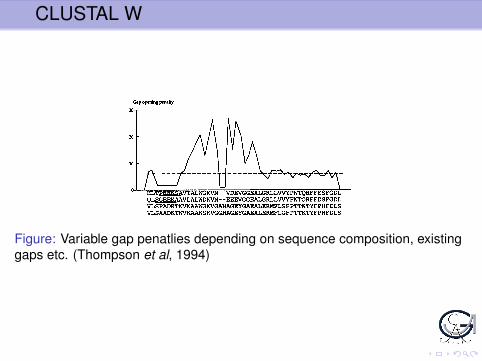
Figure: Variable gap penatlies depending on sequence composition, existinggaps etc. (Thompson et al, 1994)


















