
World Health Organization's Prequalification Program for medicines
Upload
axel-hamblinCategory
view
240download
1
Introduction to WHO Prequalification of Medicines Programme
Essential requirements
Dr Milan Smid and many team colleaguesWHO Prequalification of Medicines Programme
Amman, June 2013
Amman, June 2013
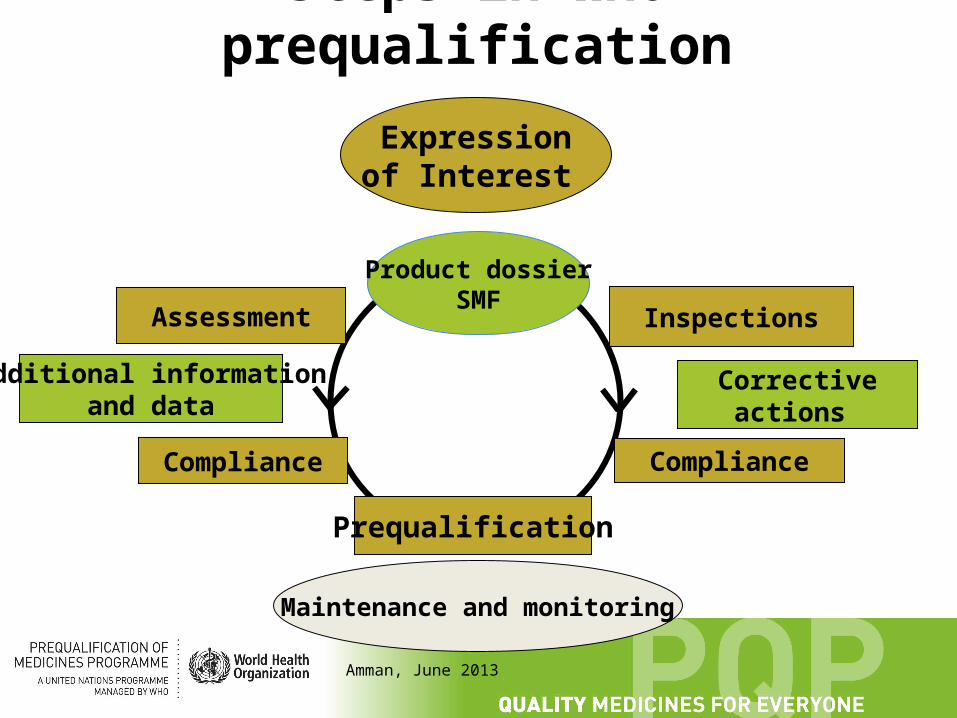
IExpression of Interest
Compliance
Additional informationand data
Corrective actions
Compliance
Assessment Inspections
Steps in WHO prequalification
Prequalification
Maintenance and monitoring
Product dossierSMF

Amman, June 20133
Application requirements - Full dossier Generic Product application
• Covering letter, in English, confirming that the information submitted in the product dossiers is true and correct.
• Product dossier (including full BE report where applicable), in English, in CTD format and the QOS-PD + QIS + BTIF/BW forms ALL completed in word format
• A product sample
• A site master file, for each manufacturing site

Amman, June 2013
How to Submit an application• Submit electronic copy ONLY of Dossier in CTD
format and Forms on CD/DVD To WHO Geneva
• After dossier has been accepted for assessment and has been allocated a WHO reference number you will be notified to proceed to step 2
• Updated or finalised Dossier and Forms (paper copies + CD/DVDs) incorporating any improvements or changes requested in step 1 and samples To UNICEF- Copenhagen
• Submit Site Master File & Contract Research Organization Master File (CROMF) (hard copies +CD/DVDs) to Geneva address
WHO/CF workshop, New Delhi, November 2012

Amman, June 2013
Data requested for prequalification
5
API
FPP
S & E
+
+
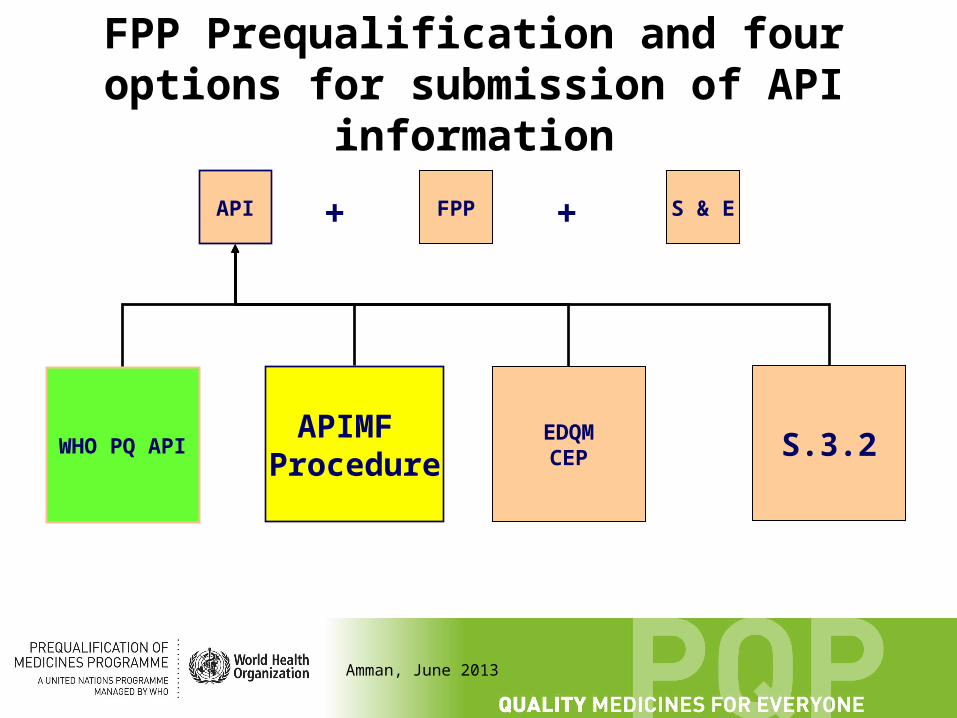
An application to prequalify an FPP typically has three parts, relevant Good Practices have to be followed during manufacture and control:
Information on the preparation and control of the API.
Information on the preparation and control of the FPP.
Safety and efficacy data, e.g. commonly replaced by demonstration of bioequivalence

Amman, June 2013
Data requested for prequalification guideline for applicants
• Consists of two parts:– Preparation of product dossiers (PDs) in Common
Technical Document (CTD) format• The preparation guideline
– Guideline on submission of documentation on multisource (Generic) Finished Pharmaceutical Product (FPP): Quality part
• Main quality guide
• Being implemented since September 2010

WHO/UNFPA workshop, Nov 2012 7
Adapting the CTD-NDS (new drug) to CTD-ANDS (generic)
RegionalAdmin
Information
Module 1
NonclinicalOverview
NonclinicalSummary
ClinicalOverview
ClinicalSummary
QualityOverall
Summary
QualityNonclinical
Study ReportsClinical
Study Reports
Module 3 Module 4 Module 5
Module 2
Not Part ofthe CTD
The CTD

Amman, June 2013
Main sub-sections of 3.2.S. Drug Substance (API)
• 3.2.S.1 General information• 3.2.S.2 Manufacture• 3.2.S.3 Characterization• 3.2.S.4 Control of the API• 3.2.S.5 Reference standards or materials• 3.2.S.6 Container closure system• 3.2.S.7 Stability

Amman, June 2013
Main sub-sections of 3.2.P. Drug Product (FPP)
• 3.2.P.1 Description and Composition of the FPP• 3.2.P.2 Pharmaceutical development• 3.2.P.3 Manufacture• 3.2.P.4 Excipients• 3.2.P.5 Control of the FPP• 3.2.P.6 Reference standards or materials• 3.2.P.7 Container closure system• 3.2.P.8 Stability

WHO/UNFPA workshop, Nov 2012 10
Specific quality documents: quality summaries (templates - QIS/QOS)
• The instructions for the QOS-PD (quality over all summary) run throughout the quality guideline
• Instructions for the QIS are in Section 3.2 and preface the QIS (quality information summary) template

Amman, June 201311

Amman, June 2013
Key aspects of quality assessment• To ensure that all future batches, throughout their shelf
life, will perform as the clinical/biobatch – Characteristics of the API batch used in the biobatch, details of
FPP manufacturing process and quality attributes of the the biobatch are critical reference information
• To ensure that processes are adequately described and controlled and that they are validated
• To ensure that the level of impurities in the API and FPP are within safe limits
• To ensure that container closure system will protect the product from chemical, physical and biological deterioration and that it's compatible with the product
• To ensure that analytical methods used at different stages are reliable and reproducible
Amman, June 2013
FPP Prequalification and four options for submission of API information
API FPP S & E
3.2.S
++
EDQMCEP
APIMFProcedure
WHO PQ API

Amman, June 2013
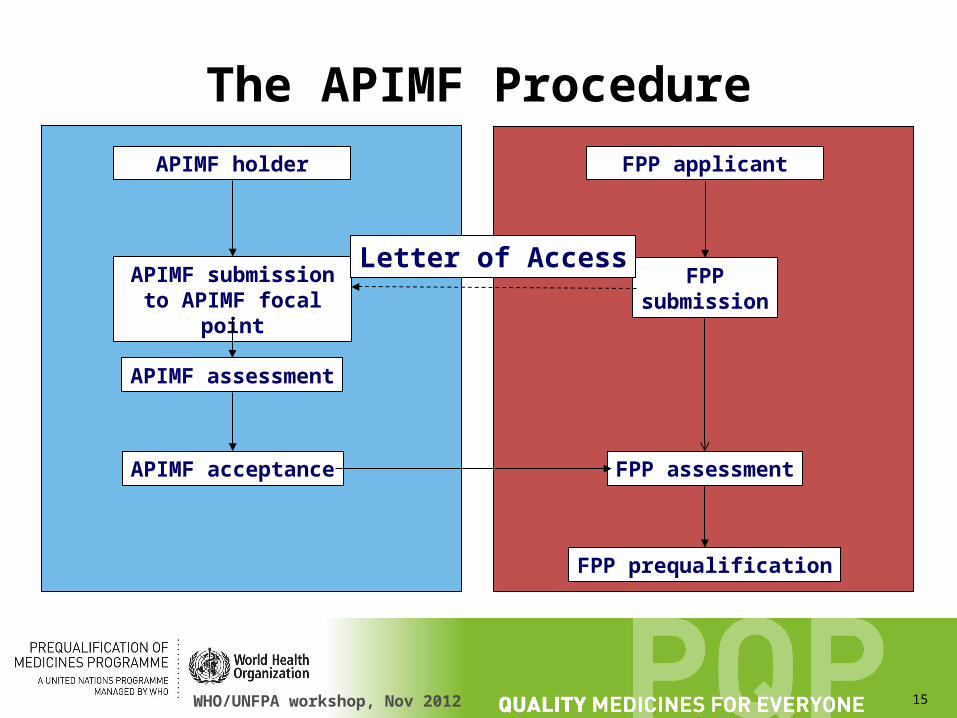
The APIMF Procedure
• The API manufacturer supplies the FPP manufacturer with the open part of their APIMF. The FPP manufacturer use this information to complete the necessary sections of their submission.
• The API manufacturer supplies the FPP with a letter of access.
• The FPP manufacturer attaches the open part of the APIMF and the letter of access to their FPP submission.
• The API manufacturer sends the open and closed sections of their APIMF directly to WHO. It is held in confidence.
WHO/UNFPA workshop, Nov 2012 15
The APIMF Procedure
APIMF holder FPP applicant
APIMF submission to APIMF focal point
FPPsubmission
APIMF assessment
APIMF acceptance FPP assessment
FPP prequalification
Letter of Access

Amman, June 2013
Advantages of the APIMF Procedure• It allows the submission of confidential information by the API
manufacturer without disclosure to the FPP applicant.• One APIMF may be used to support multiple FPP applications
without the need for repeated evaluations.• It is applicable to both pharmacopoeial and non-
pharmacopoeial APIs.• The APIMF holder prepares and maintains the APIMF
information and answers all queries directly.
• Normally all API-related changes would require the submission of a variation from associated FPP manufacturers.

Amman, June 2013
Notable Requirements
– Number of batches required to establish the FPP shelf-life
– Uniformity demonstration for the biolot instead of process validation report for pilot batches
– process validation/pharmaceutical development requirements for “established” generics
17

Amman, June 2013
Accelerated procedure for accepting RHand 2nd Line TB dossiers for assessment
• Stability Data at the time of submission only, may be reduced to no less than 3 months accelerated, plus 3 months long-term data, for not less than two primary batches of at least pilot scale. One of the primary batches should be the batch that was used for bioequivalence studies.
• In some cases, several years of long-term stability data on batches under uncontrolled storage conditions may also be accepted from Zone IV countries.
• NB all prequalification requirements for final acceptability of the dossier remain in effect, without exception, including but not limited to the submission of updated stability data during the assessment period as it becomes available, and a commitment to provide: – stability data on three production batches, and – process validation of three consecutive production batches to be
completed prior to marketing.
18

Amman, June 2013
Scope of WHO PQ guidance on Variations to a Pre-qualified medicinal product
• applies to quality part of product dossiers for an API or an FPP.
• notification requirements for API-related changes differ depending on the manner in which API information was submitted with the original FPP application.
• FPPs that rely upon the APIMF or CEP procedure have reduced reporting requirements.
• Variations for FPPs prequalified on the basis of SRA approval should be approved by the same SRA and WHO PQP notified of the approval of the changes.
19

Amman, June 2013
Requalification
Procedure for prequalification of pharmaceutical products requires holders of WHO-prequalified
products to submit a quality review after five years from the date of prequalification of the product, or when requested to do so by PQP
(whichever date is earlier)
20

21
Jakarta, March 2013
Demonstration of bioequivalence
PD studiesClinicalstudies
In vitromethods
Bioequivalence study
ONLY EXCEPTIONAL!
or

22
Jakarta, March 2013
Comparators for the Prequalification Programme
• Innovator product with established Q,S, and E sourced from well regulated market (ICH process countries). Guidance on selection and the to be provided documents should be followed.
• The comparator should be selected from the comparator list (http://apps.who.int/prequal/info_applicants/info_for_applicants_BE_comparator.htm)
• Other comparators must be justified. Recommended to consult WHO at: [email protected]
• Remember: Comparator is not always identical with a comparator required by national regulatory authority!

Amman, June 2013
Tested product¨ GMP¨ batch size
¨ pilot batch ¨ commercial batch
¨ not smaller than 100 000 units or 10 % of industrial batch size (whichever is higher)
¨ difference regarding the content of the investigative products (T and R) should preferably not be more than 5 %
¨ strength with the largest sensitivity to detect differences in the two products

Amman, June 2013
Analytical methods
• FDA Guidance for Industry– Bioanalytical method validation, May 2001
• ICH Guidance for industry– Validation of analytical methods: definitions and
terminology, June 1995 – Validation of analytical procedures: methodology,
November 1996
• EMA guideline – Bioanalytical method validation, 2011

Amman, June 2013
Quality of Bioequivalence StudiesGood Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trials subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible.

Amman, June 2013
Frequent GCP non-compliances• No informed consent, complex language• Ethics committee not independent• Dosing procedure is inadequately documented, no drug
accountability• Certificates of analysis are not consistent with study products
or not sufficiently detailed• No testing on addictive substances performed• Withdrawals are improperly documented• Meals not standardized and not documented• Storage of blood samples is not monitored• Method of calculation of PK parameters is not specified• Insufficient explanation of outliers• Chromatograms not consistent with data

Amman, June 2013
WHO BCS-based biowaiverActive substances selected for biowaiving
by WHO
HIV/AIDS:• Lamivudine (BCS 3)• Stavudine (BCS 1)• Zidovudine (BCS 1)• Abacavir sulphate
(BCS 3)• Emtricitabine (BCS 1)
TB:• Levofloxacin (BCS 1)• Ofloxacin (BCS 1)• Ethambutol ((BCS 3)• Isoniazid (BCS 3)• Pyrazinamide (BCS 3)

Amman, June 2013
International norms, standards and guidelines used in inspection activities to ensure wide applicability
• HANDBOOK FOR GOOD CLINICAL RESEARCH PRACTICE (GCP) Guidance for implementation
http://whqlibdoc.who.int/publications/2005/924159392X_eng.pdf
• Guidelines for good clinical practice (GCP) for trials on pharmaceutical products. World Health Organization, 1995 (WHO Technical Report Series, No. 850), Annex 3.
http://apps.who.int/prequal/info_general/documents/TRS850/WHO_TRS_850-Annex3.pdf
• Additional guidance for organizations performing in vivo bioequivalence studies. WHO Technical Report Series, No. 937, 2006, Annex 9
http://apps.who.int/prequal/info_general/documents/TRS937/WHO_TRS_937__annex9_eng.pdf
• Guidelines for the preparation of a contract research organization master file. World Health Organization, WHO Technical Report Series, No. 957, 2010 Annex 7, Page 271. http://www.who.int/medicines/publications/TRS957_2010.pdf
28

29
Amman, June 2013
Generics approved by SRA• http://
apps.who.int/prequal/info_applicants/Guidelines/PQProcGenericSRA_July2011.pdf
• 1. Applicants should submit the following documentation:– A letter of undertaking (template attached) with a clear statement by the responsible
person that the information submitted is true and correct.– A QA-certified copies of the Marketing and Manufacturing Authorizations issued by the
relevant SRA.– An original or certified copy of WHO-type Certificate of a Pharmaceutical Product,
issued by one of the SRAs, together with the– approved Summary of Product Characteristics (SPC), or an equivalent thereof,
including Patient Information Leaflet (PIL) and Labelling.– Assessment report(s) issued by the relevant SRA (expected to be replaced by
specifications and control methods).
• 2. Evidence of minimum five (5) years of current and continuous manufacturing experience and a copy of the last Annual Product Report. (expected to be deleted).

30
Amman, June 2013
Generics approved by SRA
• A sample of the FPP(s) in market packaging and certificate of analysis should be provided.
• Undertaking:– authorizes WHO PQP to publish "Main characteristics of the prequalified
medicinal product" on its website;– will inform WHO prequalification programme with a copy of the regulatory
acceptance letter of any change to the main characteristics of the product immediately after the variation has been approved by the relevant SRA.
– if the change affects the information in SmPC, PIL and/or container labels (immediate and outer) in electronic format
– c) has nominated a responsible employee for communication with WHO on any issues, including quality failures

Amman, June 2013
GMP Inspections by WHO
• Inspections are conducted before prequalification, on an on-going basis and in special circumstances
• Initial inspection target: <180days from dossier receipt
• Re-inspection frequency determined on a risk basis• Inspections are normally announced 1-2 months in
advance• Inspections conducted by a Stringent Regulatory
Authority are taken into account when planning inspections
31

Amman, June 2013
GMP inspections by WHO
• Inspections are conducted by a team– A WHO inspector leads the team– An inspector from another Regulatory Authority
(usually a PIC/S member) assists– The Regulatory Authority of the country of
manufacture is invited (and encouraged) to accompany the team
– Inspection report provided within target of 30 days– Closed out after review of corrective actions
32
Amman, June 2013
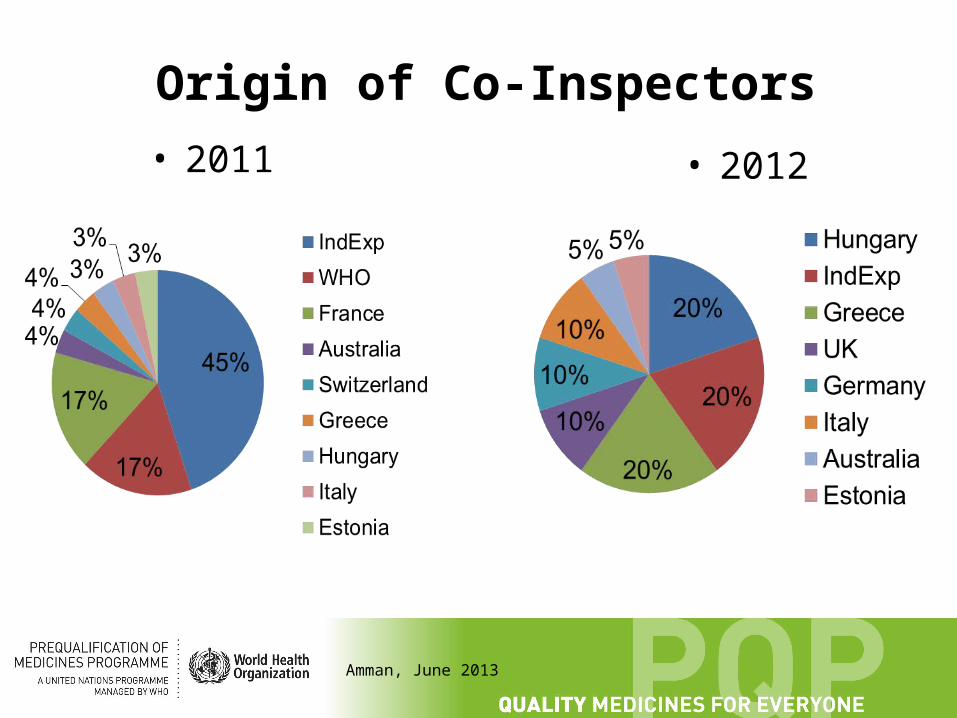
Origin of Co-Inspectors
• 2011 • 2012

Amman, June 2013
Inspections of FPP manufacturers
• Normally over 4 days• Covers all aspects of GMP
– Quality management, Quality assurance, Premises, Equipment, Documentation, Validation, Materials, Personnel
– Utilities (e.g. HVAC, water) . . .
• Also data verification (dossier) including stability data, validation (process), development batches and bio batches
• Quality control laboratory – specifications, reference standards, methods of analysis, validation and qualification
34

All FPP sites: Top 10 observation in 2011 -2012
35
0
5
10
15
20
25
30
35
40
45
50 47
41
24 2423
20 2018 18 18
Series1

Amman, June 2013
API Sites: Top 10 observation categories
0
5
10
15
20
25
30
35
40
45
50 4642
27 2624
22 21 20
13 12
Column1

37
Jakarta, March 2013
Classification of observations
Critical Observation An observation that has produced, or may result in a significant risk of producing, an API that, when used in a finished product, is harmful to the user.
Major Observation A non-critical observation that has produced or may produce a product which does not comply with its prequalification application (including variations); and/or•indicates a major deviation from the GMP guide; and/or•indicates a failure to carry out satisfactory procedures for release of batches; and/or•indicates a failure of the person responsible for QA/QC to fulfill his/her duties; and/or•consists of several other deficiencies, none of which on its own may be major, but which may together represent a major deficiency and should be explained and reported as such.
Other Observation An observation that cannot be classified as either critical or major, but indicates a departure from good manufacturing practice.
37
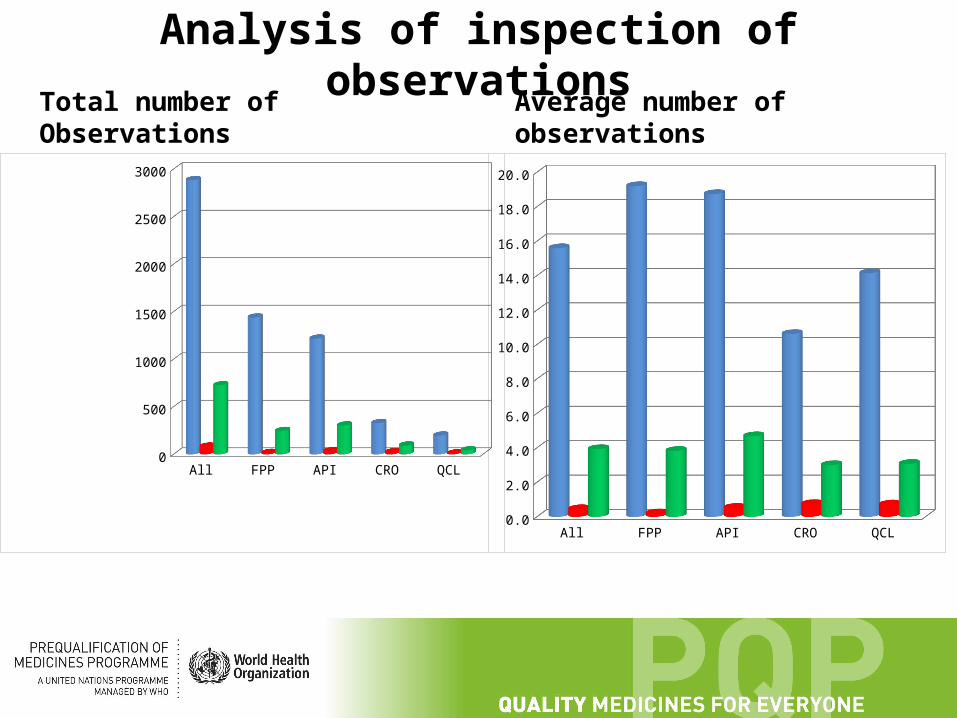
Analysis of inspection of observations
Total number of Observations Average number of observations
All FPP API CRO QCL
Aver-age all obser-va-tions
15.5945945945946
19.2 18.7384615384615
10.6129032258065
14.1428571428571
Aver-age critical
0.443243243243243
0.2 0.523076923076923
0.741935483870968
0.714285714285714
Aver-age Major
3.92972972972973
3.82666666666667
4.67692307692308
3 3.07142857142857
1.03.05.07.09.0
11.013.015.017.019.0
All FPP API CRO QCL
Total Observations 2885 1440 1218 329 198
Critical Observations 82 15 34 23 10
Major Observations 727 246 304 93 43
250
750
1250
1750
2250
2750
Amman, June 2013
Thank you for the [email protected]
39
Ask if you are not sure, PQP contacts are:
• Dossiers and assessment: [email protected]
• Inspections: [email protected]
• Quality control: [email protected]
• General: [email protected]
• Tele/videoconferences are possible


















